Co-reporter:Zhou Chen, Mark D. Leatherman, Olafs Daugulis, and Maurice Brookhart
Journal of the American Chemical Society November 8, 2017 Volume 139(Issue 44) pp:16013-16013
Publication Date(Web):October 30, 2017
DOI:10.1021/jacs.7b10281
Copolymerizations of ethylene with vinyltrialkoxysilanes using cationic (α-diimine)Ni(Me)(CH3CN)+ complexes 4a,b/B(C6F5)3 yield high molecular weight copolymers exhibiting highly branched to nearly linear backbones depending on reaction conditions and catalyst choice. Polymerizations are first-order in ethylene pressure and inverse-order in silane concentration. Microstructural analysis of the copolymers reveals both in-chain and chain-end incorporation of −Si(OR)3 groups whose ratios depend on temperature and ethylene pressure. Detailed low-temperature NMR spectroscopic investigations show that well-defined complex 3b (α-diimine)Ni(Me)(OEt2)+ reacts rapidly at −60 °C with vinyltrialkoxysilanes via both 2,1 and 1,2 insertion pathways to yield 4- and 5-membered chelates, respectively. Such chelates are the major catalyst resting states but are in rapid equilibrium with ethylene-opened chelates, (α-diimine)Ni(R)(C2H4)+ complexes, the species responsible for chain growth. Chelate rearrangement via β-silyl elimination accounts for formation of chain-end −Si(OR)3 groups and constitutes a chain-transfer mechanism. Chelate formation and coordination of the Ni center to the ether moiety, R–O–Si, of the vinylsilane somewhat decreases the turnover frequency (TOF) relative to ethylene homopolymerization, but still remarkably high TOFs of up to 4.5 × 105 h–1 and overall productivities can be achieved. Activation of readily available (α-diimine)NiBr2 complexes 2 with a combination of AlMe3/B(C6F5)3/[Ph3C][B(C6F5)4] yields a highly active and productive catalyst system for the convenient synthesis of the copolymer, a cross-linkable PE. For example, copolymers containing 0.23 mol % silane can be generated at 60 °C, 600 psig ethylene over 4 h with a productivity of 560 kg copolymer/g Ni. This method offers an alternative route to these materials, normally prepared via radical routes, which are precursors to the commercial cross-linked polyethylene, PEX-b.
Co-reporter:David BézierOlafs Daugulis, Maurice Brookhart
Organometallics January 23, 2017 Volume 36(Issue 2) pp:
Publication Date(Web):January 4, 2017
DOI:10.1021/acs.organomet.6b00850
The palladium diphosphine complex 4 converts ethylene to hyperbranched unsaturated oligomers (Mn ≈ 330 g/mol) at 22 °C. In addition to the high stability of this catalyst observed at temperatures up to 75 °C, an increase of the reaction temperature leads to a decrease in branching densities of the oligomers, in sharp contrast to the palladium and nickel diimine catalyst systems. One-pot oligomerizations/hydrogenations were carried out to form hyperbranched saturated oligomers.
Co-reporter:Andrew L. Kocen, Kristine Klimovica, Maurice Brookhart, and Olafs Daugulis
Organometallics 2017 Volume 36(Issue 4) pp:
Publication Date(Web):February 3, 2017
DOI:10.1021/acs.organomet.6b00856
In contrast to traditional diimine-palladium complexes, sterically hindered “sandwich” diimine-palladium adducts act as olefin isomerization catalysts. Terminal olefins are selectively converted to 2-olefins by a sequence of migratory insertion, β-hydride elimination, and olefin displacement. The reaction is performed at 0 °C with 1 mol % of an air-stable precatalyst and tolerates functional groups such as ketones, silyl ethers, and halogens. The isomerization may be used to produce silyl enol ethers from protected allylic alcohols.
Co-reporter:Tung Thanh Nguyen
Chemical Communications 2017 vol. 53(Issue 33) pp:4609-4611
Publication Date(Web):2017/04/20
DOI:10.1039/C7CC02063E
Aminoquinoline-directed, palladium-catalyzed sp3 C–H bond arylation in phosphonamidates and phosphinic amides is reported. The reaction employs Pd(OAc)2 as a catalyst at 10 mol% loading and cesium phosphate or potassium carbonate as a base in 1,2-dichlorobenzene as a solvent. The directing group can be removed under basic conditions to afford phosphonic acid esters. The chemistry described here is the first example of unactivated sp3 C–H bond arylation by using a phosphorus-containing directing group.
Co-reporter:Andrew L. Kocen;Maurice Brookhart
Chemical Communications 2017 vol. 53(Issue 72) pp:10010-10013
Publication Date(Web):2017/09/05
DOI:10.1039/C7CC04953F
We report a method for palladium-catalysed chain-running isomerization of terminal and internal alkenes. Using an air-stable 2,9-dimethylphenanthroline-palladium catalyst in combination with NaBAr4 promoter, olefins are converted to the most stable double bond isomer at −30 to 20 °C. Silyl enol ethers are readily formed from silylated allylic alcohols. Fluorinated substituents are compatible with the reaction conditions, allowing the synthesis of fluoroenolates. Catalyst loading as low as 0.05% can be employed on a gram scale.
Co-reporter:Andrew L. Kocen;Maurice Brookhart
Chemical Communications 2017 vol. 53(Issue 72) pp:10010-10013
Publication Date(Web):2017/09/05
DOI:10.1039/C7CC04953F
We report a method for palladium-catalysed chain-running isomerization of terminal and internal alkenes. Using an air-stable 2,9-dimethylphenanthroline-palladium catalyst in combination with NaBAr4 promoter, olefins are converted to the most stable double bond isomer at −30 to 20 °C. Silyl enol ethers are readily formed from silylated allylic alcohols. Fluorinated substituents are compatible with the reaction conditions, allowing the synthesis of fluoroenolates. Catalyst loading as low as 0.05% can be employed on a gram scale.
Co-reporter:Tung Thanh Nguyen;Liene Grigorjeva
Chemical Communications 2017 vol. 53(Issue 37) pp:5136-5138
Publication Date(Web):2017/05/04
DOI:10.1039/C7CC02062G
We report a method for cobalt-catalyzed, aminoquinoline-directed sp2 C–H bond carbonylation of sulfonamides. The reactions proceed in a dichloroethane solvent, and employ diisopropyl azodicarboxylate as a carbon monoxide source, Mn(OAc)2 as a cooxidant and potassium pivalate as a base. Halogen, ester, and amide functionalities are compatible with the reaction conditions. This method allows for a short and efficient synthesis of saccharin derivatives.
Co-reporter:James Roane
Journal of the American Chemical Society 2016 Volume 138(Issue 13) pp:4601-4607
Publication Date(Web):March 18, 2016
DOI:10.1021/jacs.6b01117
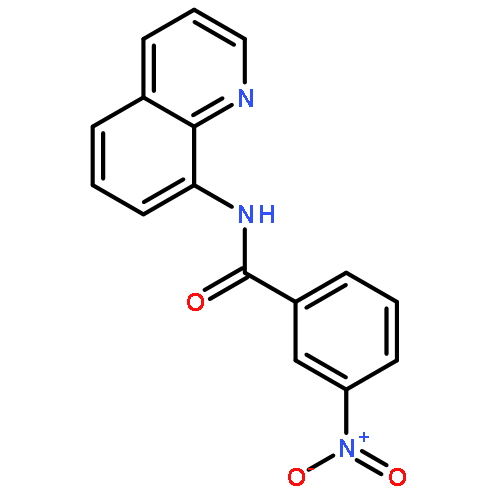
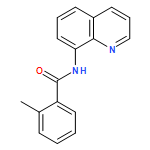
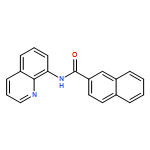
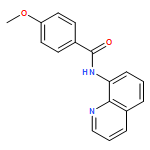
An operationally simple and general method for copper-catalyzed, aminoquinoline-assisted amination of β-C(sp2)-H bonds of benzoic acid derivatives is reported. The reaction employs Cu(OAc)2 or (CuOH)2CO3 catalysts, an amine coupling partner, and oxygen from air as a terminal oxidant. Exceptionally high generality with respect to amine coupling partners is observed. Specifically, primary and secondary aliphatic and aromatic amines, heterocycles, such as indoles, pyrazole, and carbazole, sulfonamides, as well as electron-deficient aromatic and heteroaromatic amines are competent coupling components.
Co-reporter:Zhou Chen, Weijun Liu, Olafs Daugulis, and Maurice Brookhart
Journal of the American Chemical Society 2016 Volume 138(Issue 49) pp:16120-16129
Publication Date(Web):December 1, 2016
DOI:10.1021/jacs.6b10462
Co-reporter:Tung Thanh Nguyen, Liene Grigorjeva, and Olafs Daugulis
ACS Catalysis 2016 Volume 6(Issue 2) pp:551
Publication Date(Web):December 22, 2015
DOI:10.1021/acscatal.5b02391
In this paper, we introduce arylphosphinic acid aminoquinoline amides as competent substrates for cobalt-catalyzed sp2 C–H bond functionalization. Specifically, the feasibility of their coupling with alkynes, alkenes, and allyl pivalate has been demonstrated. Reactions are catalyzed by simple Co(NO3)2 hydrate in ethanol or mixed dioxane/tBuOH solvent in the presence of Mn(OAc)3·2H2O additive, sodium pivalate, or acetate base and use oxygen from the air as an oxidant. Directing group removal affords ortho-functionalized P,P-diarylphosphinic acids.Keywords: alkenes; alkynes; cobalt; C−H functionalization; phosphinic amides
Co-reporter:Milad Mesgar and Olafs Daugulis
Organic Letters 2016 Volume 18(Issue 15) pp:3910-3913
Publication Date(Web):July 14, 2016
DOI:10.1021/acs.orglett.6b01952
Silylaryl bromides and iodides can be prepared in one step from commercially available starting materials. Arynes can be generated from these compounds under conditions nearly identical to those employed for silylaryl triflates. Three distinct transformations, ortho-arylation of N-tritylanilines, intermolecular addition of arynes to amides, and reaction of ureas with arynes, were shown to be successful for the new aryne precursors. The main advantage of silylaryl halides relative to silyl aryl triflates is their one-step preparation from commercially available starting materials.
Co-reporter:Kristine Klimovica, Kristin Kirschbaum, and Olafs Daugulis
Organometallics 2016 Volume 35(Issue 17) pp:2938-2943
Publication Date(Web):August 31, 2016
DOI:10.1021/acs.organomet.6b00487
Synthesis and full characterization of cationic isostructural “sandwich” diimine-coinage metal ethylene complexes are reported. Ethylene self-exchange kinetics proceeds by an associative exchange mechanism for Cu and Au complexes. The fastest ligand exchange was observed for Ag complex 8a. The upper limit of ΔG⧧, assuming associative ligand exchange, was found to be ca. 5.0 kcal/mol. Ethylene self-exchange in Cu complex 7b proceeds with ΔG298⧧ = 12.9 ± 0.1 kcal/mol, while the exchange is the slowest in Au complex 9, with ΔG298⧧ = 16.7 ± 0.1 kcal/mol. Copper complex 7b is unusually stable and can survive in air for years.
Co-reporter:Zhou Chen, Kate E. Allen, Peter S. White, Olafs Daugulis, and Maurice Brookhart
Organometallics 2016 Volume 35(Issue 11) pp:1756-1760
Publication Date(Web):May 6, 2016
DOI:10.1021/acs.organomet.6b00165
Traditional cationic Pd(II) and Ni(II) ethylene polymerization catalysts are supported by ortho-disubstituted aryl diimine ligands. These catalysts are capable of producing high-molecular-weight polyethylene due to positioning of bulk in the two axial sites of the square coordination plane which retards chain transfer. Similar pyridine-imine complexes bearing a single ortho-disubstituted aryl imine moiety were reported to yield very low Mn polyethylene. In earlier studies, “sandwich” diimine nickel catalysts incorporating two 8-arylnaphthylimino groups which provide exceptional shielding of the two axial sites were shown to yield ultrahigh-molecular-weight polyethylene. Here we demonstrate that 8-arylnaphthyl groups incorporated into pyridine-imine nickel catalysts that block only a single axial site are highly effective in retarding chain transfer. These catalysts produce branched polyethylene (ca. 30–90 branches per 1000 carbons) with Mn values up to 2.6 × 104 g/mol. Effects on the catalyst lifetimes and polymerization behavior as a function of substituent variations at the imine carbon and the aryl group are reported.
Co-reporter:Olafs Daugulis, James Roane, and Ly Dieu Tran
Accounts of Chemical Research 2015 Volume 48(Issue 4) pp:1053
Publication Date(Web):March 10, 2015
DOI:10.1021/ar5004626
In recent years, carbon–hydrogen bond functionalization has evolved from an organometallic curiosity to a tool used in mainstream applications in the synthesis of complex natural products and drugs. The use of C–H bonds as a transformable functional group is advantageous because these bonds are the most abundant functionality in organic molecules. One-step conversion of these bonds to the desired functionality shortens synthetic pathways, saving reagents, solvents, and labor. Less chemical waste is generated as well, showing that this chemistry is environmentally beneficial. This Account describes the development and use of bidentate, monoanionic auxiliaries for transition-metal-catalyzed C–H bond functionalization reactions. The chemistry was initially developed to overcome the limitations with palladium-catalyzed C–H bond functionalization assisted by monodentate directing groups. By the use of electron-rich bidentate directing groups, functionalization of unactivated sp3 C–H bonds under palladium catalysis has been developed. Furthermore, a number of abundant base-metal complexes catalyze functionalization of sp2 C–H bonds. At this point, aminoquinoline, picolinic acid, and related compounds are among the most used and versatile directing moieties in C–H bond functionalization chemistry. These groups facilitate catalytic functionalization of sp2 and sp3 C–H bonds by iron, cobalt, nickel, copper, ruthenium, rhodium, and palladium complexes. Exceptionally general reactivity is observed, enabling, among other transformations, direct arylation, alkylation, fluorination, sulfenylation, amination, etherification, carbonylation, and alkenylation of carbon–hydrogen bonds. The versatility of these auxilaries can be attributed to the following factors. First, they are capable of stabilizing high oxidation states of transition metals, thereby facilitating the C–H bond functionalization step. Second, the directing groups can be removed, enabling their use in synthesis and functionalization of natural products and medicinally relevant substances. While the development of these directing groups presents a significant advance, several limitations of this methodology are apparent. The use of expensive second-row transition metal catalysts is still required for efficient sp3 C–H bond functionalization. Furthermore, the need to install and subsequently remove the relatively expensive directing group is a disadvantage.
Co-reporter:Zhou Chen, Milad Mesgar, Peter S. White, Olafs Daugulis, and Maurice Brookhart
ACS Catalysis 2015 Volume 5(Issue 2) pp:631
Publication Date(Web):December 11, 2014
DOI:10.1021/cs501948d
Neutral nickel methyl complexes incorporating 2,8-diarylnaphthyl groups have been synthesized and characterized. Salicylaldiminato nickel systems 1a,b are exceptionally active neutral nickel single component catalysts for the polymerization of ethylene capable of producing lightly branched ultrahigh-molecular-weight polyethylene (UHMWPE). In addition, complex 1a shows a “quasi-living” polymerization behavior.Keywords: branched polyethylene; nickel catalysts; polymerization; salicyaldimine; UHMWPE
Co-reporter:Liene Grigorjeva and Olafs Daugulis
Organic Letters 2015 Volume 17(Issue 5) pp:1204-1207
Publication Date(Web):February 17, 2015
DOI:10.1021/acs.orglett.5b00155
A method for aminoquinoline-directed, cobalt-promoted dimerization of benzamides has been developed. Reactions proceed in ethanol solvent in the presence of Mn(OAc)2 cocatalyst and Na2CO3 base and use oxygen as a terminal oxidant. Bromo, iodo, nitro, ether, and ester moieties are compatible with the reaction conditions. Cross-coupling of electronically dissimilar aminoquinoline benzamides proceeds with modest yields and selectivities.
Co-reporter:Tulaza Vaidya ; Kristine Klimovica ; Anne M. LaPointe ; Ivan Keresztes ; Emil B. Lobkovsky ; Olafs Daugulis ;Geoffrey W. Coates
Journal of the American Chemical Society 2014 Volume 136(Issue 20) pp:7213-7216
Publication Date(Web):April 28, 2014
DOI:10.1021/ja502130w
While traditional polymerization of linear α-olefins (LAOs) typically provides amorphous, low Tg polymers, chain-straightening polymerization represents a route to semicrystalline materials. A series of α-diimine nickel catalysts were tested for the polymerization of various LAOs. Although known systems yielded amorphous or low-melting polymers, the “sandwich” α-diimines 3–6 yielded semicrystalline “polyethylene” comprised primarily of unbranched repeat units via a combination of uncommon regioselective 2,1-insertion and precision chain-walking events.
Co-reporter:Thanh Truong ; Milad Mesgar ; Ky Khac Anh Le
Journal of the American Chemical Society 2014 Volume 136(Issue 24) pp:8568-8576
Publication Date(Web):June 3, 2014
DOI:10.1021/ja504886x
A method for base-promoted arylation of arenes and heterocycles by aryl halides and aryl triflates is described. Additionally, in situ electrophilic trapping of ArLi intermediates generated in the reaction of benzyne with deprotonated arenes or heterocycles has been developed, providing rapid and easy access to a wide range of highly functionalized polyaryls. Base-promoted arylation methodology complements transition-metal-catalyzed direct arylation and allows access to structures that are not easily accessible via other direct arylation methods. The reactions are highly functional-group tolerant, with alkene, ether, dimethylamino, trifluoromethyl, ester, cyano, halide, hydroxyl, and silyl functionalities compatible with reaction conditions.
Co-reporter:Liene Grigorjeva and Olafs Daugulis
Organic Letters 2014 Volume 16(Issue 17) pp:4688-4690
Publication Date(Web):August 22, 2014
DOI:10.1021/ol502007t
A method for direct carbonylation of aminoquinoline benzamides has been developed. Reactions proceed at room temperature in trifluoroethanol solvent, use oxygen from air as an oxidant, and require Mn(OAc)3 as a cocatalyst. Benzoic and acrylic acid derivatives can be carbonylated by carbon monoxide affording imides in good yields. Halogen, nitro, ether, cyano, and ester functional groups are tolerated. The directing group can be removed under mild conditions affording phthalimides.
Co-reporter:Liene Grigorjeva and Olafs Daugulis
Organic Letters 2014 Volume 16(Issue 17) pp:4684-4687
Publication Date(Web):August 22, 2014
DOI:10.1021/ol502005g
A method for cobalt-catalyzed, aminoquinoline-directed ortho-functionalization of sp2 C–H bonds with alkenes has been developed. Reactions proceed at room temperature in trifluoroethanol solvent, use oxygen from air as an oxidant, and require Mn(OAc)3 as a cocatalyst. Benzoic, heteroaromatic, and acrylic acid aminoquinoline amides react with ethylene as well as mono- and disubstituted alkenes affording products in good yields. Excellent functional group tolerance is observed; halogen, nitro, ether, and unprotected alcohol functionalities are compatible with the reaction conditions.
Co-reporter:Dr. Liene Grigorjeva ; Olafs Daugulis
Angewandte Chemie International Edition 2014 Volume 53( Issue 38) pp:10209-10212
Publication Date(Web):
DOI:10.1002/anie.201404579
Abstract
A method for cobalt-catalyzed, aminoquinoline- and picolinamide-directed C(sp2)H bond alkenylation by alkynes was developed. The method shows excellent functional-group tolerance and both internal and terminal alkynes are competent substrates for the coupling. The reaction employs a Co(OAc)2⋅4 H2O catalyst, Mn(OAc)2 co-catalyst, and oxygen (from air) as a terminal oxidant.
Co-reporter:Dr. Liene Grigorjeva ; Olafs Daugulis
Angewandte Chemie 2014 Volume 126( Issue 38) pp:10373-10376
Publication Date(Web):
DOI:10.1002/ange.201404579
Abstract
A method for cobalt-catalyzed, aminoquinoline- and picolinamide-directed C(sp2)H bond alkenylation by alkynes was developed. The method shows excellent functional-group tolerance and both internal and terminal alkynes are competent substrates for the coupling. The reaction employs a Co(OAc)2⋅4 H2O catalyst, Mn(OAc)2 co-catalyst, and oxygen (from air) as a terminal oxidant.
Co-reporter:Thanh Truong, Kristine Klimovica, and Olafs Daugulis
Journal of the American Chemical Society 2013 Volume 135(Issue 25) pp:9342-9345
Publication Date(Web):June 12, 2013
DOI:10.1021/ja4047125
We have developed a method for direct, copper-catalyzed, auxiliary-assisted fluorination of β-sp2 C–H bonds of benzoic acid derivatives and γ-sp2 C–H bonds of α,α-disubstituted benzylamine derivatives. The reaction employs a CuI catalyst, a AgF fluoride source, and DMF, pyridine, or DMPU solvent at moderately elevated temperatures. Selective mono- or difluorination can be achieved by simply changing reaction conditions. The method shows excellent functional group tolerance and provides a straightforward way for the preparation of ortho-fluorinated benzoic acids.
Co-reporter:Thanh Truong and Olafs Daugulis
Chemical Science 2013 vol. 4(Issue 1) pp:531-535
Publication Date(Web):18 Oct 2012
DOI:10.1039/C2SC21288A
Two reactions of phenols with arynes have been developed. If LiTMP base is employed, arynes generated from aryl chlorides react with phenols to form helicenes. o-Arylation of phenols can be achieved by employing tBuONa base in the presence of AgOAc. Direct arylation of binol was achieved leading to the shortest pathway to o,o′-diarylbinols.
Co-reporter:Hung Tran-Vu and Olafs Daugulis
ACS Catalysis 2013 Volume 3(Issue 10) pp:2417
Publication Date(Web):September 25, 2013
DOI:10.1021/cs400443p
A method for carboxylation of aryl iodides with carbon dioxide has been developed. The reaction employs low loadings of copper iodide/N,N,N′,N′-tetramethylethylenediamine (TMEDA) or N,N′-dimethylethylenediamine (DMEDA) catalyst, 1 atm of CO2, dimethylsulfoxide (DMSO) or dimethylacetamide (DMA) solvent, and proceeds at 25–70 °C. Good functional group tolerance is observed, with ester, bromide, chloride, fluoride, ether, hydroxy, amino, and ketone functionalities tolerated. Additionally, hindered aryl iodides such as iodomesitylene can also be carboxylatedKeywords: arenecarboxylic acids; aryl halides; carbon dioxide; carboxylation; copper catalysis
Co-reporter:James Roane and Olafs Daugulis
Organic Letters 2013 Volume 15(Issue 22) pp:5842-5845
Publication Date(Web):November 1, 2013
DOI:10.1021/ol402904d
A method for direct, auxiliary-assisted alkoxylation and phenoxylation of β-sp2 C–H bonds of benzoic acid derivatives and γ-sp2 C–H bonds of amine derivatives is reported. The reaction employs (CuOH)2CO3 catalyst, air as an oxidant, phenol or alcohol coupling partner, DMF, pyridine, or DMPU solvent, and K2CO3, tetramethylguanidine, or K3PO4 base at 70–130 °C.
Co-reporter:Ly Dieu Tran;James Roane ;Dr. Olafs Daugulis
Angewandte Chemie 2013 Volume 125( Issue 23) pp:6159-6162
Publication Date(Web):
DOI:10.1002/ange.201300135
Co-reporter:Ly Dieu Tran;James Roane ;Dr. Olafs Daugulis
Angewandte Chemie International Edition 2013 Volume 52( Issue 23) pp:6043-6046
Publication Date(Web):
DOI:10.1002/anie.201300135
Co-reporter:Enrico T. Nadres, Gerson Ivan Franco Santos, Dmitry Shabashov, and Olafs Daugulis
The Journal of Organic Chemistry 2013 Volume 78(Issue 19) pp:9689-9714
Publication Date(Web):September 19, 2013
DOI:10.1021/jo4013628
The scope of palladium-catalyzed, auxiliary-assisted direct arylation and alkylation of sp2 and sp3 C–H bonds of amine and carboxylic acid derivatives has been investigated. The method employs a palladium acetate catalyst, substrate, aryl, alkyl, benzyl, or allyl halide, and inorganic base in tert-amyl alcohol or water solvent at 100–140 °C. Aryl and alkyl iodides as well as benzyl and allyl bromides are competent reagents in this transformation. The picolinic acid auxiliary is used for amine γ-functionalization, and the 8-aminoquinoline auxiliary is used for carboxylic acid β-functionalization. Some optimization of base, additives, and solvent is required for achieving best results.
Co-reporter:Danfeng Zhang, Enrico T. Nadres, Maurice Brookhart, and Olafs Daugulis
Organometallics 2013 Volume 32(Issue 18) pp:5136-5143
Publication Date(Web):September 10, 2013
DOI:10.1021/om400704h
Nickel(II) α-diimine dibromide complexes incorporating 8-p-tolylnaphthylimino groups have been prepared and used for ethylene polymerization by activation with modified methylalumoxane (MMAO). These catalysts possess increased axial bulk relative to standard diimine-nickel complexes, resulting in lower rates of chain transfer relative to chain propagation rates and thus higher polymer molecular weights and narrower PDIs. They yield the most highly branched PE produced by Ni catalysts seen to date. The easy synthesis of 8-aryl-1-naphthylamines provides ready access to a new class of α-diimine-based nickel catalysts in which the 8-substituent is ideally positioned to provide steric bulk at the axial sites.
Co-reporter:Ly Dieu Tran ; Ilya Popov
Journal of the American Chemical Society 2012 Volume 134(Issue 44) pp:18237-18240
Publication Date(Web):October 29, 2012
DOI:10.1021/ja3092278
An auxiliary-assisted, copper catalyzed or promoted sulfenylation of benzoic acid derivative β-C–H bonds and benzylamine derivative γ-C–H bonds has been developed. The method employs disulfide reagents, copper(II) acetate, and DMSO solvent at 90–130 °C. Application of this methodology to the direct trifluoromethylsulfenylation of C–H bonds was demonstrated.
Co-reporter:Thanh Truong and Olafs Daugulis
Organic Letters 2012 Volume 14(Issue 23) pp:5964-5967
Publication Date(Web):November 13, 2012
DOI:10.1021/ol302875x
A method for direct, transition-metal-free ortho-arylation of anilines by aryl chlorides, bromides, fluorides, and triflates has been developed. This methodology provides the most direct approach to 2-arylanilines since no protecting or directing groups on nitrogen are required. The arylation is functional-group tolerant, with alkene, ether, trifluoromethyl, dimethylamino, carbonyl, chloro, and cyano functionalities tolerated. Phenylation of enantiopure binaphthyldiamine affords a product with >99% ee.
Co-reporter:O. Daugulis
Chemistry of Heterocyclic Compounds 2012 Volume 48( Issue 1) pp:21-26
Publication Date(Web):2012 April
DOI:10.1007/s10593-012-0963-9
The use of non-activated aryl chloride electrophiles in the direct arylation of carbon–hydrogen bonds in five-membered ring heterocycles is described. Specifically, palladium-, ruthenium-, and nickel-catalyzed transformations, as well as reactions proceeding via benzyne intermediates, are discussed.
Co-reporter:Hien-Quang Do, Hung Tran-Vu, and Olafs Daugulis
Organometallics 2012 Volume 31(Issue 22) pp:7816-7818
Publication Date(Web):June 21, 2012
DOI:10.1021/om300393m
A method for copper-catalyzed oxidative dimerization of nitronates and enolates using oxygen as the terminal oxidant has been developed. Cyclization through oxidative intramolecular coupling is also feasible for both nitronates and enolates. The mild reaction conditions lead to good functional group tolerance.
Co-reporter:Ly Dieu Tran ;Dr. Olafs Daugulis
Angewandte Chemie International Edition 2012 Volume 51( Issue 21) pp:5188-5191
Publication Date(Web):
DOI:10.1002/anie.201200731
Co-reporter:Thanh Truong ;Dr. Olafs Daugulis
Angewandte Chemie International Edition 2012 Volume 51( Issue 47) pp:11677-11679
Publication Date(Web):
DOI:10.1002/anie.201206568
Co-reporter:Ly Dieu Tran ;Dr. Olafs Daugulis
Angewandte Chemie 2012 Volume 124( Issue 21) pp:5278-5281
Publication Date(Web):
DOI:10.1002/ange.201200731
Co-reporter:Thanh Truong ;Dr. Olafs Daugulis
Angewandte Chemie 2012 Volume 124( Issue 47) pp:11845-11847
Publication Date(Web):
DOI:10.1002/ange.201206568
Co-reporter:Thanh Truong
Journal of the American Chemical Society 2011 Volume 133(Issue 12) pp:4243-4245
Publication Date(Web):March 8, 2011
DOI:10.1021/ja200184b
A transition-metal-free method for arylation of heterocycle and arene carbon−hydrogen bonds by aryl chlorides and fluorides has been developed. The reactions proceed via aryne intermediates and are highly regioselective with respect to the C−H bond coupling component.
Co-reporter:Hien-Quang Do
Journal of the American Chemical Society 2011 Volume 133(Issue 34) pp:13577-13586
Publication Date(Web):August 8, 2011
DOI:10.1021/ja2047717
A general method for a highly regioselective copper-catalyzed cross-coupling of two aromatic compounds using iodine as an oxidant has been developed. The reactions involve an initial iodination of one arene followed by arylation of the most acidic C–H bond of the other coupling component. Cross-coupling of electron-rich arenes, electron-poor arenes, and five- and six-membered heterocycles is possible in many combinations. Typically, a 1/1.5 to 1/3 ratio of coupling components is used, in contrast to existing methodology that often employs a large excess of one of the arenes. Common functionalities such as ester, ketone, aldehyde, ether, nitrile, nitro, and amine are well-tolerated.
Co-reporter:Enrico T. Nadres
Journal of the American Chemical Society 2011 Volume 134(Issue 1) pp:7-10
Publication Date(Web):December 29, 2011
DOI:10.1021/ja210959p
A method for five- and six-membered heterocycle formation by palladium-catalyzed C–H/N–H coupling is presented. The method employs a picolinamide directing group, PhI(OAc)2 oxidant, and toluene solvent at 80–120 °C. Cyclization is effective for sp2 as well as aliphatic and benzylic sp3 C–H bonds.
Co-reporter:Enrico T. Nadres, Anna Lazareva, and Olafs Daugulis
The Journal of Organic Chemistry 2011 Volume 76(Issue 2) pp:471-483
Publication Date(Web):December 30, 2010
DOI:10.1021/jo1018969
The palladium-catalyzed direct arylation of indoles, pyrroles, and furans by aryl chlorides has been demonstrated. The method employs a palladium acetate catalyst, 2-(dicyclohexylphosphino)-biphenyl ligand, and an inorganic base. Electron-rich and electron-poor aryl chlorides as well as chloropyridine coupling partners can be used, and arylated heterocycles are obtained in moderate to good yields. Optimization of base, ligand, and solvent is required for achieving best results.
Co-reporter:Jason E. Kreutz ; Anton Shukhaev ; Wenbin Du ; Sasha Druskin ; Olafs Daugulis ;Rustem F. Ismagilov
Journal of the American Chemical Society 2010 Volume 132(Issue 9) pp:3128-3132
Publication Date(Web):February 11, 2010
DOI:10.1021/ja909853x
This paper uses microfluidics to implement genetic algorithms (GA) to discover new homogeneous catalysts using the oxidation of methane by molecular oxygen as a model system. The parameters of the GA were the catalyst, a cocatalyst capable of using molecular oxygen as the terminal oxidant, and ligands that could tune the catalytic system. The GA required running hundreds of reactions to discover and optimize catalyst systems of high fitness, and microfluidics enabled these numerous reactions to be run in parallel. The small scale and volumes of microfluidics offer significant safety benefits. The microfluidic system included methods to form diverse arrays of plugs containing catalysts, introduce gaseous reagents at high pressure, run reactions in parallel, and detect catalyst activity using an in situ indicator system. Platinum(II) was identified as an active catalyst, and iron(II) and the polyoxometalate H5PMo10V2O40 (POM-V2) were identified as active cocatalysts. The Pt/Fe system was further optimized and characterized using NMR experiments. After optimization, turnover numbers of approximately 50 were achieved with approximately equal production of methanol and formic acid. The Pt/Fe system demonstrated the compatibility of iron with the entire catalytic cycle. This approach of GA-guided evolution has the potential to accelerate discovery in catalysis and other areas where exploration of chemical space is essential, including optimization of materials for hydrogen storage and CO2 capture and modifications.
Co-reporter:Dmitry Shabashov
Journal of the American Chemical Society 2010 Volume 132(Issue 11) pp:3965-3972
Publication Date(Web):February 22, 2010
DOI:10.1021/ja910900p
We have developed a method for auxiliary-directed, palladium-catalyzed β-arylation and alkylation of sp3 and sp2 C−H bonds in carboxylic acid derivatives. The method employs a carboxylic acid 2-methylthioaniline- or 8-aminoquinoline amide substrate, aryl or alkyl iodide coupling partner, palladium acetate catalyst, and an inorganic base. By employing 2-methylthioaniline auxiliary, selective monoarylation of primary sp3 C−H bonds can be achieved. If arylation of secondary sp3 C−H bonds is desired, 8-aminoquinoline auxiliary may be used. For alkylation of sp3 and sp2 C−H bonds, 8-aminoquinoline auxiliary affords the best results. Some functional group tolerance is observed and amino- and hydroxy-acid derivatives can be functionalized. Preliminary mechanistic studies have been performed. A palladacycle intermediate has been isolated, characterized by X-ray crystallography, and its reactions have been studied.
Co-reporter:Thanh Truong, Joseph Alvarado, Ly Dieu Tran and Olafs Daugulis
Organic Letters 2010 Volume 12(Issue 6) pp:1200-1203
Publication Date(Web):March 1, 2010
DOI:10.1021/ol902970z
A number of first-row transition metal salts catalyze deprotonative dimerization of acidic arenes. Under the atmosphere of oxygen, nickel, manganese, cobalt, and iron chlorides have been shown to dimerize five- and six-membered ring heterocycles as well as electron-poor arenes. Both tetramethylpiperidide and dicyclohexylamide bases can be employed; however, the former afford slightly higher yields.
Co-reporter:Hien-Quang Do and Olafs Daugulis
Organic Letters 2010 Volume 12(Issue 11) pp:2517-2519
Publication Date(Web):May 4, 2010
DOI:10.1021/ol100772u
A method for regioselective cyanation of heterocycles has been developed. A number of aromatic heterocycles as well as azulene can be cyanated in reasonable to good yields by using a copper cyanide catalyst and an iodine oxidant.
Co-reporter:Ly Dieu Tran and Olafs Daugulis
Organic Letters 2010 Volume 12(Issue 19) pp:4277-4279
Publication Date(Web):September 8, 2010
DOI:10.1021/ol101684u
A method for iron-catalyzed deprotonative alkylation of arene C−H bonds by alkyl iodides and bromides has been developed. In the presence of an amide base, both primary and secondary alkyl halides can be coupled with furans, thiophenes, pyridine derivatives, and some electron-withdrawing-group containing arenes.
Co-reporter:Olafs Daugulis, Hien-Quang Do and Dmitry Shabashov
Accounts of Chemical Research 2009 Volume 42(Issue 8) pp:1074
Publication Date(Web):June 24, 2009
DOI:10.1021/ar9000058
The transition-metal-catalyzed functionalization of C−H bonds is a powerful method for generating carbon−carbon bonds. Although significant advances to this field have been reported during the past decade, many challenges remain. First, most of the methods are substrate-specific and thus cannot be generalized. Second, conversions of unactivated (i.e., not benzylic or α to heteroatom) sp3 C−H bonds to C−C bonds are rare, with most examples limited to t-butyl groups, a conversion that is inherently simple because there are no β-hydrogens that can be eliminated. Finally, the palladium, rhodium, and ruthenium catalysts routinely used for the conversion of C−H bonds to C−C bonds are expensive. Catalytically active metals that are cheaper and less exotic (e.g., copper, iron, and manganese) are rarely used. This Account describes our attempts to provide solutions to these three problems. We have developed a general method for directing-group-containing arene arylation by aryl iodides. Using palladium acetate as the catalyst, we arylated anilides, benzamides, benzoic acids, benzylamines, and 2-substituted pyridine derivatives under nearly identical conditions. We have also developed a method for the palladium-catalyzed auxiliary-assisted arylation of unactivated sp3 C−H bonds. This procedure allows for the β-arylation of carboxylic acid derivatives and the γ-arylation of amine derivatives. Furthermore, copper catalysis can be used to mediate the arylation of acidic arene C−H bonds (i.e., those with pKa values <35 in DMSO). Using a copper iodide catalyst in combination with a base and a phenanthroline ligand, we successfully arylated electron-rich and electron-deficient heterocycles and electron-poor arenes possessing at least two electron-withdrawing groups. The reaction exhibits unusual regioselectivity: arylation occurs at the most hindered position. This copper-catalyzed method supplements the well-known C−H activation/borylation methodology, in which functionalization usually occurs at the least hindered position. We also describe preliminary investigations to determine the mechanisms of these transformations. We anticipate that other transition metals, including iron, nickel, cobalt, and silver, will also be able to facilitate deprotonation/arylation reaction sequences.
Co-reporter:Hien-Quang Do
Journal of the American Chemical Society 2009 Volume 131(Issue 47) pp:17052-17053
Publication Date(Web):November 9, 2009
DOI:10.1021/ja907479j
A general method for copper-catalyzed deprotonative dimerization of arenes by employing oxygen as the terminal oxidant has been developed. Electron-rich and electron-poor heterocycles as well as electron-poor arenes are reactive. The method is tolerant to functionalities such as nitro, cyano, dialkylamino, and ester groups.
Co-reporter:Hien-Quang Do and Olafs Daugulis
Chemical Communications 2009 (Issue 42) pp:6433-6435
Publication Date(Web):28 Sep 2009
DOI:10.1039/B912890E
A highly regioselective, one-pot sequential iodination–copper-catalyzed cross-coupling of arene C–H bonds has been developed affording an efficient method for biaryl synthesis.
Co-reporter:Ilya Popov, Hien-Quang Do and Olafs Daugulis
The Journal of Organic Chemistry 2009 Volume 74(Issue 21) pp:8309-8313
Publication Date(Web):October 14, 2009
DOI:10.1021/jo9015369
A general method has been developed for in situ trapping of arylmetal intermediates by halogen, sulfur, ketone, and aldehyde electrophiles affording the functionalization of the most acidic position in arene. Pentafluorobenzene, benzothiazole, and benzoxazole can be functionalized by using K3PO4 base. For less acidic arenes, tBuOLi base is required. Arenes with DMSO pKa values of 35 or less are reactive.
Co-reporter:Olafs Daugulis Dr.;Vladimir G. Zaitsev Dr.
Angewandte Chemie International Edition 2005 Volume 44(Issue 26) pp:
Publication Date(Web):24 MAY 2005
DOI:10.1002/anie.200500589
A thousand turnovers in bi- and triaryl formation: Substituted anilides have been ortho-arylated by aryl iodides under palladium catalysis (see scheme). This method is highly tolerant of the presence of additional functional groups (FGs). Even an iodo functionality on the anilide moiety and a bromo functionality on the aryl iodide are compatible with this methodology.
Co-reporter:Olafs Daugulis Dr.;Vladimir G. Zaitsev Dr.
Angewandte Chemie 2005 Volume 117(Issue 26) pp:
Publication Date(Web):24 MAY 2005
DOI:10.1002/ange.200500589
Bi- und Triarylbildung mit hohen Umsatzzahlen: Substituierte Anilide wurden mit Aryliodiden unter Palladiumkatalyse ortho-aryliert (siehe Schema). Dieses Verfahren gelingt in Gegenwart einer Vielzahl anderer funktioneller Gruppen (FGs). Sogar ein Iodsubstituent an der Anilideinheit und ein Bromsubstituent am Aryliodid stören nicht.
Co-reporter:Ilya Popov ; Sergey Lindeman
Journal of the American Chemical Society () pp:
Publication Date(Web):May 31, 2011
DOI:10.1021/ja2041942
A general method has been developed for arylation of readily available 1H-perfluoroalkanes. The method employs aryl iodide and 1H-perfluoroalkane reagents, DMPU solvent, TMP2Zn base, and a copper chloride/phenanthroline catalyst. Preliminary mechanistic studies are reported.
Co-reporter:Thanh Truong and Olafs Daugulis
Chemical Science (2010-Present) 2013 - vol. 4(Issue 1) pp:NaN535-535
Publication Date(Web):2012/10/18
DOI:10.1039/C2SC21288A
Two reactions of phenols with arynes have been developed. If LiTMP base is employed, arynes generated from aryl chlorides react with phenols to form helicenes. o-Arylation of phenols can be achieved by employing tBuONa base in the presence of AgOAc. Direct arylation of binol was achieved leading to the shortest pathway to o,o′-diarylbinols.
Co-reporter:Hien-Quang Do and Olafs Daugulis
Chemical Communications 2009(Issue 42) pp:NaN6435-6435
Publication Date(Web):2009/09/28
DOI:10.1039/B912890E
A highly regioselective, one-pot sequential iodination–copper-catalyzed cross-coupling of arene C–H bonds has been developed affording an efficient method for biaryl synthesis.
Co-reporter:Tung Thanh Nguyen, Liene Grigorjeva and Olafs Daugulis
Chemical Communications 2017 - vol. 53(Issue 37) pp:NaN5138-5138
Publication Date(Web):2017/04/26
DOI:10.1039/C7CC02062G
We report a method for cobalt-catalyzed, aminoquinoline-directed sp2 C–H bond carbonylation of sulfonamides. The reactions proceed in a dichloroethane solvent, and employ diisopropyl azodicarboxylate as a carbon monoxide source, Mn(OAc)2 as a cooxidant and potassium pivalate as a base. Halogen, ester, and amide functionalities are compatible with the reaction conditions. This method allows for a short and efficient synthesis of saccharin derivatives.
Co-reporter:Tung Thanh Nguyen and Olafs Daugulis
Chemical Communications 2017 - vol. 53(Issue 33) pp:NaN4611-4611
Publication Date(Web):2017/04/10
DOI:10.1039/C7CC02063E
Aminoquinoline-directed, palladium-catalyzed sp3 C–H bond arylation in phosphonamidates and phosphinic amides is reported. The reaction employs Pd(OAc)2 as a catalyst at 10 mol% loading and cesium phosphate or potassium carbonate as a base in 1,2-dichlorobenzene as a solvent. The directing group can be removed under basic conditions to afford phosphonic acid esters. The chemistry described here is the first example of unactivated sp3 C–H bond arylation by using a phosphorus-containing directing group.

![(R)-(4,4',6,6'-Tetramethoxybiphenyl-2,2'-diyl) bis[bis(4-methoxy-3,5-dimeth ylphenyl)phosphine]](/data/chemimg/2238000/1365531-93-6.png)
![(R)-(4,4',6,6'-Tetramethoxybiphenyl-2,2'-diyl) bis[bis(4-methoxy-3,5-dimeth ylphenyl)phosphine]](/data/chemimg/2238000/1365531-93-6_b.png)


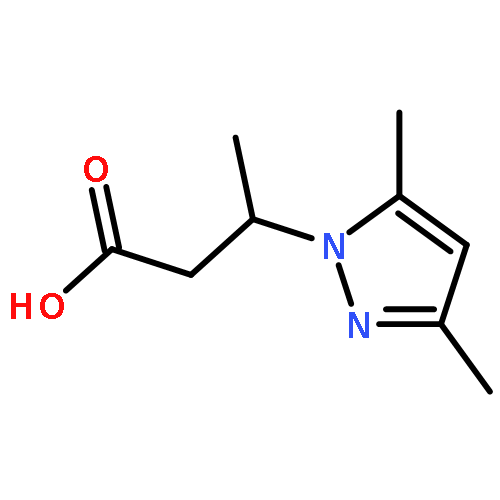






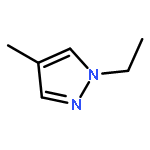
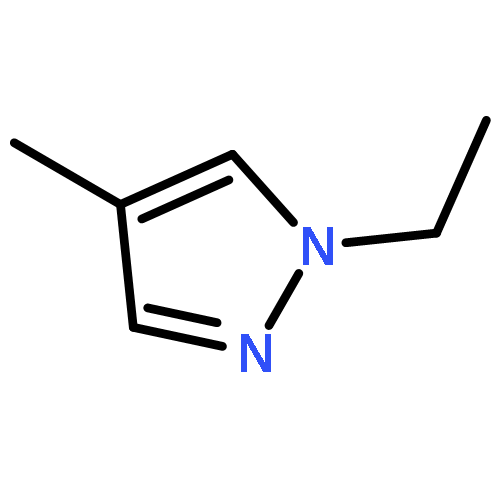


![Silane, (1,1-dimethylethyl)dimethyl[(2-methyl-2-propenyl)oxy]-](http://img.cochemist.com/ccimg/111200/111160-83-9.png)
![Silane, (1,1-dimethylethyl)dimethyl[(2-methyl-2-propenyl)oxy]-](http://img.cochemist.com/ccimg/111200/111160-83-9_b.png)
![Silane, (1,1-dimethylethyl)dimethyl[(1E)-1-propenyloxy]-](http://img.cochemist.com/ccimg/96600/96504-40-4.png)
![Silane, (1,1-dimethylethyl)dimethyl[(1E)-1-propenyloxy]-](http://img.cochemist.com/ccimg/96600/96504-40-4_b.png)
![1H-Pyrazole, 3,5-dimethyl-1-tricyclo[3.3.1.13,7]dec-1-yl-](http://img.cochemist.com/ccimg/92300/92234-55-4.png)
![1H-Pyrazole, 3,5-dimethyl-1-tricyclo[3.3.1.13,7]dec-1-yl-](http://img.cochemist.com/ccimg/92300/92234-55-4_b.png)
![SILANE, (1,1-DIMETHYLETHYL)DIMETHYL[(2-METHYL-1-PROPENYL)OXY]-](http://img.cochemist.com/ccimg/90400/90354-86-2.png)
![SILANE, (1,1-DIMETHYLETHYL)DIMETHYL[(2-METHYL-1-PROPENYL)OXY]-](http://img.cochemist.com/ccimg/90400/90354-86-2_b.png)


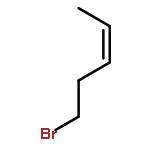












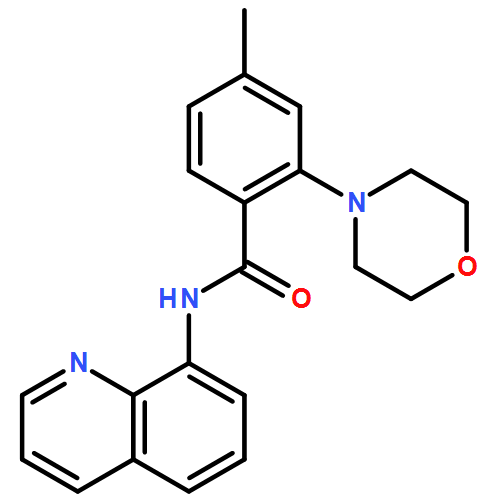
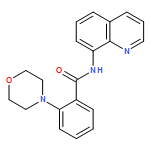
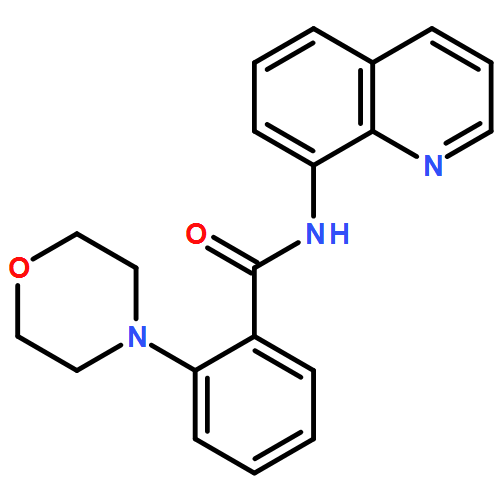
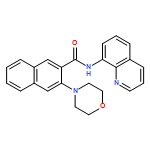
![2-(2'-fluorophenyl)benzo[b]thiophene](http://img.cochemist.com/ccimg/936800/936734-95-1.png)
![2-(2'-fluorophenyl)benzo[b]thiophene](http://img.cochemist.com/ccimg/936800/936734-95-1_b.png)






![[1,1'-Biphenyl]-2-amine, 3',5-dimethoxy-](http://img.cochemist.com/ccimg/154900/154876-31-0.png)
![[1,1'-Biphenyl]-2-amine, 3',5-dimethoxy-](http://img.cochemist.com/ccimg/154900/154876-31-0_b.png)

![Benzenamine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/122000/121942-75-4.png)
![Benzenamine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/122000/121942-75-4_b.png)



![2-(o-Tolyl)benzo[b]thiophene](http://img.cochemist.com/ccimg/55100/55084-51-0.png)
![2-(o-Tolyl)benzo[b]thiophene](http://img.cochemist.com/ccimg/55100/55084-51-0_b.png)

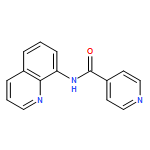
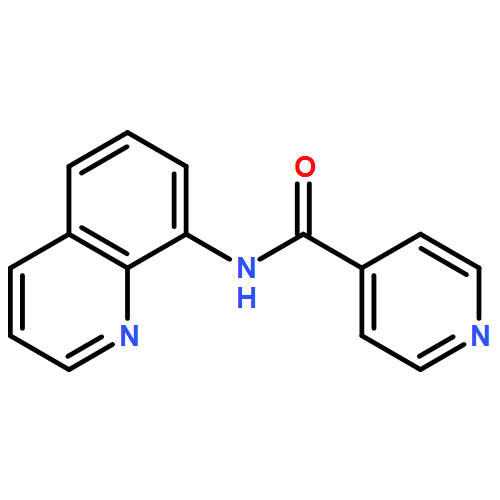
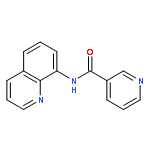
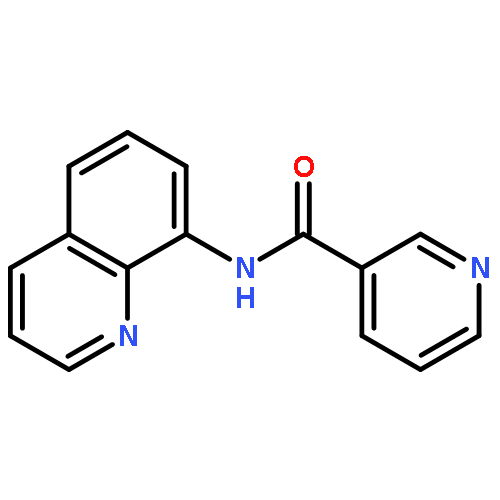










![[1,1'-Biphenyl]-2,2'-dicarboxylicacid, 5,5'-dibromo-](http://img.cochemist.com/ccimg/14000/13974-99-7.png)
![[1,1'-Biphenyl]-2,2'-dicarboxylicacid, 5,5'-dibromo-](http://img.cochemist.com/ccimg/14000/13974-99-7_b.png)
















![Benzo[b]thiophene, 2-phenyl-](http://img.cochemist.com/ccimg/1300/1207-95-0.png)
![Benzo[b]thiophene, 2-phenyl-](http://img.cochemist.com/ccimg/1300/1207-95-0_b.png)







![Benzoic acid, 4-[(8-quinolinylamino)carbonyl]-, methyl ester](/data/chemimg/3735000/878687-85-5.png)
![Benzoic acid, 4-[(8-quinolinylamino)carbonyl]-, methyl ester](/data/chemimg/3735000/878687-85-5_b.png)