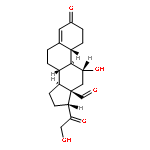
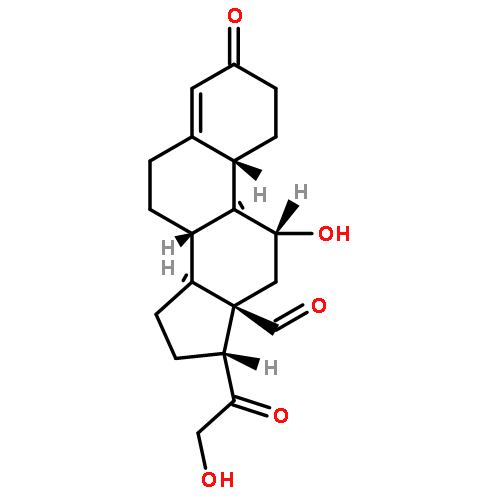
Co-reporter:Kohei Sano, Manami Ohashi, Kengo Kanazaki, Akira Makino, Ning Ding, Jun Deguchi, Yuko Kanada, Masahiro Ono, and Hideo Saji
Bioconjugate Chemistry April 19, 2017 Volume 28(Issue 4) pp:1024-1024
Publication Date(Web):February 7, 2017
DOI:10.1021/acs.bioconjchem.6b00715
Photoacoustic (PA) imaging has been considered an attractive imaging modality for sensitive and in-depth imaging of biomolecules with a high resolution in vivo. PA imaging probes utilizing fluorescence dyes, including indocyanine green (ICG), have been proposed to enhance PA signal intensity. On the other hand, nanomicelles modified with polysarcosine (PSar), a biocompatible hydrophilic polymer, on their surface have previously achieved rapid tumor uptake, suggesting active transport of PSar into tumor tissues. Thus, we hypothesized that PSar-based materials might be utilized as diagnostic probes for targeting tumors and therefore evaluated the potential of PSar labeled with an ICG derivative, ICG-PSar, as a PA imaging probe for targeting cancer. In this study, ICG-PSars with differing molecular weights (10, 20, and 30 kDa) were synthesized. In vitro cellular uptake studies using ICG-PSar demonstrated rapid uptake in colon26 tumor cells partially via macropinocytosis-mediated endocytosis. In vivo fluorescence imaging and biodistribution study indicated that ICG-PSar30k exhibited high accumulation in the tumor (8.4% dose/g), with high tumor-to-blood ratios reaching 4.6 at 24 h post injection of the probe. Finally, in vivo PA imaging studies showed that PA signal increased in tumors (251%) but not in blood vessels, achieving high contrast tumor imaging at 24 h after ICG-PSar30k probe injection. These results suggest that ICG-PSar has potential as a tumor-targeting PA imaging probe.
Co-reporter:Hiroyuki Kimura, Haruka Okuda, Masumi Ishiguro, Kenji Arimitsu, Akira Makino, Ryuichi Nishii, Anna Miyazaki, Yusuke Yagi, Hiroyuki Watanabe, Ikuo Kawasaki, Masahiro Ono, and Hideo Saji
ACS Medicinal Chemistry Letters April 13, 2017 Volume 8(Issue 4) pp:418-418
Publication Date(Web):March 20, 2017
DOI:10.1021/acsmedchemlett.6b00520
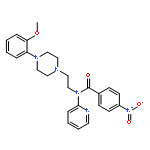
In nonsmall-cell lung carcinoma patients, L858R mutation of epidermal growth factor receptor (EGFR) is often found, and molecular target therapy using EGFR tyrosine kinase inhibitors is effective for the patients. However, the treatment frequently develops drug resistance by secondary mutation, of which approximately 50% is T790M mutation. Therefore, the ability to predict whether EGFR will undergo secondary mutation is extremely important. We synthesized a novel radiofluorinated 4-(anilino)pyrido[3,4-d]pyrimidine derivative ([18F]APP-1) and evaluated its potential as a positron emission tomography (PET) imaging probe to discriminate the difference in mutations of tumors. EGFR inhibition assay, cell uptake, and biodistribution study showed that [18F]APP-1 binds specifically to the L858R mutant EGFR but not to the L858R/T790M mutant. Finally, on PET imaging study using [18F]APP-1 with tumor-bearing mice, the H3255 tumor (L858R mutant) was more clearly visualized than the H1975 tumor (L858R/T790M mutant).Keywords: 4-(anilino)pyrido[3,4-d]pyrimidine; Epidermal growth factor receptor tyrosine kinase (EGFR-TK); fluorin-18; L858R mutant EGFR; positron emission tomography;
Co-reporter:Daiko Matsuoka, Hiroyuki Watanabe, Yoichi Shimizu, Hiroyuki Kimura, Masahiro Ono, Hideo Saji
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 21(Issue 21) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.bmcl.2017.09.037
Prostate-specific membrane antigen (PSMA), which is highly expressed in both localized and metastatic prostate cancer (PCa), is an ideal target for imaging and therapy of PCa. We previously reported radiolabeled asymmetric urea derivatives as a PSMA-targeting radiotracer for single-photon emission computed tomography (SPECT) and positron emission tomography (PET) imaging. Here, based on these radiopharmaceutical probes, we designed a novel near-infrared (NIR) fluorescent imaging probe (800CW-SCE) by chemical conjugation between IRDye 800CW-Maleimide and an asymmetric urea compound, known as PSMA inhibitor, for optical imaging. In the in vitro cellular uptake study, 800CW-SCE was internalized into PSMA-positive PCa cells (LNCaP cells) but not into PSMA-negative PCa cells (PC-3 cells). Moreover, in the in vivo imaging study, the probe was highly accumulated in LNCaP tumors but not in PC-3 tumors, and remained in LNCaP tumors until 24 h after intravenous administration. These results suggest that the potent NIR conjugate may contribute to clinical intraoperative optical imaging.Download high-res image (77KB)Download full-size image
Co-reporter:Hiroyuki Kimura, Naotaka Fujita, Kaori Kanbe, Hirokazu Matsuda, Hiroyuki Watanabe, Kenji Arimitsu, Hiroyuki Fujimoto, Keita Hamamatsu, Yusuke Yagi, Masahiro Ono, Nobuya Inagaki, Hideo Saji
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 20(Issue 20) pp:
Publication Date(Web):15 October 2017
DOI:10.1016/j.bmc.2017.09.005
A non-invasive method of pancreatic β-cell mass measurement is needed to enhance our understanding of the pathogenesis of diabetes, facilitate the early diagnosis of this disease, and promote the development of novel therapeutics. Here, we described the synthesis of a novel indium-111 (111In) exendin-4 derivative, [Lys12(In-BnDTPA-Ahx)]exendin-4, through a process involving isothiocyanate-benzyl-DTPA (BnDTPA) and 6-aminohexanoic acid (Ahx) attached to an ɛ-amino group at the lysine-12 residue. We further evaluated the potential use of this derivative as a SPECT probe for pancreatic β-cell imaging. An in vitro binding assay revealed that [Lys12(natIn-BnDTPA-Ahx)]exendin-4 has a high affinity for GLP-1 receptors (IC50 = 0.43 nM). In biodistribution experiments involving normal mice, high [Lys12(111In-BnDTPA-Ahx)]exendin-4 uptake was observed in the pancreas (21.8 ± 4.0 %ID/g) and was maintained for 2 h after injection. Pre-injection of excess exendin(9−39) markedly reduced the pancreatic uptake of [Lys12(111In-BnDTPA-Ahx)]exendin-4 (95.2%), indicating that the uptake of this tracer is specific and mediated by GLP-1 receptors. Ex vivo autoradiography experiments involving pancreatic sections from MIP-GFP mice confirmed the accumulation of [Lys12(111In-BnDTPA-Ahx)]exendin-4 in pancreatic β-cells. Finally, in mice, [Lys12(111In-BnDTPA-Ahx)]exendin-4 SPECT/CT yielded clear images of the pancreas at 30 min post-injection. In conclusion, SPECT with [Lys12(111In-BnDTPA-Ahx)]exendin-4 enables to visualize β-cells in vivo non-invasively.Download high-res image (138KB)Download full-size image
Co-reporter:Kengo Kanazaki, Kohei Sano, Akira Makino, Fumio Yamauchi, Atsushi Takahashi, Tsutomu Homma, Masahiro Ono, Hideo Saji
Journal of Controlled Release 2016 Volume 226() pp:115-123
Publication Date(Web):28 March 2016
DOI:10.1016/j.jconrel.2016.02.017
Poly(ethylene glycol) (PEG) is an artificial but biocompatible hydrophilic polymer that has been widely used in clinical products. To evaluate the feasibility of using PEG derivative itself as a tumor imaging carrier via an enhanced permeability and retention (EPR) effect, we prepared indium-111-labeled PEG (111In-DTPA-PEG) and indocyanine green (ICG)-labeled PEG (ICG-PEG) with PEG molecular weights of 5–40 kDa and investigated their in vivo biodistribution in colon26 tumor-bearing mice. Thereafter, single-photon emission computed tomography (SPECT) and photoacoustic (PA) imaging studies were performed. The in vivo biodistribution studies demonstrated increased tumor uptake and a prolongation of circulation half-life as the molecular weight of PEG increased. Although the observed differences in in vivo biodistribution were dependent on the labeling method (111In or ICG), the tumor-to-normal tissue ratios were comparable. Because PEG-based probes with a molecular weight of 20 kDa (PEG20) showed a preferable biodistribution (highest accumulation among tissues excised and relatively high tumor-to-blood ratios), an imaging study using 111In-DTPA-PEG20 and ICG-PEG20 was performed. Colon26 tumors inoculated in the right shoulder were clearly visualized by SPECT 24 h after administration. Furthermore, PA imaging using ICG-PEG20 also detected tumor regions, and the detected PA signals increased in proportion with the injected dose. These results suggest that PEG derivatives (20 kDa) serve as robust diagnostic drug carriers for tumor imaging.
Co-reporter:Hiroyuki Kimura, Sotaro Sampei, Daiko Matsuoka, Naoya Harada, Hiroyuki Watanabe, Kenji Arimitsu, Masahiro Ono, Hideo Saji
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 10) pp:2251-2256
Publication Date(Web):15 May 2016
DOI:10.1016/j.bmc.2016.03.051
Prostate-specific membrane antigen (PSMA) is expressed strongly in prostate cancers and is, therefore, an attractive diagnostic and radioimmunotherapeutic target. In contrast to previous reports of PMSA-targeting 99mTc-tricarbonyl complexes that are cationic or lack a charge, no anionic 99mTc-tricarbonyl complexes have been reported. Notably, the hydrophilicity conferred by both cationic and anionic charges leads to rapid hepatobiliary clearance, whereas an anionic charge might better enhance renal clearance relative to a cationic charge. Therefore, an improvement in rapid clearance would be expected with either cationic or anionic charges, particularly anionic charges. In this study, we designed and synthesized a novel anionic 99mTc-tricarbonyl complex ([99mTc]TMCE) and evaluated its use as a single-photon emission computed tomography (SPECT) imaging probe for PSMA detection. Direct synthesis of [99mTc]TMCE from dimethyl iminodiacetate, which contains both the asymmetric urea and succinimidyl moiety important for PSMA binding, was performed using our microwave-assisted one-pot procedure. The chelate formation was successfully achieved even though the precursor included a complicated bioactive moiety. The radiochemical yield of [99mTc]TMCE was 12–17%, with a radiochemical purity greater than 98% after HPLC purification. [99mTc]TMCE showed high affinity in vitro, with high accumulation in LNCaP tumors and low hepatic retention in biodistribution and SPECT/CT studies. These findings warrant further evaluation of [99mTc]TMCE as an imaging agent and support the benefit of this strategy for the design of other PSMA imaging probes.
Co-reporter:Manami Ohashi;Ning Ding;Yuko Kanada;Hideo Saji;Jun Deguchi;Kohei Sano;Kengo Kanazaki;Masahiro Ono
Molecular Imaging and Biology 2016 Volume 18( Issue 6) pp:
Publication Date(Web):2016/12/01
DOI:10.1007/s11307-016-0977-2
The feasibility of iron oxide nanoparticles (IONPs) conjugated with anti-epidermal growth factor receptor 2 (HER2) single-chain antibody (scFv-IONPs) as novel HER2-targeted magnetic resonance (MR) contrast agents was investigated.The scFv-IONPs were prepared and identified. For in vitro MRI, NCI-N87 (HER2 high expression) and SUIT2 (low expression) cells were incubated with scFv-IONPs. For in vivo MRI, NCI-N87 and SUIT2 tumor-bearing mice were intravenously injected with scFv-IONPs and imaged before and 24 h post-injection.The scFv-IONPs demonstrated high transverse relaxivity (296.3 s−1 mM−1) and affinity toward HER2 (KD = 11.7 nM). In the in vitro MRI, NCI-N87 cells treated with scFv-IONPs exhibited significant MR signal reduction (44.6 %) than SUIT2 cells (6.8 %). In the in vivo MRI, decrease of MR signals in NCI-N87 tumors (19.3 %) was more notable than that in SUIT2 tumors (6.2 %).The scFv-IONPs enabled HER2-specific tumor MR imaging, suggesting the potential of scFv-IONPs as a robust HER2-targeted MR contrast agent.
Co-reporter:Naoya Kondo, Takashi Temma, Jun Deguchi, Kohei Sano, Masahiro Ono, Hideo Saji
Journal of Controlled Release 2015 Volume 220(Part A) pp:476-483
Publication Date(Web):28 December 2015
DOI:10.1016/j.jconrel.2015.11.012
Co-reporter:Yusuke Yagi, Hiroyuki Kimura, Kenji Arimitsu, Masahiro Ono, Kazuya Maeda, Hiroyuki Kusuhara, Tetsuya Kajimoto, Yuichi Sugiyama and Hideo Saji
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 4) pp:1113-1121
Publication Date(Web):13 Nov 2014
DOI:10.1039/C4OB01953A
Fluorine-18 labeled radiotracers, such as [18F]fluorodeoxyglucose, can be used as practical diagnostic agents in positron emission tomography (PET). Furthermore, the properties of pharmaceuticals can be enhanced significantly by the introduction of fluorine groups into their original structures, and significant progress has been made during the last three decades towards the development of practical procedures for the introduction of fluorine. The replacement of the fluorine atoms present in pharmaceuticals with radioactive 18F atoms is a rational approach for designing novel PET tracers. As a fluorine-containing pharmaceutical agent, pitavastatin has attracted considerable interest from researchers working in the life sciences because it can act as an antihyperlipidemic agent as well as a substrate for hepatic organic anion transporting polypeptides (hOATP). With this in mind, it was envisaged that [18F]pitavastatin would be used as an excellent imaging agent for hOATP, which prompted us to investigate the synthesis of this agent. Herein, we report a practical method for the synthesis of [18F]pitavastatin by the Suzuki coupling reaction of p-iodofluorobenzene and a quinoline boronate derivative, with the desired product being formed in a radiochemical yield of 12 ± 3% (decay corrected from [18F]fluoride ions, n = 3).
Co-reporter:Kengo Kanazaki, Kohei Sano, Akira Makino, Yoichi Shimizu, Fumio Yamauchi, Satoshi Ogawa, Ning Ding, Tetsuya Yano, Takashi Temma, Masahiro Ono, Hideo Saji
Nanomedicine: Nanotechnology, Biology and Medicine 2015 Volume 11(Issue 8) pp:2051-2060
Publication Date(Web):November 2015
DOI:10.1016/j.nano.2015.07.007
Photoacoustic (PA) imaging is a promising imaging modality that provides biomedical information with high sensitivity and resolution. Iron oxide nanoparticles (IONPs) have been regarded as remarkable PA contrast agents because of their low toxicity and biodegradable properties. However, IONP delivery is restricted by its modest leakage and retention in tumors. In this study, we designed IONPs (20 nm, 50 nm, and 100 nm) conjugated with anti-HER2 moieties [whole IgG, single-chain fragment variable (scFv), and peptide] for HER2-targeted PA tumor imaging. The binding affinity, cellular uptake, and in vivo biodistribution were examined. We propose 20-nm anti-HER2 scFv-conjugated IONPs (SNP20) as a novel PA contrast agent. SNP20 demonstrated high affinity and specific binding to HER2-expressing cells; it selectively visualized HER2-positive tumors in PA imaging studies. These data indicate that SNP20 is a potential PA contrast agent for imaging of HER2-expressing tumors.From the Clinical EditorIron oxide nanoparticles have been demonstrated to be good contrast agents for tumor imaging. They may also be useful in photoacoustic (PA) imaging, which can provide high sensitivity data and image resolution. The authors here coupled iron oxide nanoparticles with anti-HER2 antibody fragment and showed significant retention of these nanoparticles in tumors. This combination may provide another option for enhanced imaging of tumors.HER2 (epidermal growth factor receptor 2)-targeted tumor diagnosis provides biomedical information on tumor malignancy. By utilizing a photoacoustic (PA) imaging technique, which detect ultrasonic waves thermoelastically induced by the absorption of photons by PA contrast agent through PA effect, iron oxide nanoparticles (IONPs) (20 nm, 50 nm, or 100 nm) conjugated with anti-HER2 moieties (Herceptin, single chain Fv (scFv), or peptide) were evaluated for HER2-targeted photoacoustic tumor imaging (left panel). The 20-nm IONPs conjugated with scFv demonstrated high affinity and specific binding to HER2, and selectively visualized HER2-positive tumors in photoacoustic imaging studies (right panel).
Co-reporter:Takashi Temma;Hayato Hisada;Masashi Ueda;Hideo Saji;Yuji Nakamoto;Hiroyuki Kimura;Masahiro Ono;Kaori Togashi;Yoichi Shimizu
Molecular Imaging and Biology 2015 Volume 17( Issue 1) pp:
Publication Date(Web):2015/02/01
DOI:10.1007/s11307-014-0769-5
We aimed to develop a gallium-68 (Ga-68)-labeled single-chain variable fragment (scFv) targeting the human epidermal growth factor receptor 2 (HER2) to rapidly and noninvasively evaluate the status of HER2 expression.Anti-HER2 scFv was labeled with Ga-68 by using deferoxamine (Df) as a bifunctional chelate. Biodistribution of [68Ga]Df-anti-HER2 scFv was examined with tumor-bearing mice and positron emission tomography (PET) imaging was performed. The changes in HER2 expression after anti-HER2 therapy were monitored by PET imaging.[68Ga]Df-anti-HER2 scFv was obtained with high radiochemical yield after only a 5-min reaction at room temperature. The probe showed high accumulation in HER2-positive xenografts and the intratumoral distribution of radioactivity coincided with HER2-positive regions. Furthermore, [68Ga]Df-anti-HER2 scFv helped visualize HER2-positive xenografts and monitor the changes in HER2 expression after anti-HER2 therapy.[68Ga]Df-anti-HER2 scFv could be a promising probe to evaluate HER2 status by in vivo PET imaging, unless trastuzumab is prescribed as part of the therapy.
Co-reporter:Hiroyuki Kimura, Tomoki Kawai, Yoshio Hamashima, Hidekazu Kawashima, Kenji Miura, Yuta Nakaya, Makoto Hirasawa, Kenji Arimitsu, Tetsuya Kajimoto, Yoshiro Ohmomo, Masahiro Ono, Manabu Node, Hideo Saji
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 1) pp:285-291
Publication Date(Web):1 January 2014
DOI:10.1016/j.bmc.2013.11.026
Improved radiopharmaceuticals for imaging cerebral acetylcholinesterase (AChE) are needed for the diagnosis of Alzheimer’s disease (AD). Thus, 11C-labeled (−)-galanthamine and its enantiomers were synthesized as novel agents for imaging the localization and activity of AChE by positron emission tomography (PET). C-11 was incorporated into (−)- and (+)-[11C]galanthamine by N-methylation of norgalanthamines with [11C]methyl triflate. Simple accumulation of 11C in the brain was measured in an in vivo biodistribution study using mice, whilst donepezil was used as a blocking agent in analogous in vivo blocking studies. In vitro autoradiography of rat brain tissue was performed to investigate the distribution of (−)-[11C]galanthamine, and confirmed the results of PET studies in mice. The radiochemical yields of N-methylation of (−)- and (+)-norgalanthamines were 13.7% and 14.4%, respectively. The highest level of accumulation of 11C in the brains of mice was observed at 10 min after administration (2.1% ID/g). Intravenous pretreatment with donepezil resulted in a 30% decrease in accumulation of (−)-[11C]galanthamine in the striatum; however, levels in the cerebellum were unchanged. In contrast, use of (+)-[11C]galanthamine led to accumulation of radioactivity in the striatum equal to that in the cerebellum, and these levels were unaffected by pretreatment with donepezil. In in vitro autoradiography of regional radioactive signals of brain sections showed that pretreatment with either (−)-galanthamine or donepezil blocked the binding of (−)-[11C]galanthamine to the striatum, while sagittal PET imaging revealed accumulation of (−)-[11C]galanthamine in the brain. These results indicate that (−)-[11C]galanthamine showed specific binding to AChE, whereas (+)-[11C]-galanthamine accumulated in brain tissue by non-specific binding. Thus, optically pure (−)-[11C]galanthamine could be a useful PET tracer for imaging cerebral AChE.Sagittal PET image of (−)-[11C]galanthamine in the ddY mouse.
Co-reporter:Satoru Onoe;Takashi Temma;Yoichi Shimizu;Masahiro Ono
Cancer Medicine 2014 Volume 3( Issue 4) pp:775-786
Publication Date(Web):
DOI:10.1002/cam4.252
Abstract
Mitochondrial membrane potential (Δψm) alteration is an important target for cancer diagnosis. In this study, we designed a series of near-infrared fluorescent cationic cyanine dyes with varying alkyl chain lengths (IC7-1 derivatives) to provide diverse lipophilicities and serum albumin-binding rates, and we evaluated the usefulness of these derivatives for in vivo Δψm imaging. IC7-1 derivatives with side chains from methyl to hexyl (IC7-1-Me to IC7-1-He) were synthesized, and their optical properties were measured. Cellular uptake and intracellular distribution were investigated with depolarized HeLa cells from carbonyl cyanine m-chlorophenylhydrazone (CCCP) treatment using a spectrofluorometer and a fluorescence microscope. Serum albumin-binding rates were evaluated using albumin-binding inhibitors. In vivo optical imaging was performed with HeLa cell xenograft mice following intravenous administration of IC7-1 derivatives with or without warfarin and CCCP as in vivo blocking agents. IC7-1 derivatives showing maximum excitation and emission wavelengths at 823 nm and ~845 nm, respectively, were synthesized. IC7-1-Me to -Bu showed fluorescence in mitochondria that decreased with CCCP treatment in a concentration-dependent manner, which showed that IC7-1-Me to -Bu successfully indicated Δψm. Tumors were clearly visualized after IC7-1-Bu administration. Treatment with warfarin or CCCP significantly decreased IC7-1-Bu fluorescence in the tumor region. In summary, IC7-1-Bu exhibited fluorescence localized to mitochondria dependent on Δψm, which enabled clear in vivo tumor imaging via serum albumin as a drug carrier for effective tumor targeting. Our data suggest that IC7-1-Bu is a promising NIR probe for in vivo imaging of the altered Δψm of tumor cells.
Co-reporter:Hiroyuki Kimura, Hirokazu Matsuda, Hiroyuki Fujimoto, Kenji Arimitsu, Kentaro Toyoda, Eri Mukai, Hiroshi Nakamura, Yu Ogawa, Mikako Takagi, Masahiro Ono, Nobuya Inagaki, Hideo Saji
Bioorganic & Medicinal Chemistry 2014 22(13) pp: 3270-3278
Publication Date(Web):
DOI:10.1016/j.bmc.2014.04.059
Co-reporter:Hiroyuki Kimura;Yusuke Yagi;Noriyuki Ohneda;Hiro Odajima;Masahiro Ono
Journal of Labelled Compounds and Radiopharmaceuticals 2014 Volume 57( Issue 12) pp:680-686
Publication Date(Web):
DOI:10.1002/jlcr.3232
Microwave technology has been successfully applied to enhance the effectiveness of radiolabeling reactions. The use of a microwave as a source of heat energy can allow chemical reactions to proceed over much shorter reaction times and in higher yields than they would do under conventional thermal conditions. A microwave reactor developed by Resonance Instrument Inc. (Model 520/521) and CEM (PETWave) has been used exclusively for the synthesis of radiolabeled agents for positron emission tomography by numerous groups throughout the world. In this study, we have developed a novel resonant-type microwave reactor powered by a solid-state device and confirmed that this system can focus microwave power on a small amount of reaction solution. Furthermore, we have demonstrated the rapid and facile radiosynthesis of 16α-[18F]fluoroestradiol, 4-[18F]fluoro-N-[2-(1-methoxyphenyl)-1-piperazinyl]ethyl-N-2-pyridinylbenzamide, and N-succinimidyl 4-[18F]fluorobenzoate using our newly developed microwave reactor.
Co-reporter:Yoichi Shimizu, Takashi Temma, Isao Hara, Akira Makino, Ryo Yamahara, Ei-ichi Ozeki, Masahiro Ono, Hideo Saji
Nanomedicine: Nanotechnology, Biology and Medicine 2014 Volume 10(Issue 1) pp:187-195
Publication Date(Web):January 2014
DOI:10.1016/j.nano.2013.06.009
Near-infrared (NIR: 800-1000 nm) fluorescent probes, which activate their fluorescence following interaction with functional biomolecules, are desirable for noninvasive and sensitive tumor diagnosis due to minimal tissue interference. Focusing on bioavailability and applicability, we developed a probe with a self-assembling polymer micelle, a lactosome, encapsulating various quantities of NIR dye (IC7-1). We also conjugated anti-HER2 single chain antibodies to the lactosome surface and examined the probe’s capacity to detect HER2 in cells and in vivo. Micelles encapsulating 20 mol% IC7-1 (hIC7L) showed 30-fold higher fluorescence (λem: 858 nm) after micelle denaturation compared to aqueous buffer. Furthermore, antibody modification allowed specific activation of the probe (HER2-hIC7L) following internalization by HER2-positive cells, with the probe concentrating in lysosomes. HER2-hIC7L intravenously administered to mice clearly and specifically visualized HER2-positive tumors by in vivo optical imaging. These results indicate that HER2-hIC7L is a potential activatable NIR probe for sensitive tumor diagnosis.From the Clinical EditorNear-infrared probes that activate their fluorescence following interaction with specific biomolecules are desirable for noninvasive and sensitive tumor detection due to minimal tissue interference. This team of authors developed a probe termed hIC7L and demonstrate its potential in HER2 tumor diagnosis.We describe an NIR fluorescence activatable probe (HER2-hIC7L) composed of a self-assembling polymer micelle encapsulating quantities of near-infrared (NIR) fluorophore (IC7-1) with a surface modification by anti-HER2 single chain antibodies. HER2-hIC7L first interacts with HER2 on tumor cells via immunoreaction, followed by probe internalization by the cells, whereupon denaturation causes probe dequenching and a 30-fold increase in fluorescence. The probe could clearly visualize HER2-positive tumors but not HER2-negative tumors in in vitro and in vivo studies. Therefore, HER2-hIC7L would be effective for in vivo detection of HER2-expressing tumors.
Co-reporter:Naoya Harada ; Hiroyuki Kimura ; Masahiro Ono
Journal of Medicinal Chemistry 2013 Volume 56(Issue 20) pp:7890-7901
Publication Date(Web):September 24, 2013
DOI:10.1021/jm400895s
Prostate-specific membrane antigen (PSMA) has been identified as a diagnostic and therapeutic target for prostate cancer. (S)-2-[3-[(R)-1-Carboxy-2-mercaptoethyl]ureido-pentanedioic acid (Cys-CO-Glu) were used to design novel PSMA targeting probes by nucleophilic conjugate addition between cysteine and maleimide based reagents. 3 ([123I]IGLCE) was synthesized by this strategy and showed high affinity for PSMA. Results of binding inhibition assays of these derivatives suggested the importance of an aromatic group and succinimide moiety for high affinity. [123I]3 was evaluated in vivo with PSMA positive LNCaP and PSMA negative PC-3 human prostate cancer xenograft bearing mice. [125I]3 accumulated in LNCaP tumors but not in PC-3 tumors, and the accumulation was inhibited by 2-(phosphonomethyl)pentanedioic acid (2-PMPA). Use of [123I]3 provided positive images of LNCaP tumors in single photon emission tomography scans. These results warrant further evaluation of [123I]3 and its derivatives as radiolabeled probes for the diagnosis of prostate cancer.
Co-reporter:Kenji Arimitsu;Hiroyuki Kimura;Tetsuya Kajimoto;Masahiro Ono;Yoshiro Ohmomo;Masayuki Yamashita;Manabu Node
Journal of Labelled Compounds and Radiopharmaceuticals 2013 Volume 56( Issue 11) pp:562-572
Publication Date(Web):
DOI:10.1002/jlcr.3063
Guided by the known molecular recognition interactions between N-acetylglucosaminyltransferase V (GnT-V) and certain synthetic substrates, we synthesized a radiolabeled double-stranded glycolipid composed of a long-chain alkyl unit and a radioiodinated phenylalkyl unit, [125I]-2-[N-(2-hydroxy-3-hexadecyloxy)propyl-15-(4-iodophenyl)pentadecanecarboxamido]ethyl 2-acetamido-2-deoxy-β-d-glucopyranosyl-(12)-α-d-mannopyranosyl-(16)-β-d-glucopyranoside ([125I]2), as a novel intravital glycolipid mimic substrate of GnT-V. The radioactive iodine (125I) was incorporated via iododestannylation of the phenyltributyltin derivative, 2-[N-(2-acetoxy-3-hexadecyloxy)propyl-15-(4-tributylstannylphenyl)pentadecanecarboxamido]ethyl 3,4,6-tri-O-acetyl-2-acetamido-2-deoxy-β-d-glucopyranosyl-(12)-3,4,6-O-acetyl-α-d-mannopyranosyl-(16)-2,3,4-tri-O-acetyl-β-d-glucopyranoside (26). Subsequent deacetylation at the final step afforded [125I]2.
Co-reporter:Masashi Ueda;Azusa Miyano;Shinae Kizaka-Kondoh;Kei Ogawa;Masahiro Ono;Hideo Saji
Molecular Imaging and Biology 2013 Volume 15( Issue 6) pp:
Publication Date(Web):2013/12/01
DOI:10.1007/s11307-013-0647-6
We aimed to develop a radiolabeled peptide probe for the imaging of hypoxia-inducible factor-1 (HIF-1)-active tumors.We synthesized the peptide probes that contain or lack an essential sequence of the oxygen-dependent degradation of HIF-1α in proteasomes (123/125I-DKOP30 or 125I-mDKOP, respectively). The degradation of probes was evaluated in vitro using cell lysates containing proteasomes. In vivo biodistribution study, planar imaging, autoradiography, and comparison between probe accumulation and HIF-1 transcriptional activity were also performed.The 125I-DKOP30 underwent degradation in a proteasome-dependent manner, while 125I-mDKOP was not degraded. Biodistribution analysis showed 125I-DKOP30 accumulation in tumors. The tumors were clearly visualized by in vivo imaging, and intratumoral distribution of 125I-DKOP30 coincided with the HIF-1α-positive hypoxic regions. Tumoral accumulation of 125I-DKOP30 was significantly correlated with HIF-1-dependent luciferase bioluminescence, while that of 125I-mDKOP was not.123I-DKOP30 is a useful peptide probe for the imaging of HIF-1-active tumors.
Co-reporter:Yoichi Shimizu;Takashi Temma;Isao Hara;Ryo Yamahara
Journal of Fluorescence 2012 Volume 22( Issue 2) pp:719-727
Publication Date(Web):2012 March
DOI:10.1007/s10895-011-1007-z
Optical imaging with near-infrared (NIR) fluorescent probes is a useful diagnostic technology for in vivo tumor detection. Our plan was to develop novel NIR fluorophore-micelle complex probes. IC7-1 and IC7-2 were synthesized as novel lipophilic NIR fluorophores, which were encapsulated in an amphiphilic polydepsipeptide micelle “lactosome”. The fluorophore-micelle complexes IC7-1 lactosome and IC7-2 lactosome were evaluated as NIR fluorescent probes for in vivo tumor imaging. IC7-1 and IC7-2 were synthesized and then encapsulated in lactosomes. The optical properties of IC7-1, IC7-2, IC7-1 lactosome and IC7-2 lactosome were measured. IC7-1 lactosome and IC7-2 lactosome were administered to tumor-bearing mice, and fluorescence images were acquired for 48 h. IC7-1 and IC7-2 were successfully synthesized in 12% and 6.3% overall yield, and maximum emission wavelengths in chloroform were observed at 858 nm and 897 nm, respectively. Aqueous buffered solutions of IC7-1 lactosome and IC7-2 lactosome showed similar fluorescence spectra in chloroform and higher or comparable quantum yields and higher photostability compared with ICG. Both lactosome probes specifically visualized tumor tissue 6 h post-administration. IC7-1 lactosome and IC7-2 lactosome could be promising NIR probes for in vivo tumor imaging.
Co-reporter:Mengchao Cui ; Masahiro Ono ; Hiroyuki Kimura ; Boli Liu
Journal of Medicinal Chemistry 2011 Volume 54(Issue 7) pp:2225-2240
Publication Date(Web):March 21, 2011
DOI:10.1021/jm101404k
A new and extensive set of dibenzylideneacetone derivatives was synthesized and screened for affinity toward Aβ1−42 aggregates. Structure−activity relationships revealed the binding of dibenzylideneacetones to be affected by various substituents. The introduction of a substituent group in the ortho position reduced or abolished the binding. However, the para position was highly tolerant of sterically demanding substitutions. Three radioiodinated ligands (6, 70, and 71) and two 18F fluoro-pegylated (FPEG) ligands (83 and 85) were prepared, all of which displayed high affinity for Aβ1−42 aggregates (Ki ranging from 0.9 to 7.0 nM). In biodistribution experiments, they exhibited good initial penetration (1.59, 4.68, 4.56, 4.13, and 5.15% ID/g, respectively, at 2 min) of and fast clearance from the brain. Autoradiography with sections of postmortem AD brain and transgenic mouse brain confirmed the high affinity of these tracers. These preliminary results strongly suggest the dibenzylideneacetone structure to be a potential new scaffold for β-amyloid imaging probes.
Co-reporter:Masahiro Ono ; Yan Cheng ; Hiroyuki Kimura ; Mengchao Cui ; Shinya Kagawa ; Ryuichi Nishii
Journal of Medicinal Chemistry 2011 Volume 54(Issue 8) pp:2971-2979
Publication Date(Web):March 23, 2011
DOI:10.1021/jm200057u
In vivo imaging of β-amyloid plaques in the brain may lead to the early diagnosis of Alzheimer’s disease (AD) and monitoring of the progression and effectiveness of treatment. In the present study, we report on the development of two potential PET probes, [18F]FPYBF-2 ([18F]10) and [18F]FPHBF-2 ([18F]21), for imaging of β-amyloid plaques in AD brain. In experiments in vitro, 10 and 21 displayed high affinity for Aβ(1−42) aggregates (Ki = 2.41 and 3.85 nM, respectively). In biodistribution experiments using normal mice, they displayed high uptake in the brain (7.38 and 8.18% ID/g at 2 min postinjection, respectively), and the radioactivity washed out from the brain rapidly (3.15 and 3.87% ID/g at 60 min postinjection, respectively), which is highly desirable for β-amyloid imaging agents. In vivo, they clearly labeled β-amyloid plaques in Tg2576 mice. Furthermore, the specific labeling of β-amyloid plaques by 10 and 21 was observed in autoradiographs of sections of autopsied AD brain. These new fluorinated benzofuran derivatives are promising PET probes for imaging cerebral β-amyloid plaques.
Co-reporter:Masahiro Ono, Shun Hayashi, Kenji Matsumura, Hiroyuki Kimura, Yoko Okamoto, Masafumi Ihara, Ryosuke Takahashi, Hiroshi Mori, and Hideo Saji
ACS Chemical Neuroscience 2011 Volume 2(Issue 5) pp:269
Publication Date(Web):March 21, 2011
DOI:10.1021/cn200002t
A novel series of rhodanin (RH) and thiohydantoin (TH) derivatives were designed and synthesized for detecting tau pathology in the brains of patients with Alzheimer’s disease (AD). In experiments in vitro using tau and β-amyloid (Aβ) aggregates, the TH derivative, TH2, showed high specific binding to tau aggregates. In hippocampal sections obtained from AD patients, TH2 intensely stained neurofibrillary tangles. In experiments using normal mice, [125I]TH2 showed good uptake (1.54%ID/g, 2 min postinjection) into and a rapid washout (0.25%ID/g, 60 min postinjection) from the brain. [123I]TH2 should be further investigated as a potential imaging agent for detecting tau pathology.Keywords: Alzheimer’s disease; imaging; rhodanine; tau; thiohydantoin
Co-reporter:Mengchao Cui, Masahiro Ono, Hiroyuki Kimura, Boli Liu, Hideo Saji
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 13) pp:4148-4153
Publication Date(Web):1 July 2011
DOI:10.1016/j.bmc.2011.04.049
A series of benzofuran-2-yl(phenyl)methanone derivatives were synthesized and evaluated as novel probes for β-amyloid plaques. These derivatives were produced by a Rap–Stoermer condensation reaction. Compounds with a N,N-dimethylamino group displayed high affinity for Aβ1–42 aggregates with Ki values in the nanomolar range. Autoradiography with brain sections of AD model mice (APP/PS1) revealed that a radioiodinated probe, [125I]10, labeled β-amyloid plaques selectively and displayed good brain uptake (3.53% ID/g) at 2 min. The results suggest that benzofuran-2-yl(phenyl)methanone derivatives should be investigated further as potential probes for detecting β-amyloid plaques in the AD brain.
Co-reporter:Takahiro Mukai, Masayori Hagimori, Kenji Arimitsu, Takahiro Katoh, Misa Ukon, Tetsuya Kajimoto, Hiroyuki Kimura, Yasuhiro Magata, Eiji Miyoshi, Naoyuki Taniguchi, Manabu Node, Hideo Saji
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 14) pp:4312-4321
Publication Date(Web):15 July 2011
DOI:10.1016/j.bmc.2011.05.047
N-acetylglucosaminyltransferase V (GnT-V) is one of the most relevant glycosyltransferases to tumor invasion and metastasis. Based on previous findings of molecular recognition between GnT-V and synthetic substrates, we designed and synthesized a p-iodophenyl-derivatized trisaccharide, 2-(4-iodophenyl)ethyl 6-O-[2-O-(2-acetamido-2-deoxy-β-d-glucopyranosyl)-α-d-mannopyranosyl]-β-d-glucopyranoside (IPGMG, 1) and its radiolabeled form, [125I]IPGMG ([125I]1), for use in assays of GnT-V activity in vitro. The tributyltin derivative, 2-[4-(n-tributylstannyl)phenyl]ethyl 6-O-[2-O-(3,4,6-tri-O-acetyl-2-acetamido-2-deoxy-β-d-glucopyranosyl)-3,4,6-tri-O-acetyl-α-d-mannopyranosyl]-2,3,4-tri-O-acetyl-β-d-glucopyranoside (21), was synthesized as a precursor for the preparation of [125I]1. The iododestannylation of 21 using hydrogen peroxide as an oxidant followed by deacetylation yielded [125I]1. When [125I]1 was incubated in GnT-V-expressing cells with a UDP-GlcNAc donor, the production of β1–6GlcNAc-bearing IPGMG (IPGGMG, 2) was confirmed by radio-HPLC. In kinetic analysis, 1 was found to be a good substrate with a Km of 23.7 μM and a Vmax of 159 pmol/h. μg protein. [125I]1 would therefore be a useful synthetic substrate for the quantitative determination of GnT-V activity.
Co-reporter:Kenji Matsumura, Masahiro Ono, Shun Hayashi, Hiroyuki Kimura, Yoko Okamoto, Masafumi Ihara, Ryosuke Takahashi, Hiroshi Mori and Hideo Saji
MedChemComm 2011 vol. 2(Issue 7) pp:596-600
Publication Date(Web):26 Apr 2011
DOI:10.1039/C1MD00034A
This paper describes the synthesis and biological evaluation of novel phenyldiazenyl benzothiazole (PDB) derivatives as probes for imaging neurofibrillary tangles (NFTs) in patients with Alzheimer's disease (AD). We successfully synthesized three PDB derivatives using a diazo coupling reaction. In binding experiments in vitro, the compounds displayed higher affinity for tau aggregates than for Aβ aggregates. In fluorescent staining experiments using AD brain sections, 9 visualized NFTs clearly. No-carrier-added radioiodinated PDB derivatives were successfully prepared through an iododestannylation reaction from the corresponding tributyltin derivatives. [125I]9 labeled NFTs in sections of brain tissue from a patient with AD, but not a control. In biodistribution experiments using normal mice, the PDB derivatives displayed an uptake into the brain, sufficient for imaging NFTs, ranging from 0.94 to 3.2% ID g−1, but a relatively slow washout. Although further modifications are necessary to improve the pharmacokinetics in the brain, PDB with high affinity for tau aggregates may be useful as a backbone structure to develop agents for imaging NFTs in AD brains.
Co-reporter:Mengchao Cui, Masahiro Ono, Hiroyuki Kimura, Boli Liu, Hideo Saji
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 14) pp:4193-4196
Publication Date(Web):15 July 2011
DOI:10.1016/j.bmcl.2011.05.079
In a search for new probes to detect β-amyloid plaques in the brain of patients with Alzheimer’s disease (AD), we have synthesized and evaluated a series of quinoxaline derivatives containing a ‘6+6−6’ ring system. These quinoxaline derivatives showed excellent affinity for Aβ1–42 aggregates with Ki values ranging from 2.6 to 10.7 nM. Autoradiography with sections of brain tissue from an animal model of AD mice (APP/PS1) and AD patients revealed that [125I]5 labeled β-amyloid plaques specifically. In biodistribution experiments using normal mice, [125I]5 displayed high uptake (6.03% ID/g at 2 min) into and a moderately fast washout from the brain. Although additional refinements are needed to decrease the lipophilicity and improve the washout rate, the quinoxaline scaffold may be useful as a backbone structure to develop novel β-amyloid imaging agents.
Co-reporter:Mengchao Cui, Masahiro Ono, Hiroyuki Kimura, Bo Li Liu, Hideo Saji
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 3) pp:980-982
Publication Date(Web):1 February 2011
DOI:10.1016/j.bmcl.2010.12.045
A series of chaclone derivatives containing an indole moiety were evaluated in competitive binding assays with Aβ1-42 aggregates versus [125I]IMPY. The affinity of these compounds ranged from 4.46 to >1008 nM, depending on the substitution on the phenyl ring. Fluorescent staining in vitro showed that one compound with a N,N-dimethylamino group intensely stained Aβ plaques within brain sections of AD transgenic mice. The radioiodinated probe [125I]-(E)-3-(1H-indol-5-yl)-1-(4-iodophenyl)prop-2-en-1-one, [125I]4, was prepared and autoradiography in sections of brain tissue from an animal model of AD showed that it labeled Aβ plaques specifically. However, experiments with normal mice indicated that [125I]4 exhibited a low uptake into the brain in vivo (0.41% ID/g at 2 min). Additional chemical modifications of this indole-chalcone structure may lead to more useful imaging agents for detecting β-amyloid plaques in the brains of AD patients.A series of novel indole-chalcone derivatives were synthesized and evaluated as Aβ imaging probes.
Co-reporter:Naoya Harada, Hiroyuki Kimura, Masahiro Ono, Daisuke Mori, Yoshiro Ohmomo, Tetsuya Kajimoto, Hidekazu Kawashima, Hideo Saji
Journal of Organometallic Chemistry 2011 696(23) pp: 3745-3749
Publication Date(Web):
DOI:10.1016/j.jorganchem.2011.08.033
Co-reporter:Yuji Kuge;Hideo Saji;Kohei Sano;Ryusuke Nakai;Takashi Azuma;Takashi Temma;Michiko Narazaki
Molecular Imaging and Biology 2011 Volume 13( Issue 6) pp:1196-1203
Publication Date(Web):2011/12/01
DOI:10.1007/s11307-010-0463-1
We aimed to establish a magnetic resonance imaging (MRI) protocol for the sensitive and specific imaging of functional molecules with a pre-targeting strategy utilizing the streptavidin–biotin interaction. Membrane type-1 matrix metalloproteinase (MT1-MMP) was selected as the target molecule.The biotinylated polyamidoamine dendrimer (PAMAM)-based contrast agent (Bt-PAMAM-DTPA(Gd)) was prepared, and its proton relaxivity (r1) and affinity to streptavidin were evaluated. Tumor-bearing mice were pre-targeted with streptavidin-conjugated anti-MT1-MMP monoclonal antibody (mAb), streptavidin-conjugated negative control IgG, or saline and 3 days later were injected with Bt-PAMAM-DTPA(Gd) followed immediately by MRI for a period of 3 h.High r1 (15.5 L mmol−1 s−1) and 1.9-fold higher affinity than d-biotin were obtained. Significantly higher relative tumor signals were observed in mice pre-targeted with streptavidin-conjugated anti-MT1-MMP mAb (165% at 3 h vs. pre-administration) than with saline or streptavidin-conjugated negative control IgG (P < 0.0001).This pre-targeting approach can accomplish sensitive and specific in vivo MRI of functional molecules.
Co-reporter:M Ueda;Y Iida;A Tominaga;T Yoneyama;M Ogawa;Y Magata;H Nishimura;Y Kuge;H Saji
British Journal of Pharmacology 2010 Volume 159( Issue 6) pp:1201-1210
Publication Date(Web):
DOI:10.1111/j.1476-5381.2009.00613.x
Background and purpose: Much interest is currently being focused on the anti-nociceptive effects mediated by nicotinic acetylcholine (nACh) receptors, including their location and mechanism of action. The purpose of this study was to elucidate these issues using 5-iodo-3-(2(S)-azetidinylmethoxy)pyridine (5IA), a nACh receptor agonist, and [125I]5IA.
Experimental approach: We partially ligated the sciatic nerve of Sprague-Dawley rat to induce neuropathic pain [Seltzer's partial sciatic nerve ligation (PSL) model]. We then examined the changes in nACh receptor density in the CNS using [125I]5IA autoradiography and the involvement of nACh receptors in anti-nociceptive effects in the region where changes occurred.
Key results: Autoradiographic studies showed that the accumulation of [125I]5IA and the number of nACh receptors in the thalamus of PSL rats were increased about twofold compared with those in the sham-operated rats. No change was observed in other brain regions. Rats injected in the ventral posterolateral thalamic nucleus (VPL) with 5IA demonstrated a significant and dose-dependent anti-allodynic effect and this effect was completely antagonized by mecamylamine, injected with 5IA, into the VPL. The blockade of nACh receptors in the VPL by mecamylamine decreased by 70% the anti-allodynic effect of 5IA, given i.c.v. Moreover, mecamylamine given intra-VPL by itself, induced significant hyperalgesia.
Conclusions and implications: Our findings suggest that the nACh receptors expressed in the VPL play an important role in the anti-allodynic effects produced by exogenous and endogenous agonists.
Co-reporter:Yan Cheng, Masahiro Ono, Hiroyuki Kimura, Shinya Kagawa, Ryuichi Nishii, Hidekazu Kawashima, and Hideo Saji
ACS Medicinal Chemistry Letters 2010 Volume 1(Issue 7) pp:321
Publication Date(Web):July 11, 2010
DOI:10.1021/ml100082x
A series of fluorinated benzofuran derivatives as potential tracers for positron emission tomography (PET) targeting β-amyloid plaques in the brains of patients with Alzheimer's disease (AD) were synthesized and evaluated. The derivatives were produced using an intramolecular Wittig reaction. In experiments in vitro, all displayed high affinity for Aβ(1−42) aggregates with Ki values in the nanomolar range. Radiofluorinated 17, [18F]17, in particular labeled β-amyloid plaques in sections of Tg2576 mouse brain and displayed high uptake (5.66% ID/g) at 10 min postinjection, sufficient for PET imaging. In addition, in vivo β-amyloid plaque labeling can be clearly demonstrated with [18F]17 in Tg2576 mice. In conclusion, [18F]17 may be useful for detecting β-amyloid plaques in patients with AD.Keywords (keywords): Alzheimer's disease; benzofuran; fluorine-18; positron emission tomography (PET)
Co-reporter:Masahiro Ono, Manami Ishikawa, Hiroyuki Kimura, Shun Hayashi, Kenji Matsumura, Hiroyuki Watanabe, Yoichi Shimizu, Yan Cheng, Mengchao Cui, Hidekazu Kawashima, Hideo Saji
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 13) pp:3885-3888
Publication Date(Web):1 July 2010
DOI:10.1016/j.bmcl.2010.05.027
The imaging of β-amyloid (Aβ) aggregates in the brain may lead to the early detection of Alzheimer’s disease (AD) and monitoring of the progression and effectiveness of treatment. The purpose of this study was to develop dual modality SPECT and fluorescent probes based on boron dipyrromethane (BODIPY) as a core structure. We designed and synthesized an 125I-labeled derivative of BODIPY (BODIPY7). BODIPY7 had a Ki value of 108 nM for Aβ(1–42) aggregates and exhibited peaks of absorption/emission at 606/613 nm. It detected Aβ plaques in sections of brain tissue from an animal model of AD and displayed low uptake in the brain and high uptake in the liver in normal mice. Although additional modifications of the BODIPY scaffold are necessary to improve brain uptake, these results should aid the development of dual functional SPECT/fluorescent probes for the imaging of Aβ plaques in the brain.
Co-reporter:Yan Cheng, Masahiro Ono, Hiroyuki Kimura, Shinya Kagawa, Ryuichi Nishii, Hideo Saji
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 20) pp:6141-6144
Publication Date(Web):15 October 2010
DOI:10.1016/j.bmcl.2010.08.016
A potential probe for PET targeting β-amyloid plaques in Alzheimer’s disease (AD) brain, FPYBF-1 (5-(5-(2-(2-(2-fluoroethoxy)ethoxy)ethoxy)benzofuran-2-yl)-N,N-dimethylpyridin-2-amine), was synthesized and evaluated. In experiments in vitro, FPYBF-1 displayed high affinity for Aβ(1–42) aggregates (Ki = 0.9 nM), and substantial labeling of β-amyloid plaques in sections of postmortem AD brains but not control brains. In experiments in vivo, [18F]FPYBF-1 displayed good initial uptake (5.16%ID/g at 2 min postinjection) and rapid washout from the brain (2.44%ID/g at 60 min postinjection) in normal mice, and excellent binding to β-amyloid plaques in a murine model of AD. Furthermore, the specific labeling of plaques labeling was observed in autoradiographs of autopsied AD brain sections. [18F]FPYBF-1 may be a useful probe for imaging β-amyloid plaques in living brain tissue.This Letter describes a novel 18F-labeled pyridyl benzofuran derivative ([18F]FPYBF-1) as a potential PET imaging probe for β-amyloid plaques in Alzheimer’s brains.
Co-reporter:Kazuki Aita;Takashi Temma;Yuji Kuge;Koh-ichi Seki
Luminescence 2010 Volume 25( Issue 1) pp:19-24
Publication Date(Web):
DOI:10.1002/bio.1136
Abstract
We have developed a new NIR fluorescent probe based on an ytterbium(III) (E)-1-(pyridin-2-yl-diazenyl)naphthalen-2-ol (PAN) complex. This probe emits near-infrared luminescence derived from the Yb ion through excitation of the PAN moiety with visible light (λex = 530 nm, λem = 975 nm). The results support the possible utility of the probe for in vivo fluorescence molecular imaging. Copyright © 2009 John Wiley & Sons, Ltd.
Co-reporter:Takashi Kudo;Yuji Kuge;Masashi Ueda;Takahiro Mukai;Shotaro Tanaka;Hiroaki Konishi;Azusa Miyano;Masahiro Ono;Shinae Kizaka-Kondoh;Masahiro Hiraoka;Hideo Saji
European Journal of Nuclear Medicine and Molecular Imaging 2010 Volume 37( Issue 8) pp:1566-1574
Publication Date(Web):2010/08/01
DOI:10.1007/s00259-010-1467-4
Hypoxia-inducible factor-1 (HIF-1) plays an important role in malignant tumour progression. For the imaging of HIF-1-active tumours, we previously developed a protein, POS, which is effectively delivered to and selectively stabilized in HIF-1-active cells, and a radioiodinated biotin derivative, (3-123I-iodobenzoyl)norbiotinamide (123I-IBB), which can bind to the streptavidin moiety of POS. In this study, we aimed to investigate the feasibility of the pretargeting method using POS and 123I-IBB for rapid imaging of HIF-1-active tumours.Tumour-implanted mice were pretargeted with POS. After 24 h, 125I-IBB was administered and subsequently, the biodistribution of radioactivity was investigated at several time points. In vivo planar imaging, comparison between 125I-IBB accumulation and HIF-1 transcriptional activity, and autoradiography were performed at 6 h after the administration of 125I-IBB. The same sections that were used in autoradiographic analysis were subjected to HIF-1α immunohistochemistry.125I-IBB accumulation was observed in tumours of mice pretargeted with POS (1.6%ID/g at 6 h). This result is comparable to the data derived from 125I-IBB-conjugated POS-treated mice (1.4%ID/g at 24 h). In vivo planar imaging provided clear tumour images. The tumoral accumulation of 125I-IBB significantly correlated with HIF-1-dependent luciferase bioluminescence (R=0.84, p<0.01). The intratumoral distribution of 125I-IBB was heterogeneous and was significantly correlated with HIF-1α-positive regions (R=0.58, p<0.0001).POS pretargeting with 123I-IBB is a useful technique in the rapid imaging and detection of HIF-1-active regions in tumours.
Co-reporter:Masahiro Ono, Shun Hayashi, Hiroyuki Kimura, Hidekazu Kawashima, Morio Nakayama, Hideo Saji
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 19) pp:7002-7007
Publication Date(Web):1 October 2009
DOI:10.1016/j.bmc.2009.08.032
We synthesized push–pull benzothiazole derivatives and evaluated their potential as β-amyloid imaging probes. In binding experiments in vitro, the benzothiazoles showed excellent affinity for synthetic Aβ(1-42) aggregates. β-Amyloid plaques in the mouse and human brain were clearly visualized with the benzothiazoles, reflecting the results in vitro. These compounds may be a useful scaffold for the development of novel PET/SPECT and fluorescent tracers for detecting β-amyloid in Alzheimer’s brains.
Co-reporter:Kazuma Ogawa;Takahiro Mukai;Keiichi Kawai;Norito Takamura;Hirofumi Hanaoka;Kazuyuki Hashimoto;Hideo Saji;Kazuhiro Shiba;Hirofumi Mori
European Journal of Nuclear Medicine and Molecular Imaging 2009 Volume 36( Issue 1) pp:115-121
Publication Date(Web):2009/01/01
DOI:10.1007/s00259-008-0925-8
We have developed a 186Re-mercaptoacetylglycylglycylglycine complex-conjugated bisphosphonate (186Re-MAG3-HBP) for the treatment of painful bone metastases. We assumed competitive inhibitors of protein binding to be useful for procuring a favorable biodistribution of 186Re-MAG3-HBP for the palliation of bone pain because it has been reported that the concurrent administration of 99mTc-MAG3 and drugs with high affinity for serum protein produced competitive displacement at specific binding sites and enhanced total clearance and tissue distribution.The displacement effects of several protein-binding inhibitors on the protein binding of 186Re-MAG3-HBP were investigated. Biodistribution experiments were performed by intravenously administering 186Re-MAG3-HBP into rats with ceftriaxone as a competitive protein-binding inhibitor or saline.The protein binding of 186Re-MAG3-HBP in rat serum, human serum, and a human serum albumin solution was significantly decreased by the addition of ceftriaxone, which has high affinity for binding site I on serum albumin. In the biodistribution experiments, pretreatment with ceftriaxone enhanced the clearance of the radioactivity of 186Re-MAG3-HBP in blood and nontarget tissues but had no effect on accumulation in bone.The findings suggested that the use of protein-binding competitive inhibitors would be effective in improving the pharmacokinetics of radiopharmaceuticals with high affinity for serum protein.
Co-reporter:Yasushi Kiyono;Tomoko Yamashita;Hisako Doi;Yuji Kuge;Toshiya Katsura;Ken-Ichi Inui;Hideo Saji
European Journal of Nuclear Medicine and Molecular Imaging 2007 Volume 34( Issue 4) pp:448-452
Publication Date(Web):2007/04/01
DOI:10.1007/s00259-006-0256-6
Radionuclide therapy with 131I-labelled meta-iodobenzylguanidine ([131I]MIBG) is effective in cases where it is difficult to carry out surgical resection or debulking of neuroendocrine tumours (NETs). However, it has recently been reported that P-glycoprotein (P-gp) is expressed in these NETs. Therefore, it is important to clarify whether MIBG is a substrate of P-gp or not. In this study, using a human cell line which overexpresses P-gp, LLC-GA5-COL150, we investigated this question.The transcellular transport and accumulation of [125I]MIBG were measured using monolayer cultures grown in Transwell chambers. [125I]MIBG was added to either the basolateral or the apical side, aliquots of the incubation medium on the other side were taken at specified times, and the radioactivity was measured. For accumulation experiments, the cells on the filters were solubilised and the radioactivity in aliquots was measured.There were no significant differences in the transport of MIBG between LLC-PK1 and LLC-GA5-COL150 monolayers in either direction until 60 min. With respect to the accumulation of MIBG, there were no significant differences between LLC-PK1 and LLC-GA5-COL150 cells in either direction.MIBG is not a substrate of P-gp. Therefore, radionuclide therapy with MIBG would be useful in the treatment of NETs expressing P-gp.
Co-reporter:Kazuki Aita;Yuji Kuge;Takashi Temma
Luminescence 2007 Volume 22(Issue 5) pp:455-461
Publication Date(Web):4 JUL 2007
DOI:10.1002/bio.984
We developed a novel fluorescent probe that contains the neodymium(III) complex moiety and fluorescein moiety. This probe can emit long-lived near-infrared luminescence derived from a Nd ion through excitation of the fluorescein moiety with visible light (λex = 488 nm, λem = 880 nm, lifetime = 2.3 µs). These results indicate the possibility of the probe as a candidate for in vivo fluorescence molecular imaging. Copyright © 2007 John Wiley & Sons, Ltd.
Co-reporter:Hiroshi Nishimura;Takahiro Mukai;Kazuyuki Hashimoto;Yasushi Arano;Hirofumi Hanaoka;Kazuma Ogawa
Journal of Labelled Compounds and Radiopharmaceuticals 2004 Volume 47(Issue 11) pp:753-761
Publication Date(Web):19 AUG 2004
DOI:10.1002/jlcr.864
To develop a radiopharmaceutical for the palliation of painful bone metastases based on the concept of bifunctional radiopharmaceuticals, we designed a bisphosphonate derivative attached to a stable 186Re-monoaminemonoamidedithiol (MAMA) chelate (186Re-MAMA-BP) to improve the instability of 186Re-HEDP. The precursor (Tr-MAMA-BP) of 186Re-MAMA-BP was synthesized by coupling the carboxyl group of the Tr-MAMA derivative with the amino group of the bisphosphonate derivative. This 186Re-labeled compound was prepared by a ligand exchange reaction using 186Re-glucoheptonate with a radiochemical yield of 32.0 ± 4.1%. In the incubation study in buffered solution (pH 7.0), 186Re-MAMA-BP was more stable than 186Re-HEDP. This suggests that 186Re-MAMA-BP is a potential radiopharmaceutical for the palliation of painful bone metastases. Copyright © 2004 John Wiley & Sons, Ltd.
Co-reporter:Akira Makino, Takahiro Arai, Masahiko Hirata, Masahiro Ono, ... Hideo Saji
Nuclear Medicine and Biology (January 2016) Volume 43(Issue 1) pp:101-107
Publication Date(Web):1 January 2016
DOI:10.1016/j.nucmedbio.2015.09.008
Phosphatidylinositol 3-kinase (PI3K) activity and protein expression levels are often increased in tumor regions. Since PI3K plays a crucial role in regulating cell growth and proliferation, inhibiting PI3K-dependent pathways could be a promising approach for cancer treatment. In clinical practice, however, evaluation of PI3K expression levels is limited to immunohistochemistry of patient samples, which requires invasive biopsies.Here we report the synthesis of three candidate compounds, FMTA-1, 2 and 3, and evaluate their capacity to detect PI3K expression levels with positron emission tomography (PET). Among the three candidates, FMTA-2 showed a lower IC50 value for PI3K. 18F Radiolabeling of FMTA-2 to produce [18F]FMTA-2 was accomplished and its capacity for detecting PI3K expression levels was evaluated in vitro and in vivo. Cell uptake of [18F]FMTA-2 correlated well with cellular PI3K expression levels, and was suppressed by the ATP-competitive PI3K inhibitor ZSTK474. In an in vivo experiment using tumor-transplanted model mice, a higher signal-to-noise ratio (S/N) was seen with [18F]FMTA-2 in animals transplanted with DMS114 cells (expressing high PI3K levels) relative to DU145 cells (expressing low PI3K levels). However, in vivo pharmacokinetics of [18F]FMTA-2 was undesirable and the absolute amount of this compound that accumulated at the tumor region was low. To the best of our knowledge, this study represents the first trial of a PET tracer for detecting PI3K. Although further improvement of the probe is required prior to clinical application, these results should encourage future work.
Co-reporter:Takashi Temma, Kantaro Nishigori, Satoru Onoe, Sotaro Sampei, ... Hideo Saji
Nuclear Medicine and Biology (February 2015) Volume 42(Issue 2) pp:184-191
Publication Date(Web):1 February 2015
DOI:10.1016/j.nucmedbio.2014.10.006
IntroductionCancer-associated adipocytes metabolically interact with adjacent cancer cells to promote tumor proliferation and metastasis. Fatty acid binding protein 4 (FABP4) participates in this interaction, and is gathering attention as a therapeutic and diagnostic target. Positron emission tomography (PET) is a useful diagnostic method that enables noninvasive in vivo quantitative imaging of biofunctional molecules with probes labeled with positron-emitting radioisotopes. Here a novel 18F labeled probe for PET FABP4 imaging developed through dedicated drug design from a radioiodinated probe we recently reported is evaluated in vitro and in vivo.MethodsWe designed the [18F]-labeled FTAP1 and FTAP3 probe, composed of a single or triple oxyethylene linker and a triazolopyrimidine scaffold derived from an FABP4 inhibitor. FABP4 binding affinities for chemically synthesized FTAP1 and FTAP3 were measured using FABP4 and 8-anilino-1-naphthalene sulfonic acid. Cell membrane permeability was measured using a commercially available plate assay system. After radiosynthesis, [18F]FTAP1 affinity and selectivity were evaluated using immobilized FABP3, FABP4, and FABP5. Cell uptake was investigated using differentiated adipocytes expressing FABP4 with inhibitor treatment. Following biodistribution studies in C6 glioblastoma-bearing mice, ex vivo autoradiography and immunohistochemistry were performed using thin sliced tumor sections. PET/CT imaging was then performed on C6 tumor bearing mice.ResultsFTAP1 showed high FABP4 affinity (Ki = 68 ± 8.9 nM) and adequate cell permeability. [18F]FTAP1 with ≥ 98% radiochemical purity was shown to selectively bind to FABP4 (16.3- and 9.3-fold higher than for FABP3 and FABP5, respectively). [18F]FTAP1 was taken up by FABP4 expressing cells, and this uptake could be blocked by an inhibitor, indicating very low non-specific cell binding. [18F]FTAP1 showed high tumor accumulation, which demonstrates its potential use for in vivo tumor PET imaging, and the intratumoral radioactivity distribution corresponded to the FABP4 expression profile.Conclusion[18F]FTAP1 is a promising PET probe to target FABP4.
Co-reporter:Takashi Temma, Yuji Kuge, Kohei Sano, Junko Kamihashi, Naoyuki Obokata, Hidekazu Kawashima, Yasuhiro Magata, Hideo Saji
Brain Research (30 May 2008) Volume 1212() pp:18-24
Publication Date(Web):30 May 2008
DOI:10.1016/j.brainres.2008.03.033
Hypertension is a major stroke risk factor and is correlated with worse outcome after stroke. Thus, the effects of hypertension on cerebral hemodynamics and metabolism within an hour after stroke must be evaluated in detail. Cerebral blood flow (CBF), oxygen extraction fraction (OEF), cerebral metabolic rate for oxygen (CMRO2) and cerebral metabolic rate for glucose (CMRglc) were measured 1 h after the occlusion of the right middle cerebral artery (MCA) in male spontaneously hypertensive rats (SHR) and male normotensive Wistar Kyoto rats (WKY). Physiological responses were determined by positron emission tomography (PET) using 15O-H2O and radiolabeled 15O-O2 blood (methodology previously developed in this laboratory) and by autoradiography (ARG) using 18F-FDG. The right hemisphere of SHR showed lower CBF values than the left hemisphere after stroke (right: 0.17 ± 0.07 mL/min/g; left: 0.29 ± 0.08 mL/min/g), CMRO2 (right: 2.55 ± 0.80 mL/min/100 g; left: 4.11 ± 0.84 mL/min/100 g) and CMRglc (right: 52.4 ± 16.2 mg/min/100 g; left: 65.6 ± 10.2 mg/min/100 g). WKY rats exhibited significant decreases only in CBF and CMRO2. These results suggest greater underlying physiologic disturbances in SHR. Also, the occlusion significantly reduced CBF in both hemispheres of SHR compared with WKY, suggesting a disturbance of the autoregulatory mechanism in SHR. In summary, our results indicate that hypertension intensifies metabolic disturbances after the onset of stroke, at least in the first hour. Therefore, we suggest that hypertension not only increases the incidence of stroke but also exacerbates stroke-mediated damage.
Co-reporter:Hiroyuki Kimura, Hirokazu Matsuda, Yu Ogawa, Hiroyuki Fujimoto, Kentaro Toyoda, Naotaka Fujita, Kenji Arimitsu, Keita Hamamatsu, Yusuke Yagi, Masahiro Ono, Nobuya Inagaki, Hideo Saji
Bioorganic & Medicinal Chemistry (15 February 2017) Volume 25(Issue 4) pp:
Publication Date(Web):15 February 2017
DOI:10.1016/j.bmc.2016.12.051
Insulinoma is a tumor derived from pancreatic β-cells, and the resulting hyperinsulinemia leads to characteristic hypoglycemia. Recent studies have reported the frequent overexpression of glucagon-like peptide-1 receptor (GLP-1R) in human insulinomas, suggesting that the binding of a radiolabeled compound to GLP-1R is useful for the imaging of such tumors. Exendin(9-39), a fragment peptide of exendin-3 and -4, binds GLP-1R with high affinity and acts as an antagonist. Accordingly, radiolabeled exendin(9-39) derivatives have also been investigated as insulinoma imaging probes that might be less likely to induce hypoglycemia. In this study, we synthesized a novel indium-111 (111In)-benzyl-diethylenetriaminepentaacetic acid (111In-BnDTPA)-conjugated exendin(9-39), 111In-BnDTPA-exendin(9-39), and evaluated its utility as a probe for the SPECT imaging of insulinoma. natIn-BnDTPA-exendin(9-39) exhibited a high affinity for GLP-1R (IC50 = 2.5 nM), stability in plasma, and a specific activity that improved following reactions with a solvent and solubilizer. Regarding the in vivo biodistribution of 111In-BnDTPA-exendin(9-39) in INS-1 tumor-bearing mice, high uptake levels were observed in tumors (14.6% ID/g at 15 min), with corresponding high tumor-to-blood (T/B), tumor-to-muscle (T/M), and tumor-to-pancreas (T/P) ratios (T/B = 2.55, T/M = 22.7, T/P = 2.7 at 1 h). The pre-administration of excess nonradioactive exendin(9-39) significantly reduced accumulation in both the tumor and pancreas (76% and 68% inhibition, respectively) at 1 h after 111In-BnDTPA-exendin(9-39) injection, indicating that the GLP-1R mediated a majority of 111In-BnDTPA-exendin(9-39) uptake in the tumor and pancreas. Finally, 111In-BnDTPA-exendin(9-39) SPECT/CT studies in mice yielded clear images of tumors at 30 min post-injection. These results suggest that 111In-BnDTPA-exendin(9-39) could be a useful SPECT molecular imaging probe for the detection and exact localization of insulinomas.
Co-reporter:Yusuke Yagi, Hiroyuki Kimura, Kenji Arimitsu, Masahiro Ono, Kazuya Maeda, Hiroyuki Kusuhara, Tetsuya Kajimoto, Yuichi Sugiyama and Hideo Saji
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 4) pp:NaN1121-1121
Publication Date(Web):2014/11/13
DOI:10.1039/C4OB01953A
Fluorine-18 labeled radiotracers, such as [18F]fluorodeoxyglucose, can be used as practical diagnostic agents in positron emission tomography (PET). Furthermore, the properties of pharmaceuticals can be enhanced significantly by the introduction of fluorine groups into their original structures, and significant progress has been made during the last three decades towards the development of practical procedures for the introduction of fluorine. The replacement of the fluorine atoms present in pharmaceuticals with radioactive 18F atoms is a rational approach for designing novel PET tracers. As a fluorine-containing pharmaceutical agent, pitavastatin has attracted considerable interest from researchers working in the life sciences because it can act as an antihyperlipidemic agent as well as a substrate for hepatic organic anion transporting polypeptides (hOATP). With this in mind, it was envisaged that [18F]pitavastatin would be used as an excellent imaging agent for hOATP, which prompted us to investigate the synthesis of this agent. Herein, we report a practical method for the synthesis of [18F]pitavastatin by the Suzuki coupling reaction of p-iodofluorobenzene and a quinoline boronate derivative, with the desired product being formed in a radiochemical yield of 12 ± 3% (decay corrected from [18F]fluoride ions, n = 3).













![2-bromobenzo[d]thiazole-6-carbonitrile](http://img.cochemist.com/ccimg/864300/864265-77-0.png)
![2-bromobenzo[d]thiazole-6-carbonitrile](http://img.cochemist.com/ccimg/864300/864265-77-0_b.png)


![6-Benzothiazolecarbonitrile, 2-[4-(dimethylamino)phenyl]-](/data/chemimg/1853900/566170-01-2.png)
![6-Benzothiazolecarbonitrile, 2-[4-(dimethylamino)phenyl]-](/data/chemimg/1853900/566170-01-2_b.png)




![Benzoxazole, 6-bromo-2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/537100/537029-52-0.png)
![Benzoxazole, 6-bromo-2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/537100/537029-52-0_b.png)

















![Propanedinitrile, [[5-(dimethylamino)-2-thienyl]methylene]-](http://img.cochemist.com/ccimg/160900/160893-15-2.png)
![Propanedinitrile, [[5-(dimethylamino)-2-thienyl]methylene]-](http://img.cochemist.com/ccimg/160900/160893-15-2_b.png)









![Propanedinitrile, [(2E)-3-[4-(dimethylamino)phenyl]-2-propenylidene]-](http://img.cochemist.com/ccimg/129200/129179-87-9.png)
![Propanedinitrile, [(2E)-3-[4-(dimethylamino)phenyl]-2-propenylidene]-](http://img.cochemist.com/ccimg/129200/129179-87-9_b.png)














![6H-Benzofuro[3a,3,2-ef][2]benzazepin-6-ol,4a,5,9,10,11,12-hexahydro-3-methoxy-, (4aS,6R,8aS)-](http://img.cochemist.com/ccimg/41400/41303-74-6.png)
![6H-Benzofuro[3a,3,2-ef][2]benzazepin-6-ol,4a,5,9,10,11,12-hexahydro-3-methoxy-, (4aS,6R,8aS)-](http://img.cochemist.com/ccimg/41400/41303-74-6_b.png)




![(+/-)-(4aS,6R,8aS)-4a,5,9,10,11,12-hexahydro-3-methoxy-11-methyl-6H-benzofuro[3a,3,2-ef][2]benzazepin-6-ol](http://img.cochemist.com/ccimg/23200/23173-12-8.png)
![(+/-)-(4aS,6R,8aS)-4a,5,9,10,11,12-hexahydro-3-methoxy-11-methyl-6H-benzofuro[3a,3,2-ef][2]benzazepin-6-ol](http://img.cochemist.com/ccimg/23200/23173-12-8_b.png)
![2,4-Pentadienal, 5-[4-(dimethylamino)phenyl]-, (2E,4E)-](http://img.cochemist.com/ccimg/20500/20432-36-4.png)
![2,4-Pentadienal, 5-[4-(dimethylamino)phenyl]-, (2E,4E)-](http://img.cochemist.com/ccimg/20500/20432-36-4_b.png)
![2-Propenal, 3-[4-(dimethylamino)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/20500/20432-35-3.png)
![2-Propenal, 3-[4-(dimethylamino)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/20500/20432-35-3_b.png)





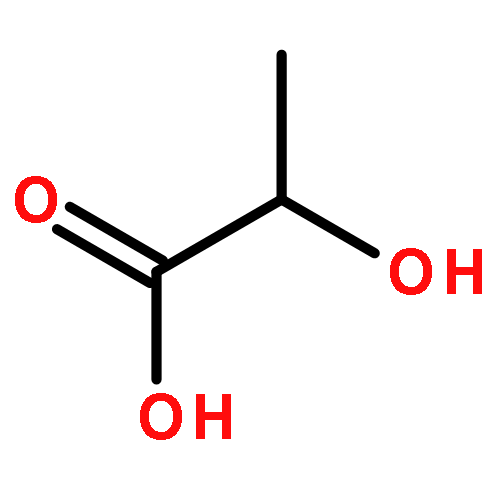
![2-Propenoic acid, 3-[4-(dimethylamino)phenyl]-, (2E)-](/data/chemimg/1851700/5676-65-3.png)
![2-Propenoic acid, 3-[4-(dimethylamino)phenyl]-, (2E)-](/data/chemimg/1851700/5676-65-3_b.png)
![Propanedinitrile,2-[[4-(dimethylamino)phenyl]methylene]-](http://img.cochemist.com/ccimg/2900/2826-28-0.png)
![Propanedinitrile,2-[[4-(dimethylamino)phenyl]methylene]-](http://img.cochemist.com/ccimg/2900/2826-28-0_b.png)


![[(3-chlorophenyl)hydrazono]malononitrile](http://img.cochemist.com/ccimg/600/555-60-2.png)
![[(3-chlorophenyl)hydrazono]malononitrile](http://img.cochemist.com/ccimg/600/555-60-2_b.png)