Co-reporter:Henri Brunner, Hikaru Kitamura, and Takashi Tsuno
Organometallics July 10, 2017 Volume 36(Issue 13) pp:2424-2424
Publication Date(Web):June 22, 2017
DOI:10.1021/acs.organomet.7b00311
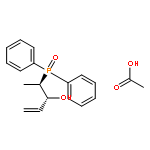
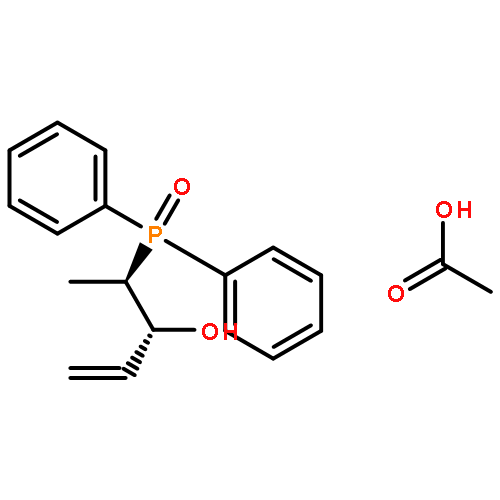
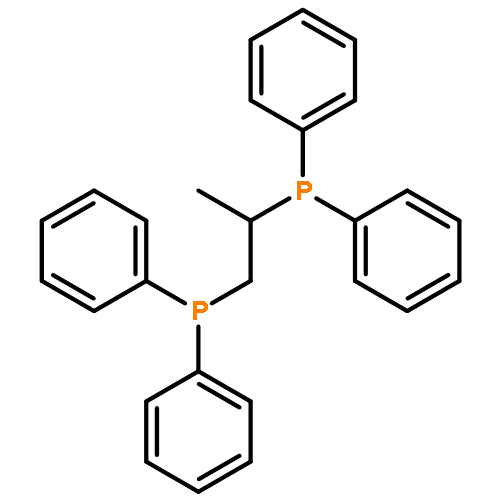
The complexes [CpFe{Ph2P(CH2)nPPh2}NCMe]PF6, [CpFe(PPh2Me)2NCMe]PF6, and [CpFe{Ph2P(CH2)nPPh2}PPh2(OR)]PF6, R = Me, Et, and iPr, with chelate ring sizes between 4 and 7 were synthesized and characterized by spectroscopy and X-ray analysis. In these complexes, the monodentate ligands acetonitrile and PPh2(OR) tend to dissociate. The kinetics of the ligand exchanges MeCN/P(OMe)3 and PPh2(OR)/P(OMe)3 was measured. In the acetonitrile series, the first-order reaction of the five-membered chelate complex [CpFe(dppe)NCMe]PF6 had a half-life of 549 min in CDCl3 at 293 K. The other chelate complexes [CpFe(P-P)NCMe]PF6 and the nonchelate analogue [CpFe(PPh2Me)2NCMe]PF6 reacted faster by factors of 20–50. The PPh2(OR) series revealed a dramatic difference between the complexes [CpFe(P-P)PPh2(OR)]PF6 with five- and six-membered chelate rings. The PPh2(OR)/P(OMe)3 exchange in the dppp complex (six-membered chelate ring) was 500 times faster than in the dppe complex (five-membered chelate ring). This is due to the increase of the P–Fe–P angle in the dppp chelate ring, which diminishes the binding pocket of the PPh2(OR) ligand. In the nonchelate complex [CpFe(PPh2Me)2NCMe]PF6, a novel and unexpected bimolecular PPh2Me/PPh2(OMe) substitution was observed.
Co-reporter:Henri Brunner;Takashi Tsuno
Dalton Transactions 2017 vol. 46(Issue 15) pp:5103-5109
Publication Date(Web):2017/04/10
DOI:10.1039/C7DT00474E
In half-sandwich compounds of the type [Cp*ML1L2PPh3] the PPh3 propeller is stabilized by attractive CH/π interactions in which Co–H bonds specifically interact with the Ci and Co atoms of neighbouring phenyl rings, as in the T-shaped benzene dimer (i/o = ipso/ortho). This stabilization was not taken into account in a recent conformational analysis based on van der Waals energy calculations and minimization of steric compression (Dalton Trans., 2015, 44, 5451–5466). It is shown that in all 116 structures discussed in this analysis the CoH–Ci/o distances fall below the sum of the van der Waals radii, establishing attractive CH/π interactions, although the short contacts could easily be avoided by phenyl rotation to relieve steric strain. In 53 of the described structures there are acyl substituents which form conformation-determining Co–H⋯O(acyl) hydrogen bonds that are not taken into account in the recent analysis. The steric-only model is not an adequate description of M–PPh3 complexes.
Co-reporter:Henri Brunner, Takashi Tsuno
Inorganica Chimica Acta 2016 Volume 446() pp:132-142
Publication Date(Web):1 May 2016
DOI:10.1016/j.ica.2016.02.039
•The architecture of the triphenylphosphine propeller in transition metal complexes is controlled by weak CH/π interactions comparable to the archetypal T-shaped benzene dimer and not by steric-only interactions as claimed in the literature.•ortho-CH bonds bind to ipso- and ortho-carbon atoms of adjacent phenyl rings.•In the trans-[MCl2(PPh3)2] complexes it is the Cl/π interaction which determines the orientation of the PPh3 propeller within the molecule.The architecture of the triphenylphosphine propeller in Cr-PPh3 complexes and in the compounds trans-[MCl2(PPh3)2], M = Ni, Pd, and Pt, is analyzed on the basis of a CSD search. The three phenyl rings interact with each other by formation of weak CH/π bonds comparable to the archetypal T-shaped benzene dimer. ortho-CH bonds from inside the propeller bind to ipso- and ortho-carbon atoms of adjacent phenyl rings. In the broad energy minimum A/B there is a discontinuity in the transition from A to B. Binding switches from inside to outside ortho-carbon atoms. ortho-CH bonds from outside the propeller establish similar weak bonds with unsaturated ligands (and the metal atom) which control the arrangement of the PPh3 propeller within the molecule. In the trans-[MCl2(PPh3)2] complexes it is the Cl/π interaction which determines the orientation of the PPh3 propeller in the molecule.The architecture of the triphenylphosphine propeller in Cr-PPh3 complexes and trans-[MCl2(PPh3)2], M = Ni, Pd, and Pt, compounds is analyzed on the basis of a CSD search. The three phenyl rings interact with each other by formation of weak CH/π bonds. ortho-CH bonds bind to ipso- and ortho-carbon atoms of adjacent phenyl rings. In the trans-[MCl2(PPh3)2] complexes it is the Cl/π interaction which determines the orientation of the PPh3 propeller within the molecule.
Co-reporter:Henri Brunner and Takashi Tsuno
Organometallics 2015 Volume 34(Issue 7) pp:1287-1293
Publication Date(Web):March 18, 2015
DOI:10.1021/acs.organomet.5b00022
In the solid state and also in solution CpM–L–E–Ph compounds adopt preferential conformations, in which C–H bonds of the Cp ligand bind to the π system of the phenyl substituent. This holds for CpMo(CO)2–amidinato and −thioamidato as well as for CpM–P(OPh)3 complexes. In the solid state this is demonstrated by short contacts CH–Csp2, retrieved in a CSD search, comparable to those in the archetypal T-shaped benzene dimer. In solution the CH/π stabilization shows up in a typical high-field shift of the CpH signal, due to the anisotropy beam of the phenyl substituent, and in the thermodynamic stability of major and minor diastereomers in diastereomer equilibria. A special motif is the unsymmetrical bonding of two CpH bonds to the phenyl substituent, reminiscent of the new tilted T-shaped benzene dimer.
Co-reporter:Henri Brunner, Takashi Tsuno, and Michael Bodensteiner
Organometallics 2014 Volume 33(Issue 9) pp:2257-2265
Publication Date(Web):April 23, 2014
DOI:10.1021/om500175u
The Cambridge Structural Database comprises 89 structures with M–Prosphos chelate rings. On this basis a compilation analysis of the conformation of the M–Prophos chelate ring has been carried out. Typical conformational features, such as the puckering of the chelate ring and the arrangement of the phenyl rings, important for the transmission of the chiral information in enantioselective catalysts, are attributed to CH/π interactions within the M–Prophos system, although intramolecular interactions, in particular with Cp and Cp* ligands, also play a role. Distances below the van der Waals radii indicate weak bonding, provided the overlap angles are large enough. Normally, intermolecular interactions and packing effects are not structure-determining. Otherwise, recurrent motifs would not be as dominant, as established in the present analysis. The most important contribution comes from the interaction of the methyl group at the asymmetric center in the equatorial position of the five-membered chelate ring with the axial phenyl ring of the adjacent PPh2 group. The distances from the methyl group to the ipso and ortho carbon atoms of the axial phenyl ring are much shorter than those to Ci and Co of the equatorial phenyl ring. In addition, the overlap angles are better. In an active process the methyl group attracts one of the two sides of the phenyl rings to establish an effective CH/π interaction with one of the ortho carbon atoms. Within the PPh2 groups the interactions of the o-CH bond of one phenyl ring with the ipso and ortho carbon atoms of the other phenyl ring vary from strong to weak. Weak attractions are also observed between the hydrogen atoms of the CHMeCH2 backbone and the neighboring phenyl rings. The fact that the methyl group at the asymmetric center overwhelmingly occupies the equatorial position in the chelate ring is attributed to the missing phenyl–phenyl stabilization, when the methyl group switches into the exceptional axial position. Decreasing CH/π intensity, based on distance and overlap angle, is visualized by broad, normal, thin, and dashed arrows.
Co-reporter:Henri Brunner, Takashi Tsuno, Gábor Balázs, and Michael Bodensteiner
The Journal of Organic Chemistry 2014 Volume 79(Issue 23) pp:11454-11462
Publication Date(Web):November 5, 2014
DOI:10.1021/jo5020454
In 1,2-Me,Ph substitution patterns of organic compounds the methyl group attracts one of the phenyl sides to establish a CH/π bond with one of the ortho carbon atoms (the Co side), leading to a characteristic tilting of the phenyl ring around its Ci–Cp axis. This phenyl rotation shortens the CMe–Co distances to bonding contacts between the methyl hydrogen atoms and the ortho carbon atom Co well below the van der Waals distance of 3.70 Å. On the other hand, it elongates the CMe–Co′ distances outside of the reach of any CH/π interaction (>3.70 Å). Our study is based on a search in the Cambridge Structural Database for substructures Me–C═C–Ph, Me–C–C–Ph, and Me–C–N–Ph with 1,2-Me,Ph substitution patterns. In the 1,2-Me,Ph substitution motif the torsion angle CMe–C–C–Ci determines the length of the CMe–Ci and CMe–Co distances. For aromatic compounds these torsion angles are close to 0°, but in five- and six-membered ring compounds and in open-chain compounds the torsion angles vary considerably. Universally, for torsion angles up to 80° CH/π bonds were found, whereas the long CMe–Ci and CMe–Co distances for torsion angles >80° do not allow a CH/π interaction. The results of the present CSD analysis are supported by calculations.
Co-reporter:Henri Brunner, Takaki Kurosawa, Manfred Muschiol, Takashi Tsuno, Gábor Balázs, and Michael Bodensteiner
Organometallics 2013 Volume 32(Issue 17) pp:4904-4911
Publication Date(Web):August 21, 2013
DOI:10.1021/om400631m
The compounds (RFe,RC)-/(SFe,RC)-[CpFe(Prophos)PPh(OMe)2]PF6 and (RFe,RC)-/(SFe,RC)-[CpFe(Prophos)PPh2(OR)]PF6 (R = Me, Et, iPr, tBu) were synthesized, starting from (RFe,RC)-/(SFe,RC)-[CpFe(Prophos)NCMe]PF6, and characterized, including X-ray analyses. Bubbling a stream of N2 through the solution speeded up the slow substitution reactions by removing the acetonitrile formed in the rate-determining cleavage of the Fe–NCMe bond. Due to their acceptor ligands P(OMe)3 and PPh(OMe)2 the complexes [CpFe(Prophos)P(OMe)3]PF6 and [CpFe(Prophos)PPh(OMe)2]PF6 are configurationally stable at the metal atom. In contrast, [CpFe(Prophos)PPh3]PF6 does not form, due to the large cone angle of the ligand PPh3. Ideally, the electronic and steric effects of the ligands PPh2(OR) (R = Me, Et, iPr, tBu) are such that they tend to dissociate from the congested complexes (RFe,RC)- and (SFe,RC)-[CpFe(Prophos)PPh2(OR)]PF6. In the series (RFe,RC)- and (SFe,RC)-[CpFe(Prophos)PPh2(OR)]PF6, (R = Me, Et, iPr, tBu) electron donation and the cone angle of the ligands increase. Thus, the rates of the ligand exchange with P(OMe)3 increased, initiated by the slow dissociation of the Fe–PPh2(OR) bond. For the RFe,RC/SFe,RC diastereomers of [CpFe(Prophos)PPh2(OR)]PF6 the half-lives of the first-order reactions were 125/350 h (R = Me), 75/275 h (R = Et), and 12/34 h (R = iPr) in CDCl3 at 60 °C.
Co-reporter:Henri Brunner;Michael Bodensteiner;Takashi Tsuno
Chirality 2013 Volume 25( Issue 10) pp:663-667
Publication Date(Web):
DOI:10.1002/chir.22199
ABSTRACT
Salicylidenimine palladium(II) complexes trans-Pd(O,N)2 adopt step and bowl arrangements. A stereochemical analysis subdivides 52 compounds into 41 step and 11 bowl types. Step complexes with chiral N-substituents and all the bowl complexes induce chiral distortions in the square planar system, resulting in Δ/Λ configuration of the Pd(O,N)2 unit. In complexes 1, 2, 3, 4, 5, 6 with enantiomerically pure N-substituents ligand chirality entails a specific square chirality and only one diastereomer assembles in the lattice. Dimeric Pd(O,N)2 complexes with bridging N-substituents in trans-arrangement are inherently chiral. For dimers 7, 8, 9, 10, 11 different chirality patterns for the Pd(O,N)2 square are observed. The crystals contain racemates of enantiomers. In complex 12 two independent molecules form a tight pair. The (RC) configuration of the ligand induces the same Δ chirality in the Pd(O,N)2 units of both molecules with varying square chirality due to the different crystallographic location of the independent molecules. In complexes 13 and 14 atrop isomerism induces specific configurations in the Pd(O,N)2 bowl systems. The square chirality is largest for complex 15 [(Diop)Rh(PPh3)Cl)], a catalyst for enantioselective hydrogenation. In the lattice of 15 two diastereomers with the same (RC,RC) configuration in the ligand Diop but opposite Δ and Λ square configurations co-crystallize, a rare phenomenon in stereochemistry. Chirality 25:663–667, 2013. © 2013 Wiley Periodicals, Inc.
Co-reporter:Henri Brunner, Takaki Kurosawa, Manfred Muschiol, Takashi Tsuno, and Hayato Ike
Organometallics 2012 Volume 31(Issue 8) pp:3395-3401
Publication Date(Web):April 6, 2012
DOI:10.1021/om3001789
The compounds (RFe,RC)-/(SFe,RC)-[CpFe(Prophos)NCMe]X (X = I, PF6), configurationally labile at the metal center, were used in the MeCN/ligand exchange reactions with cyclohexyl isocyanide (CyNC) and tert-butyl isocyanide (tBuNC). Kinetic measurements showed that the MeCN/CyNC exchange in diastereomerically pure (SFe,RC)-[CpFe(Prophos)NCMe]X proceeded via the slow SN1-type dissociation of the Fe–NCMe bond, already observed in the MeCN/phosphite exchange reactions. The product (RFe,RC)-/(SFe,RC)-[CpFe(Prophos)CNCy]X (X = I, PF6) was formed in diastereomer ratios between 40:60 and 60:40. However, specific for the MeCN/CyNC exchange in (SFe,RC)-[CpFe(Prophos)NCMe]PF6, in some of the samples a fast initial reaction interfered, initiated by traces of oxygen, which oxidized the cation in (SFe,RC)-[CpFe(Prophos)NCMe]PF6 to (SFe,RC)-[CpFe(Prophos)NCMe]2+. This dipositive cation started an electrocatalytic chain reaction, producing (RFe,RC)-/(SFe,RC)-[CpFe(Prophos)CNCy]PF6 with a high stereoselectivity of 2:98 in favor of (SFe,RC)-[CpFe(Prophos)CNCy]PF6. Deactivation processes terminated the chain reaction, depending on the varying amounts of (SFe,RC)-[CpFe(Prophos)NCMe]2+ present in the system. Larger amounts of oxygen or oxidants, such as I2 and AgPF6, caused immediate complete conversion to (RFe,RC)/(SFe,RC)-[CpFe(Prophos)CNR]PF6 in a diastereomer ratio of 2:98. In contrast to the hexafluorophosphate salt, addition of a crystal of iodine did not initiate the chain reaction in the iodide salt [CpFe(Prophos)NCMe]I, because I2 added to I– to form I3–, which did not oxidize the cation of [CpFe(Prophos)NCMe]I. Instead, there was slow conversion according to the dissociative pathway. The correlation between the configuration of (RFe,RC)- and (SFe,RC)-[CpFe(Prophos)CNCy]X and the conformation of the Fe-Prophos chelate ring on the one hand and the correlation with the P–P coupling constants of the Prophos ligand on the other hand was corroborated.
Co-reporter: Henri Brunner;Manfred Muschiol; Takashi Tsuno;Hyato Ike;Takaki Kurosawa;Kazuhito Koyama
Angewandte Chemie 2012 Volume 124( Issue 4) pp:1092-1095
Publication Date(Web):
DOI:10.1002/ange.201104960
Co-reporter: Henri Brunner;Manfred Muschiol; Takashi Tsuno;Hyato Ike;Takaki Kurosawa;Kazuhito Koyama
Angewandte Chemie International Edition 2012 Volume 51( Issue 4) pp:1067-1070
Publication Date(Web):
DOI:10.1002/anie.201104960
Co-reporter:Henri Brunner, Hayato Ike, Manfred Muschiol, Takashi Tsuno, Naohisa Umegaki, and Manfred Zabel
Organometallics 2011 Volume 30(Issue 3) pp:414-421
Publication Date(Web):January 20, 2011
DOI:10.1021/om100276t
The compounds [CpFe(Prophos)Cl] and [CpFe(Prophos)I] were prepared in photochemical reactions of [CpFe(CO)2Cl] and [CpFe(CO)2I] with (R)-Prophos. They consist of pairs of RFe,RC and SFe,RC diastereomers which only differ in the configuration at the metal atom. The diastereomerically pure compounds (SFe,RC)-[CpFe(Prophos)Cl] and (RFe,RC)-[CpFe(Prophos)I], which have the same relative configurations, were isolated. They epimerize via change of the Fe configuration and approach the equilibria (RFe,RC)-/(SFe,RC)-[CpFe(Prophos)Cl] = 5/95 and (RFe,RC)-/(SFe,RC)-[CpFe(Prophos)I] = 95/5 in first-order reactions with half-lives of 43 min at 20 °C and 50 min at 50 °C in C6D6, respectively. The reaction of (RFe,RC)-/(SFe,RC)-[CpFe(Prophos)I] = 95/5 with KCN afforded the cyano complex [CpFe(Prophos)CN] in the diastereomer ratio RFe,RC/SFe,RC = 50/50. Both diastereomers (RFe,RC)- and (SFe,RC)-[CpFe(Prophos)CN] could be isolated diastereomerically pure. The compounds (RFe,RC)- and (SFe,RC)-[CpFe(Prophos)CN] are configurationally stable at the metal center. There is no diastereomer interconversion, not even at higher temperatures. The carbonyl complexes [CpFe(Prophos)CO]I, [CpFe(Prophos)CO]PF6, and [IndFe(Prophos)CO]I were prepared in thermal reactions of [CpFe(CO)2I] and [IndFe(CO2)I] with (R)-Prophos or in an autoclave reaction of [CpFe(Prophos)I]/NH4PF6 with CO under pressure. All the carbonyl complexes are configurationally stable at the metal center. Seven diastereomers were characterized by X-ray crystallography. Including the two diastereomers (RFe,RC)-[CpRu(Prophos)Br] and (RFe,RC)-[CpRu(Prophos)I], a conformational analysis of the M-Prophos chelate ring was carried out, resulting in characteristic differences between major and minor diastereomers.
Co-reporter:Henri Brunner, Hayato Ike, Manfred Muschiol, Takashi Tsuno, Kazuhiro Koyama, Takaki Kurosawa, and Manfred Zabel
Organometallics 2011 Volume 30(Issue 13) pp:3666-3676
Publication Date(Web):June 14, 2011
DOI:10.1021/om2003893
The diastereomers (RFe,RC)/(SFe,RC)-[CpFe(Prophos)NCMe]X (X = I, PF6), 5:95, differing only in the metal configuration, were prepared from (RFe,RC)/(SFe,RC)-[CpFe(Prophos)I] (95:5) in acetonitrile in the absence or presence of NH4PF6. The diastereomers interconverted by change of the Fe configuration in first-order reactions in CDCl3 at 293 K with half-lives of 216 min (iodide) and 96 min (hexafluorophosphate) in an SN1-type dissociation of the MeCN ligand, followed by pyramidal inversion of the 16-electron intermediates (RFe,RC)- and (SFe,RC)-[CpFe(Prophos)]+ and recombination with MeCN. In the presence of phosphite ligands there was MeCN/ligand exchange, the kinetics of which was measured. The rates of the MeCN/phosphite exchange decreased with increasing cone angle for P(OCH2)3CMe (101°), P(OMe)3 (107°), and P(OPh)3 (128°). PPh3 (145°) did not enter the vacant coordination site in the intermediates (RFe,RC)- and (SFe,RC)-[CpFe(Prophos)]+. Phosphines such as Ph2PCH2CH2PPh2 and PBu3 bound only loosely to the intermediates. The phosphite complexes (RFe,RC)/(SFe,RC)-[CpFe(Prophos)P(OR)3]X were configurationally stable at the metal atom. In the MeCN/phosphite exchange reactions the diastereomeric ratio of the products was constant, explained by an equilibrium between the intermediates in an energy diagram in which the barrier for the unimolecular pyramidal inversion was lower than the barriers for the bimolecular reactions of the intermediates with the phosphite ligands. A correlation between the major diastereomers (SFe,RC)-[CpFe(Prophos)NCMe]X (X = I, PF6) and the diastereomers of the phosphite complexes with the same relative configuration and the favored envelope conformation of the Fe-Prophos chelate ring was corroborated, and a new correlation with the P–P coupling constants of the Prophos ligand was established, including all the compounds of a former study.
Co-reporter:Henri Brunner, Manfred Muschiol, Takashi Tsuno, Takemoto Takahashi and Manfred Zabel
Organometallics 2010 Volume 29(Issue 2) pp:428-435
Publication Date(Web):December 29, 2009
DOI:10.1021/om900830a
This paper reports the synthesis, isomer separation, and X-ray characterization of the compounds (SRu,SC)-/(RRu,SC)-[CpRu(Chairphos)Cl], Chairphos = (S)-1,3-bis(diphenylphosphanyl)butane, and cis-/trans-[CpRu(Dppm-Me)Cl], Dppm-Me = 1,1-bis(diphenylphosphanyl)ethane. The Cl/I exchange reactions proceeded with predominant retention of the metal configuration, accompanied by some inversion, except for trans-[CpRu(Dppm-Me)Cl], which was stereospecifically converted to trans-[CpRu(Dppm-Me)I]. Temperature-dependent kinetic measurements afforded rates and activation parameters of the Cl/I exchange and epimerization reactions that follow basilica-type energy profiles. Dissociation of Cl− from [CpRu(Chairphos)Cl] and [CpRu(Dppm-Me)Cl] gives pyramidal intermediates [CpRu(Chairphos)]+ and [CpRu(Dppm-Me)]+, which maintain the metal configuration. The 16-electron intermediates can react with excess I− to form the iodo complexes with retention of the metal configuration, or they can change the metal configuration by pyramidal inversion, leading to formation of iodo complexes with inverted metal configuration. The kinetic measurements show that the pyramidal inversion via planar transition states depends on the P−Ru−P′ angles. It increases with decreasing chelate ring size, because small P−Ru−P′ angles resist planarization in the transition, which requires larger P−Ru−P′ angles.
Co-reporter:Henri Brunner and Takashi Tsuno
Accounts of Chemical Research 2009 Volume 42(Issue 10) pp:1501
Publication Date(Web):July 15, 2009
DOI:10.1021/ar900029t
In organometallic chemistry, ligand dissociation is a key intermediate step in many useful processes. The dissociation of halide from an 18-electron half-sandwich complex (that is, with a single cyclopentadienyl ligand) of the type [CpRu(P−P′)Hal] leaves an unsaturated 16-electron intermediate [CpRu(P−P′)]+, which is then ready for subsequent addition reactions. Does the intermediate maintain its structure with a vacant site in place of the dissociated ligand, or does it rearrange, either concurrent with its formation or subsequently? In other words, are the 16-electron species planar or pyramidal? The outcome is relevant for chiral-at-metal compounds [CpML1L2L3] because an intermediate [CpML1L2] retains its chirality with respect to the metal atom as long as it is pyramidal, whereas it loses its chiral information if it becomes planar. In this Account, we address experimental results and theoretical calculations that help illuminate the energetics of structural rearrangements after halide dissociation. The rate-determining step in the halide exchange and racemization reactions of (RRu)- and (SRu)-[CpRu(P−P′)Cl] is the cleavage of the Ru−Cl bond to give pyramidal intermediates (RRu)- and (SRu)-[CpRu(P−P′)]+, which have kept the original metal configurations. These unsaturated intermediates can react with added ligands, such as Br− or I− (k2 paths). The substitution products form with retention of the metal configuration. However, the pyramidal intermediates (RRu)- and (SRu)-[CpRu(P−P′)]+ can also invert to their mirror images (k3 paths). For (RRu)- and (SRu)-[CpRu(P−P′)]+, the barrier of the pyramidal inversion (k3) is much higher than that of the halide addition (k2). The competition ratio k3/k2 determines how much racemization occurs in a ligand exchange reaction. The competition ratio k3/k2 can be determined from the ratio of the (RRu)- and (SRu)-products, which is constant throughout the course of the reaction. For compounds like [CpRu(P−P′)Hal], k3/k2 is much smaller than 1, resulting in an energy profile that resembles a basilica. These results, established with chiral-at-metal compounds, are supported by calculations that show that 16-electron half-sandwich intermediates with σ-donating ligands, such as [CpM(NH3)2], adopt planar structures, whereas strongly π-bonding ligands, as in [CpM(CO)2], lead to pyramidal intermediates. The computed activation energies for the pyramidal inversion are on the order of 10 kcal/mol, with the planar species being transition states. The ligand dissociation behavior of 18-electron transition metal complexes is compared with nucleophile dissociation in main group compounds with octet configurations; here we include new computational results. Without exception, unsaturated main group intermediates, such as the carbenium ions formed in SN1 reactions, are planar. Our results and analysis help put transition metal chemistry on a firmer mechanistic foundation, and chiral-at-metal compounds are invaluable to this end.
Co-reporter:Henri Brunner, Andreas Köllnberger, Arshad Mehmood, Takashi Tsuno, Manfred Zabel
Journal of Organometallic Chemistry 2004 Volume 689(Issue 24) pp:4244-4262
Publication Date(Web):29 November 2004
DOI:10.1016/j.jorganchem.2004.07.057
PN ligands 3 and 4, derived from 2-diphenylphosphanylmethylpyridine 2a, were synthesized, to which in the backbone a tether to a cyclopentadiene system and for comparison an iPr substituent were attached. The chiral compounds were resolved by introduction of a menthoxy substituent into the 2-position of the pyridine system and/or palladium complexes with enantiomerically pure co-ligands. The tripod ligand 3b contains three different binding sites (Cp, P, N) connected by a resolved chiral carbon atom. (SC)-configuration of this tripod ligand enforces (RRh)-configuration at the metal atom in the halfsandwich rhodium complex (LMent,SC,RRh)-7b. The opposite metal configuration is inaccessible. Substitution of the chloro ligand in (LMent,SC,RRh)-7b by halide (Br, I) or pseudohalide (N3, CN, SCN) ligands occurs with retention of configuration to give complexes 8b–11b. However, in the reaction of (LMent,SC,RRh)-7b with PPh3 the pyridine arm of the tripod ligand in compound 13b becomes detached from the metal atom. In the Cp*Rh and CpRh compounds of the bidentate PN ligands 4a and 4b both metal configurations are accessible and in complexes 14a–17a and 14b–17b they equilibrate fast. The stereochemical assignments are corroborated by 9 X-ray analyses.In contrast to bidentate ligands, e.g. PN, tripod ligands CpPN− predetermine a single metal configuration preventing formation of the opposite metal configurations.
Co-reporter:H Brunner, M.R Arndt, B Treittinger
Inorganica Chimica Acta 2004 Volume 357(Issue 6) pp:1649-1669
Publication Date(Web):20 April 2004
DOI:10.1016/S0020-1693(03)00479-1
Forty porphyrin platinum conjugates were synthesized, which exhibited a photodynamic effect due to the porphyrin system and a cytostatic effect due to the platinum fragment present in the same molecule. The porphyrin ligands for the platinum complexes were synthesized starting from hematoporphyrin and deuteroporphyrin. The platinum complexes are of the (diamine)PtCl2, (diamine)Pt(phthalato), (NH3)2Pt(dicarboxylato) and (diamine)Pt(dicarboxylato) type. Their antitumor activity was tested with the MDA-MB-231 mammary carcinoma cell line with and without irradiation.Forty porphyrin platinum conjugates were synthesized, which exhibited a photodynamic effect due to the porphyrin system and a cytostatic effect due to the platinum fragment present in the same molecule. The porphyrin ligands for the platinum complexes were synthesized starting from hematoporphyrin and deuteroporphyrin. The platinum complexes are of the (diamine)PtCl2, (diamine)Pt(phthalato), (NH3)2Pt(dicarboxylato) and (diamine)Pt(dicarboxylato) type. Their antitumor activity was tested with the MDA-MB-231 mammary carcinoma cell line with and without irradiation.
Co-reporter:Henri Brunner, Nick Gruber
Inorganica Chimica Acta 2004 Volume 357(Issue 15) pp:4423-4451
Publication Date(Web):1 December 2004
DOI:10.1016/j.ica.2004.03.061
Tetraarylporphyrins of the Ar:Ar′ = 3:1-type were synthesized from pyrrole, 4-hydroxybenzaldehyde and benzaldehydes substituted with ethyleneglycol, hydroxy and quaternary ammonium substituents for solubilization in DMF and, in particular, in water. After etherification with the tosylate of diethyl cyclobutanedicarboxylate and subsequent ester hydrolysis, the resulting carboxylic acid groups were used to bind platinum fragments bearing two ammonia and (RR/SS)-trans-1,2-diaminocyclohexane ligands, respectively, as non-leaving groups. In comparison to hematoporphyrin–platinum complexes, the title compounds show a 30 nm bathochromic shift of their absorption bands increasing the penetration depth of the red light used for irradiation in photodynamic tumor therapy. The antiproliferative activity of 24 new platinum complexes differing in the porphyrin ligands and the platinum fragments were studied in tests with J82 bladder cancer cells. The compounds showed the cytotoxic effect of the platinum moiety and after irradiation the phototoxic effect of the porphyrin system.Tetraarylporphyrins of the Ar:Ar′ = 3:1-type were synthesized substituted with ethyleneglycol, hydroxy and quaternary ammonium substituents for solubilization in DMF and, in particular, in water. The antiproliferative activity was studied in tests with J82 bladder cancer cells. The compounds showed the cytotoxic effect of the platinum moiety and, in addition, after irradiation the phototoxic effect of the porphyrin system.
Co-reporter:Günther Bernhardt, Henri Brunner, Nick Gruber, Christian Lottner, Simi K. Pushpan, Takashi Tsuno, Manfred Zabel
Inorganica Chimica Acta 2004 Volume 357(Issue 15) pp:4452-4466
Publication Date(Web):1 December 2004
DOI:10.1016/j.ica.2004.03.060
A series of new carboplatin derivatives was synthesized by introducing fluoro, chloro, bromo and hydroxy substituents into the cyclobutane ring. The carboxylic acid groups were used for the complexation with platinum(II) fragments bearing two ammonia and (RR/SS)-trans-1,2-diaminocyclohexane ligands, respectively, as non-leaving groups. The antiproliferative activity of the new carboplatin analogues differing in the cyclobutanedicarboxylate ligands and the type of platinum fragment were studied in tests with J82 bladder cancer cells and SK-OV-3 as well as cisplatin-resistant NIH:OVCAR-3 ovarian cancer cells. The most active compounds were the 3-fluoro, 3-chloro and 3,3-difluoro derivatives of carboplatin. NMR spectroscopy showed that cis-diammine(3-chloro-1,1-cyclobutanedicarboxylato)platinum(II) was hydrolyzed much faster than carboplatin explaining its higher cytostatic activity.New carboplatin derivatives were synthesized by introducing fluoro, chloro, bromo and hydroxy substituents into the cyclobutane ring. The carboxylic acid groups were used for complexation to the Pt(NH3)2 fragment as non-leaving group. The antiproliferative activity of the new carboplatin analogues were studied in tests with J82, SK-OV-3 and NIH:OVCAR-3 cancer cells. The best compounds were two times as active as carboplatin itself.
Co-reporter:Henri Brunner Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 21) pp:
Publication Date(Web):12 MAY 2004
DOI:10.1002/anie.200301742
One up in limiting reactivity: The monosubstitution of multifunctional molecules is a general problem in synthetic chemistry. The catalyst [Cp*(PiPr3)(H)2RuSi(H)Ph⋅Et2O][B(C6F5)4] (Cp*=η5-C5Me5) can be used for the alkylation of phenylsilane PhSiH3 with simple olefins. The reaction stops at the monoalkylation level to give PhSi(Alk)H2 (see scheme; R=H, C6H5, C4H9).
Co-reporter:Henri Brunner Dr.
Angewandte Chemie 2004 Volume 116(Issue 21) pp:
Publication Date(Web):12 MAY 2004
DOI:10.1002/ange.200301742
Einfach selektiv: Die Monosubstitution von mehrfach funktionellen Molekülen ist ein generelles Problem in der Synthesechemie. Mit dem Katalysator [Cp*(PiPr3)(H)2RuSi(H)Ph⋅Et2O][B(C6F5)4] (Cp*=η5-C5Me5) kann die Alkylierung von Phenylsilanen PhSiH3 mit einfachen Olefinen auf der Stufe des Monoalkylierungsprodukts PhSi(Alk)H2 angehalten werden (siehe Schema, R=H, C6H5, C4H9).
Co-reporter:Henri Brunner;Markus A. Baur
European Journal of Organic Chemistry 2003 Volume 2003(Issue 15) pp:
Publication Date(Web):15 JUL 2003
DOI:10.1002/ejoc.200300206
The methodology of enantioselective decarboxylation was applied to 2-aminomalonic acid derivatives in order to obtain enantio-enriched amino acid derivatives. Full conversion was achieved stirring racemic N-acetylated 2-aminomalonic hemiesters in THF at 70 °C with 10 mol % of a chiral base for 24 h. The catalyst may be recycled. Whereas the commercially available cinchona alkaloids gave poor results, benzamide and benzenesulfonamide derivatives of 9-amino(9-deoxy)epicinchonine turned out to be effective catalysts. The best result was obtained with 2-N-acetylamino-2-ethoxycarbonyl-3-phenylpropionic acid as the starting material and N-(9-deoxyepicinchonine-9-yl)-4-methoxybenzamide as the chiral base to give ethyl N-acetyl-L-phenylalaninate in 70% ee. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2003)
Co-reporter:Henri Brunner;Karl-Peter Ittner;Dirk Lunz;Stefan Schmatloch;Thomas Schmidt;Manfred Zabel
European Journal of Organic Chemistry 2003 Volume 2003(Issue 5) pp:
Publication Date(Web):10 FEB 2003
DOI:10.1002/ejoc.200390129
The stereoselective Pd-catalyzed allylation of MBA [5-(2′-hex-3′-ynyl)-1-methylbarbituric acid] gives the commercial injection narcotic methohexital, which exists as four isomers: two diastereomeric pairs of enantiomers. The isomer composition produced depends on three stereochemical parameters: catalyst control, substrate control, and kinetic resolution. Judicious choice of these parameters allowed the synthesis of methohexital samples with greatly differing isomer compositions, and these samples were investigated with respect to their anaesthetic doses in rats. Some isomer compositions obtained were much more active than the commercially used drug and showed fewer side effects. As a consequence of the determination of the absolute configuration of the methohexital (SbRh) isomer, the unknown configuration of the trade product, the so-called α-racemate, can be established as (RbSh) and (SbRh). (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2003)
Co-reporter:H. Brunner;F. Hausmann;R. Knuechel
Photochemistry and Photobiology 2003 Volume 78(Issue 5) pp:481-486
Publication Date(Web):1 MAY 2007
DOI:10.1562/0031-8655(2003)0780481NAAEPP2.0.CO2
Photodetection (PD) and photodynamic therapy (PDT) with 5-aminolevulinic acid (ALA)–induced protoporphyrin IX (PPIX) accumulation are approaches to detect and treat dysplasia and early cancer in the gastrointestinal tract and in the urinary bladder. Because ALA-induced PPIX production is limited, we synthesized ALA ester hydrochlorides 3–22 and tested them in two different in vitro models (gastrointestinal tract: HT29–CCD18; urinary bladder: J82–UROTSA). PPIX accumulation after incubation with 0.12 mmol/L for 3 h and PPIX accumulation as a function of different incubation times were measured using flow cytometry. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assays were performed to check cellular dark toxicity. Phototoxicity after irradiation was tested. ALA nonafluorohexylester hydrochloride 11, ALA thiohexylester hydrochloride 13 and ALA dibenzyldiester dihydrochloride 19 induced appreciably increased PPIX levels and showed improved phototoxicity compared with the references ALA hydrochloride 1, ALA hexylester hydrochloride 3 and ALA benzylester hydrochloride 4. Thus, the new compounds 11, 13 and 19 are promising compounds for PD and PDT.
Co-reporter:Henri Brunner, Matthias Weber, Manfred Zabel
Journal of Organometallic Chemistry 2003 Volume 684(1–2) pp:6-12
Publication Date(Web):1 November 2003
DOI:10.1016/S0022-328X(03)00224-9
The trinuclear complex [Pt2Rh(μ3-S)2{(−)-diop}2(cod)]Cl (3) was synthesized starting from the chiral “ligand” [Pt2(μ-S)2{(−)-diop}] (2) and [Rh(cod)Cl]2, and characterized by X-ray crystallography. Compound 3 was used as a catalyst in the hydrosilylation of acetophenone with diphenylsilane and in the hydrogenation of ketopantolactone.The trinuclear complex [Pt2Rh(μ3-S)2{(−)-diop}2(cod)]Cl (3) was synthesized starting from the chiral “ligand” [Pt2(μ-S)2{(−)-diop}] (2) and [Rh(cod)Cl]2, and characterized by X-ray crystallography. Compound 3 was used as a catalyst in the hydrosilylation of acetophenone with diphenylsilane and in the hydrogenation of ketopantolactone.
Co-reporter:Henri Brunner, Maximilian Schönherr, Manfred Zabel
Tetrahedron: Asymmetry 2003 Volume 14(Issue 9) pp:1115-1122
Publication Date(Web):2 May 2003
DOI:10.1016/S0957-4166(03)00093-4
The synthesis of new hydrolytically stable oxime ether ligands by condensation of various aldehydes with O-β-d-glucopyranosylhydroxylamine 1 or O-β-d-galactopyranosylhydroxylamine 4 is described. After peracetylation of the hydroxyl groups, ligands soluble in organic solvents were obtained. The ligands have been tested in transition metal-catalysed reactions. Phosphorus-containing ligands gave high yields in palladium-catalysed allylic alkylation and rhodium-catalysed hydrosilylation reactions although the enantioselectivities were low. A 1,3-diphenylallyl–palladium(II) complex of ligand 2b was prepared and its structure was established by X-ray diffraction analysis.GraphicO-β-d-GalactopyranosylhydroxylamineC6H13NO6[α]D25=+68.0 (c 3, H2O)Source of chirality: galactose (chiral pool)O-(β-d-Galactopyranosyl)-2-diphenylphosphanylbenzaldoximeC25H26NO6P[α]D25=+15.8 (c 1, CH2Cl2)Source of chirality: galactose (chiral pool)O-(2,3,4,6-Tetra-O-acetyl-β-d-galactopyranosyl)-2-diphenylphosphanylbenzaldoximeC33H34NO10P[α]D25=+5.7 (c 1, CH2Cl2)Source of chirality: galactose (chiral pool)O-(β-d-Galactopyranosyl)pyridine-2-carbaldoximeC12H16N2O6[α]D25=−15.0 (c 3, H2O)Source of chirality: galactose (chiral pool)O-(2,3,4,6-Tetra-O-acetyl-β-d-galactopyranosyl)pyridine-2-carbaldoximeC20H24N2O10[α]D25=+9.6 (c 2, CH2Cl2)Source of chirality: galactose (chiral pool)O-β-d-GlucopyranosylsalicylaldoximeC13H17NO7[α]D25=−20.7 (c 2, MeOH)Source of chirality: glucose (chiral pool)O-β-d-GalactopyranosylsalicylaldoximeC13H17NO7[α]D25=−10.7 (c 2, MeOH)Source of chirality: galactose (chiral pool)Bis-O-(β-d-glucopyranosyl)benzene-1,2-dicarbaldoximeC20H28N2O12[α]D25=−32.8 (c 3, H2O)Source of chirality: glucose (chiral pool)Bis-O-(2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl)benzene-1,2-dicarbaldoximeC36H44N2O20[α]D25=−31.9 (c 2, CH2Cl2)Source of chirality: glucose (chiral pool)Bis-O-(β-d-glucopyranosyl)ethane-1,2-dicarbaldoximeC14H24N2O12[α]D25=−32.0 (c 3, H2O)Source of chirality: glucose (chiral pool)C30H40N2O20Bis-O-(2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl)ethane-1,2-dicarbaldoxime[α]D25=−29.0 (c 1, CH2Cl2)Source of chirality: glucose (chiral pool)Bis-O-(β-d-glucopyranosyl)pyridine-2,6-dicarbaldoximeC19H27N3O12[α]D25=+27.4 (c 3, H2O)Source of chirality: glucose (chiral pool)Bis-O-(2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl)pyridine-2,6-dicarbaldoximeC35H43N3O20[α]D25=−31.5 (c 1, CH2Cl2)Source of chirality: glucose (chiral pool)
Co-reporter:Henri Brunner Dr.;Matthias Weber Dr.;Manfred Zabel Dr.;Thomas Zwack Dr.
Angewandte Chemie 2003 Volume 115(Issue 16) pp:
Publication Date(Web):23 APR 2003
DOI:10.1002/ange.200250181
Molekulares Mobiliar: Halbsandwichkomplexe vom Typ [(Ar)MXY(NR*)] (siehe Strukturformel; X, Y=elektronegative Substituenten; M=Ru, Os, Rh, Ir) bilden im festen Zustand bevorzugt Inversionspaare, die durch kleine M⋅⋅⋅M-Abstände und CH⋅⋅⋅X/Y-Wasserstoffbrücken charakterisiert sind. Dabei bilden zwei Diastereomere mit gegebener Chiralität der NR*-Gruppe und unterschiedlichen Metallkonfigurationen mit ihren „racemischen Seiten“ ein Molekülpaar, was zu einer ungewöhnlich bevorzugten 1:1-Cokristallisation von Diastereomeren im selben Einkristall führt.
Co-reporter:Henri Brunner, Henri B. Kagan, Georg Kreutzer
Tetrahedron: Asymmetry 2003 Volume 14(Issue 15) pp:2177-2187
Publication Date(Web):1 August 2003
DOI:10.1016/S0957-4166(03)00433-6
Promoted by catalytic amounts of Ni complexes tertiary α-hydroxyketones 1a, 3a–5a undergo rearrangement, forming chiral isomers 1b, 3b–5b. The best enantioselection was obtained with the model system 1-benzoylcyclopentanol 4a/2-hydroxy-2-phenylcyclohexanone 4b. In a ligand screening 2-[4-(S)-tert-butyloxazolin-2-yl]pyridine gave the highest enantiomeric excess of 46% (S)-4b. The analogous isomerisation reactions of α-hydroxyimines 6a, 7a forming chiral α-aminoketones 6b, 7b were established.Graphic2-[4-(S)-Isopropyloxazolin-2-yl]-3-methylpyridineC12H16N2O[α]D=−82 (c 0.93, CHCl3)Source of chirality: (S)-valineAbsolute configuration: S(S)-(+)-N,N,N′,N′-Tetramethyl-1,2-diamino-3-methylbutaneC9H22N2[α]D=178 (c 2.61, CHCl3)Source of chirality: (S)-valineAbsolute configuration: S
Co-reporter:Henri Brunner Dr.;Matthias Weber Dr.;Manfred Zabel Dr.;Thomas Zwack Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 16) pp:
Publication Date(Web):23 APR 2003
DOI:10.1002/anie.200250181
Half-sandwich complexes of the type [(Ar)MXY(NR*)] (see structure; X, Y=electronegative substituents; M=Ru, Os, Rh, Ir) tend to form inversion pairs in the solid state, which are characterized by short M⋅⋅⋅M separations and CH⋅⋅⋅X/Y hydrogen bonds between the pairs of molecules. As a consequence, two diastereomers with a given chirality in the Z substituent, usually an NR* substituent, and different metal configurations approach each other with their “racemic sides” resulting in an unusually preferred 1:1 diastereomer cocrystallization in the same single crystal.
Co-reporter:Henri Brunner;Stephan Dormeier;Manfred Zabel
European Journal of Inorganic Chemistry 2002 Volume 2002(Issue 10) pp:
Publication Date(Web):30 AUG 2002
DOI:10.1002/1099-0682(200210)2002:10<2594::AID-EJIC2594>3.0.CO;2-T
The (+)-9-phenyldeltacyclanyl moiety as a chiral phosphorus substituent contains eight configurationally fixed asymmetric carbon atoms. It is easily obtained from the reaction of norbornadiene and phenylacetylene via a Co-catalyzed enantioselective homo-Diels−Alder reaction, to give (+)-8-phenyldeltacyclene. In the reaction of (+)-8-phenyldeltacyclene with 1,2-(H2P)2C6H4 the trisubstitution product P,P,P′-tris[(+)9-phenyldeltacyclan-8-yl)-1,2-bis(phosphanyl)benzene, (+)δ-LH, is obtained, in which a single P−H bond at the stereogenic phosphorus atom is left. (+)δ-LH crystallizes as a pure diastereomer, which isomerizes in solution with respect to the secondary phosphorus atom at −20 °C resulting in a 64:36 diastereomer mixture. Substitution of the P−H hydrogen atom in (+)δ-LH by benzyl, 3-bromobenzyl, 6-bromo-n-hexyl and 3-(diphenylphosphanyl)phenyl gave the bis(tertiary) diphosphanes (+)δ-LR1−(+)δ-LR4. NiBr2, PdHal2 (Hal = Cl, Br, I) and PtHal2 (Hal = Cl, Br) complexes of (+)δ-LH and (+)δ-LR1−(+)δ-LR4have been synthesized. The complexes form two diastereomers differing in the configuration of the chiral P atom. X-ray structure analyses have been carried out with (+)δ-LH, [(+)δ-LR2]NiBr2, [(+)δ-LH]PdI2 and [(+)δ-LCl]PdI2. Interestingly, for [(+)δ-LH]PdI2 both diastereomers are found in the same crystal lattice in a 1:1 ratio. In [(+)δ-LH]PdI2 and [(+)δ-LCl]PdI2the configuration at the secondary phosphorus atom is stable in solution at room temperature, whereas in the free ligand (+)δ-LH it is configurationally labile. (© Wiley-VCH Verlag GmbH, 69451 Weinheim, Germany, 2002)
Co-reporter:Henri Brunner;Stephan Dormeier;Ilias Grau;Manfred Zabel
European Journal of Inorganic Chemistry 2002 Volume 2002(Issue 10) pp:
Publication Date(Web):30 AUG 2002
DOI:10.1002/1099-0682(200210)2002:10<2603::AID-EJIC2603>3.0.CO;2-A
The synthesis of seven dinuclear complexes [(+)δ-LMHal]2 (M = Ni, Pd, Pt; Hal = Cl, Br, I) is described, in which (+)δ-L− represents the deprotonated chiral chelate ligand P,P,P′tris[(+)-9-phenyldeltacyclan-8-yl]-1,2-bis(phosphanyl)benzene, easily accessible in a highly enantioselective homo-Diels−Alder reaction. All the dinuclear complexes are bent at the phosphido bridges which connect the two square-planar halves of the molecules. The phosphorus atoms of the phosphido bridges are stereogenic centers. For [(+)δ-LNiBr]2 and [(+)δ-LPdCl]2 both diastereomers with (SP,SP) and (RP,RP) configuration were observed, whereas for [(+)δ-LPdBr]2, [(+)δ-LPdI]2, and [(+)δ-LPtCl]2 only the (SP,SP) isomers could be obtained. Furthermore, the synthesis of eight trinuclear complexes [(+)δ-L2M3Hal4] (M = Pd, Pt; Hal = Cl, Br) is reported, which contain unusual three-square arrays. In the trinuclear complexes all the halogens are oriented to one side. Halogeno and phosphido bridges connect the three squares leading to chair configurations (opposite scaffold chirality possible) and boat configurations. For [(+)δ-L2Pd3Cl4], [(+)δ-L2Pd3Br4], and [(+)δ-L2Pt3Cl4] the two diastereomers with (SP,SP) and (RP,RP) configuration and for [(+)δ-L2Pt3Br4] the diastereomers with (SP,SP) and (SP,RP) configuration could be isolated. In addition, the dinuclear complex [(+)δ-LPt2Br3(cod)] (cod = 1,5-cyclooctadiene) containing a single phosphido bridge was obtained. (© Wiley-VCH Verlag GmbH, 69451 Weinheim, Germany, 2002)
Co-reporter:Henri Brunner;Jürgen Klankermayer;Manfred Zabel
European Journal of Inorganic Chemistry 2002 Volume 2002(Issue 9) pp:
Publication Date(Web):7 AUG 2002
DOI:10.1002/1099-0682(200209)2002:9<2494::AID-EJIC2494>3.0.CO;2-7
The synthesis of a series of 18-electron chiral-at-metal (cycloheptatrienyl)molybdenum complexes [(η7-C7H7)Mo(prophos)X] [prophos = (R)-1,2-bis(diphenylphosphanyl)propane; X = NCMeBF4, Cl, I, CN, H, Me] is reported, which consist of diastereomers differing only in the metal configuration. Diastereomer enrichments have been achieved and the configurational stability of the compounds [(η7-C7H7)Mo(prophos)X] has been studied and compared with the isoelectronic ruthenium complexes [(η5-C5H5)Ru(prophos)X]. The stereochemistry of the substitution reactions has been investigated, and X-ray analysis and NMR spectroscopy were used to determine the absolute configuration of the complexes. (© Wiley-VCH Verlag GmbH, 69451 Weinheim, Germany, 2002)
Co-reporter:Henri Brunner;Jürgen Klankermayer;Manfred Zabel
European Journal of Inorganic Chemistry 2002 Volume 2002(Issue 11) pp:
Publication Date(Web):14 OCT 2002
DOI:10.1002/1099-0682(200211)2002:11<3047::AID-EJIC3047>3.0.CO;2-A
The synthesis of a series of 18-electron chiral-at-metal (cycloheptatrienyl)molybdenum complexes [(η7-C7H7)Mo(prophos)X] [prophos = (R)-1,2-bis(diphenylphosphanyl)propane; X = NCMeBF4, Cl, I, CN, H, Me] is reported, which consist of diastereomers differing only in the metal configuration. Diastereomer enrichments have been achieved and the configurational stability of the compounds [(η7-C7H7)Mo(prophos)X] has been studied and compared with the isoelectronic ruthenium complexes [(η5-C5H5)Ru(prophos)X]. The stereochemistry of the substitution reactions has been investigated, and X-ray analysis and NMR spectroscopy were used to determine the absolute configuration of the complexes. (© Wiley-VCH Verlag GmbH, 69451 Weinheim, Germany, 2002)
Co-reporter:Henri Brunner, Frauke Henning, Matthias Weber
Tetrahedron: Asymmetry 2002 Volume 13(Issue 1) pp:37-42
Publication Date(Web):13 February 2002
DOI:10.1016/S0957-4166(02)00029-0
A series of bidentate and tridentate (S)-NOBIN derivatives was synthesised and tested as chiral ligands in the Ru-catalysed enantioselective transfer hydrogenation of acetophenone with 2-propanol. Despite the similarities in chemical structure, only the imine derived from (S)-NOBIN and 2-pyridinecarbaldehyde and the corresponding amine (obtained by NaBH4 reduction of the imine) afforded (S)-1-phenylethanol in nearly quantitative yields and outstanding enantioselectivities of up to 97% e.e.Graphic(S)-2-(N,N-Dimethylamino)-2′-methoxy-1,1′-binaphthylC23H21NOE.e.=99–100%[α]D21=−154 (c 0.88, benzene)Source of chirality: asymmetric synthesis (lit.)Absolute configuration: S(S)-2-(2-Pyridinylmethylamino)-2′-hydroxy-1,1′-binaphthylC26H20N2OE.e.=99–100%[α]D21=−155 (c 0.99, benzene)Source of chirality: asymmetric synthesis (lit.)Absolute configuration: S(S)-2-(2-Pyridinylmethyleneamino)-2′-methoxy-1,1′-binaphthylC27H20N2OE.e.=99–100%[α]D21=−224 (c 1.10, benzene)Source of chirality: asymmetric synthesis (lit.)Absolute configuration: S(S)-2-(2-Hydroxybenzylideneamino)-2′-methoxy-1,1′-binaphthylC28H21NO2E.e.=99–100%[α]D21=+40 (c 1.03, benzene)Source of chirality: asymmetric synthesis (lit.)Absolute configuration: S(S)-2-(2-Diphenylphosphanylbenzylideneamino)-2′-methoxy-1,1′-binaphthylC40H30NOPE.e.=99–100%[α]D21=−177 (c 1.01, benzene)Source of chirality: asymmetric synthesis (lit.)Absolute configuration: S(S)-2-(2-Pyridinylmethyleneamino)-2′-hydroxy-1,1′-binaphthylC26H18N2OE.e.=99–100%[α]D21=+275.5 (c 1.12, benzene)Source of chirality: asymmetric synthesis (lit.)Absolute configuration: S
Co-reporter:Henri Brunner
European Journal of Inorganic Chemistry 2001 Volume 2001(Issue 4) pp:
Publication Date(Web):20 FEB 2001
DOI:10.1002/1099-0682(200104)2001:4<905::AID-EJIC905>3.0.CO;2-V
Half-sandwich compounds with a three-legged piano-stool geometry are prominent examples of optically active chiral-at-metal complexes. In these compounds, the configuration at the metal atom may be stable or labile in solution. Configurationally stable compounds can be used for the elucidation of the stereochemical course of substitution reactions and for organic synthesis in ligand transformation reactions. With configurationally labile compounds, the rate of change of the metal configuration can be measured, which sets an upper limit with regard to the handling of the compounds in solution. Surprisingly, in a number of recent papers the lability of the metal configuration has been overlooked resulting in misinterpretations and wrong conclusions.
Co-reporter:H. Brunner;J. Schölmerich;F. Hausmann;R. Knuechel;E. Endlicher;H. Messmann;R. C. Krieg
Photochemistry and Photobiology 2001 Volume 74(Issue 5) pp:721-725
Publication Date(Web):1 MAY 2007
DOI:10.1562/0031-8655(2001)0740721TEOAAE2.0.CO2
Photodynamic diagnosis (PDD) and photodynamic therapy (PDT) using 5-aminolevulinic acid (ALA)–induced protoporphyrin IX (PPIX) is an interesting approach to detect and treat dysplasia and early cancers in the gastrointestinal tract. Because of low lipophilicity resulting in poor penetration across cell membranes, high doses of ALA should be administered in order to reach clinically relevant levels of PPIX. One way of increasing PPIX accumulation is derivatization of ALA into a more lipophilic molecule. In our in vitro study, different esterifications of ALA were investigated to analyze the effects on PPIX accumulation in human adenocarcinoma cell lines. For systematic analysis of cell type–specific PPIX accumulation, three human adenocarcinoma cell lines (SW480, HT29 and CaCo2) and a fibroblast cell line (CCD18) were tested. 3-(4,5-Dimethylthiazole-2-yl)-2,5-biphenyl tetrazolium bromide (MTT) assays were performed to ensure that the ALA esters showed no cellular dark toxicity. Different concentrations (ranging from 0.012 to 0.6 mmol/L, 3 h) and incubation times (5, 10, 30, 180 min; 0.12 mmol/L) were examined. PPIX accumulation was measured using flow cytometry. ALA esters, especially ALA-hexylester and ALA-benzylester, induced significant higher PPIX levels in adenocarcinoma cell lines when compared with ALA and may be promising candidates for PDT and PDD.
Co-reporter:Henri Brunner, Andrea Hollman, Bernhard Nuber, Manfred Zabel
Journal of Organometallic Chemistry 2001 Volume 633(1–2) pp:1-6
Publication Date(Web):10 August 2001
DOI:10.1016/S0022-328X(01)01086-5
The reaction of [Cp2MoH2] and AgBF4 with the pseudohalide ligands SCN−, OCN−, CN− and SCF3− afforded the complexes [(Cp2MoH2)2AgSCN] (1), [(Cp2MoH2)2AgNCO]·CH2Cl2 (2), [(Cp2MoH2)2Ag(C3N3H2O3)]·3CH2Cl2 (3), [(Cp2MoH2)2Ag]n[(Ag2(CN)3)n] (4) and [(Cp2MoH2)2AgSCF3]·CH2Cl2 (5). 1, 2 and 5 have a [Cp2MoH2]:Ag:ligand stoichiometry of 2:1:1. Complex 3 contains the anion of the ligand (HNCO)3, formed by trimerization of HNCO. The three-dimensional structure of 4 is built up of polymeric [Ag2(CN)3−]n strings and [(Cp2MoH2)2Ag]+ units. The compounds 1–5 were characterized analytically and spectroscopically. All the structures were determined by single crystal X-ray analysis.Reaction of [Cp2MoH2] and AgBF4 with the pseudohalide ligands SCN−, OCN−, CN− and SCF3− afforded the complexes [(Cp2MoH2)2AgSCN], [(Cp2MoH2)2AgNCO]·CH2Cl2, [(Cp2MoH2)2Ag]n[(Ag2(CN)3)n] and [(Cp2MoH2)2AgSCF3]·CH2Cl2, whereas reaction of [Cp2MoH2] and AgNCO gave a mixture of the complexes [(Cp2MoH2)2AgNCO]·CH2Cl2 and [(Cp2MoH2)2Ag(C3N3H2O3)]·3CH2Cl2. All the complexes were characterized by X-ray crystallography.
Co-reporter:Henri Brunner, Maximilian Schönherr, Manfred Zabel
Tetrahedron: Asymmetry 2001 Volume 12(Issue 19) pp:2671-2675
Publication Date(Web):30 October 2001
DOI:10.1016/S0957-4166(01)00451-7
Co-reporter:Henri Brunner
Chirality 2001 Volume 13(Issue 8) pp:420-424
Publication Date(Web):16 JUL 2001
DOI:10.1002/chir.1054
The narcotic drug methohexital 1 contains two asymmetric carbon atoms and, thus, consists of four isomers, two diastereomeric pairs of enantiomers. The commercial drug is the so-called α-racemate, one pair of diastereomers only. A method was developed to prepare differently enriched mixtures of methohexital isomers without resorting to lengthy and expensive optical resolutions. A model reaction for the synthesis of methohexital is the palladium-catalyzed allylation of 1,5-dimethyl-barbituric acid 3, which is optimized and checked by molecular modeling. Catalysts with the best ligands are used in the allylation of the methohexital precursor 7, which contains the C6 sidechain at the tetrahedral center of the barbiturate skeleton. The product stereochemistry was determined by the contribution of the enantioselective Pd catalysts and by the fact that the allylation is a kinetic resolution. The methohexital isomer mixtures obtained were evaluated with the corneal stimulus test of rats. Methohexital compositions were found, which are superior to the commercially used α-racemate (Brevimytal®). Chirality 13:420–424, 2001. © 2001 Wiley-Liss, Inc.
Co-reporter:Henri Brunner, Henri B Kagan, Georg Kreutzer
Tetrahedron: Asymmetry 2001 Volume 12(Issue 3) pp:497-499
Publication Date(Web):5 March 2001
DOI:10.1016/S0957-4166(01)00045-3
Using catalytic amounts of Ni complexes the tertiary α-hydroxyketones 1-benzoylcyclobutanol 1 and 1-benzoylcyclopentanol 3 undergo α-ketol rearrangement. The use of the chiral ligand 2,6-bis[(4S)-isopropyl-2-oxazolin-2-yl]pyridine gave an enantiomeric excess of about 34% for both systems, forming (−)-2-hydroxy-2-phenylcyclopentanone 2 and (R)-(−)-2-hydroxy-2-phenylcyclohexanone 4. 1-Benzoylcyclohexanol 5 could not be catalytically rearranged to 6 under these conditions.Graphic
Co-reporter:Henri Brunner;Peter Schmidt
European Journal of Organic Chemistry 2000 Volume 2000(Issue 11) pp:
Publication Date(Web):14 JUN 2000
DOI:10.1002/1099-0690(200006)2000:11<2119::AID-EJOC2119>3.0.CO;2-5
A new catalytic method to synthesize the important anti-inflammatory agent naproxen [(S)-1] which has to be used as the (S) enantiomer, involves the enantioselective decarboxylation of the 6-methoxynaphth-2-yl derivative 2 of 2-cyanopropionic acid. Compound 2 was stirred in THF at 15 °C with catalytic amounts of chiral bases, which abstracted the carboxyl proton. After decarboxylation, reprotonation of the anion of 6 afforded the enantiomerically enriched naproxen nitrile 6, which may be hydrolyzed to naproxen. A variety of bases were screened, and cinchona alkaloids were found to give the best enantioselectivities. Thus, with quinidine 10, up to 34% ee was obtained for (S)-6. The enantiomeric excess could be increased by turning to amides of 9-amino-9-deoxyepicinchona alkaloids. The most successful 2-ethoxybenzamide 31a of 9-amino-9-deoxyepicinchonine 11 gave up to 71.9% ee (S)-6. Cyclic ethers like THF were suitable solvents, and at a temperature of 15 °C, conversion was quantitative within 24 h in most cases. For high enantioselectivities, 5−10 mol-% of chiral base was sufficient, and the catalyst could be fully recycled after decarboxylation. The model compound 2-cyano-2-phenylpropionic acid (40) was decarboxylated with base 31a to the (S) enantiomer of the corresponding nitrile 41 with 60% ee.
Co-reporter:Henri Brunner;Frank Stöhr
European Journal of Organic Chemistry 2000 Volume 2000(Issue 15) pp:
Publication Date(Web):17 JUL 2000
DOI:10.1002/1099-0690(200008)2000:15<2777::AID-EJOC2777>3.0.CO;2-0
Promoted by catalytic amounts of transition-metal complexes, the tertiary α-hydroxy ketones 1, 3, 5/6 undergo α-ketol rearrangements to afford equilibrium mixtures of isomers with a reorganization of the carbon skeleton. The range of metal complexes catalyzing the isomerizations is large; the best results were obtained with the catalyst systems NiCl2/TMEDA, Ni(acac)2, and Ni(acac)2/TMEDA (TMEDA = N,N,N′,N′-tetramethyl-1,2-diaminoethane). The catalytic rearrangements were performed at 130 °C in the absence of solvent, with a NiII/ligand/substrate ratio of 1:2:100. The equilibrium composition of the model system 1/2 is 12.5:87:5. The conversion of the achiral substrates 1 and 3 into the chiral products 2 and 4 can be used for kinetic resolution. However, the reverse reactions 2 1 and 4 3 in the equilibrations narrow the window for asymmetric induction with enantioselective catalysts of the metal component/optically active ligand type. In system 1, the highest enantiomeric excess was achieved with the catalyst systems NiCl2/pybox [18.9% (S)-2] and Ni(acac)2/pybox [19.3% (R)-2] {pybox = 2,6-bis[(S)-4-isopropyl(oxazolin-2′-yl)]pyridine}. The α-ketol rearrangement of 3 with the Ni(acac)2/pybox catalyst resulted in a maximum enantiomeric excess of 37.1% (S)-4.
Co-reporter:Henri Brunner, Andrea Hollman, Manfred Zabel, Bernhard Nuber
Journal of Organometallic Chemistry 2000 Volume 609(1–2) pp:44-52
Publication Date(Web):8 September 2000
DOI:10.1016/S0022-328X(00)00163-7
The reaction of [Cp2MoH2] and AgBF4 with the dithio-ligands Na(S2CNR2) (R=Et, Ph), Na(S2CMe), NH4(S2P(OEt)2) and K(S2COiPr) gives the complexes [(Cp2MoH2AgS2CNR2)2] (R=Et 1, Ph 2), [(Cp2MoH2AgS2CMe)2] (3), [(Cp2MoH2AgS2P(OEt)2)2] (4) and [(Cp2MoH2AgS2COiPr)n] (6) with probably n=2, all with a [Cp2MoH2]:Ag:dithio-ligand stoichiometry of 1:1:1. 1–4 form dimers in which two dithio-ligands bridge two silver atoms. In 1 and 2 one sulphur atom is coordinated to one silver atom, whereas the other is coordinated to both silver atoms. In 3 and 4 each sulphur atom is bonded to one silver atom only. In 1–3 there is a silversilver bond. However, in 4 the Ag⋯Ag distance is too long for an AgAg bond. Crystallization of the compound 6 afforded a polymer [(Cp2MoH2Ag4(S2COiPr)4)n] (7) with a stoichiometry [Cp2MoH2]:Ag:dithio-ligand=1:4:4. The polymer forms sheets with rings surrounding a [Cp2MoH2] ligand bonded to one of the four silver atoms. The monomer [(Cp2MoH2)2AgS2COEt] (5) with a stoichiometry [Cp2MoH2]:Ag:dithio-ligand=2:1:1 was prepared by addition of Na(S2COEt) to [Cp2MoH2] and AgBF4. In 5 the xanthogenate ligand is bonded with both sulphur atoms to the silver atom. The compounds 1–6 were characterized analytically and spectroscopically. The structures of 1–5 and 7 were determined by single crystal X-ray analyses.
Co-reporter:Henri Brunner, Jürgen Klankermayer, Manfred Zabel
Journal of Organometallic Chemistry 2000 Volume 601(Issue 2) pp:211-219
Publication Date(Web):28 April 2000
DOI:10.1016/S0022-328X(00)00064-4
The phosphorus(III)-bridged [1]ferrocenophanes 1,1′-ferrocenediylphenylphosphine (1), (−)-1,1′-ferrocenediylmenthylphosphine (2) and (−)-bornyl-1,1′-ferrocenediylphosphine (3) have been synthesized via the reaction of 1,1′-dilithioferrocene (TMEDA adduct) and Cl2PR (R=Ph, Men, Bor). Compounds 1 and 2 have been used as ligands in the preparation of the complexes Cp*Mn(CO)2[Fe(η5-C5H4)2PPh] (4) and (−)-trans-PdCl2[Fe(η5-C5H4)2PMen]2 (5). The new compounds 2–5 were characterized by multinuclear NMR, by MS, and 2, 4 and 5 by single-crystal X-ray diffraction. Remarkably, the cyclic dimer anti-exo,exo-1,12-dimenthyl-1,12-diphospha[1.1]ferrocenophane (6) could be isolated and structurally characterized. The thermal ring-opening polymerization of 1, 2 and 3 yielded the poly(ferrocenediylphosphines) 7, 8 and 9. Compounds 2 and 8 were used as chiral ligands in the Rh-catalyzed diastereoselective hydrogenation of folic acid.
Co-reporter:Henri Brunner, Peter Schmidt, Markus Prommesberger
Tetrahedron: Asymmetry 2000 Volume 11(Issue 7) pp:1501-1512
Publication Date(Web):April 2000
DOI:10.1016/S0957-4166(00)00091-4
Amides of 9-amino(9-deoxy)epicinchonine have been used as chiral base catalysts to induce asymmetry in enantioselective decarboxylation reactions. To understand the reaction mechanism and to optimize the bases by variation of the amide substituents, a detailed knowledge of their preferred conformation is necessary. The conformations were investigated by 1H NMR spectroscopy, X-ray analysis and semi-empirical molecular orbital calculations. Principally, cinchona alkaloids may adopt four different conformations (Fig. 1), of which openb and openb′ are preferred by 2-ethoxy-N-(9-deoxyepicinchonine-9-yl)benzamide 2 in solution. AM1 Calculations confirm these results, and the small energy barrier between the two open conformations explains their simultaneous existence in solution. Crystal structure analyses of various amides show open conformations in the solid state. Local minimum energy conformations were found for open conformations of the compound 2 protonated at the quinuclidine system, which is the active species in the enantioselective decarboxylation reaction.
Co-reporter:Henri Brunner;Irmgard Deml;Wolfgang Dirnberger;Karl-Peter Ittner;Walter Reißer;Markus Zimmermann
European Journal of Inorganic Chemistry 1999 Volume 1999(Issue 1) pp:
Publication Date(Web):23 DEC 1998
DOI:10.1002/(SICI)1099-0682(199901)1999:1<51::AID-EJIC51>3.0.CO;2-Y
Allylation of 1-methyl-5-(1′-methylpent-2′-ynyl)barbituric acid (MBS) with allyl acetate using in situ catalysts of palladium(II) acetylacetonate and chiral phosphane imine ligands resulted in the enantioselective formation of 5-allyl-1-methyl-5-(1′-methylpent-2′-ynyl)barbituric acid (Methohexital), an important anesthetic drug. Both, MBS and Methohexital contain two stereogenic carbon atoms. In MBS, the asymmetric centre in the barbiturate system is labile due to enolization. The asymmetric centre in the hexyne side chain is stable and racemic. The two asymmetric centres of Methohexital are stable and give rise to four stereoisomers, two diastereomeric racemates. An analysis of the isomers of MBS and Methohexital was established on the basis of 1H NMR and, in particular, GC including a base-line separation of the four stereoisomers of Methohexital. The stereoselectivity of the allylation is difficult to control, because the new quaternary asymmetric centre in the barbiturate ring of Methohexital is formed within the nucleophile, attacking the η3-allyl ligand of the catalyst from the side opposite to the palladium atom. Classical optically active ligands, such as diop or norphos, give only 2–6 % ee. Chiral phosphane imine ligands are a successful class of compounds, synthesized by Schiff base condensation of (2-formylphenyl)diphenylphosphane with optically active primary amines. The most efficient ligands have a hydroxymethyl and a bulky alkyl substituent at the asymmetric centre in the imine part, e.g. the L-iso-leucinol and the L-tert-leucinol derivatives 5 and 7. In the Pd-catalyzed allylation of MBS a kinetic resolution and the effect of the enantioselective catalyst interplay, the contributions of which are separated. For MBS the best stereoselectivity factor of the kinetic resolution s = kR/kS was 2.6 and 83 % “ee” were achieved. The corresponding values for Methohexital were s = 3.5 and 80 % ee in the α-dl pair. For 10 mixtures of Methohexital stereoisomers the anesthetic doses for rats were determined. With 9.1 mg/kg body weight of the animal the sample obtained from the catalysis with the D-α-phenylglycinol derivative 8 gave a much lower anesthetic dose than the widely used narcotic Brevimytal®Natrium, the sodium salt of the α-dl racemate of Methohexital, with 13.0 mg/kg body weight.
Co-reporter:Henri Brunner
Angewandte Chemie 1999 Volume 111(Issue 9) pp:
Publication Date(Web):28 APR 1999
DOI:10.1002/(SICI)1521-3757(19990503)111:9<1248::AID-ANGE1248>3.0.CO;2-K
Chirale Übergangsmetallatome treten nicht nur in Tris(chelat)komplexen [M(LL)3]n+ auf, die bereits von Alfred Werner in die Enantiomere gespalten wurden, sondern auch in metallorganischen Halbsandwichkomplexen wie 1 mit Dreibein- oder Vierbein-Klavierstuhl-Struktur. Die Verbindungen haben sich bei der Aufklärung des räumlichen Ablaufs von Reaktionen und in der organischen Synthese bewährt. Anwendungen in der enantioselektiven Katalyse beginnen sich abzuzeichnen.
Co-reporter:Henri Brunner
Angewandte Chemie International Edition 1999 Volume 38(Issue 9) pp:
Publication Date(Web):12 MAY 1999
DOI:10.1002/(SICI)1521-3773(19990503)38:9<1194::AID-ANIE1194>3.0.CO;2-X
Chiral transition metal atoms are not only present in tris-chelate complexes [M(LL)3]n+, which were already resolved into the enantiomers by Alfred Werner, but also in organometallic half-sandwich complexes such as 1 with three- or four-legged piano-stool structure. These complexes have been tools in the elucidation of the spatial course of reactions and in organic syntheses. Applications in enantioselective catalysis are beginning to show up.
Co-reporter:Henri Brunner;Josef Breu;Peter Faustmann
European Journal of Inorganic Chemistry 1998 Volume 1998(Issue 12) pp:
Publication Date(Web):23 DEC 1998
DOI:10.1002/(SICI)1099-0682(199812)1998:12<1871::AID-EJIC1871>3.0.CO;2-D
The bond lengths Co–C(CO), Co–N(NO) and angles L–Co–C(CO), L–Co–N(NO) in six tetrahedral complexes, determined by X-ray crystallography, were analysed and compared with the calculated compound Co(CO)2(NO)PH3. Distinct differences were found which allow a differentiation of the two similar ligands CO and NO. These differences are used to assign CO and NO ligands in cases where this has not been possible before including a structure in which the two independent molecules in the unit cell are diastereomers. The relationship seems to hold not only for tetrahedral compounds but also for other polyhedral coordination types.
Co-reporter:Henri Brunner;Thomas Neuhierl;Bernhard Nuber
European Journal of Inorganic Chemistry 1998 Volume 1998(Issue 12) pp:
Publication Date(Web):23 DEC 1998
DOI:10.1002/(SICI)1099-0682(199812)1998:12<1877::AID-EJIC1877>3.0.CO;2-E
Condensation of (R)-2-aminobutanol with salicylaldehyde and 2-pyrrolecarbaldehyde gave the chiral chelate ligands HLL1* and HLL2*, respectively. The diastereomeric complexes (RRu,RC)- and (SRu,RC)-[(η6-arene)Ru(LL1*)Cl], η6-arene = p-cymene (1a/1b), η6-arene = benzene (2a/2b), and (RRu,RC)- and (SRu,RC)-[(η6-arene)Ru(LL2*)Cl], η6-arene = p-cymene (3a/3b), η6-arene = benzene (4a/4b), which only differ in the ruthenium configuration, were prepared by the reaction of [(η6-arene)RuCl2]2 with the anion of the corresponding ligand HLL*. X-ray analyses of 1a/1b and 3a/3b showed a structural peculiarity. The unit cell of these complexes contained diastereomers with the same configuration at the carbon atoms but opposite configuration at the metal centers in a 1:1 ratio. Weak intramolecular O–H···Cl hydrogen bridges were formed in all the complexes. 1H-NMR studies demonstrated the configurational lability at the Ru center. The iodo complexes (RRu,RC)- and (SRu,RC)-[(η6-p-cymene)Ru(LL*)I], LL* = LL1* (5a/5b) and LL* = LL2* (6a/6b), were synthesized by halogen exchange.
Co-reporter:Henri Brunner;Reinhard Störiko
European Journal of Inorganic Chemistry 1998 Volume 1998(Issue 6) pp:
Publication Date(Web):16 JUN 1999
DOI:10.1002/(SICI)1099-0682(199806)1998:6<783::AID-EJIC783>3.0.CO;2-M
Novel N′-methylated 2-(oxazolin-2-yl)-4,4′-bipyridinium salts, bearing a chiral oxazoline moiety, were tested in the Rh(I)-catalysed enantioselective hydrosilylation. After coordination to rhodium these electron-attracting ligands are supposed to exhibit charge-transfer effects with electron-donating substrates. Therefore, a new catalytic hydrosilylation reaction with 2,5-dimethoxyacetophenone as an electron-rich substrate was developed. The results were compared with those of the non-methylated 2-(oxazolin-2-yl)-4,4′-bipyridine and related ligands. In addition, the new ligands and Rh(I)-complexes were tested in the hydrosilylation of acetophenone.
Co-reporter:Henri Brunner;Reinhard Störiko;Frank Rominger
European Journal of Inorganic Chemistry 1998 Volume 1998(Issue 6) pp:
Publication Date(Web):16 JUN 1999
DOI:10.1002/(SICI)1099-0682(199806)1998:6<771::AID-EJIC771>3.0.CO;2-Y
Novel 2-(4,4′-bipyridin-2-yl)oxazolines, bearing a chiral oxazoline moiety, were synthesised starting from 4,4′-bipyridine and selectively monomethylated in the N′-position. After coordination to rhodium these electron-poor ligands are supposed to exhibit charge-transfer effects with electron-donating substrates in the Rh(I)-catalysed enantioselective hydrosilylation (see next publication). Similar effects were expected from 4,4′-bipyridine- and pyrazine-bisoxazolines after complexation with rhodium. For comparison 2-(4-phenylpyridin-2-yl)oxazoline ligands were synthesised. Rh(I)-complexes of selected ligands were prepared and characterised, including an X-ray structure analysis.
Co-reporter:Henri Brunner;Manfred Muschiol;Thomas Neuhierl;Bernhard Nuber
Chemistry - A European Journal 1998 Volume 4(Issue 1) pp:
Publication Date(Web):14 DEC 1998
DOI:10.1002/(SICI)1521-3765(199801)4:1<168::AID-CHEM168>3.0.CO;2-N
The two hydride ligands in [Cp2MoH2] and [Cp2WH2] are directed towards the coordination center in silver(I) halide complexes such as 1. X-ray crystal structure analyses of several of these complexes with the [Cp2MoH2] ligand illustrate the diverse range of structures available.
Co-reporter:Henri Brunner and Takashi Tsuno
Dalton Transactions 2017 - vol. 46(Issue 15) pp:NaN5109-5109
Publication Date(Web):2017/03/27
DOI:10.1039/C7DT00474E
In half-sandwich compounds of the type [Cp*ML1L2PPh3] the PPh3 propeller is stabilized by attractive CH/π interactions in which Co–H bonds specifically interact with the Ci and Co atoms of neighbouring phenyl rings, as in the T-shaped benzene dimer (i/o = ipso/ortho). This stabilization was not taken into account in a recent conformational analysis based on van der Waals energy calculations and minimization of steric compression (Dalton Trans., 2015, 44, 5451–5466). It is shown that in all 116 structures discussed in this analysis the CoH–Ci/o distances fall below the sum of the van der Waals radii, establishing attractive CH/π interactions, although the short contacts could easily be avoided by phenyl rotation to relieve steric strain. In 53 of the described structures there are acyl substituents which form conformation-determining Co–H⋯O(acyl) hydrogen bonds that are not taken into account in the recent analysis. The steric-only model is not an adequate description of M–PPh3 complexes.