Co-reporter:Yubo Cui, Raghavan Balachandran, Billy W. Day, and Paul E. Floreancig
The Journal of Organic Chemistry 2012 Volume 77(Issue 5) pp:2225-2235
Publication Date(Web):February 13, 2012
DOI:10.1021/jo2023685
The synthesis of neopeltolide analogues that contain variations in the oxazole-containing side chain and in the macrolide core are reported along with the GI50 values for these compounds against MCF-7, HCT-116, and p53 knockout HCT-116 cell lines. Although biological activity is sensitive to changes in the macrocycle and the side chain, several analogues displayed GI50 values of <25 nM. Neopeltolide and several of the more potent analogues were significantly less potent against p53 knockout cells, suggesting that p53 plays an auxiliary role in the activity of these compounds.
Co-reporter:Shuangyi Wan ; Fanghui Wu ; Jason C. Rech ; Michael E. Green ; Raghavan Balachandran ; W. Seth Horne ; Billy W. Day ;Paul E. Floreancig
Journal of the American Chemical Society 2011 Volume 133(Issue 41) pp:16668-16679
Publication Date(Web):September 8, 2011
DOI:10.1021/ja207331m
The potent cytotoxins pederin and psymberin have been prepared through concise synthetic routes (10 and 14 steps in the longest linear sequences, respectively) that proceed via a late-stage multicomponent approach to construct the N-acyl aminal linkages. This route allowed for the facile preparation of a number of analogs that were designed to explore the importance of the alkoxy group in the N-acyl aminal and functional groups in the two major subunits on biological activity. These analogs, including a pederin/psymberin chimera, were analyzed for their growth inhibitory effects, revealing several new potent cytotoxins and leading to postulates regarding the molecular conformational and hydrogen bonding patterns that are required for biological activity. Second generation analogs have been prepared based on the results of the initial assays and a structure-based model for the binding of these compounds to the ribosome. The growth inhibitory properties of these compounds are reported. These studies show the profound role that organic chemistry in general and specifically late-stage multicomponent reactions can play in the development of unique and potent effectors for biological responses.
Co-reporter:Wei Zhu ; María Jiménez ; Won-Hyuk Jung ; Daniel P. Camarco ; Raghavan Balachandran ; Andreas Vogt ; Billy W. Day ;Dennis P. Curran
Journal of the American Chemical Society 2010 Volume 132(Issue 26) pp:9175-9187
Publication Date(Web):June 14, 2010
DOI:10.1021/ja103537u
The dictyostatins are a promising class of potential anti-cancer drugs because they are powerful microtubule-stabilizing agents, but the complexity of their chemical structures is a severe impediment to their further development. On the basis of both synthetic and medicinal chemistry analyses, 16-desmethyl-25,26-dihydrodictyostatin and its C6 epimer were chosen as potentially potent yet accessible dictyostatin analogues, and three new syntheses were developed. A relatively classical synthesis involving vinyllithium addition and macrocyclization gave way to a newer and more practical approach based on esterification and ring-closing metathesis reaction. Finally, aspects of these two approaches were combined to provide a third new synthesis based on esterification and Nozaki−Hiyama−Kishi reaction. This was used to prepare the target dihydro analogues and the natural product. All of the syntheses are streamlined because of their high convergency. The work provided several new analogues of dictyostatin, including a truncated macrolactone and a C10 E-alkene, which were 400- and 50-fold less active than (−)-dictyostatin, respectively. In contrast, the targeted 16-desmethyl-25,26-dihydrodictyostatin analogues retained almost complete activity in preliminary biological assays.
Co-reporter:Yun Fan, Emanuel M. Schreiber and Billy W. Day
Journal of Natural Products 2009 Volume 72(Issue 10) pp:1748-1754
Publication Date(Web):September 23, 2009
DOI:10.1021/np900245k
The polyketide natural product (+)-discodermolide is a potent microtubule stabilizer that has generated considerable interest in its synthetic, medicinal, and biological chemistry. It progressed to early clinical oncology trials, where it showed some efficacy in terms of disease stabilization but also some indications of causing pneumotoxicity. Remarkably, there are no reports of its metabolism. Here, we examined its fate in mixed human liver microsomes. Due to limited availability of the agent, we chose a nanoflow liquid chromatography-electrospray ionization-mass spectrometry analytical approach employing quadrupolar ion trap and quadrupole−quadrupole-time-of-flight instruments for these studies. (+)-Discodermolide was rapidly converted to eight metabolites, with the left-side lactone (net oxidation) and the right-side diene (epoxidation followed by hydrolysis, along with an oxygen insertion product) being the most metabolically labile sites. Other sites of metabolism were the allylic and pendant methyl moieties in the C12−C14 region of the molecule. The results provide information on the metabolic soft spots of the molecule and can be used in further medicinal chemistry efforts to optimize discodermolide analogues.
Co-reporter:Miranda J. Sarachine, Jelena M. Janjic, Peter Wipf, Billy W. Day
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 9) pp:2404-2408
Publication Date(Web):1 May 2009
DOI:10.1016/j.bmcl.2009.03.075
The C-cyclopropylalkylamide scaffold was previously identified as a new structural framework for antiestrogens. A second generation library provided three compounds that bind estrogen receptor (ER)α. Further screening of this library identified an ERβ hit and inspired another round of SAR. A new focused library was tested for binding to the ERs, and for effects on the growth of breast cancer cell lines and protein levels of common cell cycle regulators.
Co-reporter:Hikmat N. Daghestani, David G. Fernig, Billy W. Day
Biosensors and Bioelectronics 2009 Volume 25(Issue 1) pp:136-141
Publication Date(Web):15 September 2009
DOI:10.1016/j.bios.2009.06.014
Dual polarization interferometry (DPI) and resonant mirror (RM) methods were used to characterize the growth of microtubules (MTs) on biosensor surfaces. The structure and dynamics of MTs play an important role in cell division and are a target for many anti-cancer drugs. Evidence from DPI demonstrated the growth of MTs on streptavidin–biotinylated-tubulin surfaces from the increase in mass and thickness, with a simultaneous decrease in density. The initial increase in thickness of 0.236 nm/min suggested the elongation of protofilaments before they join laterally to form the MT, where the rate of growth increased to 0.436 nm/min. Continuous mass increases were also observed when tubulin was added to a similar underlying RM surface. Tubulin binding to these surfaces was also temperature dependent, increasing the absolute response with MT stabilizers, while inhibiting binding with destabilizers when temperature was changed from 15 to 37 °C. Finally, the initial rates of tubulin assembly (mean ± SD, n = 3) with MT-stabilizer agents were significantly higher at 1.50 ± 0.27 and 1.04 ± 0.13 arcseconds/s, respectively, compared to 0.37 ± 0.11 arcseconds/s for tubulin containing GTP only. In the presence of the MT destabilizers, colchicine and dolastatin 10, the slopes of initial rates were lower than in their absence at 0.05 ± 0.01 and 0.27 ± 0.08 arcseconds/s, respectively. This provides evidence for the ability of surface-based optical sensors to distinguish between MT stabilizers and destabilizers, while also paving the path to develop other methods to screen for MT-perturbing agents using the same underlying surface engineering.
Co-reporter:Raghavan Balachran;Tamara D. Hopkins;Catherine A. Thomas;Peter Wipf
Chemical Biology & Drug Design 2008 Volume 71( Issue 2) pp:117-124
Publication Date(Web):
DOI:10.1111/j.1747-0285.2007.00616.x
Several natural and synthetic naphthoquinone spiroketals are potent inhibitors of the thioredoxin–thioredoxin reductase redox system. Based on the antimitotic and weak antitubulin actions noted for SR-7 ([8-(furan-3-ylmethoxy)-1-oxo-1,4-dihydronaphthalene-4-spiro-2′-naphtho[1″,8″-de][1′,3′][dioxin]), a library of related compounds was screened for tubulin-perturbing properties. Two compounds, TH-169 (5′-hydroxy-4′H-spiro[1,3-dioxolane-2,1′-naphthalen]-4′-one) and TH-223 (5′-methoxy-4′H-spiro[1,3-dioxane-2,1′-naphthalen]-4′-one), had substantial effects on tubulin assembly and were antiproliferative at low micromolar concentrations. TH-169 was the most potent at blocking GTP-dependent polymerization of 10 μm tubulin in vitro with a remarkable 50% inhibitory concentration of ca. 400 nm. It had no effect on paclitaxel-induced microtubule assembly and did not cause microtubule hypernucleation. TH-169 failed to compete with colchicine for binding to β-tubulin. The 50% antiproliferative concentration of TH-169 against human cancer cells was at or slightly below 1 μm. Flow cytometry showed that 1 μm TH-169 caused an increase in G2/M and hypodiploid cells. TH-169 eliminated the PC-3 cells’ polyploid population and increased their expression of p21WAF1 and Hsp70 in a concentration-dependent manner. The antiproliferative effect of TH-169 was irreversible and independent of changes in caspases, actin, tubulin, glyceraldehyde phosphate dehydrogenase or Bcl-xS/L. This structurally simple naphthoquinone spiroketal represents a small molecule, tubulin-interactive agent with a novel apoptotic pathway and attractive biological function.
Co-reporter:Jelena M. Janjic, Ying Mu, Christopher Kendall, Corey R.J. Stephenson, Raghavan Balachandran, Brianne S. Raccor, Ying Lu, Guangyu Zhu, Wen Xie, Peter Wipf, Billy W. Day
Bioorganic & Medicinal Chemistry 2005 Volume 13(Issue 1) pp:157-164
Publication Date(Web):3 January 2005
DOI:10.1016/j.bmc.2004.09.048
A new structural scaffold for antiestrogens was identified from the cell-based screening of transcriptional regulation properties of a 67-member library of homoallylic amides, allylic amides, and C-cyclopropylalkylamides. C-Cyclopropylalkylamide 3a (O-ethyl-N-{2-[(1S*,2R*)-2-{(R*)-[(diphenylphosphinoyl)amino](phenyl)methyl}cyclopropyl]ethyl}-N-[(4-methylphenyl)sulfonyl]carbamate) had antagonistic activity similar to that of tamoxifen and was further evaluated. Compound 3a inhibited estradiol-induced proliferation of the ER-positive MCF-7 cells but had no effect on ER-negative MDA-MB231 human breast cancer cells. Furthermore, high micromolar concentrations of 3a exhibited minimal cytotoxicity to the ER-negative line. The biological activities of the enantiomers of 3a did not differ from one another nor from that of racemic 3a.A new structural scaffold for antiestrogens was identified from the cell-based screening of a 67-member library of homoallylic amides, allylic amides, and C-cyclopropylalkylamides. Several derivatives had activity comparable to that of tamoxifen.
Co-reporter:Raghavan Balachandran;Manda J Welsh;Billy W Day
Oncogene 2003 22(55) pp:8924-8930
Publication Date(Web):2003-12-04
DOI:10.1038/sj.onc.1207060
Two paclitaxel(Ptx)-resistant ovarian cancer cell lines, 1A9/Ptx-10 and 1A9/Ptx-22, isolated from the 1A9 cell line (a clone of the A2780 line) by continuous exposure to Ptx and verapamil, have point mutations in their major -tubulin gene and in one or both alleles of their TP53 gene. These cells were examined for alterations in cell cycle regulators and the tubulin-binding protein stathmin. Unlike parental cells, neither 1A9/Ptx-10 nor 1A9/Ptx-22 expressed detectable levels of p21WAF1/Cip1, a putative transcriptional regulator of stathmin, but did overexpress stathmin and Bcl2. No differences were noted in the expression levels of proliferative cell nuclear antigen or tyrosine-phosphorylated p34Cdc2. Ptx treatment altered little the expression of stathmin in the parental cell line, although it increased p21WAF1/Cip1 levels several-fold. Infection of Ptx-resistant lines with a wild-type TP53-bearing adenovirus (AdWTp53) changed cell cycle distribution and increased the levels of p21WAF1/Cip1, but caused no changes in stathmin levels. Microtubule drug resistance in ovarian carcinoma may be associated with altered p53/21WAF1/Cip1 regulatory pathways for stathmin expression and function.
Co-reporter:Billy W. Day, Cyrous O. Kangani, Kwasi S. Avor
Tetrahedron: Asymmetry 2002 Volume 13(Issue 11) pp:1161-1165
Publication Date(Web):21 June 2002
DOI:10.1016/S0957-4166(02)00261-6
Two new highly stereoselective routes to (2R,3S,4R)-3-(tert-butyldimethylsilanyloxy)-2,4-dimethyl-5-oxopentanoic acid methoxymethylamide, an important intermediate in natural product synthesis, are described. Both schemes are considerably shorter and less expensive than methods previously reported. The title compound was then converted to direct precursors of C1–C7 and C17–24 fragments of the potent microtubule stabilizer (+)-discodermolide.Graphic(3R,4S,5S)-4-(tert-Butyldimethylsilanyloxy)-3,5-dimethyltetrahydropyran-2-oneC13H26O3SiMp=55–55.5°C (pentane)[α]D18=+20.4 (c 0.22, CHCl3)Source of chirality: Evans’ auxiliaryAbsolute configuration: 3R,4S,5S(2R,3S,4S,5Z)-3-(tert-Butyldimethylsilanyloxy)-6-iodo-2,4-dimethylhex-5-enoic acid methoxymethylamideC16H32NO3SiI[α]D18=+65.9 (c 1.0, CHCl3)Source of chirality: Evans’ auxiliaryAbsolute configuration: 2R,3S,4S,5Z(2R,3S,4S,5Z)-3-(tert-Butyldimethylsilanyloxy)-2,4-dimethylocta-5,7-dienoic acid methoxymethylamideC18H35NO3Si[α]D18=+53.2 (c 0.01, CHCl3)Source of chirality: Evans’ auxiliaryAbsolute configuration: 2R,3S,4S,5Z(2R,3S,4S,5Z)-3-(tert-Butyldimethylsilanyloxy)-2,4-dimethylocta-5,7-dienalC16H30O2Si[α]D18=−16.7 (c 1.30, CHCl3)Source of chirality: Evans’ auxiliaryAbsolute configuration: 2R,3S,4S,5Z

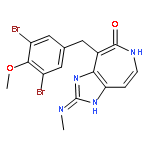
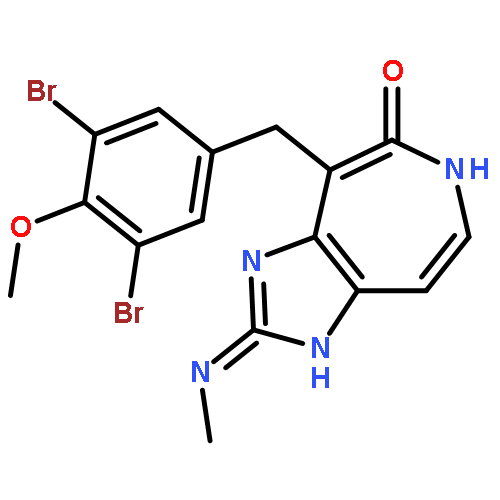
![4-[(3,5-dibromo-4-methoxyphenyl)methyl]-6-methyl-2-(methylamino)imidazo[4,5-d]azepin-5-one](http://img.cochemist.com/ccimg/634200/634151-15-8.png)
![4-[(3,5-dibromo-4-methoxyphenyl)methyl]-6-methyl-2-(methylamino)imidazo[4,5-d]azepin-5-one](http://img.cochemist.com/ccimg/634200/634151-15-8_b.png)






![Benzonitrile, 4-[(E)-(1,3-dihydro-1-oxo-2H-inden-2-ylidene)methyl]-](http://img.cochemist.com/ccimg/59200/59120-86-4.png)
![Benzonitrile, 4-[(E)-(1,3-dihydro-1-oxo-2H-inden-2-ylidene)methyl]-](http://img.cochemist.com/ccimg/59200/59120-86-4_b.png)
![1H-Inden-1-one, 2-[(4-fluorophenyl)methylene]-2,3-dihydro-, (2E)-](http://img.cochemist.com/ccimg/59100/59082-20-1.png)
![1H-Inden-1-one, 2-[(4-fluorophenyl)methylene]-2,3-dihydro-, (2E)-](http://img.cochemist.com/ccimg/59100/59082-20-1_b.png)
![1H-Inden-1-one, 2-[(4-chlorophenyl)methylene]-2,3-dihydro-, (2E)-](http://img.cochemist.com/ccimg/17500/17434-26-3.png)
![1H-Inden-1-one, 2-[(4-chlorophenyl)methylene]-2,3-dihydro-, (2E)-](http://img.cochemist.com/ccimg/17500/17434-26-3_b.png)
![1H-Inden-1-one, 2,3-dihydro-2-[(4-methoxyphenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/17500/17434-24-1.png)
![1H-Inden-1-one, 2,3-dihydro-2-[(4-methoxyphenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/17500/17434-24-1_b.png)
![1H-Inden-1-one, 2,3-dihydro-2-[(4-methylphenyl)methylene]-, (2E)-](http://img.cochemist.com/ccimg/17500/17434-22-9.png)
![1H-Inden-1-one, 2,3-dihydro-2-[(4-methylphenyl)methylene]-, (2E)-](http://img.cochemist.com/ccimg/17500/17434-22-9_b.png)



