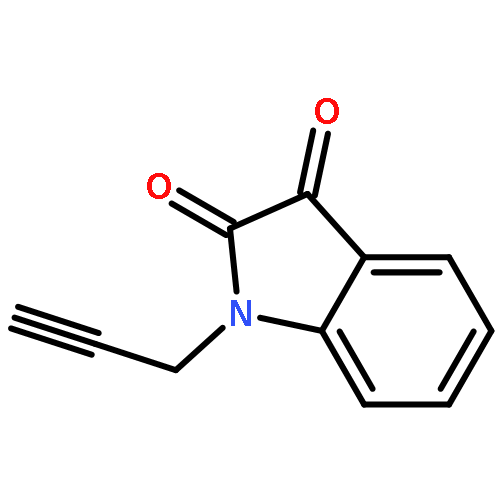
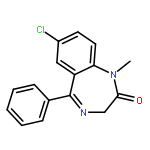
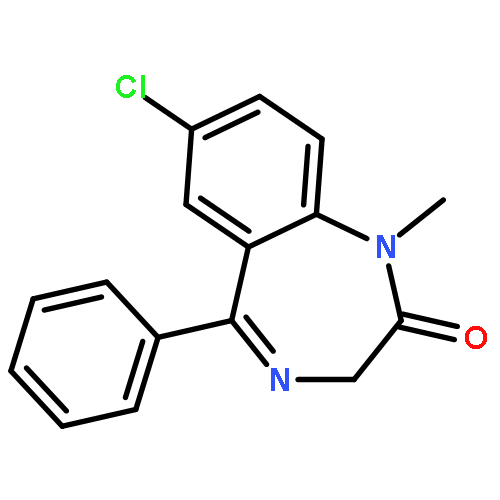
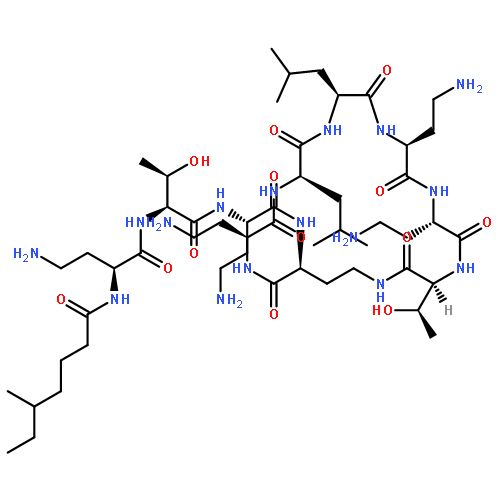
Co-reporter:Zhong-Hua Li, Xue-Qi Liu, Peng-Fei Geng, Feng-Zhi Suo, Jin-Lian Ma, Bin Yu, Tao-Qian Zhao, Zhao-Qing Zhou, Chen-Xi Huang, Yi-Chao Zheng, and Hong-Min Liu
ACS Medicinal Chemistry Letters April 13, 2017 Volume 8(Issue 4) pp:384-384
Publication Date(Web):March 6, 2017
DOI:10.1021/acsmedchemlett.6b00423
Lysine specific demethylase 1 (LSD1) plays a pivotal role in regulating the lysine methylation. The aberrant overexpression of LSD1 has been reported to be involved in the progression of certain human malignant tumors. Abrogation of LSD1 with RNAi or small molecule inhibitors may lead to the inhibition of cancer proliferation and migration. Herein, a series of [1,2,3]triazolo[4,5-d]pyrimidine derivatives were synthesized and evaluated for their LSD1 inhibitory effects. The structure–activity relationship studies (SARs) were conducted by exploring three regions of this scaffold, leading to the discovery of compound 27 as potent LSD1 inhibitor (IC50 = 0.564 μM). Compound 27 was identified as a reversible LSD1 inhibitor and showed certain selectivity to LSD1 over monoamine oxidase A/B (MAO-A/B). When MGC-803 cells were treated with compound 27, the activity of LSD1 can be significantly inhibited, and the cell migration ability was also suppressed. Docking studies indicated that the hydrogen interaction between the nitrogen atom in the pyridine ring and Met332 could be responsible for the improved activity of 2-thiopyridine series. The [1,2,3]triazolo[4,5-d]pyrimidine scaffold can be used as the template for designing new LSD1 inhibitors.Keywords: LSD1 inhibitor; migration inhibition; molecular docking; [1,2,3]Triazolo[4,5-d]pyrimidine;
Co-reporter:Zhong-Hua Li;Xue-Qi Liu;Peng-Fei Geng;Jin-Lian Ma;Tao-Qian Zhao;Hao-Ming Wei;Bin Yu
MedChemComm (2010-Present) 2017 vol. 8(Issue 8) pp:1655-1658
Publication Date(Web):2017/08/16
DOI:10.1039/C7MD00165G
A series of thiazolo[5,4-d]pyrimidine derivatives were synthesized and evaluated for their antiproliferative activities against several human cancer cell lines. Structure–activity relationship studies were carried out, showing that most of the target compounds had good inhibition against the tested cell lines. Among them, compound 7i exhibited potent inhibition against human gastric cancer cells MGC-803 and HGC-27 with IC50 values of 4.64 and 5.07 μM, respectively and around 12-fold selectivity between MGC-803 and GES-1, indicating a relatively low toxicity to normal cells. The potency and low toxicity of compound 7i make the thiazolo[5,4-d]pyrimidine an attractive scaffold for designing new derivatives selectively targeting MGC-803 cells.
Co-reporter:Ling Fu, Yu-Qing Wang, Bing-Kai Han, Xiao-Rui Li, Xiao-Jing Shi, Fen Yin, Jun-Wei Wang, Pei-Rong Zhao, Yu Ke, Hong-Min Liu
European Journal of Pharmacology 2017 Volume 815(Volume 815) pp:
Publication Date(Web):15 November 2017
DOI:10.1016/j.ejphar.2017.08.004
Jaridon 6, a novel ent-kaurene diterpenoid derived from Rabdosia rubescens (Hemsl.) Hara, possesses strong anti-tumor activity in esophageal cancer cells. In this study, we explored the underlying molecular events of the anti-tumor activity of Jaridon 6. Cell viability and apoptosis results obtained by flow cytometry confirmed the tumor inhibitory effect of Jaridon 6 in esophageal cancer cells. A cDNA microarray was performed and the observations were validated using quantitative reverse transcription polymerase chain reaction. The microarray data showed that 151 genes were differentially expressed between the untreated group and the Jaridon 6-treated group, among these were 57 upregulated genes, and 94 downregulated genes (P < 0.01, fold change threshold: 2). These included genes such as Wnt, peroxisome, and genes involved in chemokine signaling pathways. In addition, Western blot analysis demonstrated that Jaridon 6 regulated the expression of Wnt pathway proteins, including reduced levels of Dvl 2, survivin and cyclin D1, and increased levels of p-β-catenin, and AXIN2 in EC109 and EC9706 esophageal cancer cells. In addition, recombinant murine Wnt3a could change the regulation of Jaridon 6 on Wnt pathway proteins. Immunohistochemical analysis indicated that the anti-tumor activity of Jaridon 6 was closely related to the Wnt signaling pathway in esophageal cancer cells.Download high-res image (220KB)Download full-size image
Co-reporter:Zhong-Hua Li, Xue-Qi Liu, Peng-Fei Geng, Ji Zhang, Jin-Lian Ma, Bo Wang, Tao-Qian Zhao, Bing Zhao, Xin-Hui Zhang, Bin Yu, Hong-Min Liu
European Journal of Medicinal Chemistry 2017 Volume 138(Volume 138) pp:
Publication Date(Web):29 September 2017
DOI:10.1016/j.ejmech.2017.07.039
•The thiazolo[5,4-d]pyrimidine derivatives showed moderate to good growth inhibition against the tested cancer cells.•Among them, compound 22 exerted the most antiproliferative activity against HGC-27 cell line.•Compound 22 inhibited the cell colony formation and migration of HGC-27.•Compound 22 induced the apoptosis of HGC-27 cells, and led to the expression changes of key proteins related to apoptosis.•The atom replacement strategy would be viable for designing new anticancer agents.A series of thiazolo[5,4-d]pyrimidine derivatives were designed through the atom replacement strategy based on biologically validated scaffolds and then evaluated for their antiproliferative activities on cancer cell lines. The structure-activity relationship studies were conducted, leading to the identification of compound 22, which exhibited good antiproliferative activity against HGC-27 with an IC50 value of 1.22 μM and low toxicity against GES-1 cells. Mechanistic studies showed that compound 22 inhibited the colony formation and migration of HGC-27 as well as induced apoptosis. The western blot experiments proved that compound 22 up-regulated expression of Bax, down-regulated expression levels of Bcl-2 and cleaved caspased-3/9. These findings indicate that compound 22 may serve as a template for designing new agents for the treatment of human gastric cancers. The atom replacement strategy could be viable strategy for designing new anticancer drugs and may find its applications in drug design.Download high-res image (175KB)Download full-size image
Co-reporter:Dong-Jun Fu, Jian Song, Yu-Hui Hou, Ruo-Han Zhao, Jia-Huan Li, Ruo-Wang Mao, Jia-Jia Yang, Ping Li, Xiao-Lin Zi, Zhong-Hua Li, Qing-Qing Zhang, Fei-Yan Wang, Sai-Yang Zhang, Yan-Bing Zhang, Hong-Min Liu
European Journal of Medicinal Chemistry 2017 Volume 138(Volume 138) pp:
Publication Date(Web):29 September 2017
DOI:10.1016/j.ejmech.2017.07.011
•SAR for this 5,6-diaryl-1,2,4-triazine scaffold is explored.•11E inhibited colony formation and arrested cell cycle at G2/M phase.•11E caused morphological changes and decreased mitochondrial membrane potential.•5,6-Diaryl-1,2,4-triazine hybrids were discovered as novel apoptosis inducers.A series of 5,6-diaryl-1,2,4-triazines hybrids bearing a 1,2,3-triazole linker were synthesized by molecular hybridization strategy and evaluated for antiproliferative activity against three selected cancer cell lines (MGC-803, EC-109 and PC-3). The first structure-activity relationship (SAR) for these 5,6-diaryl-1,2,4-triazines is explored in this report with evaluation of 15 variants of the structural class. Among these chemical derivatives, 3-(((1-(4-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-5,6-diphenyl-1,2,4-triazine (11E) showed the more potent inhibitory effect against three cell lines than 5-Fu. Cellular mechanism studies in MGC-803 cells elucidated 11E inhibited colony formation and arrested cell cycle at G2/M phase. Furthermore, compound 11E caused morphological changes, decreased mitochondrial membrane potential, and induced apoptosis through the apoptosis-related proteins in MGC-803 cells. It was the first time, to our knowledge, that 5,6-diaryl-1,2,4-triazines bearing a 1,2,3-triazole linker were used as potential apoptosis inducers.Download high-res image (263KB)Download full-size image
Co-reporter:Zhong-Hua Li, Ji Zhang, Xue-Qi Liu, Peng-Fei Geng, Jin-Lian Ma, Bo Wang, Tao-Qian Zhao, Bing Zhao, Hao-Ming Wei, Chao Wang, Dong-Jun Fu, Bin Yu, Hong-Min Liu
European Journal of Medicinal Chemistry 2017 Volume 135(Volume 135) pp:
Publication Date(Web):28 July 2017
DOI:10.1016/j.ejmech.2017.04.056
•The thiazolo[5,4-d]pyrimidine derivatives showed potent inhibition against the cancer cells.•Compound 24 exerted the most antiproliferative activity against MGC-803 cells.•Compound 24 inhibited the cell colony formation and migration of MGC803.•Compound 24 induced the apoptosis of MGC-803 cells.A series of thiazolo[5,4-d]pyrimidine derivatives were synthesized and evaluated for their antiproliferative activities on three cancer cell lines. The structure-activity relationship studies were conducted through the variation in the three regions of the thiazolo-pyrimidine core. Substitution with morpholine led to compound 24, which exerted the most potent antiproliferative activity as well as good selectivity between cancer and normal cells (IC50 values of 1.03 μM against MGC803 and 38.95 μM against GES-1). In addition, compound 24 inhibited the colony formation and migration of MGC803 as well as induced apoptosis. Western blot experiments indicated the expression changes of apoptosis-related proteins, including up-regulation of Bax and caspase-3/9, as well as down-regulation of Bcl-2.A series of thiazolo[5,4-d]pyrimidine derivatives were designed through the drug repurposing strategy. Compound 24 displayed high potency and selectivity against MGC-803 cells, and induced the apoptosis of MGC-803 cells.Download high-res image (170KB)Download full-size image
Co-reporter:Ying-Chao Duan, Yuan-Yuan Guan, Xiao-Yu Zhai, Li-Na Ding, Wen-Ping Qin, Dan-Dan Shen, Xue-Qi Liu, Xu-Dong Sun, Yi-Chao Zheng, Hong-Min Liu
European Journal of Medicinal Chemistry 2017 Volume 126(Volume 126) pp:
Publication Date(Web):27 January 2017
DOI:10.1016/j.ejmech.2016.11.035
•A series of resveratrol derivatives were discovered as novel LSD1 inhibitors.•Most compounds exhibited potent inhibitory activities against recombinant LSD1.•Compounds 4e and 4m elevated the level of H3K4Me2 significantly in MGC-803 cells.•Compounds 4e and 4m increased the expression level of CD86 mRNA in MGC-803 cells.Inhibition of lysine-specific demethylase 1 (LSD1) has recently emerged as an attractive therapeutic target for treating cancer and other diseases. As a continuity of our ongoing effort to identify novel small-molecule LSD1-inhibitors, we designed and synthesized a series of resveratrol derivatives, which were shown to be potent inhibitors of LSD1. Among them, compounds 4e and 4m displayed the most potent LSD1-inhibitory activities in enzyme assays, with IC50 values of 121 nM and 123 nM, respectively. Biochemistry study and docking analysis indicated that compounds 4e and 4m were reversible LSD1 inhibitors. High content analysis showed that 4e and 4m induced a dose-dependent increase of dimethylated Lys4 of histone H3 and had no impact on the expression of LSD1 in MGC-803 cells. Furthermore, 4e or 4m could remarkably increase the mRNA level of CD86, a surrogate cellular biomarker for LSD1 activity, in MGC-803 cells, suggesting that they are likely to exhibit LSD1-inhibitory activities intracellularly. These findings should encourage further modification of these compounds to produce more potent LSD1 inhibitors with potential anticancer activity.Download high-res image (162KB)Download full-size image
Co-reporter:Kai Sun, Jia-Di Peng, Feng-Zhi Suo, Ting Zhang, Yun-Dong Fu, Yi-Chao Zheng, Hong-Min Liu
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 22(Issue 22) pp:
Publication Date(Web):15 November 2017
DOI:10.1016/j.bmcl.2017.10.003
Lysine specific demethylase 1 (LSD1), the first identified histone demethylase, plays an important role in epigenetic regulation of gene activation and repression, has been reported to be up-regulated and involved in numbers of solid malignant tumors. In this study, we identified a series of phenylalanyl hydrazones based LSD1 inhibitors, and the most potent one, compound 4q, can inactivate LSD1 with IC50 = 91.83 nM. In cellular level, compound 4q can induce the accumulation of CD86 as well as H3K4me2, and inhibit gastric cancer cell proliferation by inactivating LSD1. Our findings indicated that compound 4q may serve as a potential leading compound to target LSD1 overexpressed gastric cancer.Download high-res image (96KB)Download full-size image
Co-reporter:Zhong-Hua Li, Xue-Qi Liu, Tao-Qian Zhao, Peng-Fei Geng, Ying Liu, Bing Zhao, Wen-Ge Guo, Bin Yu, Hong-Min Liu
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 18(Issue 18) pp:
Publication Date(Web):15 September 2017
DOI:10.1016/j.bmcl.2017.08.021
•The diheteroaryl thioether derivatives showed potent inhibition against the cancer cells.•Compound 5 exerted the most potent and broad-spectrum antiproliferative activity.•Compound 5 induced G2/M arrest of MKN-45 cells.•Compound 5 induced apoptosis of MKN-45 through the intrinsic apoptotic pathway.A series of structurally new diheteroaryl thioether analogs was designed, prepared and screened toward MGC-803, MKN-45, EC-109 and H1650. Most of the target compounds displayed moderate to potent antiproliferative activities. Among them, compound 5 showed the best antiproliferative activity against the tested cell lines with the half maximal inhibitory concentration (IC50) values below 10 μM. In addition, flow cytometry analysis showed that compound 5 increased Bax expression, down-regulated expression of Bcl-2, cleaved caspases-3/9, finally inducing apoptosis of MKN-45 cells as well as arrested the cell cycle at G2/M phase. This study suggests that the diheteroaryl thioethers are a class of emerging chemotypes for developing antitumor agents or biological probes, and compound 5 could serve as a good starting point to design new apoptosis inducers.Download high-res image (58KB)Download full-size image
Co-reporter:Lina Ding, Zhi-Zheng Wang, Xu-Dong Sun, Jing Yang, Chao-Ya Ma, Wen Li, Hong-Min Liu
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 15(Issue 15) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.bmcl.2017.05.065
Recently, Histone Lysine Specific Demethylase 1 (LSD1) was regarded as a promising anticancer target for the novel drug discovery. And several small molecules as LSD1 inhibitors in different structures have been reported. In this work, we carried out a molecular modeling study on the 6-aryl-5-cyano-pyrimidine fragment LSD1 inhibitors using three-dimensional quantitative structure–activity relationship (3D-QSAR), molecular docking and molecular dynamics simulations. Comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) were used to generate 3D-QSAR models. The results show that the best CoMFA model has q2 = 0.802, r2ncv = 0.979, and the best CoMSIA model has q2 = 0.799, r2ncv = 0.982. The electrostatic, hydrophobic and H-bond donor fields play important roles in the models. Molecular docking studies predict the binding mode and the interactions between the ligand and the receptor protein. Molecular dynamics simulations results reveal that the complex of the ligand and the receptor protein are stable at 300 K. All the results can provide us more useful information for our further drug design.Download high-res image (64KB)Download full-size image
Co-reporter:Panpan Song;Fei Cui;Na Li;Jingchao Xin;Qisheng Ma;Xiangchuan Meng;Chaojie Wang;Qinpo Cao;Yifei Gu;Yu Ke;Qiurong Zhang;Hongmin Liu
Chinese Journal of Chemistry 2017 Volume 35(Issue 10) pp:1633-1639
Publication Date(Web):2017/10/01
DOI:10.1002/cjoc.201700005
A series of novel 1,2,3-triazole-quinazoline derivatives were synthesized in five steps starting from anthranilamide by conventional methods. All the title compounds 10a—10r were evaluated for cytotoxic activity against four human cancer cell lines (MGC-803, EC-109, MCF-7 and HGC-27) using MTT assay in vitro. Some of the synthesized compounds exhibited moderate to potent activity against tested cancer cell lines. Among them, compounds 10 h and 10q exhibited excellent growth inhibition against HGC-27 and compound 10 m also possessed excellent activity against MCF-7, with IC50 values less than 1 µmol/L. Especially, compound 10 h was more cytotoxic than 5-fluorouracil against all tested four human cancer cell lines.
Co-reporter:Biao Hu;Bo Wang;Bing Zhao;Qian Guo;Zhong-Hua Li;Xin-Hui Zhang;Guang-Yao Liu;Ying Liu;Ying Tang;Fan Luo;Ya Du;Ya-Xin Chen;Li-Ying Ma
MedChemComm (2010-Present) 2017 vol. 8(Issue 12) pp:2173-2180
Publication Date(Web):2017/12/14
DOI:10.1039/C7MD00353F
A series of novel thiosemicarbazone derivatives were synthesized and evaluated for their antiproliferative activity against several selected tumor cell lines of different origins using the MTT assay. The preliminary results indicated that the MGC-803 cell line was remarkably sensitive to all the synthesized compounds. Among this series, compound 5n showed the best inhibitory activity with an IC50 value of 0.93 μM (about 10-fold more potent than 3-AP) against MGC-803. Further mechanism studies revealed that compound 5n could obviously inhibit the proliferation of MGC-803 cells by inducing apoptosis and arresting the cell cycle at the S phase. Compound 5n also showed marked inhibition of cell migration and invasion, without significant cytotoxicity against gastric epithelial immortalized GES-1 cells.
Co-reporter:Cong Wang, Dongxiao Yang, Liping Jiang, Saiqi Wang, Junwei Wang, Kairui Zhou, Xiaoli Shi, Liming Chang, Ying Liu, Yu Ke, Hongmin Liu
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 9(Issue 9) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.bmcl.2017.02.008
•A combination of paclitaxel and jesridonin in esophageal carcinoma was explored.•Paclitaxel combined with jesridonin showed enhanced anti-tumor activity.•This effect was via the mitochondrial-mediated apoptotic pathway.The novel compound jesridonin, has extensive anti-tumor activity. In this study, we aim to investigate the cytotoxic effects of jesridonin in combination with paclitaxel. Our results showed that jesridonin in combination with paclitaxel had synergistic cytotoxic effects on human esophageal carcinoma both in vitro and in vivo. Hoechst 33258 staining and the Annexin-V FITC assay demonstrated that paclitaxel synergized with jesridonin in a stronger induction of apoptosis than treatment with paclitaxel or jesridonin alone. Western blotting results revealed that the synergistic apoptosis-induction effects of paclitaxel and jesridonin were mediated by the mitochondrial pathway. This may provide a novel strategy to overcome drug resistance for esophageal cancer therapy.Download high-res image (38KB)Download full-size image
Co-reporter:Bo Wang, Bing Zhao, Lu-Ping Pang, Yuan-Di Zhao, Qian Guo, Jun-Wei Wang, Yi-Chao Zheng, Xin-Hui Zhang, Ying Liu, Guang-Yao Liu, Wen-Ge Guo, Chao Wang, Zhong-Hua Li, Xue-Jing Mao, Bin Yu, Li-Ying Ma, Hong-Min Liu
Pharmacological Research 2017 Volume 122(Volume 122) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.phrs.2017.05.025
Histone lysine specific demethylase 1 (LSD1) plays an important role in epigenetic modifications, and aberrant expression of LSD1 predicts tumor progression and poor prognosis in human esophageal cancers. In this study, a series of LSD1 inhibitors were synthesized and proved to be highly potent against human esophageal squamous cell carcinoma (ESCC). Our data showed that these LSD1 inhibitors selectively suppressed the viability of esophageal cancer cell line (EC-109) bearing overexpressed LSD1. Among these, compound LPE-1 (LSD1 IC50 = 0.336 ± 0.003 μM) significantly suppressed proliferation, induced apoptosis, arrested cell cycle of EC109 cells at G2/M phase, and caused changes of the associated protein markers correspondingly. We also found that compound LPE-1 potently inhibited the migration and invasion of EC-109 cells. Docking studies showed that the cyano group formed hydrogen bonds with Val811 and Thr810. Additionally, the thiophene moiety formed arene-H interaction with Trp761 residue. In vivo studies showed that compound LPE-1 inhibited tumor growth of xenograft models bearing EC-109 without obvious toxicity. Collectively, our findings indicate that LSD1 may be a potential therapeutic target in ESCC, and compound LPE-1 could serve as a lead compound for further development for anti-ESCC drug discovery.Download high-res image (242KB)Download full-size image
Co-reporter:Zhong-Hua Li, Xue-Qi Liu, Tao-Qian Zhao, Peng-Fei Geng, Wen-Ge Guo, Bin Yu, Hong-Min Liu
European Journal of Medicinal Chemistry 2017 Volume 139(Volume 139) pp:
Publication Date(Web):20 October 2017
DOI:10.1016/j.ejmech.2017.08.042
•The triazole-pyrimidine/thiourea hybrids inhibited proliferation of cancer cells.•Compound 5r exerted the most potent inhibition against the tested cancer cells.•Compound 5r induced the apoptosis and G2/M arrest of H1650 cells.•Compound 5r led to the expression changes of key proteins related to apoptosis.A series of new [1,2,3]triazolo[4,5-d]pyrimidine/thiourea hybrids were designed and synthesized through the scaffold replacement/ring cleavage strategy. SARs studies revealed that the N-heteroarene moiety attached to the thiourea is preferred over the phenyl ring for the R2 substituents, while the hydrophobic aromatic group is beneficial for improving the activity. Among these compounds, compound 5r significantly inhibited cell growth of lung cancer cell lines H1650 and A549 (IC50 = 1.91, 3.28 μM, respectively), but was less toxic against the normal cell line GES-1 (IC50 = 27.43 μM). Mechanistic studies showed that compound 5r could remarkably inhibit the colony formation of H1650 cells, induced apoptosis possibly through the intrinsic apoptotic pathways, and arrested the cell cycle at G2/M phase. Our studies suggest that the [1,2,3]triazolo[4,5-d]pyrimidine/thiourea hybrids are a new class of chemotypes possessing interesting antiproliferative activity against lung cancer cells and could be potentially utilized for designing new antitumor agents.Download high-res image (192KB)Download full-size image
Co-reporter:Zhong-Hua Li, Dong-Xiao Yang, Peng-Fei Geng, Ji Zhang, Hao-Ming Wei, Biao Hu, Qian Guo, Xin-Hui Zhang, Wen-Ge Guo, Bing Zhao, Bin Yu, Li-Ying Ma, Hong-Min Liu
European Journal of Medicinal Chemistry 2016 Volume 124() pp:967-980
Publication Date(Web):29 November 2016
DOI:10.1016/j.ejmech.2016.10.022
•The hydrazone-triazole-pyrimidines showed moderate to good growth inhibition against the tested cancer cells.•Some of compounds with IC50 below 10 μM showed less toxic to GES-1.•Compound 43 exerted the most antiproliferative activity against MGC-803 cells.•Compound 43 induced apoptosis of MGC-803 cells.•Compound 43 led to the decrease of MMP and the change of expression level of key proteins related to apoptosis.A series of [1,2,3]triazolo[4,5-d]pyrimidine derivatives bearing a hydrazone moiety were designed, synthesized and evaluated for their antiproliferative activity against several cancer cell lines of different origins by MTT assay. Most of the synthesized compounds demonstrated moderate to good activity against the cancer cell lines selected. Especially, compound 43 showed the most potent antiproliferative activity as well as good selectivity between cancer and normal cells (IC50 values of 0.85 μM against MGC-803 and 56.17 μM against GES-1). In addition, compound 43 evidently inhibited the colony formation of MGC-803 cells at 0.8 μM. Further mechanism studies revealed that compound 43 could induce apoptosis of MGC-803 cells probably through the mitochondrial pathway accompanied with decrease of the mitochondrial membrane potential (MMP), activations of caspase-9/3, up-regulation of the expression of Bax, Bak and PUMA, as well as down-regulation of that of Bcl-2 and Mcl-1.A series of hydrazone-triazole-pyrimidine analogues were designed and synthesized. Among them, compound 43 displayed the most inhibitory potency against MGC-803 cells and induced the apoptosis of MGC-803 cells.
Co-reporter:Bin Yu, Sai-Qi Wang, Ping-Ping Qi, Dong-Xiao Yang, Kai Tang, Hong-Min Liu
European Journal of Medicinal Chemistry 2016 Volume 124() pp:350-360
Publication Date(Web):29 November 2016
DOI:10.1016/j.ejmech.2016.08.065
•The conjugates showed moderate to good growth inhibition against the tested cancer cells.•These hybrids exerted selective inhibition towards MGC-803 cells.•These compounds were less toxic to normal cells HL-7702 and GES-1.•Compound 5a induced apoptosis and cell cycle arrest.•Compound 5a inhibited migration of MGC-803 cells.A series of new isatin/triazole conjugates were designed based on the hypothesis that the ester-linked compounds could be enzymatically hydrolyzed by cellular esterases inside the cells. These compounds showed moderate to good growth inhibition toward the tested cancer cells, exerted selective inhibition toward MGC-803 cells and were less toxic to normal cells HL-7702 and GES-1. Of these compounds, compound 5a showed the best inhibitory activity against MGC-803 cells (IC50 = 9.78 μM), induced apoptosis through multiple mechanisms, as well as inhibited migration of MGC-803 cells.The conjugates showed moderate growth inhibition against several cancer cells and were less toxic to normal cells. Compound 5a potently inhibited growth of MGC-803 cells, induced apoptosis, and inhibited migration.
Co-reporter:Bin Yu, Ping-Ping Qi, Xiao-Jing Shi, Ruilei Huang, Hao Guo, Yi-Chao Zheng, De-Quan Yu, Hong-Min Liu
European Journal of Medicinal Chemistry 2016 Volume 117() pp:241-255
Publication Date(Web):19 July 2016
DOI:10.1016/j.ejmech.2016.04.024
•Steroidal hybrids 12a–x were synthesized and showed varied cytotoxicity against the tested cancer cell lines.•Compounds with terminal isatin scaffold were sensitive to SH-SY5Y cells possibly through LSD1 inactivation.•Compound 12g potently inhibited growth of SH-SY5Y cells (IC50 = 4.06 μM).•Compound 12g arrested cell cycle at G2/M phase, induced apoptosis and decreased MMP.•Docking simulations were performed to show the binding models of compound 12g in the active site of LSD1.A series of steroidal hybrids with different terminal bioactive scaffolds were synthesized using the molecular hybridization approach and further evaluated for their antiproliferative activity against several cancer cell lines of different origins using the MTT assay. The preliminary results indicated that compounds 12a–h with the terminal isatin motif were remarkably sensitive to SH-SY5Y cells, thereby exerting potent growth inhibition in vitro. This selectivity is possibly attributed to LSD1 inactivation (IC50 = 3.18 μM). Besides, we also found that the chloro atom at the 7-position on the isatin core was beneficial for the activity through the SARs studies. Among this series, compound 12g showed the best inhibitory activity (IC50 = 4.06 μM) against SH-SY5Y cells, which was comparable to that of 5-FU. Compound 12g arrested cell cycle at G2/M phase, induced apoptosis accompanied with decrease of mitochondrial membrane potential, and inhibited LSD1 potently (IC50 = 3.18 μM). Docking studies showed that compound 12g formed interactions with surrounding amino acid residues and the steroid nucleus occupied the tubular hydrophobic cavity of the active site. Compounds 13–18 represented weak to moderate activity against the tested cancer cell lines. The steroidal dimer 20 and the structurally simplified non-steroidal dimer 21 were found to be devoid of the inhibitory activity.Steroidal hybrids were synthesized and evaluated for their antiproliferative activity. Compound 12g potently inhibited growth of SH-SY5Y cells possibly through the inactivation of LSD1, arrested cell cycle at G2/M phase, induced apoptosis and decreased MMP. Docking simulations were performed to rationalize the potency toward LSD1.
Co-reporter:Bin Yu, Zhiqiang Yu, Ping-Ping Qi, De-Quan Yu, Hong-Min Liu
European Journal of Medicinal Chemistry 2016 Volume 124() pp:248
Publication Date(Web):29 November 2016
DOI:10.1016/j.ejmech.2016.08.040
Co-reporter:Dong-Jun Fu, Sai-Yang Zhang, Ying-Chao Liu, Xiao-Xin Yue, Jun-Ju Liu, Jian Song, Ruo-Han Zhao, Feng Li, Hui-Hui Sun, Yan-Bing Zhang and Hong-Min Liu
MedChemComm 2016 vol. 7(Issue 8) pp:1681-1681
Publication Date(Web):04 Aug 2016
DOI:10.1039/C6MD90036D
Correction for ‘Design, synthesis and antiproliferative activity studies of 1,2,3-triazole–chalcones’ by Dong-Jun Fu et al., Med. Chem. Commun., 2016, DOI: 10.1039/c6md00169f.
Co-reporter:Dong-Jun Fu, Sai-Yang Zhang, Ying-Chao Liu, Xiao-Xin Yue, Jun-Ju Liu, Jian Song, Ruo-Han Zhao, Feng Li, Hui-Hui Sun, Yan-Bing Zhang and Hong-Min Liu
MedChemComm 2016 vol. 7(Issue 8) pp:1664-1671
Publication Date(Web):20 Jun 2016
DOI:10.1039/C6MD00169F
A series of 1,2,3-triazole–chalcone hybrids were designed, synthesized and evaluated for their antiproliferative activity against three selected cancer cell lines (SK-N-SH, HepG-2 and MGC-803). Most of the synthesized compounds exhibited moderate to good activity against all the cancer cell lines selected. Particularly, compound 12k showed the most excellent antiproliferative activity with an IC50 value of 1.53 μM against SK-N-SH cancer cells. The mechanism studies revealed that compound 12k inhibited the proliferation of SK-N-SH cancer cells by inducing apoptosis and arresting the cell cycle at the G1 phase.
Co-reporter:Tao Guo, Shu-Lei Han, Ying-Chao Liu, Yu Liu, Hong-Min Liu
Tetrahedron Letters 2016 Volume 57(Issue 10) pp:1097-1099
Publication Date(Web):9 March 2016
DOI:10.1016/j.tetlet.2016.01.093
A convenient and efficient approach for the synthesis of 2,3-dihydro-2,3′-bisindoles has been developed in good yields via dimerization of N-H indole derivatives. The synthetic utility of this approach was demonstrated by the rapid construction of bisindoles in gram-scale. Biological evaluation revealed that some of the obtained 2,3-dihydro-2,3′-bisindoles exhibited significant in vitro antiproliferative activities on human-derived lung, liver, stomach, and breast cancer cell lines.
Co-reporter:Tao Guo, Yu Liu, Yun-Hui Zhao, Pan-Ke Zhang, Shu-Lei Han, Hong-Min Liu
Tetrahedron Letters 2016 Volume 57(Issue 35) pp:3920-3923
Publication Date(Web):31 August 2016
DOI:10.1016/j.tetlet.2016.07.058
•Palladium-catalyzed external-oxidant-free coupling reactions.•A range of 1-alkenylisoquinolines and 2-alkenylquinolines are provided.•Some of the derivatives exhibit good antiproliferative activity.A convenient and efficient approach for the synthesis of 1-alkenylisoquinolines and 2-alkenylquinolines was developed via palladium-catalyzed coupling reactions between isoquinoline/quinoline N-oxides with olefins under external-oxidant-free conditions. Biological evaluation revealed that some of the obtained 1-alkenylisoquinolines exhibited in vitro antiproliferative activities on human-derived prostate, breast, esophageal, and stomach cancer cell lines.
Co-reporter:En Zhang, Peng-Yan Bai, Wei Sun, Shang Wang, Ming-Ming Wang, Shuai-Min Xu, Hong-Min Liu
Carbohydrate Research 2016 Volume 434() pp:33-36
Publication Date(Web):3 November 2016
DOI:10.1016/j.carres.2016.08.003
•A new azasugar was synthesized using one-pot reaction as a key step.•Our method plays an important role in the research of new azasugars.•The glycosidase inhibitory activity of the target molecule were not ideal.A new azasugar (3S,4S)-3-((R)-1,2-dihydroxyethyl)pyrrolidine-3,4-diol (1) was obtained from commercially available d-glucose using one-pot reductive cyclization as a key step. The target product, i.e., the iminosugar isomer, was obtained in 10 steps and 24.3% overall yield. Only three column chromatography purifications were needed in this synthesis. The biological activity of the target molecule as glycosidase inhibitor was studied, but the inhibitory activity against four glycosidases was not good (IC50 > 100 μM).A new azasugar isomer (3S,4S)-3-((R)-1,2-dihydroxyethyl)pyrrolidine-3,4-diol was obtained from D-glucose in ten steps and 24.3% overall yield using one-pot reduction cyclization as the key step. Only three column chromatography purifications were needed in this synthesis. Biological activity evaluation as inhibitor against glycosidase were studied but the results were not ideal.
Co-reporter:Cong Wang;Ran Wang;Kairui Zhou;Saiqi Wang;Junwei Wang;Hongge Shi;Yinhui Dou;Dongxiao Yang;Liming Chang;Xiaoli Shi;Ying Liu;Xiaowei Xu;Xiujuan Zhang;Yu Ke;Hongmin Liu
Cancer Chemotherapy and Pharmacology 2016 Volume 78( Issue 5) pp:971-982
Publication Date(Web):2016/11/01
DOI:10.1007/s00280-016-3149-9
Gastric cancer is the third most common cause of cancer mortality worldwide, and paclitaxel (PTX) is one of the most widely used traditional drugs in gastric cancer therapy. However, the response to traditional therapy is limited by acquired chemo-resistance and side effects. Here, we establish a newly designed combination therapy consisting of a compound that is a structural variant of oridonin, i.e. Jesridonin (JD), and low-dose PTX for gastric cancer cells (MKN45) to investigate whether the anti-tumour activity of low-dose PTX could be enhanced when combined with JD.The interaction of JD and low-dose PTX was detected in MKN45 cells using the median-effect analysis method. The synergistic effect on cell viability and apoptosis was measured by MTT assay, colony formation assay, transient transfection, flow cytometry and Western blotting. The synergistic in vivo effect of JD plus low-dose PTX was evaluated in nude mouse xenograft models using H&E and TUNEL staining and Western blotting.JD plus low-dose PTX showed a synergistic effect, as the combination indexes were less than 1. Additionally, a synergistic anti-proliferative and pro-apoptotic effect was detected for the combination of JD and low-dose PTX. The apoptotic mechanism induced by JD plus PTX revealed that the combination therapy synergistically activated the mitochondrial pathway.Our findings suggest that JD enhances the anti-tumour effect of low-dose PTX on gastric carcinoma cancer cells in both vitro and in vivo, accompanied by activation of the mitochondrial pathway, which may present a more effective therapeutic strategy in gastric cancer treatment.
Co-reporter:Li-Ying Ma; Yi-Chao Zheng; Sai-Qi Wang; Bo Wang; Zhi-Ru Wang; Lu-Ping Pang; Miao Zhang; Jun-Wei Wang; Lina Ding; Juan Li; Cong Wang; Biao Hu; Ying Liu; Xiao-Dan Zhang; Jia-Jia Wang; Zhi-Jian Wang; Wen Zhao
Journal of Medicinal Chemistry 2015 Volume 58(Issue 4) pp:1705-1716
Publication Date(Web):January 22, 2015
DOI:10.1021/acs.jmedchem.5b00037
Histone lysine specific demethylase 1 (LSD1) was reported to be overexpressed in several human cancers and recognized as a promising anticancer drug target. In the current study, we designed and synthesized a novel series of pyrimidine–thiourea hybrids and evaluated their potential LSD1 inhibitory effect. One of the compounds, 6b, containing a terminal alkyne appendage, was shown to be the most potent and selective LSD1 inhibitor in vitro and exhibited strong cytotoxicity against LSD1 overexpressed gastric cancer cells. Compound 6b also showed marked inhibition of cell migration and invasion as well as significant in vivo tumor suppressing and antimetastasis role, without significant side effects by oral administration. Our findings indicate that the pyrimidine–thiourea-based LSD1 inactivator may serve as a leading compound targeting LSD1 overexpressed cancers.
Co-reporter:Jing-Li Ren, Xu-Yao Zhang, Bin Yu, Xi-Xin Wang, Kun-Peng Shao, Xiao-Ge Zhu, Hong-Min Liu
European Journal of Medicinal Chemistry 2015 Volume 93() pp:321-329
Publication Date(Web):26 March 2015
DOI:10.1016/j.ejmech.2015.02.026
•Three series of novel AHLs were synthesized and evaluated for their cytotoxicity.•Some of these AHLs showed better cytotoxicity than 5-Fu and OdDHL.•4-Amino chalcone scaffold was first discovered with promising cytotoxicity.•Compound 11e was found to be able to induce apoptosis of MCF-7 cells.•Compound 11e arrested cell cycle of MCF-7 cells at G2/M phase.Three series of novel AHL analogs were synthesized and evaluated for their in vitro cytotoxic activity against four human cancer cell lines. The SARs investigation indicated that AHLs with a terminal phenyl group, especially those with the chalcone scaffold had remarkably enhanced cytotoxicity than those with the hydrophobic side chains. Besides, some of these compounds were much more potent than 5-Fu and natural OdDHL. Through the detailed SARs discussions, we found that compounds 10a-k and 14 with the 4-amino chalcone scaffold showed excellent inhibition against all the tested cancer cell lines and were much more potent than 5-Fu and AHLs. Such scaffold may act as a template for further lead optimization. Compound 10i with a 3, 4, 5-trimethoxy group was the most potent one against all the tested cancer cell lines. Flow cytometry analysis indicated that analog 11e induced the cellular apoptosis and cell cycle arrest of MCF-7 cells at G2/M phase in a concentration-and time-dependent manner.Extensive SARs studies of AHLs have led to the identification of compound 11e with potent cytotoxicity, which can induce the cellular apoptosis and cell cycle arrest of MCF-7 cells at G2/M phase in a concentration-and time-dependent manner.
Co-reporter:Bin Yu, Zhiqiang Yu, Ping-Ping Qi, De-Quan Yu, Hong-Min Liu
European Journal of Medicinal Chemistry 2015 Volume 95() pp:35-40
Publication Date(Web):5 May 2015
DOI:10.1016/j.ejmech.2015.03.020
•Spirooxindoles are promising scaffolds in drug discovery.•CFI-400945 has entered phase І clinical trials for cancer therapy.•The kinase selectivity of CFI-4009345 needs to be further explored.The identification of novel anticancer agents with high efficacy and low toxicity has always been an intriguing topic in medicinal chemistry. The unique structural features of spirooxindoles together with diverse biological activities have made them promising structures in new drug discovery. Among spirooxindoles, CFI-400945 holds its promise as the first potent PLK4 inhibitor, the fumarate of CFI-400945 has entered phase I clinical trials for the treatment of solid tumors. However, questions remain as to whether PLK4 is the only relevant therapeutic target for CFI-400945. To highlight this significant progress of CFI-400945 in last two years, this review centers on the identification from a focused kinase library, structural optimizations and strategies involved, structure-activity relationships, modes of action, target validation, chemical synthesis and, more importantly, the kinase selectivity between PLK4 and other targets.The ELISA assay of a focused kinase library yielded the indolinone as PLK4 inhibitor. Extensive modifications generated CFI-400945, which has entered phase I clinical trials for cancer therapy. However, the kinase selectivity is the major concern that needs to be further investigated.
Co-reporter:Li-Ying Ma, Bo Wang, Lu-Ping Pang, Miao Zhang, Sai-Qi Wang, Yi-Chao Zheng, Kun-Peng Shao, Deng-Qi Xue, Hong-Min Liu
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 5) pp:1124-1128
Publication Date(Web):1 March 2015
DOI:10.1016/j.bmcl.2014.12.087
A series of novel 1,2,3-triazole–pyrimidine–urea hybrids were designed, synthesized and evaluated for anticancer activity against four selected cancer cell lines (MGC-803, EC-109, MCF-7 and B16-F10). Majority of the synthesized compounds exhibited moderate to potent activity against all the cancer cell lines assayed. Particularly, compounds 26, 30 and 38 exhibited excellent growth inhibition against B16-F10 with IC50 values of 32 nM, 35 nM and 42 nM, respectively. Flow cytometry analysis demonstrated that compound 26 induced the cellular apoptosis in a concentration-dependent manner.Compounds 26, 30 and 38 exhibited excellent growth inhibition against B16-F10 with IC50 values of 32 nM, 35 nM and 42 nM, respectively.
Co-reporter:Tao Guo, Ying-Chao Liu, Bo Li, Hong-Min Liu
Tetrahedron Letters 2015 Volume 56(Issue 19) pp:2469-2471
Publication Date(Web):6 May 2015
DOI:10.1016/j.tetlet.2015.03.103
A convenient and efficient approach for the synthesis of α,α-(bisallyl and bispropargyl)-substituted amines has been developed in good yields via Zn-promoted aza-Barbier-type reactions of N-sulfonyl imidates with various allyl and propargyl reagents. The synthetic utility of this approach was demonstrated by the rapid construction of pyrrolidine derivatives.
Co-reporter:Shi-Yao Meng, Yong-Kang Jia, Ming Li, Wei-Wei Han, Fu-Qiang Wang, Shi-Qing Zhu, Shang-Shang Qin, Hui-Ying Song, Yu Hou, Xiu-Fang Shi, Feng-Wu Liu, Hong-Min Liu
Tetrahedron 2015 Volume 71(Issue 50) pp:9420-9428
Publication Date(Web):16 December 2015
DOI:10.1016/j.tet.2015.10.054
A versatile method for synthesis of chiral furopyran-β-one analogues starting from 1,4:3,6-dianhydrofructose has been developed. The target compounds were acquired by treatment of 1,4:3,6-dianhydrofructose with benzaldehydes in the presence of KF/Al2O3 and subsequent ring expansion of furanones induced by TMSOTf (trimethylsilyl triflate). The ring expansion involves an unprecedented nucleophilic 1,2-migration of a keto-carbonyl as a key step. In vitro antifungal activity of all compounds obtained were evaluated against five human pathogenic fungi (Candida parapsilosis ATCC22019, Candida albicans ATCC10231, Trichophyton rubrum CMCC(F)T1f, Epidermophyton floccosum CMCC(F)E1d, and Microsporum canis CMCC(F)M3d). We found that the furopyran-β-one analogues bearing chloro or fluoro group showed significant inhibitory activity against dermatophytes, especially M. canis.
Co-reporter:Deng-Qi Xue, Xu-Yao Zhang, Chao-Jie Wang, Li-Ying Ma, Nan Zhu, Peng He, Kun-Peng Shao, Peng-Ju Chen, Yi-Fei Gu, Xiao-Song Zhang, Cai-Feng Wang, Cong-Hui Ji, Qiu-Rong Zhang, Hong-Min Liu
European Journal of Medicinal Chemistry 2014 Volume 85() pp:235-244
Publication Date(Web):6 October 2014
DOI:10.1016/j.ejmech.2014.07.031
•Two series of novel 1,2,4-triazolo[3,4-a]phthalazine derivatives were synthesized.•Most of these compounds exhibited broad-spectrum cytotoxic activity.•Ph at C-3 was most active in all series.•One of them (Ph at C-3) was more potent than 5-Fu against the four cancer cell lines.•One of them (Ph at C-3) showed cell cycle arrest at G2/M phase and induced apoptosis.Trying to develop potent and selective anticancer agents, two series of novel 1,2,4-triazolo[3,4-a]phthalazine derivatives were designed and synthesized. Their antitumor activities were evaluated by MTT method against four selected human cancer cell lines (MGC-803, EC-9706, HeLa and MCF-7). Our results showed that compound 11h exhibited good anticancer activities compared to 5-fluorouracil against the four tested cell lines, with IC50 values ranging from 2.0 to 4.5 μM. Flow cytometry analysis indicated that compound 11h induced the cellular early apoptosis and cell cycle arrest at G2/M phase in EC-9706.The compound 11h exhibited IC50 values against the four tested human cancer cell lines ranging from 2.0 to 4.5 μM.
Co-reporter:Li-Ying Ma, Lu-Ping Pang, Bo Wang, Miao Zhang, Biao Hu, Deng-Qi Xue, Kun-Peng Shao, Bao-Le Zhang, Ying Liu, En Zhang, Hong-Min Liu
European Journal of Medicinal Chemistry 2014 Volume 86() pp:368-380
Publication Date(Web):30 October 2014
DOI:10.1016/j.ejmech.2014.08.010
•A series of novel 1,2,3-triazole-pyridimine hybrids were synthesized.•Several compounds were more potent than 5-fluorouracil against EC-109, MCF-7 and MGC-803.•Compound 17 was highly selective in its cytotoxicity activity.•Compound 17 showed cell cycle arrest at G2/M phase and induced apoptosis.A series of novel 1,2,3-triazole-pyrimidine hybrids were designed, synthesized and evaluated for their anticancer activity against four selected cancer cell lines (MGC-803, EC-109, MCF-7 and B16-F10). Most of the synthesized compounds exhibited moderate to good activity against all the cancer cell lines selected. Compound 17 showed the most excellent anticancer activity with single-digit micromolar IC50 values ranging from 1.42 to 6.52 μM. Further mechanism studies revealed that compound 17 could obviously inhibit the proliferation of EC-109 cancer cells by inducing apoptosis and arresting the cell cycle at G2/M phase.Novel 1,2,3-triazole-pyridimine hybrids 17 exhibited about 7 fold more potent than 5-Fu against EC-109.
Co-reporter:Bei-Xi Jia, Xian-Lei Zeng, Feng-Xiao Ren, Lu Jia, Xiao-Qing Chen, Jie Yang, Hong-Min Liu, Qiang Wang
Food Chemistry 2014 Volume 155() pp:31-37
Publication Date(Web):15 July 2014
DOI:10.1016/j.foodchem.2014.01.022
•Baeckeins F–I were isolated from the roots of Baeckea frutescens.•The structures were elucidated by analysis of the 1D/2D-NMR and HR-ESI-MS spectra.•The absolute stereochemistry was assigned by quantum chemical CD calculations.•The anti-inflammatory activities for the isolates were also evaluated.Baeckea frutescens is an aromatic shrub used in South China as an ornamental and as a spice. Four unusual C-methylated biflavonoids named baeckeins F–I (1–4) were isolated from the roots of B. frutescens. The baeckeins F–I possessed a unique carbon skeleton, a flavonol conjugated with a coumaronochromone molecule via the unusual linkages of C-2–C-8∗ and C-3–O–C-7∗. Their structures were elucidated by analysis of the 1D (1H/13C) and 2D NMR (HSQC/HMBC/NOESY) and HR-ESI-MS spectroscopic data, and the absolute stereochemistry for chiral carbons of C-2 and C-3 was established by CD spectrometry combined with quantum chemical calculations. Baeckeins F–I (1–4) were also evaluated for their anti-inflammatory activities by detecting the NO production of LPS-induced RAW264.7 murine macrophage cell line; baeckein I (4) with the β-d-glucose unit and configuration of (2R,3R) exhibited the highest NO inhibitory activity (IC50 = 15.2 μM), which was similar to that of the positive control indomethacin.
Co-reporter:Juan Li;Xiaoming Ding;Jiaxin Zheng;Dan Liu;Fei Guo;Hongmin Liu;Yanbing Zhang
Journal of Separation Science 2014 Volume 37( Issue 17) pp:2439-2445
Publication Date(Web):
DOI:10.1002/jssc.201400349
A sensitive and efficient method was developed for the simultaneous determination of eight synthetic dyes (Chrysoidin, Auramine O, Sudan(I–IV), Para Red, and Rhodamine B) in bean and meat products using high-performance liquid chromatography with tandem mass spectrometry. A simple extraction procedure using acetonitrile has been applied for the extraction of these dyes from spiked bean and meat samples. Chromatographic separation was achieved on a Waters XTerra C18 column (2.1 × 150 mm, 5 μm) with a multistep gradient elution. Detection and quantification were performed using mass spectrometry in multiple reaction monitoring mode. Linear calibrations were obtained with correlation coefficients R2 > 0.99. The limits of detection and quantification for the eight dyes were in the ranges of 0.03–0.75 and 0.1–2.0 μg/kg depending on matrices, respectively. The recoveries of these dyes in different food matrices were between 71.2 and 116.9% with relative standard deviations <15.2%, suggesting that the developed method is promising for the accurate quantification of the eight dyes at trace levels in bean and meat products.
Co-reporter:Kunpeng Shao;Xuyao Zhang;Xiaosong Zhang;Dengqi Xue;Liying Ma;Qiurong Zhang;Hongmin Liu
Chinese Journal of Chemistry 2014 Volume 32( Issue 5) pp:443-447
Publication Date(Web):
DOI:10.1002/cjoc.201400095
Abstract
A series of 4-anilino-6-phenylpyrimidines containing urea moiety were synthesized and the structures of all products were confirmed by 1H NMR, 13C NMR and HRMS. The antiproliferative activities of these compounds were evaluated against three human tumor cell lines (MGC-803, MCF-7 and EC-109) by applying the MTT assay method. compounds 4a, 4b and 6a showed the most effective activity, among which, 6a was more cytotoxic than 5-fluorouracil against all tested human cancer cell lines with IC50 values ranging from 1.80 to 2.72 µmol·L−1.
Co-reporter:Li-Hua Huang, Hong-De Xu, Zhen-Yu Yao, Yan-Guang Wang, Hong-Min Liu
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 3) pp:973-975
Publication Date(Web):1 February 2014
DOI:10.1016/j.bmcl.2013.12.056
Novel C6-amino substituted purine nucleoside analogues (2–12) bearing a modified pyranose-like D ring of the 4-azasteroid moiety were efficiently synthesized through nucleophilic substitution at C6 position of the steroidal nucleoside precursors (1a, b) with versatile amines. All the synthesized new compounds were evaluated for their anticancer activity in vitro against Hela, PC-3 and MCF-7 cell lines. Among them, compounds 4b, 7b and 9b exhibited significant cytotoxicity with the IC50 values of 2.99 μM (PC-3), 2.84 μM, (PC-3) and 2.69 μM (Hela), respectively.
Co-reporter:Qiu-Rong Zhang, Deng-Qi Xue, Peng He, Kun-Peng Shao, Peng-Ju Chen, Yi-Fei Gu, Jing-Li Ren, Li-Hong Shan, Hong-Min Liu
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 4) pp:1236-1238
Publication Date(Web):15 February 2014
DOI:10.1016/j.bmcl.2013.12.010
A series of novel 1,2,4-triazolo [3,4-a] phthalazine derivatives were synthesized in five steps from a common precursor, phthalic anhydride. Most of synthesized phthalazine derivatives showed inhibitory activity against Staphylococcus aureus. One of phthalazine derivatives 5l showed inhibitory activity against all tested bacterial and fungal strains.
Co-reporter:Hai-Wei Xu, Ling-Jie Zhao, Huan-Fei Liu, Dan Zhao, Jiao Luo, Xiao-Ping Xie, Wen-Sheng Liu, Jia-Xin Zheng, Gui-Fu Dai, Hong-Min Liu, Long-Hua Liu, Yi-Bo Liang
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 10) pp:2388-2391
Publication Date(Web):15 May 2014
DOI:10.1016/j.bmcl.2014.03.012
In this study we report the synthesis and activity against bovine viral diarrhea virus (BVDV) of a novel series of bicycle δ-sultones containing γ-lactones. BVDV is responsible for major losses in cattle. Some of the synthesized δ-sultones showed pronounced anti-BVDV activity with EC50 values of 0.12–1.0 μM and no significant cytotoxicity. Among them, the ortho bromosubstituted derivative 4f (EC50 = 0.12 μM) showed better antiviral activity than other derivatives and was 10 fold more that of than positive control ribavirin (EC50 = 1.3 μM). BVDV is also considered to be a valuable surrogate for the hepatitis C virus (HCV) in antiviral drug studies. The above results provided a novel candidate for the development of anti-HCV agents.
Co-reporter:Peng-Ju Chen, Ang Yang, Yi-Fei Gu, Xiao-Song Zhang, Kun-Peng Shao, Deng-Qi Xue, Peng He, Teng-Fei Jiang, Qiu-Rong Zhang, Hong-Min Liu
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 12) pp:2741-2743
Publication Date(Web):15 June 2014
DOI:10.1016/j.bmcl.2014.04.037
A series of novel 4-substituted-2-{[(1H-benzo[d]imidazol-2-yl)methyl] thio}-6-methylpyrimidine derivatives were designed, synthesized and evaluated for their cytotoxic activities against four human cancer cell lines and inhibitory activities against five type culture strains in vitro. Some of synthetic pyrimidine–benzimidazol combinations showed good inhibitory activities against Stenotrophomonas maltophilia, especially compounds 7b and 7c. Compounds 7a and 7d exhibited enhanced activities against MGC-803 in vitro, when compared to 5-Fu.Compounds 7a and 7b exhibited enhanced cytotoxic activities against MGC-803 in vitro, when compared to 5-Fu. The activities of compounds 7b and 7c against Stenotrophomonas maltophilia, which is rather drug resistant species, with the MIC of 2 μg/ml and 4 μg/ml.
Co-reporter:Kun-Peng Shao, Xu-Yao Zhang, Peng-Ju Chen, Deng-Qi Xue, Peng He, Li-Ying Ma, Jia-Xin Zheng, Qiu-Rong Zhang, Hong-Min Liu
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 16) pp:3877-3881
Publication Date(Web):15 August 2014
DOI:10.1016/j.bmcl.2014.06.050
A series of pyrimidine–benzimidazol hybrids was synthesized and evaluated for anticancer activity on four human cancer cell lines including MCF-7, MGC-803, EC-9706 and SMMC-7721. Some of the synthesized compounds exhibited moderate to potent activity against MGC-803 and MCF-7. Among them, compounds 5a–b and 6a–b showed most effective activity. Compounds 5b and 6b were more cytotoxic than 5-fluorouracil against all tested four human cancer cell lines, with IC50 values ranging from 2.03 to 10.55 μM and 1.06 to 12.89 μM, respectively. Flow cytometry analysis demonstrated that treatment of MGC-803 with 6b led to cell cycle arrest at G2/M phase accompanied by an increase in apoptotic cell death.Compounds 5b and 6b were more cytotoxic than 5-fluorouracil against all tested four human cancer cell lines, with IC50 values ranging from 2.03 to 10.55 μM and 1.06 to 12.89 μM, respectively. Flow cytometry analysis demonstrated that treatment of MGC-803 with 6b led to cell cycle arrest at G2/M phase accompanied by an increase in apoptotic cell death.
Co-reporter:Xian-Wei Ye, Yi-Chao Zheng, Ying-Chao Duan, Meng-Meng Wang, Bin Yu, Jing-Li Ren, Jin-Lian Ma, En Zhang and Hong-Min Liu
MedChemComm 2014 vol. 5(Issue 5) pp:650-654
Publication Date(Web):03 Mar 2014
DOI:10.1039/C4MD00031E
Two series of coumarin–1,2,3-triazole–dithiocarbamate hybrids were designed, synthesized and evaluated for their inhibitory activity towards lysine specific demethylase 1 (LSD1). Compounds 8a, 8d–8f, 8i–8l presented potent activity against lysine specific demethylase 1. Among them, compound 8k showed potent and reversible inhibition against lysine specific demethylase 1 with an IC50 value of 0.39 μM, which was 74-fold more potent than that of tranylcypromine (2-PCPA). Besides, compound 8k displayed excellent selectivity against lysine specific demethylase 1 without inhibition against monoamine oxidases (MAOs) A and B. Further investigation revealed that compound 8k was active at both recombinant and cell levels by upregulating the expression of H3K4me1, H3K4me2 and H3K9me2.
Co-reporter:Jin-Mei Xu, En Zhang, Xiao-Jing Shi, Yan-Chao Wang, Bin Yu, Wei-Wei Jiao, Ya-Zhuo Guo, Hong-Min Liu
European Journal of Medicinal Chemistry 2014 80() pp: 593-604
Publication Date(Web):
DOI:10.1016/j.ejmech.2014.03.022
Co-reporter:Juan Li, Dan-Dan Liu, Yu Ke, Fei Guo, Xiao-Ming Ding, Shuai-Liang Wang, Yu Chen, Hong-Min Liu, Ying-Qian Su, Yan-Yang Nan
Journal of Chromatography B 2014 960() pp: 14-18
Publication Date(Web):
DOI:10.1016/j.jchromb.2014.04.010
Co-reporter:Yi-Chao Zheng ; Ying-Chao Duan ; Jin-Lian Ma ; Rui-Min Xu ; Xiaolin Zi ; Wen-Lei Lv ; Meng-Meng Wang ; Xian-Wei Ye ; Shun Zhu ; David Mobley ; Yan-Yan Zhu ; Jun-Wei Wang ; Jin-Feng Li ; Zhi-Ru Wang ; Wen Zhao
Journal of Medicinal Chemistry 2013 Volume 56(Issue 21) pp:8543-8560
Publication Date(Web):October 16, 2013
DOI:10.1021/jm401002r
Lysine specific demethylase 1 (LSD1), the first identified histone demethylase, plays an important role in epigenetic regulation of gene activation and repression. The up-regulated LSD1’s expression has been reported in several malignant tumors. In the current study, we designed and synthesized five series of 1,2,3-triazole–dithiocarbamate hybrids and screened their inhibitory activity toward LSD1. We found that some of these compounds, especially compound 26, exhibited the most specific and robust inhibition of LSD1. Interestingly, compound 26 also showed potent and selective cytotoxicity against LSD1 overexpressing gastric cancer cell lines MGC-803 and HGC-27, as well as marked inhibition of cell migration and invasion, compared to 2-PCPA. Furthermore, compound 26 effectively reduced the tumor growth bared by human gastric cancer cells in vivo with no signs of adverse side effects. These findings suggested that compound 26 deserves further investigation as a lead compound in the treatment of LSD1 overexpressing gastric cancer.
Co-reporter:Gai-Zhi Liu, Hai-Wei Xu, Peng Wang, Zong-Tao Lin, Ying-Chao Duan, Jia-Xin Zheng, Hong-Min Liu
European Journal of Medicinal Chemistry 2013 Volume 65() pp:323-336
Publication Date(Web):July 2013
DOI:10.1016/j.ejmech.2013.04.062
•Series of novel 5-substituted-3,4-diphenylfuran-2-ones were synthesized.•Some compounds showed good anti-proliferative effects on prostate cancer.•Compounds 13p exhibited potent anticancer activity with IC50 value was 5 μM.•Compound 13p showed cell cycle arrest at G0/G1 phase and may induced apoptosis.•These compounds are easy to synthesize.Series of 5-substituted-3,4-diphenylfuran-2-ones were stereoselectively prepared. Their potential anti-proliferative effects on prostate cancer and some of their cyclooxygenases (COXs) inhibitory activities were evaluated. Structure–activity relationship (SAR) data, acquired by substituent modification at the para-position and ortho-position of the C-3 phenyl ring and 5-substituted modification of the central furanone, showed that 3-(2-chloro-phenyl)-4-(4-methanesulfonyl-phenyl)-5-(1-methoxy-ethyl)-5H-furan-2-one (13p) was the most potent compound and could effectively reduce the proliferation of prostate cancer cells (PC3 cell IC50 = 20 μM; PC3 PCDNA cell IC50 = 5 μM; PC3 SKP2 cell IC50 = 5 μM; DU145 cell IC50 = 25 μM). The cell cycle analysis for 13p in DU145 indicated that 13p may induce G1 phase arrest.The most potent compound 13p showed good anti-proliferative effects on prostate cancer (IC50 = 5 μM). The cell cycle analysis for 13p in DU145 indicated that 13p may induce G1 arrest.
Co-reporter:Song-Lin Zhu, Ya Wu, Cong-Jun Liu, Chang-Yong Wei, Jing-Chao Tao, Hong-Min Liu
European Journal of Medicinal Chemistry 2013 Volume 65() pp:70-82
Publication Date(Web):July 2013
DOI:10.1016/j.ejmech.2013.04.044
•Two series of novel heterocycle fused with isosteviol derivatives were synthesized.•Cytotoxic activities of sixty analogs of isosteviol were tested in vitro.•Compound 11t showed the highest cytotoxities (IC50: 2.71, 3.18, 1.09 and 13.52 μM).•The structure–activity relationships of the isosteviol analogs were discussed.Two series of novel isosteviol-fused pyrazoline and pyrazole derivatives were facilely synthesized via intramolecular 1,3-dipolar cycloaddition and condensation reaction, respectively. All compounds were characterized by NMR, IR and HRMS spectra. The stereochemistry of compounds 9b, 10, 11a and 11v were further confirmed by X-ray crystallographic analysis. The antiproliferative activities of the structurally related pyrazoline and pyrazole derivatives were tested in vitro on four human malignant cell lines (SGC 7901, A549, Raji and HeLa): Our results revealed that isosteviol-fused pyrazole derivatives exhibited noteworthy cytotoxic activities. Among them, 2,4-di-Cl-phenylpyrazole derivative 11t displayed better cytotoxities with IC50 values: 2.71, 3.18, 1.09 and 13.52 μM against SGC 7901, A549, Raji and HeLa, respectively, compared to cisplatin (IC50 values: 7.56, 17.78, 17.32 and 14.31 μM, respectively).Novel isosteviol-fused pyrazoline and pyrazole derivatives were facilely prepared. Compounds 11t displayed noteworthy cytotoxities (IC50 = 2.71, 3.18, 1.09 and 13.52 μM) against SGC 7901, A549, Raji and HeLa, respectively.
Co-reporter:Bin Yu, Xiao-Jing Shi, Jing-li Ren, Xiao-Nan Sun, Ping-Ping Qi, Yuan Fang, Xian-Wei Ye, Meng-Meng Wang, Jun-Wei Wang, En Zhang, De-Quan Yu, Hong-Min Liu
European Journal of Medicinal Chemistry 2013 Volume 66() pp:171-179
Publication Date(Web):August 2013
DOI:10.1016/j.ejmech.2013.05.035
•Two series of novel steroidal dienamides were synthesized.•All these compounds exhibited broad-spectrum cytotoxic activities.•Most of them were more potent than 5-Fu against five human cancer cell lines.•Compounds 4j and 4k were highly selective in their cytotoxic activities.•Compound 4c showed cell cycle arrest at G2/M phase and induced early apoptosis.Two series of steroidal dienamides 4a–q and 5a–f were designed, synthesized and evaluated for cytotoxic activities against five human cancer cell lines (MGC-803, EC109, PC-3, SMMC-7721 and MCF-7). The protocol developed efficiently achieved the construction of carbon–carbon double bond and selective conversion of nitrile group into carboxamide in one-pot procedure. Besides, compounds 4a–q and 5a–f showed moderate to excellent cytotoxic activities with the IC50 values ranging from 0.1 to 40 μM and most of them were more potent than 5-fluorouracil. Particularly, four compounds 4d, 4e, 4q and 5a showed excellent selectivity against MGC-803 with the IC50 values less than 1 μM. Flow cytometry analysis demonstrated that compound 4c caused the cellular early apoptosis and cell cycle arrest in G2/M phase in a concentration-independent manner.All the compounds showed moderate to excellent cytotoxic activities against five human cancer cell lines. Four compounds showed excellent selectivity against MGC-803 (IC50 < 1 μM). Compound 4c induced G2/M arrest and early apoptosis.
Co-reporter:Bin Yu, Xiao-Jing Shi, Yong-Fei Zheng, Yuan Fang, En Zhang, De-Quan Yu, Hong-Min Liu
European Journal of Medicinal Chemistry 2013 Volume 69() pp:323-330
Publication Date(Web):November 2013
DOI:10.1016/j.ejmech.2013.08.029
•Compound 3 was efficiently synthesized in two steps.•Compound 3 exhibited broad-spectrum cytotoxic activity and was more potent than 5-Fu.•Compound 3 showed low toxicity against L-02.•Compound 3 showed cell cycle arrest at G2/M phase and induced apoptosis.A novel [1,2,4] triazolo [1,5-a] pyrimidine-based phenyl-linked steroid dimer was designed, synthesized and evaluated for its cytotoxic activity against five human cancer cell lines and the cytotoxicity against human normal liver cell L-02. Compound 3 showed excellent cytotoxic activity and good selectivity between cancer and normal cells. Further mechanistic studies revealed that treatment of EC109 cells with compound 3 caused an obvious G2/M arrest in a concentration- and time-dependent manner and induced apoptosis probably through the mitochondrial pathway accompanied with the decrease of mitochondrial membrane potential, activations of caspase-9/-3, cleavage of MDM2 as well as up-regulation of the expressions of p53 and Bax.Compound 3 showed excellent cytotoxic activity against five human cancer cell lines (IC50 < 3.2 μM) and low toxicity against L-02 and induced G2/M arrest and apoptosis probably through the mitochondrial pathway.
Co-reporter:Song-Lin Zhu, Ya Wu, Cong-Jun Liu, Chang-Yong Wei, Jing-Chao Tao, Hong-Min Liu
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 5) pp:1343-1346
Publication Date(Web):1 March 2013
DOI:10.1016/j.bmcl.2012.12.091
Two series of novel carbothioamide-substituted pyrazole and isoxazolidine derivatives were facilely prepared by functional interconversions in ring D of the tetracyclic diterpene isosteviol. The in vitro cytotoxic activities against four human tumor cell lines were evaluated. Our results indicated that carbothioamide-substituted pyrazole derivatives exhibited noteworthy cytotoxic activities. Specifically, compound 12p (IC50 = 6.51 μM) had the most potent cytotoxicity against Raji cell, which may be exploitable as a lead compound for the development of potent antitumor agents.Two series of novel carbothioamide-substituted pyrazole and isoxazolidine derivatives were facilely prepared. Within all compounds, 12p (IC50 = 6.51 μM) showed the highest cytotoxic activity against Raji cell.
Co-reporter:Jing-Li Ren, En Zhang, Xian-Wei Ye, Meng-Meng Wang, Bin Yu, Wen-Hua Wang, Ya-Zhuo Guo, Hong-Min Liu
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 14) pp:4154-4156
Publication Date(Web):15 July 2013
DOI:10.1016/j.bmcl.2013.05.035
Two series of novel AHL analogues were designed, synthesized and evaluated for antibacterial activity under cell membrane conditions in vitro. Analogues 4a–c and 4g–m presented potent activity against Gram-positive bacteria. Especially the analogue 4l exerted the most potent inhibition against Bacillus subtilis with MIC50 value of 1.443 μg/ml. To our surprise, analogues 6a–c and 6g showed weak inhibition against Gram-negative bacteria with MIC50 values ranging from 17.589 to 67.840 μg/ml. This was the first report about synthesis and antibacterial evaluation in vitro of AHL analogues containing dithioester linkage.
Co-reporter:Hai-Wei Xu, Chao Xu, Zi-qi Fan, Ling-Jie Zhao, Hong-Min Liu
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 3) pp:737-739
Publication Date(Web):1 February 2013
DOI:10.1016/j.bmcl.2012.11.090
Pulvinone and several 3-fluoro-4-morpholino substituted pulvinone derivatives were synthesized in five steps from a common precursor, phenyl acetic acid. Most of synthetic morpholine substituted pulvinones showed inhibitory activity against Esherichia coli. For the first time, the inhibition of pulvinone and its derivatives against Gram-negative bacteria was reported.
Co-reporter:Juan Li, Xiao-Ming Ding, Dan-Dan Liu, Fei Guo, Yu Chen, Yan-Bing Zhang, Hong-Min Liu
Journal of Chromatography B 2013 Volumes 942–943() pp:46-52
Publication Date(Web):30 December 2013
DOI:10.1016/j.jchromb.2013.10.010
•LC–MS/MS was used for the determination of eight banned dyes in complex food.•A simple sample treatment procedure without further cleanup was developed.•Most of the analytes have satisfactory LODs and LOQs depending on matrices.A sensitive and accurate method based on the use of liquid chromatography–tandem mass spectrometry (LC–MS/MS) was developed for the simultaneous determination of eight illegal synthetic dyes (Sudan (I–IV), Para Red, Rhodamine B, Chrysoidin and Auramine O) in chili products. A simple sample treatment procedure entailing the use of an extraction step with acetonitrile/H2O (9/1) without further cleanup was developed. HPLC was performed on a C18 column using a multistep gradient elution with 5 mM ammonium acetate (pH 3.0 with formic acid) and methanol as the mobile phase. Mass spectral acquisition was done in multiple reaction monitoring (MRM) mode using positive electrospray ionization (ESI). Linear calibrations were obtained with correlation coefficients R2 > 0.99. Limit of detection (LOD) and limit of quantification (LOQ) for the studied dyes were in the ranges of 0.05–0.6 μg kg−1 and 0.3–3.0 μg kg−1 depending on matrices, respectively. The recoveries of the eight synthetic dyes in five matrices ranged from 70.5% to 119.2%. The intra- and inter-day precisions (RSDs) were between 2.3–15.8% and 5.7–15.6%, respectively. The applicability of the method to the determination of eight banned dyes in chili products was demonstrated.
Co-reporter:Ming-Li Zhao, En Zhang, Jie Gao, Zhao Zhang, Yu-Tao Zhao, Wen Qu, Hong-Min Liu
Carbohydrate Research 2012 Volume 351() pp:126-129
Publication Date(Web):1 April 2012
DOI:10.1016/j.carres.2012.01.013
A formal synthesis of Jaspine B was completed in 42.4% overall yield with only three purification steps (one by crystallization and two by column chromatography). The key step in the synthesis involves a regio- and stereoselective epoxide ring-opening reaction and the configuration inversion of the C3-hydroxyl group through oxidation and reduction. All of the reagents and materials used were quite common and inexpensive.
Co-reporter:Lin Yan;Haiwei Xu;Fengwu Liu;Jin Zhao;Hongmin Liu
Chinese Journal of Chemistry 2012 Volume 30( Issue 4) pp:914-918
Publication Date(Web):
DOI:10.1002/cjoc.201100179
Abstract
A series of new andrographolide C-glycoside derivatives were synthesized by a facile route. The new compounds showed higher potency than the parent andrographolide evaluated as α-glycosidase inhibitors in the preliminary study.
Co-reporter:Yue-Feng Bi, Zhen-Ji Wang, Ruo-Fei Guan, Yu-Ting Ye, Yuan-Yuan Chen, Yan-Bing Zhang, Hong-Min Liu
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 15) pp:5141-5143
Publication Date(Web):1 August 2012
DOI:10.1016/j.bmcl.2012.05.014
A new series of tanshinone IIA (DIIA) derivatives were synthesized through the reaction of brominated tanshinone IIA (1-Br DIIA) and aromatic acids in the presence of K2CO3. Twenty compounds were synthesized, and all of them were novel. Vasodilative activities for synthesized compounds were valuated in vitro on the contractile response of vascular thoracic aorta smooth muscle from Wistar rats. The results showed that most compounds exhibited a concentration-dependent inhibition on the contractile response of norepinephrine. Four prepared compounds, 4, 5, 8 and 13 revealed relatively remarkable vasodilative activity.In this study, tanshione IIA derivatives were synthesized by the reactions of brominated tanshinone IIA with some aromatic acids for the first time. Vasodilative activities in vitro of tanshinone IIA derivatives were valuated on the contractile response of vascular thoracic aorta smooth muscle from Wistar rats. Most of them exhibited a concentration-dependent inhibition of the contractile response of norepinephrine. The structure–activity relationship showed that vascular relaxation was significantly different by aromatic acids with different substituent groups.
Co-reporter:Pei Wang;En Zhang;Peng Zhao;Qing-Hua Ren;Yuan-Yuan Guan
Chirality 2012 Volume 24( Issue 12) pp:1013-1017
Publication Date(Web):
DOI:10.1002/chir.22089
ABSTRACT
The separation of rac-o-chloromandelic acid 1 with enantiopure aryloxypropylamine via diastereomeric salt formation was investigated. (R)-o-chloromandelic acid (R)-1, a key intermediate for the antithrombotic agent clopidogrel, was obtained in 65% yield and 98% ee by Dutch resolution of rac-1 with (S)-2-hydroxyl-3-(p-chlorophenoxy) propylamine (S)-5 as resolving agent and (S)-2-hydroxyl-3-(o-nitrophenoxy) propylamine (S)-4 as nucleation inhibitor. Chirality 24:1013–1017, 2012. © 2012 Wiley Periodicals, Inc.
Co-reporter:Pei Wang, En Zhang, Jian-Feng Niu, Qing-Hua Ren, Peng Zhao, Hong-Min Liu
Tetrahedron: Asymmetry 2012 Volume 23(Issue 14) pp:1046-1051
Publication Date(Web):31 July 2012
DOI:10.1016/j.tetasy.2012.06.023
Chiral mandelic acid (S)-1, which is an important precursor for stereoselective transformations and a versatile intermediate for pharmaceuticals, was resolved with the Pope and Peachey method. Enantiopure 1-amino-3-phenoxypropan-2-ol (S)-2, a key intermediate for pharmaceuticals, was used to resolve rac-mandelic acid rac-1 successfully for the first time. The less soluble salt (S)-1·(S)-2·H2O could be obtained in 77% yield and 98% de (E 75%) using (S)-2 and LiOH in water. The crystal structure of the less soluble salt (S)-1·(S)-2·H2O showed that the water molecule played a key role in forming the crystals.Chiral mandelic acid (S)-1, which is an important precursor for stereoselective transformations and a versatile intermediate for pharmaceuticals, was resolved with the Pope and Peachey method using (S)-2 as resolving agent. The less soluble salt (S)-1·(S)-2·H2O could be obtained in 77% yield and 98% de (E 75%).(S)-Mandelic acid·(S)-1-amino-3-phenoxypropan-2-ol·H2OC17H23NO6Enantiomeric excess of liberated mandelic acid: ee = 98% [by HPLC][α]D23=+27.0 (c 1.0, ethanol)Source of chirality of mandelic acid: resolved by (S)-1-amino-3-phenoxypropan-2-olAbsolute configuration of liberated mandelic acid: (S)(S)-1-Amino-3-phenoxypropan-2-olC9H13NO2Enantiomeric excess: ee = 100% [by HPLC][α]D24=-10.5 (c 1.0, ethanol)Source of chirality: (S)-epichlorohydrinAbsolute configuration: (S)(S)-N-Methyl-1-amino-3-phenoxypropan-2-olC10H15NO2Enantiomeric excess: ee = 100% [by HPLC][α]D25=-16.0 (c 1.0, ethanol)Source of chirality: (S)-epichlorohydrinAbsolute configuration: (S)(S)-N-Ethyl-1-amino-3-phenoxypropan-2-olC11H17NO2Enantiomeric excess: ee = 100% [by HPLC][α]D19=-13.0 (c 1.0, ethanol)Source of chirality: (S)-epichlorohydrinAbsolute configuration: (S)
Co-reporter:Xiao-Juan Wang, Hai-Wei Xu, Lin-Lin Guo, Jia-Xin Zheng, Bo Xu, Xiao Guo, Chen-Xin Zheng, Hong-Min Liu
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 10) pp:3074-3077
Publication Date(Web):15 May 2011
DOI:10.1016/j.bmcl.2011.03.029
Three series of butenolide-containing dithiocarbamates were designed and synthesized. Their anti-tumor activity in vitro was evaluated. Among them compound I-14 exhibited broad spectrum anti-cancer activity against five human cancer cell lines with IC50 <30 μM. Structure–activity relationship analysis showed that the introduction of dithiocarbamate side chains on the C-3 position of butenolide was crucial for anti-tumor activity.Three series of butenolide-containing dithiocarbamates were designed and synthesized. Their anti-tumor activity in vitro was evaluated. Among them compound I-14 exhibited excellent anti-cancer activity against five human cancer cell lines with IC50 <10 μg/mL. Structure–activity relationship analysis showed that the introduction of dithiocarbamate side chains on the C-3 position of butenolide was crucial for anti-tumor activity.
Co-reporter:Li-Hua Huang, Yan-Guang Wang, Gong Xu, Xiang-Hua Zhang, Yong-Fei Zheng, Hui-Li He, Wen-Zheng Fu, Hong-Min Liu
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 20) pp:6203-6205
Publication Date(Web):15 October 2011
DOI:10.1016/j.bmcl.2011.07.049
A series of novel N-glycoside analogues with 4-azasteroid moiety bearing sugar-like D ring were conveniently synthesized by constructing the core dihydropyran ring embedded in 4-azasteroidal skeleton which was prepared from 4-aza-5α-androst-3,17-dione 1 in four steps. The structure of 6b were unambiguously proved by the appropriate X-ray structural analysis. Anticancer activity was found for all of the analogues with purinyl moiety against breast cancer (MCF-7), human neuroblastoma (SK-N-SH), cervical cancer cell (HeLa) and prostatic cancer (PC-3), while the analogue 7 containing 1,2,4-triazole heterocycle as the nucleobase was inactive against all of the tested cancer cell lines. The biology results showed the purinyl moiety attached to the pyran ring of 6a–d, substituent at 6′-position of purine base and introduction of a halogen atom at 2′-position of 6′-chloropurine had obviously effect on the evaluated anticancer activity.
Co-reporter:Shao-Min Wang, Wei-Guo Zhu, Jian-Xun Kang, Hong-Min Liu, Jun-Miao Chen, Cui-Ping Li, Kai Zhang
Carbohydrate Research 2011 Volume 346(Issue 2) pp:203-209
Publication Date(Web):1 February 2011
DOI:10.1016/j.carres.2010.11.018
The reaction process for the selective deprotection of acetylated glucosides by dibutyltin oxide in methanol is investigated by using methyl 2,3,4,6-tetra-O-acetyl-α-d-glucopyranoside as a model substrate with ESIMS and NMR techniques. According to the results, it is inferred that at first, dimeric 1,3-dimethoxytetrabutyldistannoxane is formed by the reaction of dibutyltin oxide with methanol, and then the tetraorganodistannoxane reacts with the acetylated glucoside to produce glucoside–organotin complex intermediates. Finally, the complex intermediates are hydrolyzed leading to the free-OH glucoside and organotin acetate derivatives. The reaction is affected by neighboring group participation and steric hindrance, which allow for high selectivities among different acetyl groups in acetylated glucosides.
Co-reporter:Haiwei Xu;Pingjuan Jiang;Weiyi Li;Junfeng Wang;Hongmin Liu
Chinese Journal of Chemistry 2011 Volume 29( Issue 10) pp:2114-2118
Publication Date(Web):
DOI:10.1002/cjoc.201180366
Abstract
In this study, a novel route to (−)-limonidilactone analogues 10, 11 by way of stereoselective reaction of andrographolide (5) from 8, 9 was reported. The anti-tumor and α-glucosidsae inhibition activities of some synthetic compounds were screened.
Co-reporter:Wei Liu;Bing Zhao;Yin-Chao Li
Magnetic Resonance in Chemistry 2011 Volume 49( Issue 9) pp:611-615
Publication Date(Web):
DOI:10.1002/mrc.2770
Abstract
Complexations between three oridonin derivatives and β-cyclodextrin (βCD) were studied by nuclear magnetic resonance (NMR) method. Job's plots for complexes were depicted by 1H NMR spectra chemical shifts, which proved the 1:1 stoichiometry inclusion complex formation between each derivative and βCD. Two-dimensional rotating frame overhauser effect spectroscopy (2D ROESY) support the above conclusion and also proved that ring A of each oridonin derivative deeply enters into hydrophobic cavity from the wider rim and the other parts are outside the cavity. Apparent formation constants (Ka) of complexes between three oridonin derivatives and two CDs are calculated according to Scott's equation. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:Juan Li, Zu-Yue Tu, Jun-Yuan Yin, Guo-Xia Yue, Hong-Min Liu
Journal of Chromatography B 2011 Volume 879(Issue 22) pp:2107-2112
Publication Date(Web):15 July 2011
DOI:10.1016/j.jchromb.2011.05.046
A rapid, sensitive and specific liquid chromatography–tandem mass spectrometry (LC–MS/MS) method has been developed for identification of potassium dehydroandrographolidi succinas and its metabolites in rat urine. Five male rats were administrated a single dose (100 mg/kg) of potassium dehydroandrographolidi succinas by i.v. injection. The urine were sampled from 0 to 24 h and purified by using Oasis® HLB extraction cartridge, then the purified urine samples were separated on a reversed-phase C18 column with a linear gradient and detected by an on-line MS detector. Identification and structural elucidation of the metabolites were performed by comparing their changes in molecular mass (Δm) and MS/MS spectra with those of the parent drug. Seven metabolites and the parent drug were found in rat urine. All these metabolites were reported for the first time.
Co-reporter:Guiqin Hou;Zhaoming Lu;Mingyue Liu;Hongmin Liu;Lexun Xue
Digestive Diseases and Sciences 2011 Volume 56( Issue 5) pp:1315-1322
Publication Date(Web):2011/05/01
DOI:10.1007/s10620-010-1474-0
Esophageal squamous cell carcinoma (ESCC) is one of the most frequently diagnosed cancers in China, but the etiology and mode of carcinogenesis of this disease remain poorly understood. The phosphatase and tensin homolog deleted from chromosome 10 (PTEN) with putative tumor suppressing is frequently mutated in many cancers.The aim of this study was to investigate whether there exists a mutation in the PTEN gene of the ESCC cells, and the effects of the wild type and mutated PTEN genes on the proliferation and apoptosis of the ESCC cells.The wild type and mutated PTEN genes were cloned from human placenta and ESCC cells, respectively, and their effects on the proliferation and apoptosis of the ESCC cells were investigated. Also, the relationship between the PTEN gene status and sensitivity of the EC9706 cells to cisplatin was determined in the xenografts of nude mice.There were mutations in the PTEN gene from ESCC cells. The proliferation of the EC9706 cells was clearly inhibited by the wild type PTEN gene, but not by the mutated PTEN gene in vitro. Furthermore, the wild type PTEN gene inhibited the growth of transplantable tumor, induced cell apoptosis, and improved the sensitivity of the EC9706 cells to cisplatin in vivo.The findings of the present study demonstrate that there are mutations in the PTEN gene of the ESCC cells and that the wild type PTEN gene has important effects on the ESCC cells in vitro and in vivo.
Co-reporter:Gai-Zhi Liu, Hai-Wei Xu, Guang-Wei Chen, Peng Wang, Ya-Na Wang, Hong-Min Liu, De-Quan Yu
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 4) pp:1626-1632
Publication Date(Web):15 February 2010
DOI:10.1016/j.bmc.2009.12.061
A series of desloratadine derivatives were stereoselectively synthesized and evaluated for H1 antihistamine activity. For the evaluation of H1 antihistamine activity, the in vitro histamine-induced contraction of the guinea-pig ileum assay (HC) was used. The synthesized desloratadine derivatives 7, 8 and 9 are structurally related to rupatadine and were generated by replacement of the 5-methyl-3-pyridine group of rupatadine with γ-alkylidene butenolide. Their H1 antihistamine activities have shown a high dependence on the exact nature of the substituent in the lactone ring. Optimum structures 7, 8a and 8g display potent activity inhibiting histamine-induced effects.A novel family of desloratadine derivatives as antagonist of histamine were prepared. Above three optimum structures display potent activity inhibiting histamine-induced effects.
Co-reporter:Yue-Feng Bi, Hai-Wei Xu, Xiao-Qing Liu, Xiao-Juan Zhang, Zhen-Ji Wang, Hong-Min Liu
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 16) pp:4892-4894
Publication Date(Web):15 August 2010
DOI:10.1016/j.bmcl.2010.06.076
A series of 2,2′-(substituted methylene)bis-(1,6,6-trimethyl-6,7,8,9-tetrahydrophenanthro[1,2-b]furan-10,11-dione) derivatives were synthesized by the reaction of tanshinone IIA (D1) and aromatic aldehyde in the presence of p-TsOH. Bromination derivative of D1 and hydrolysis product of cryptotanshinone (D2) were also prepared in this work. Vasodilation activity in vitro of them was valuated on the contractile response of vascular thoracic aorta smooth muscle from Wistar rats for the first time. Most of them exhibited a concentration-dependent inhibition on the contractile response of norepinephrine.Vasodilation activity in vitro of them was valuated on the contractile response of vascular thoracic aorta smooth muscle from Wistar rats for the first time. Most of them exhibited a concentration-dependent inhibition of the contractile response of norepinephrine.
Co-reporter:Fuyi Zhang, Hong Liu, Yong-Feng Li, Hong-Min Liu
Carbohydrate Research 2010 Volume 345(Issue 6) pp:839-843
Publication Date(Web):19 April 2010
DOI:10.1016/j.carres.2010.01.004
Novel methyl 4,6-O-benzylidenespiro[2-deoxy-α-d-arabino-hexopyranoside-2,2′-imidazolidine] and its homologue methyl 4,6-O-benzylidene-3′,4′,5′,6′-tetrahydro-1′H-spiro[2-deoxy-α-d-arabino-hexopyranoside-2,2′-pyrimidine] have been synthesized in good yields by reaction of methyl 4,6-O-benzylidene-α-d-arabino-hexopyranosid-2-ulose with 1,2-diaminoethane and 1,3-diaminopropane. The results are completely different from the reaction with arylamines or alkylamines. One-pot synthesis of novel (E)-methyl 4-[hydroxy (methoxy)methylene]-5-oxo-1-alkyl-(4,6-O-benzylidene-2-deoxy-α-d-glucopyranosido)[3,2-b]pyrrolidines has been achieved by the reaction of alkylamines with the butenolide-containing sugar, derived from the aldol condensation of methyl 4,6-O-benzylidene-α-d-arabino-hexopyranosid-2-ulose with diethyl malonate. These sugar-γ-butyrolactam derivatives are potential GABA receptor ligands.
Co-reporter:Fu Yi Zhang, Chun Li Wu, Cui Zhang, Hong Min Liu
Chinese Chemical Letters 2010 Volume 21(Issue 7) pp:798-801
Publication Date(Web):July 2010
DOI:10.1016/j.cclet.2010.02.020
The building block of N-alkyl derivative of allosamidin (chitinase inhibitor), 4,6-O-benzylidene-N-octyl-d-allosamine hydrochloride was stereoselectively synthesized in two steps under mild conditions. Nucleophilic addition of octylamine to 2-oxoglucopyranoside gave a ‘carbonyl group transfer’ product in 62% yield. Subsequent stereoselective reduction of newly formed CO with NaBH4 produced title compound in 75% yield. X-ray diffraction analysis indicates the title compound adopts syn 1, 2, 3 stereochemistry and chair–chair conformation. The crystal structure is stabilized by hydrogen bonds.
Co-reporter:Yong-Hong Li;Li-Hua Huang;Shu-Sheng Zhang
Chromatographia 2010 Volume 71( Issue 11-12) pp:987-991
Publication Date(Web):2010 June
DOI:10.1365/s10337-010-1615-9
An OJ-H chiral column has been used for direct resolution of the enantiomers of betaxolol and related intermediates in the preparation of (S)-betaxolol. The enantiomers can be excellently resolved (RS > 2) within 9.4 min with high peak symmetry. The enantiomers of some acetylated intermediates, which cannot be resolved on an OD-H column, can also be resolved. The method is simple and suitable for routine determination of ee values in the preparation of (S)-betaxolol.
Co-reporter:Li-Hong Shan, Hong-Min Liu, Ke-Xue Huang, Gui-Fu Dai, Chen Cao, Rui-Jing Dong
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 23) pp:6637-6639
Publication Date(Web):1 December 2009
DOI:10.1016/j.bmcl.2009.10.019
Co-reporter:Feng-Wu Liu, Zhen-Ji Wang, Xiao-Ping Song, Sai-Yang Zhang, Hong-Min Liu
Carbohydrate Research 2009 Volume 344(Issue 18) pp:2439-2443
Publication Date(Web):14 December 2009
DOI:10.1016/j.carres.2009.09.028
Henry reactions of a novel higher sugar derivative, (1R)-(1,4:3,6-dianhydro-d-mannitol-2-yl)-1,4:3,6-dianhydro-d-fructose 5,5′-dinitrate (Alternate nomenclature: (1R)-(isomannid-2-yl)-1,4:3,6-dianhydro-d-fructose 5,5′-dinitrate), with nitromethane and nitroethane were studied. The kinetic and thermodynamic reactions with nitromethane under different conditions were carried out to afford (2S)- and (2R)-β-nitroalcohols, respectively. But when using nitroethane the reaction gave a (2S)-β-nitroalcohol with an inverted configuration at vicinal carbon C-1. Two stereogenic centers were generated, and one was altered in the reaction.
Co-reporter:Jing-Yu Zhang, Hong-Min Liu, Hai-Wei Xu, Li-Hong Shan
Tetrahedron: Asymmetry 2008 Volume 19(Issue 4) pp:512-517
Publication Date(Web):4 March 2008
DOI:10.1016/j.tetasy.2008.01.039
An efficient non-enzymatic kinetic resolution strategy capable of accessing optically active β-adrenergic antagonists intermediates is reported. The C-12 higher carbon sugar derived from naturally occurring sucrose was employed to probe the kinetic resolution. Excellent enantiomeric excesses (ee >99%) and high yields were obtained under very mild conditions. The chiral auxiliary could be recovered in a high reclaimed ratio (>95%) and reusable form without any decrease of the resolving ability.(2R,3R,3aS,6R,6aR,3′R,3′aS,6′R,6′aR,5″R)-Spiro-[6,3′,6′-trihydroxy-octahydro[2,3′]bi[furo[3,2-b]furan]-3,2″-[5″-phenoxymethyl-1″,3″-oxazolidine]C21H27NO9[α]D20=+58.0 (c 1.00, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (5″R)(2R,3R,3aS,6R,6aR,3′R,3′aS,6′R,6′aR,5″R)-Spiro-[6,3′,6′-trihydroxy-octahydro[2,3′]bi[furo[3,2-b]furan]-3,2″-[5″-[4″′-[2″″-hydroxyethyl]]phenoxymethyl-1″,3″-oxazolidine]C23H31NO10[α]D20=+67.3 (c 1.00, CH3OH)Source of chirality: asymmetric synthesisAbsolute configuration: (5″R)(2R,3R,3aS,6R,6aR,3′R,3′aS,6′R,6′aR,5″R)-Spiro-[6,3′,6′-trihydroxy-octahydro[2,3′]bi[furo[3,2-b]furan]-3,2″-[5″-[4‴-[2‴′-methoxyethyl]]phenoxymethyl-1″,3″-oxazolidine]C24H33NO10[α]D20=+25.6 (c 1.00, CH3OH)Source of chirality: asymmetric synthesisAbsolute configuration: (5″R)(R)-1-Amino-3-[4-[2-hydroxyethyl]phenoxy]propan-2-olC11H17NO3[α]D20=+4.0 (c 1.00, CH3OH)Source of chirality: kinetic resolutionAbsolute configuration: (2R)
Co-reporter:Hai-Wei Xu, Jun-Feng Wang, Gai-Zhi Liu, Guang-Feng Hong and Hong-Min Liu
Organic & Biomolecular Chemistry 2007 vol. 5(Issue 8) pp:1247-1250
Publication Date(Web):16 Mar 2007
DOI:10.1039/B701051F
In this paper, a novel route to γ-alkylidenebutenolides (γ-AIBs) by way of stereoselective vinylogous aldol reaction of the unactivated butenolide in simple and general conditions is reported.
Co-reporter:Shun Xu, Gang Wang, Hong-Min Liu, Lin-Jiang Wang, Hui-Fang Wang
Journal of Molecular Structure: THEOCHEM 2007 Volume 809(1–3) pp:79-85
Publication Date(Web):14 May 2007
DOI:10.1016/j.theochem.2007.01.036
The DMol3 calculations, based on the density functional theory (DFT), have been used to study the structure–activity relationship of trans-resveratrol in the reaction of scavenging free radicals. The geometries of trans-resveratrol and its three phenoxy radicals were obtained at the Generalized Gradient Approximation (GGA) level, with the spin unrestricted approach and BLYP/DND method. Three possible reactions of scavenging hydroperoxyl radicals, in terms of H-atom-transfer mechanism, were designed. The transition state for each reaction was determined, and the intrinsic reaction coordinate was analyzed. The activation energy and thermodynamic state functions were calculated. The differences of the BDE and structure properties of stationary points show that 4′-radical is stabler and 4′-hydroxyl is more reactive. And the results also show that the reaction of 4′-hydroxyl scavenging hydroperoxyl radicals is more competitive in view of thermodynamics and kinetics. Thus, the dissimilarity of antioxidant activity among O–H groups of trans-resveratrol, which had been reported by many experimental studies, is explained.
Co-reporter:Hong-Min Liu;Da-Peng Zou;Fuyi Zhang;Wei-Guo Zhu;Tao Peng
European Journal of Organic Chemistry 2004 Volume 2004(Issue 10) pp:
Publication Date(Web):27 APR 2004
DOI:10.1002/ejoc.200300763
A simple, one-pot multi-step route for the synthesis of a higher carbon sugar 3 by the HDA reaction of a α,β-unsaturated ketone prepared in situ from protected D-xylose with PDC in C6H6 or CH3CN, followed by the reduction to the C10 higher carbon sugar derivatives 4 and 5, is described. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2004)
Co-reporter:Hong-Min Liu, Fuyi Zhang and Da-Peng Zou
Chemical Communications 2003 (Issue 16) pp:2044-2045
Publication Date(Web):04 Jul 2003
DOI:10.1039/B306227A
2,2-Bis(C-branched-chain)glucopyranosid-3-uloses, designed for the preparation of biologically active natural product iridoid derivatives, are synthesized selectively by the new reaction of butenolide-containing sugar with active methylene compounds, and the new reaction is clarified as autoxidation followed by Michael addition of carbanion.
Co-reporter:Bin Yu, Xiao-Nan Sun, Xiao-Jing Shi, Ping-Ping Qi, Yi-Chao Zheng, De-Quan Yu, Hong-Min Liu
Steroids (October 2015) Volume 102() pp:92-100
Publication Date(Web):1 October 2015
DOI:10.1016/j.steroids.2015.08.003
•Spirooxindoles are privileged scaffolds in drug discovery.•A series of novel steroidal spirooxindoles were synthesized.•Most of these compounds exhibited moderate to good antiproliferative activity.•Some of them were more potent than 5-FU.•Compound 3d caused cell cycle arrest at G2/M phase and early apoptosis.A series of novel steroidal spirooxindoles 3a–h were synthesized from pregnenolone in a high regioselective manner using the 1,3-dipolar cycloaddition as the key step. This protocol resulted in the formation of two C–C bonds, one C–N bond and the creation of one pyrrolidine ring and three contiguous stereocenters in a single operation. Biological evaluation showed that these synthesized steroidal spirooxindoles exhibited moderate to good antiproliferative activity against the tested cell lines and some of them were more potent than 5-FU. Among them, compounds 3e and 3f displayed the best antiproliferative activity against MCF-7 cells with the IC50 values of 4.0 and 3.9 μM, respectively. Flow cytometry analysis demonstrated that compound 3d caused the cellular apoptosis and cell cycle arrest at G2/M phase in a concentration-dependent manner. Docking results indicated that compound 3d fitted well into the MDM2 active site 1RV1 by interacting with Lys94 and Thr101 residues.Download full-size image
Co-reporter:Bin Yu, Ping-Ping Qi, Xiao-Jing Shi, Li-Hong Shan, De-Quan Yu, Hong-Min Liu
Steroids (October 2014) Volume 88() pp:44-52
Publication Date(Web):1 October 2014
DOI:10.1016/j.steroids.2014.05.022
•A new class of steroidal pyran–oxindole hybrids were efficiently synthesized in one pot.•These compounds were more sensitive to MCF-7 and MGC-803 cells.•Compounds 4f and 4i were more potent than 5-Fu against T24 and MGC-803 cells.•Compound 4f showed excellent cytotoxicity against T24 cells (IC50 = 4.43 μM).•Compound 4i showed cell cycle arrest at G2/M phase and induced early apoptosis.A series of novel steroidal pyran–oxindole hybrids were efficiently synthesized in a single operation through the vinylogous aldol reaction of vinyl malononitrile 3 with substituted isatins involving the construction of C–C and C–O bonds. Some compounds displayed moderate to good cytotoxicity against T24, SMMC-7721, MCF-7 and MGC-803 cells. Compounds 4f and 4i were more potent than 5-Fu against T24 and MGC-803 cells with the IC50 values of 4.43 and 8.45 μM, respectively. Further mechanism studies indicated that compound 4i induced G2/M arrest and early apoptosis in a concentration- and time-dependent manner.Graphical abstractDownload full-size image
Co-reporter:Bin Yu, Xiao-Nan Sun, Xiao-Jing Shi, Ping-Ping Qi, Yuan Fang, En Zhang, De-Quan Yu, Hong-Min Liu
Steroids (November 2013) Volume 78(Issue 11) pp:1134-1140
Publication Date(Web):1 November 2013
DOI:10.1016/j.steroids.2013.08.001
•The steroidal dienamides were synthesized stereoselectively through a cascade aldol/cyclization process.•These compounds had moderate to excellent cytotoxic activities.•Some of these compounds were more potent than 5-fluorouracil.•Compound 3c represented excellent inhibition against MCF-7 (IC50 = 0.76 μM).The stereoselective and metal-free protocol involving a cascade aldol/cyclization process for the synthesis of steroidal (E, E) dienamides from steroidal α, α-dicyanoalkene was reported. This protocol efficiently achieved the construction of CC bond and selective conversion of cyano group into carboxamide in one-pot procedure under mild condition. Further biological evaluation showed that some of these compounds had moderate to excellent cytotoxic activities against all the tested cancer cell lines and were more potent than well-known drug 5-fluorouracil. Particularly, compound 3c represented excellent inhibitory effect against MCF-7 (IC50 = 0.76 μM), which was about 10-fold more potent than 5-fluorouracil.Download full-size image
Co-reporter:Yan-Ling Zhang, Ya-Fei Li, Jun-Wei Wang, Bin Yu, Yun-Kai Shi, Hong-Min Liu
Steroids (May 2016) Volume 109() pp:22-28
Publication Date(Web):1 May 2016
DOI:10.1016/j.steroids.2016.03.005
•Multicomponent assembly of steroidal dihydropyridinyl spirooxindoles is described.•The protocol formed two C–C bonds, two C–N bonds, and an all-carbon quaternary stereogenic center in a single operation.•Some of these compounds were more potent than 5-FU.•Compound 5o represented excellent inhibitory effect toward EC-109 (IC50 = 0.32 μM).Multicomponent assembly of steroidal dihydropyridinyl spirooxindoles from pregnenolone, isatins, malononitrile, and ammonium acetate is described, which involves the formation of two C–C bonds, two C–N bonds, and an all-carbon quaternary stereogenic center in a single operation. MTT assays showed that some of these compounds had moderate to excellent cytotoxicity against the tested cancer cell lines and were more potent than 5-FU. Particularly, compound 5o represented excellent inhibitory effect toward EC-109 (IC50 = 0.3 μM), being about 33-fold more potent than 5-FU.Download full-size image
Co-reporter:Li-Hua Huang, Yong-Fei Zheng, Yong-Zheng Lu, Chuan-Jun Song, Yan-Guang Wang, Bin Yu, Hong-Min Liu
Steroids (May 2012) Volume 77(Issue 6) pp:710-715
Publication Date(Web):1 May 2012
DOI:10.1016/j.steroids.2012.03.002
The preparation of steroidal[17,16-d][1,2,4]triazolo[1,5-a]pyrimidines and their biological evaluation as potential anticancer agents are herein reported. These novel heterosteroids (2, 4) were prepared through the condensation reaction of 3-amino-1,2,4-triazole with 16-arylidene-17-ketosteroids (1, 3). All the synthesized compounds were evaluated for their anticancer activity in vitro against PC-3 (human prostatic carcinoma), MCF-7 (human breast carcinoma) and EC9706 (human esophageal carcinoma) cell lines. Among the screened compounds, 2i, 2n and 4f showed significant inhibitory activity against all the three human cell lines.Graphical abstractDownload full-size imageHighlights► A novel class of steroidal[17,16-d]triazolopyrimidines was synthesized. ► These heterosteroids contain the 1,2,4-triazolo[1,5-a]pyrimidine moiety. ► Some of the synthesized compounds showed promising anticancer activity.
Co-reporter:Hai-Wei Xu, Gai-Zhi Liu, Song-Lin Zhu, Guang-Feng Hong, Hong-Min Liu, Qiong Wu
Steroids (June 2010) Volume 75(Issue 6) pp:419-423
Publication Date(Web):1 June 2010
DOI:10.1016/j.steroids.2010.02.007
A series of digoxin derivatives containing the γ-alkylidene butenolide moiety were synthesised by way of stereoselective vinylogous aldol reaction of the unactivated butenolide in simple conditions. The structures of compounds synthesised were characterised by infrared (IR), nuclear magnetic resonance (NMR) and HR-MS. Preliminary bioassay shows that some of them have cardiac functions, especially compound 2g that induced a marked increase in myocardial contractility at 10 ng ml−1 and 20 ng ml−1 concentrations without digitalis toxicity.
Co-reporter:Hong-Min Liu, Wenzhong Ge, Heping Li, Jian Wu
Steroids (June 2007) Volume 72(Issues 6–7) pp:509-513
Publication Date(Web):1 June 2007
DOI:10.1016/j.steroids.2006.12.009
Fermentation of 5α,6α-epoxy-3β-hydroxy-16-pregnen-20-one (4) with Trichoderma viride under aerobic condition yielded 3β,5α,6β-trihydroxy-16-pregnen-20-one (5) and 3β,5α,6β,15β-tetrahydroxy-16-pregnen-20-one (6). Each microbial metabolite was characterized by spectroscopic methods. Compounds 6 and 3β,5α,15β-trihydroxy-16-pregnen-6,20-dione (7) are reported for the first time.
Co-reporter:Li-Hua Huang, Yang Li, Hong-De Xu, Yong-Fei Zheng, Hong-Min Liu
Steroids (July 2014) Volume 85() pp:13-17
Publication Date(Web):1 July 2014
DOI:10.1016/j.steroids.2014.03.017
•Novel C6-cyclo secondary amine substituted purine nucleoside analogues was synthesized.•These compounds contain the modified pyranose-like D ring of the 4-azasteroid moiety.•Compounds 5c and 6b exhibited significant cytotoxicity on PC-3 cell lines.Novel C6-cyclo secondary amine substituted purine steroid-nucleoside analogues (2–9) were efficiently synthesized through displacement of the C6 chloro on the purine ring of series 1 with versatile cyclic secondary amines, including pyrrolidines, piperidine, morpholine, and piperazines. All the newly-synthesized compounds were evaluated for their anticancer activity in vitro against Hela, PC-3 and MCF-7 cell lines. Among them, compounds 5c and 6b exhibited significant cytotoxicity on PC-3 cell lines.Download full-size image
Co-reporter:Shao-Min Wang, Yan-Bing Zhang, Hong-Min Liu, Guo-Bin Yu, Ke-Rang Wang
Steroids (January 2007) Volume 72(Issue 1) pp:26-30
Publication Date(Web):1 January 2007
DOI:10.1016/j.steroids.2006.10.004
Dibutyltin oxide (DBTO) was first utilized for the deacetylation of steroid and diterpene esters. The results showed the deprotection of acetylated steroids and diterpenes separately with moderate catalysis dibutyltin oxide in methanol selectively removed part acetyl groups of these substrates, whereas several functional groups of the steroids and diterpenes were retained and neither isomerization nor degradation of these substrates was observed. It seems that the acetyl groups with lower steric hindrance or near carbonyl, alkoxy, or hydroxyl groups can be cleaved by the reaction, whereas the acetyl groups with higher steric hindrance or without carbonyl, alkoxy, or hydroxyl groups neighboring were retained under the same conditions. One of the interesting results obtained was the selective hydrolysis of the 3β-O-acetyl group in the presence of the 6β group in 3β,6β-Di-O-acetyl-5α-hydroxypregn-16-en-20-one. This allows for subsequent introduction of one unit at C-3 and the other unit at C-6. This procedure is useful for the synthesis of a series of closely related isomers of 3β,5α,6β-trihydroxypregn-16-en-20-one and other widespread polyhydroxysteroids in marine organisms and some terrestrial species.
Co-reporter:Yi-Chao Zheng, Dan-Dan Shen, Meng Ren, Xue-Qi Liu, Zhi-Ru Wang, Ying Liu, Qian-Na Zhang, Li-Juan Zhao, Li-Jie Zhao, Jin-Lian Ma, Bin Yu, Hong-Min Liu
Bioorganic Chemistry (December 2016) Volume 69() pp:
Publication Date(Web):December 2016
DOI:10.1016/j.bioorg.2016.10.004
•It is the first time to characterize baicalin as a LSD1 inhibitor.•Baicalin does not only inactive LSD1 in recombinant level, but also in cells.•Baicalin may inhibit gastric cancer cell proliferation and migration.Baicalin is one of the active ingredients in the skullcap, with a variety of pharmacological effects, such as blood pressure reduction, sedation, liver-protection, gallbladder-protection, anti-bacteria, and anti-inflammation. In our study, baicalin was first characterized as a LSD1 inhibitor with an IC50 of 3.01 μM and showed strong LSD1 inhibitory effect in cells. Baicalin may serve as a template for designing flavone-based LSD1 inhibitors.
Co-reporter:Li-Hua Huang, Hong-De Xu, Zhuo-Ya Yang, Yong-Fei Zheng, Hong-Min Liu
Steroids (April 2014) Volume 82() pp:1-6
Publication Date(Web):1 April 2014
DOI:10.1016/j.steroids.2013.12.004
•A novel class of C6-piperazine substituted purine nucleoside analogues was synthesized.•These compounds contain the modified pyranose-like D ring of the 4-azasteroid moiety.•Compounds 8b and 9b exhibited significant cytotoxicity on PC-3 cell lines.Novel C6-piperazine substituted purine nucleoside analogues (2–9) bearing a modified pyranose-like D ring of the 4-azasteroid moiety were efficiently synthesized through nucleophilic substitution at C6 position of the steroid–nucleoside precursors (1) with versatile piperazines. All newly-synthesized compounds were evaluated for their anticancer activity in vitro against Hela, PC-3 and MCF-7 cell lines. Among them, compounds 8b and 9b exhibited significant cytotoxicity on PC-3 cell lines.Download full-size image
Co-reporter:Yong-Cheng Ma, Nan Su, Xiao-Jing Shi, Wen Zhao, Yu Ke, Xiaolin Zi, Ning-Min Zhao, Yu-Hua Qin, Hong-Wei Zhao, Hong-Min Liu
Toxicology and Applied Pharmacology (15 January 2015) Volume 282(Issue 2) pp:227-236
Publication Date(Web):15 January 2015
DOI:10.1016/j.taap.2014.11.003
•Jaridonin induced G2/M phase arrest through induction of redox imbalance.•Jaridonin increased the level of ROS through depleting glutathione in cell.•ATM–Chk1/2–Cdc25C were involved in Jaridonin-induced cell cycle arrest.•Jaridonin selectively inhibited cancer cell viability and cell cycle progression.Jaridonin, a novel diterpenoid from Isodon rubescens, has been shown previously to inhibit proliferation of esophageal squamous cancer cells (ESCC) through G2/M phase cell cycle arrest. However, the involved mechanism is not fully understood. In this study, we found that the cell cycle arrest by Jaridonin was associated with the increased expression of phosphorylation of ATM at Ser1981 and Cdc2 at Tyr15. Jaridonin also resulted in enhanced phosphorylation of Cdc25C via the activation of checkpoint kinases Chk1 and Chk2, as well as in increased phospho-H2A.X (Ser139), which is known to be phosphorylated by ATM in response to DNA damage. Furthermore, Jaridonin-mediated alterations in cell cycle arrest were significantly attenuated in the presence of NAC, implicating the involvement of ROS in Jaridonin's effects. On the other hand, addition of ATM inhibitors reversed Jaridonin-related activation of ATM and Chk1/2 as well as phosphorylation of Cdc25C, Cdc2 and H2A.X and G2/M phase arrest. In conclusion, these findings identified that Jaridonin-induced cell cycle arrest in human esophageal cancer cells is associated with ROS-mediated activation of ATM–Chk1/2–Cdc25C pathway.Download high-res image (105KB)Download full-size image
Co-reporter:Hai-Wei Xu, Jun-Feng Wang, Gai-Zhi Liu, Guang-Feng Hong and Hong-Min Liu
Organic & Biomolecular Chemistry 2007 - vol. 5(Issue 8) pp:NaN1250-1250
Publication Date(Web):2007/03/16
DOI:10.1039/B701051F
In this paper, a novel route to γ-alkylidenebutenolides (γ-AIBs) by way of stereoselective vinylogous aldol reaction of the unactivated butenolide in simple and general conditions is reported.

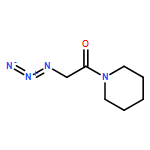
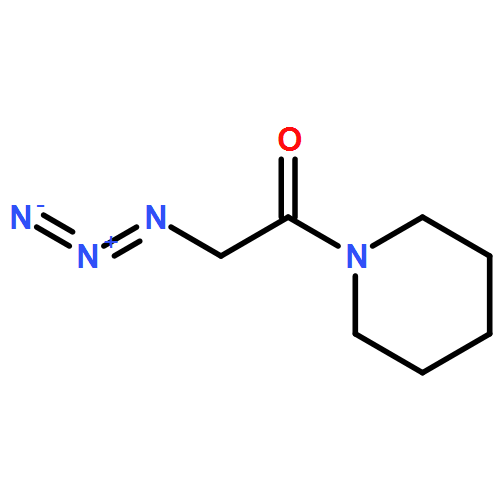
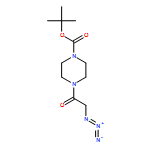

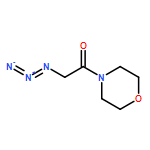
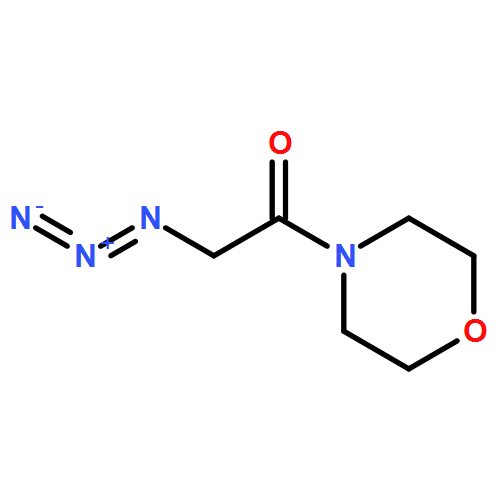
![Boronic acid,B-[4-[(2E)-3-(4-iodophenyl)-1-oxo-2-propen-1-yl]phenyl]-](http://img.cochemist.com/ccimg/562900/562823-84-1.png)
![Boronic acid,B-[4-[(2E)-3-(4-iodophenyl)-1-oxo-2-propen-1-yl]phenyl]-](http://img.cochemist.com/ccimg/562900/562823-84-1_b.png)













![4(1H)-Pyrimidinone, 6-hydroxy-2-[(phenylmethyl)thio]-](http://img.cochemist.com/ccimg/148100/148020-95-5.png)
![4(1H)-Pyrimidinone, 6-hydroxy-2-[(phenylmethyl)thio]-](http://img.cochemist.com/ccimg/148100/148020-95-5_b.png)






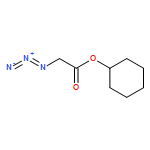

![Ethanone, 1-[4-(2-propynylamino)phenyl]-](http://img.cochemist.com/ccimg/109800/109702-67-2.png)
![Ethanone, 1-[4-(2-propynylamino)phenyl]-](http://img.cochemist.com/ccimg/109800/109702-67-2_b.png)
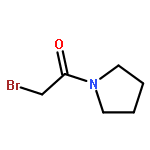
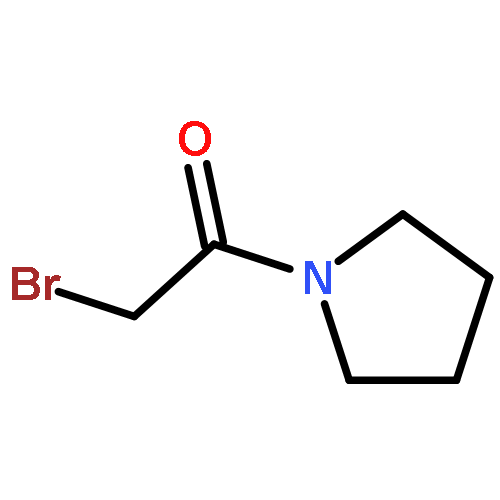


![[1,2,4]Triazolo[1,5-a]pyrimidine, 5-methyl-7-(4-morpholinyl)-2-[(phenylmethyl)thio]-](/data/chemimg/1256700/51646-39-0.png)
![[1,2,4]Triazolo[1,5-a]pyrimidine, 5-methyl-7-(4-morpholinyl)-2-[(phenylmethyl)thio]-](/data/chemimg/1256700/51646-39-0_b.png)
![[1,2,4]Triazolo[1,5-a]pyrimidine, 7-chloro-5-methyl-2-[(phenylmethyl)thio]-](/data/chemimg/1259300/51646-34-5.png)
![[1,2,4]Triazolo[1,5-a]pyrimidine, 7-chloro-5-methyl-2-[(phenylmethyl)thio]-](/data/chemimg/1259300/51646-34-5_b.png)
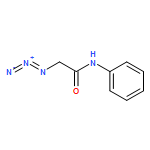
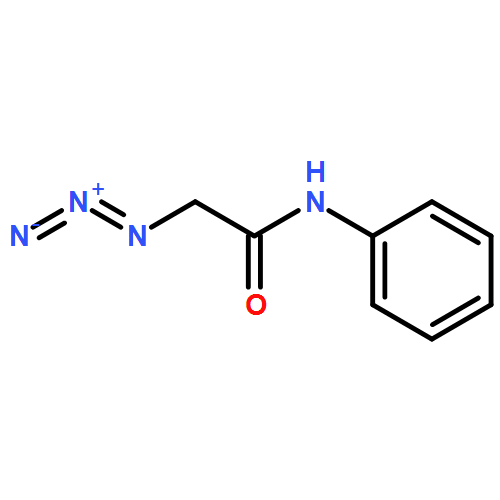










![(3aR,5R,6aS)-5-((R)-2,2-Dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyldihydrofuro[2,3-d][1,3]dioxol-6(3aH)-one](http://img.cochemist.com/ccimg/2900/2847-00-9.png)
![(3aR,5R,6aS)-5-((R)-2,2-Dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyldihydrofuro[2,3-d][1,3]dioxol-6(3aH)-one](http://img.cochemist.com/ccimg/2900/2847-00-9_b.png)





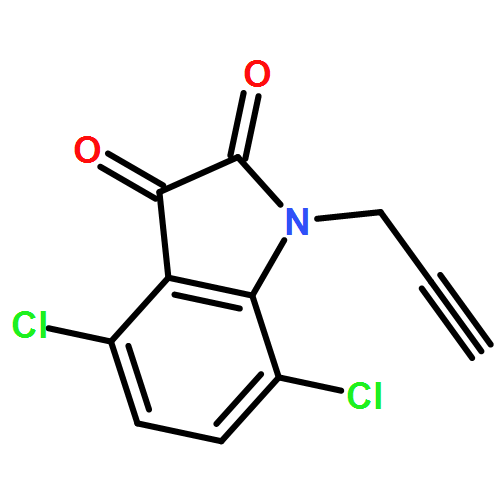




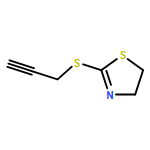
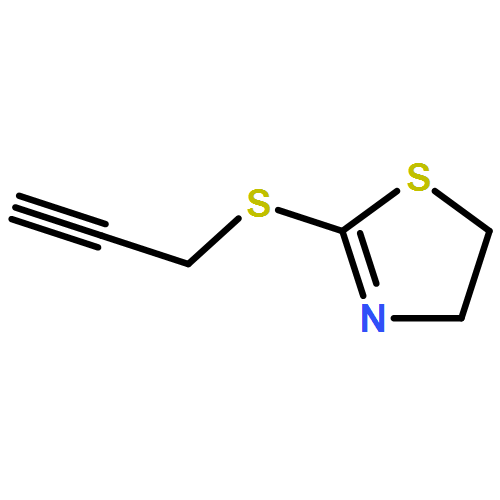


![2-Propen-1-one, 1-phenyl-3-[4-(2-propyn-1-yloxy)phenyl]-, (2E)-](/data/chemimg/3723400/1620336-56-2.png)
![2-Propen-1-one, 1-phenyl-3-[4-(2-propyn-1-yloxy)phenyl]-, (2E)-](/data/chemimg/3723400/1620336-56-2_b.png)




![2-(3,4-dihydroxyphenyl)-3,5,11,12-tetrahydroxy-6-methyl-4H,8H-[2]benzopyrano[3,4-h]-1-benzopyran-4,8-dione](http://img.cochemist.com/ccimg/1357900/1357865-67-8.png)
![2-(3,4-dihydroxyphenyl)-3,5,11,12-tetrahydroxy-6-methyl-4H,8H-[2]benzopyrano[3,4-h]-1-benzopyran-4,8-dione](http://img.cochemist.com/ccimg/1357900/1357865-67-8_b.png)
![2-(3,4-dihydroxyphenyl)-3,5,10,11-tetrahydroxy-6-methyl-4H,8H-[2]benzopyrano[3,4-h]-1-benzopyran-4,8-dione](http://img.cochemist.com/ccimg/1357900/1357865-66-7.png)
![2-(3,4-dihydroxyphenyl)-3,5,10,11-tetrahydroxy-6-methyl-4H,8H-[2]benzopyrano[3,4-h]-1-benzopyran-4,8-dione](http://img.cochemist.com/ccimg/1357900/1357865-66-7_b.png)
![(1aR,7aR)-4,6-dihydroxy-1a-(3-hydroxy-4-((2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yloxy)phenyl)-7a-(2-hydroxy-4-(3,5,7-trihydroxy-6-methyl-4-oxo-4H-chromen-2-yl)phenoxy)-5-methyl-1aH-oxireno[2,3-b]chromen-7(7aH)-one](http://img.cochemist.com/ccimg/1346600/1346521-74-1.png)
![(1aR,7aR)-4,6-dihydroxy-1a-(3-hydroxy-4-((2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yloxy)phenyl)-7a-(2-hydroxy-4-(3,5,7-trihydroxy-6-methyl-4-oxo-4H-chromen-2-yl)phenoxy)-5-methyl-1aH-oxireno[2,3-b]chromen-7(7aH)-one](http://img.cochemist.com/ccimg/1346600/1346521-74-1_b.png)
![(1aS,7aS)-4,6-dihydroxy-1a-(3-hydroxy-4-((2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yloxy)phenyl)-7a-(2-hydroxy-4-(3,5,7-trihydroxy-6-methyl-4-oxo-4H-chromen-2-yl)phenoxy)-5-methyl-1aH-oxireno[2,3-b]chromen-7(7aH)-one](http://img.cochemist.com/ccimg/1346600/1346521-73-0.png)
![(1aS,7aS)-4,6-dihydroxy-1a-(3-hydroxy-4-((2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yloxy)phenyl)-7a-(2-hydroxy-4-(3,5,7-trihydroxy-6-methyl-4-oxo-4H-chromen-2-yl)phenoxy)-5-methyl-1aH-oxireno[2,3-b]chromen-7(7aH)-one](http://img.cochemist.com/ccimg/1346600/1346521-73-0_b.png)






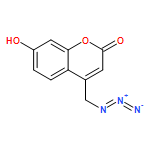





![Acetamide, 2-chloro-N-[(2-pyridinylamino)carbonyl]-](http://img.cochemist.com/ccimg/653000/652992-39-7.png)
![Acetamide, 2-chloro-N-[(2-pyridinylamino)carbonyl]-](http://img.cochemist.com/ccimg/653000/652992-39-7_b.png)

















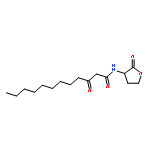
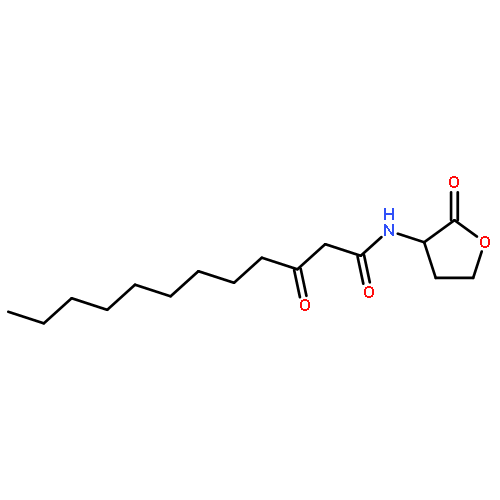













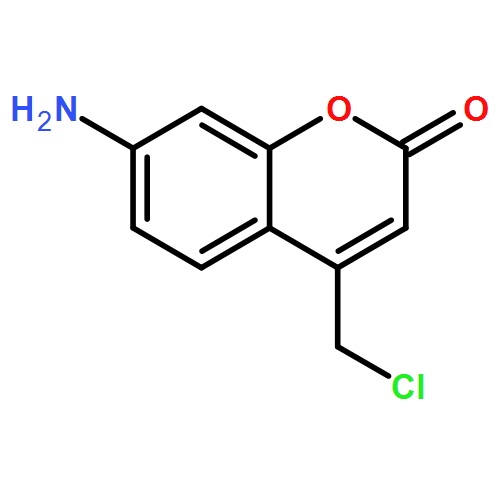






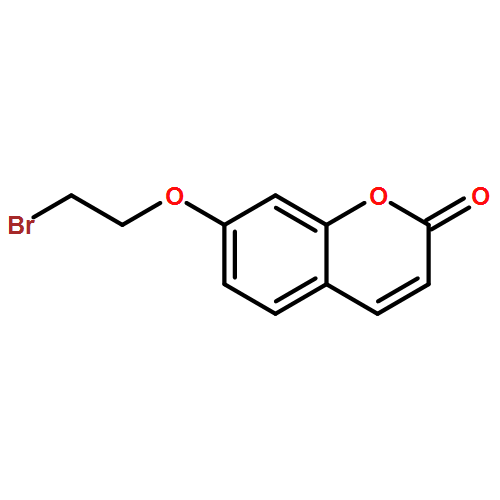
![6-morpholin-4-yl-3-phenyl[1,2,4]triazolo[3,4-a]phthalazine](http://img.cochemist.com/ccimg/87600/87539-85-3.png)
![6-morpholin-4-yl-3-phenyl[1,2,4]triazolo[3,4-a]phthalazine](http://img.cochemist.com/ccimg/87600/87539-85-3_b.png)








![1-[3-(TRIFLUOROMETHYL)BENZYL]-1H-INDOLE-2,3-DIONE](http://img.cochemist.com/ccimg/79200/79183-40-7.png)
![1-[3-(TRIFLUOROMETHYL)BENZYL]-1H-INDOLE-2,3-DIONE](http://img.cochemist.com/ccimg/79200/79183-40-7_b.png)










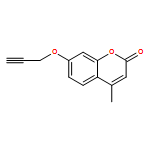
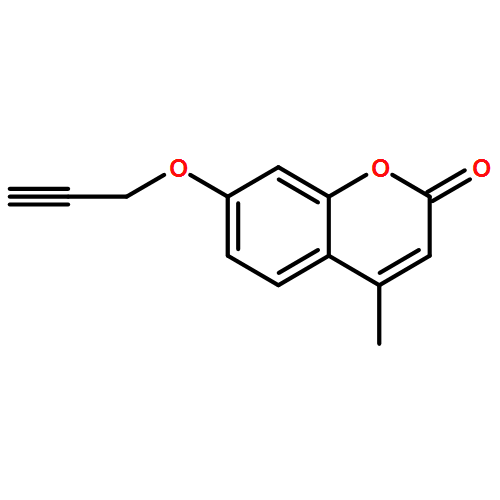

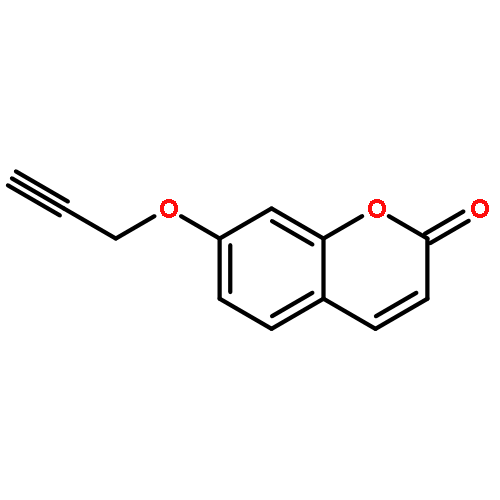

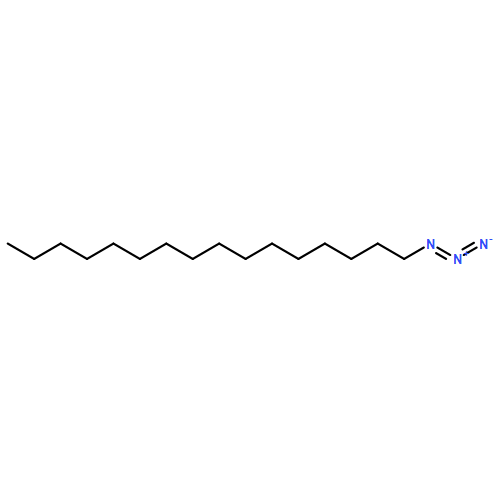














![Ethanone,1-[4-(2-propyn-1-yloxy)phenyl]-](http://img.cochemist.com/ccimg/34300/34264-14-7.png)
![Ethanone,1-[4-(2-propyn-1-yloxy)phenyl]-](http://img.cochemist.com/ccimg/34300/34264-14-7_b.png)


![1H-Indole-2,3-dione,1-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/27000/26960-66-7.png)
![1H-Indole-2,3-dione,1-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/27000/26960-66-7_b.png)




![5-Chloro-2-(chloromethyl)-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/20500/20443-38-3.png)
![5-Chloro-2-(chloromethyl)-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/20500/20443-38-3_b.png)

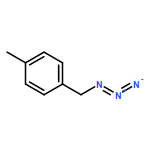

![1-[2-(2-chlorophenyl)ethenyl]isoquinoline](http://img.cochemist.com/ccimg/14200/14174-76-6.png)
![1-[2-(2-chlorophenyl)ethenyl]isoquinoline](http://img.cochemist.com/ccimg/14200/14174-76-6_b.png)






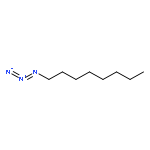





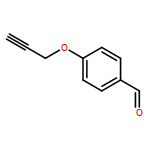
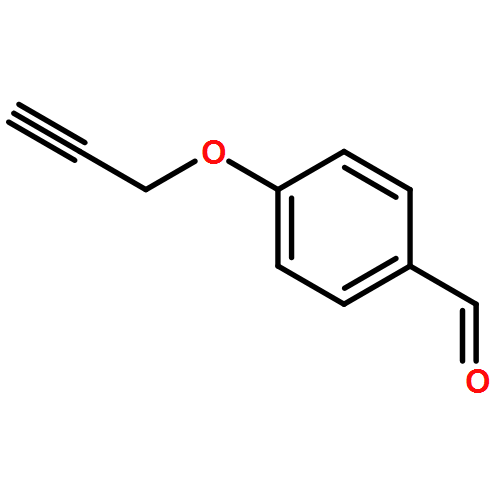
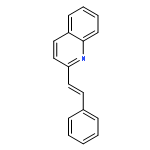
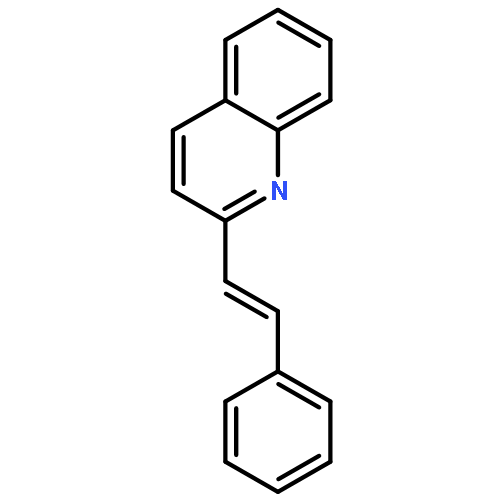







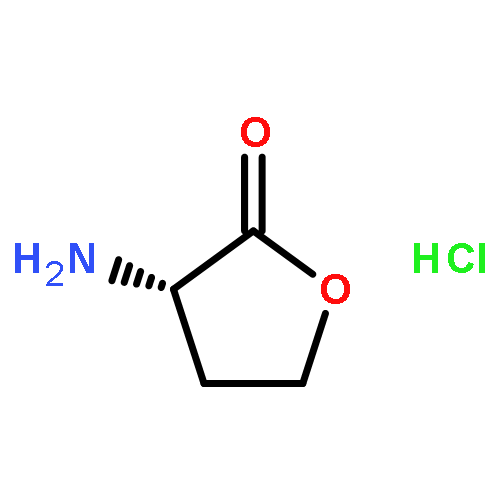




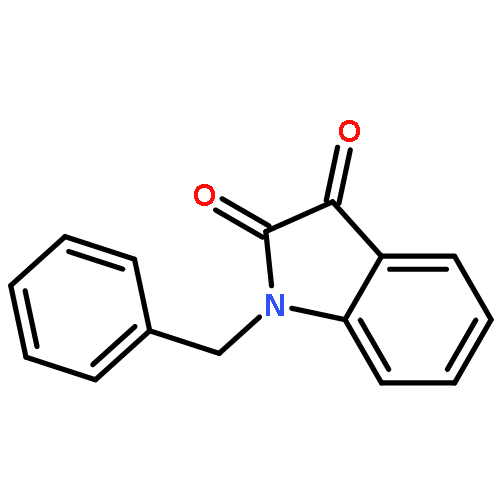





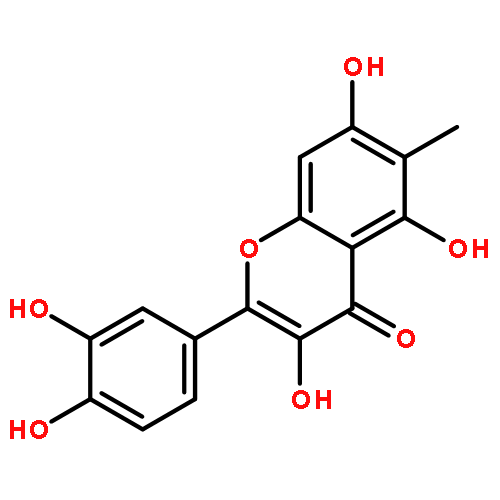










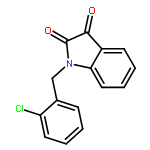



![(2R,5R,6R)-6-{[{[(4-ethyl-2,3-dioxopiperazin-1-yl)carbonyl]amino}(phenyl)acetyl]amino}-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid - (2S,3S,5R)-3-methyl-7-oxo-3-(1H-1,2,3-triazol-1-ylmethyl)-4-thia-1-azabicyclo[3.2.0]heptane-2-c](/data/chemimg/105600/123683-33-0.png)
![(2R,5R,6R)-6-{[{[(4-ethyl-2,3-dioxopiperazin-1-yl)carbonyl]amino}(phenyl)acetyl]amino}-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid - (2S,3S,5R)-3-methyl-7-oxo-3-(1H-1,2,3-triazol-1-ylmethyl)-4-thia-1-azabicyclo[3.2.0]heptane-2-c](/data/chemimg/105600/123683-33-0_b.png)

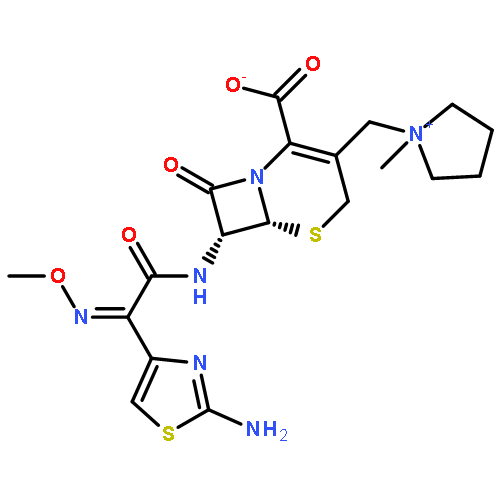

![6-Chloro-3-methyl-[1,2,4]triazolo[3,4-a]-phthalazine](http://img.cochemist.com/ccimg/67500/67458-38-2.png)
![6-Chloro-3-methyl-[1,2,4]triazolo[3,4-a]-phthalazine](http://img.cochemist.com/ccimg/67500/67458-38-2_b.png)
![6-CHLORO-3-PHENYL-[1,2,4]TRIAZOLO[3,4-A]PHTHALAZINE](http://img.cochemist.com/ccimg/56900/56813-54-8.png)
![6-CHLORO-3-PHENYL-[1,2,4]TRIAZOLO[3,4-A]PHTHALAZINE](http://img.cochemist.com/ccimg/56900/56813-54-8_b.png)
![1,2,4-Triazolo[3,4-a]phthalazine,6-chloro-](http://img.cochemist.com/ccimg/52500/52494-53-8.png)
![1,2,4-Triazolo[3,4-a]phthalazine,6-chloro-](http://img.cochemist.com/ccimg/52500/52494-53-8_b.png)