Co-reporter:Tyler A. Chavez, Yonglin Liu, and John P. Toscano
The Journal of Organic Chemistry 2016 Volume 81(Issue 15) pp:6320-6328
Publication Date(Web):June 30, 2016
DOI:10.1021/acs.joc.6b00949
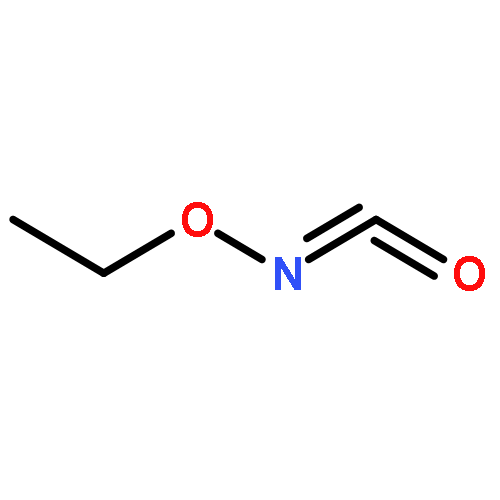
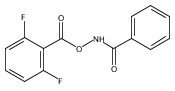
N-Ethyloxycarbonyl-S,S-dibenzothiphene sulfilimine and N-t-butyloxycarbonyl-S,S-dibenzothiphene sulfilimine have been utilized as precursors to ethoxycarbonylnitrene and t-butyloxycarbonylnitrene. B3LYP/6-31G(d) calculations predict triplet ground states for both oxycarbonylnitrenes, albeit by small margins. Triplet ethoxycarbonylnitrene and triplet t-butyloxycarbonylnitrene have been observed following photolysis of these sulfilimine precursors by time-resolved infrared (TRIR) spectroscopy. Kinetic studies show that ethoxycarbonylnitrene reacts with solvents such as acetonitrile and cyclohexane, while t-butyloxycarbonylnitrene undergoes an intramolecular insertion reaction to produce 5,5-dimethyl oxazolidinone. Product analysis following photolysis of N-t-butyloxycarbonyl-S,S-dibenzothiphene sulfilimine confirms that the oxazolidinone is the major product with an estimated yield of 90%. The products from these two nitrenes are derived from the corresponding singlet nitrene, either directly or via thermal repopulation of the singlet from the lower-energy triplet nitrene.
Co-reporter:Saghar Nourian, Robert P. Lesko, Daryl A. Guthrie, John P. Toscano
Tetrahedron 2016 Volume 72(Issue 40) pp:6037-6042
Publication Date(Web):6 October 2016
DOI:10.1016/j.tet.2016.08.016
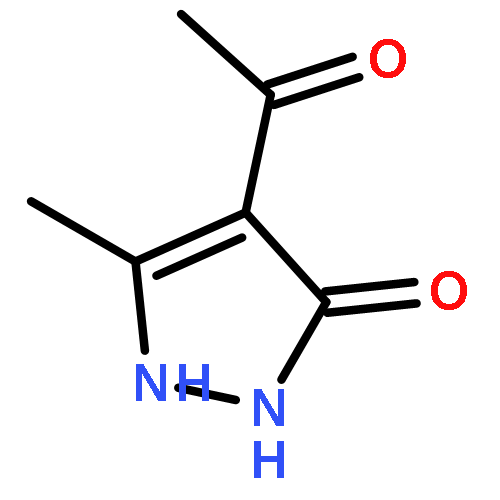
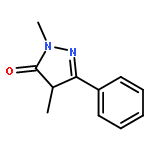
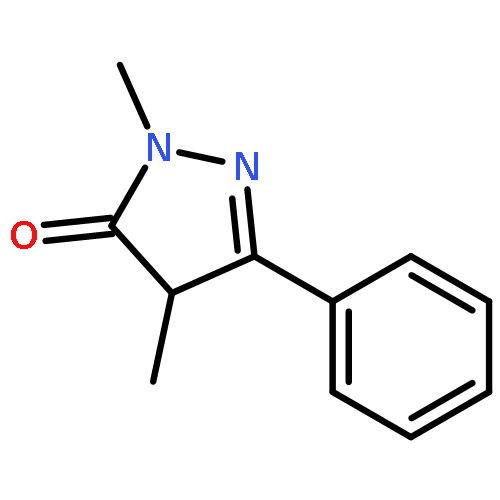
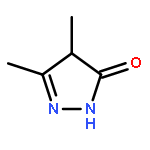
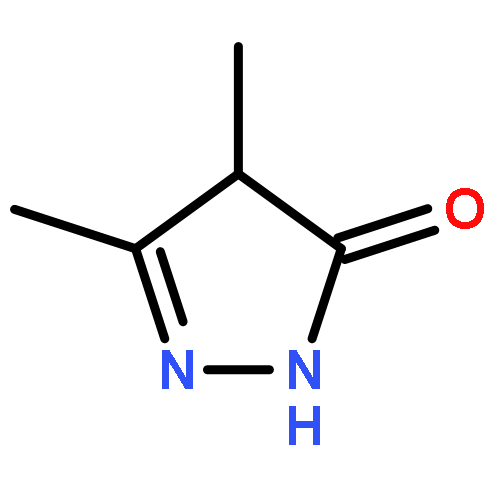
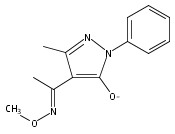
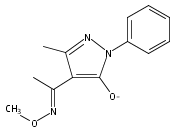
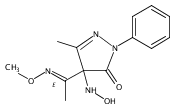
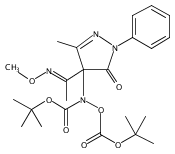
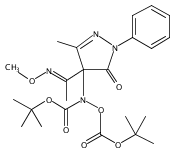
A new class of nitrosocarbonyl precursors, O-substituted hydroxamic acids with pyrazolone leaving groups (OHPY), is described. These compounds generate nitrosocarbonyl intermediates, which upon hydrolysis release nitroxyl (azanone, HNO) under physiologically relevant conditions. Pyrazolones have been used to confirm the generation of nitrosocarbonyls by competitive trapping to form isomeric N-substituted hydroxamic acids (NHPY) via an N-selective nitrosocarbonyl aldol reaction. The rate of nitrosocarbonyl release from OHPY donors is impacted by donor substituents, including the pyrazolone leaving group.
Co-reporter:Saghar Nourian, Zachary A. Zilber, and John P. Toscano
The Journal of Organic Chemistry 2016 Volume 81(Issue 19) pp:9138-9146
Publication Date(Web):September 12, 2016
DOI:10.1021/acs.joc.6b01705
A novel class of nitrosocarbonyl precursors, N-substituted hydroxamic acids with pyrazolone leaving groups (NHPY), has been synthesized. Under physiological conditions, these compounds generate nitrosocarbonyl intermediates, which upon hydrolysis release nitroxyl (azanone, HNO) in excellent yields. The amount and rate of nitrosocarbonyl generation are dependent on the nature of the pyrazolone leaving groups and significantly on the structural properties of the NHPY donors. Pyrazolones have been found to be efficient nitrosocarbonyl traps, undergoing an N-selective nitrosocarbonyl aldol reaction. This trapping reaction has been used to confirm the involvement of nitrosocarbonyl intermediates in NHPY aqueous decomposition. In addition, NHPY compounds are shown to generate nitrosocarbonyls efficiently under mild basic conditions in organic solvent and may therefore also enjoy synthetic utility.
Co-reporter:Daryl A. Guthrie, Saghar Nourian, Cyrus G. Takahashi, and John P. Toscano
The Journal of Organic Chemistry 2015 Volume 80(Issue 3) pp:1349-1356
Publication Date(Web):January 13, 2015
DOI:10.1021/jo5023316
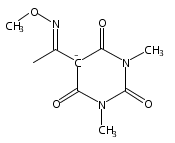
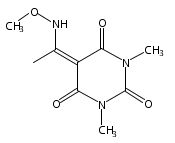
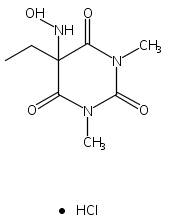
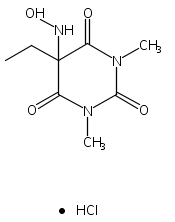
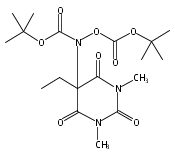
Due to its inherent reactivity, HNO must be generated in situ through the use of donor compounds. One of the primary strategies for the development of new HNO donors has been modifying hydroxylamines with good leaving groups. A recent example of this strategy is the (hydroxylamino)barbituric acid (HABA) class of HNO donors. In this case, however, an undesired intramolecular rearrangement pathway to the corresponding hydantoin derivative competes with HNO formation, particularly in the absence of chemical traps for HNO. This competitive non-HNO-producing pathway has restricted the development of the HABA class to examples with fast HNO release profiles at physiological pH and temperature (t1/2 < 1 min). Herein, the factors that favor the rearrangement pathway have been examined and two independent strategies that protect against rearrangement to favor HNO generation have been developed. The timecourse and stoichiometry for the in vitro conversion of these compounds to HNO (trapped as a phosphine aza-ylide) and the corresponding barbituric acid (BA) byproduct have been determined by 1H NMR spectroscopy under physiologically relevant conditions. These results confirm the successful extension of the HABA class of pure HNO donors with half-lives at pH 7.4, 37 °C ranging from 19 to 107 min.
Co-reporter:Daryl A. Guthrie, Anthony Ho, Cyrus G. Takahashi, Anthony Collins, Matthew Morris, and John P. Toscano
The Journal of Organic Chemistry 2015 Volume 80(Issue 3) pp:1338-1348
Publication Date(Web):January 16, 2015
DOI:10.1021/jo502330w
A new and versatile class of HNO donors, the (hydroxylamino)pyrazolone (HAPY) series of HNO donors utilizing pyrazolone (PY) leaving groups, is described. HNO, the smallest N-based aldehyde equivalent, is used as a reagent along with a variety of PY compounds to synthesize the desired HAPY donors in what can be considered an N-selective HNO-aldol reaction in up to quantitative yields. The bimolecular rate constant of HNO with PY in pH 7.4 phosphate buffer at 37 °C can reach 8 × 105 M–1 s–1. In 1H NMR experiments, the HAPY compounds generate HNO quantitatively (trapped as a phosphine aza-ylide) with half-lives spanning 3 orders of magnitude (minutes to days) under physiologically relevant conditions. B3LYP/6-31G* calculations confirm the energetically favorable reactions between HNO and the PY enol and enolate, whereas HNO release is expected to occur through the oxyanion (OHN-PY) of each HAPY compound. HNO has been shown to provide functional support to failing hearts.
Co-reporter:Gizem Keceli and John P. Toscano
Biochemistry 2014 Volume 53(Issue 22) pp:
Publication Date(Web):May 12, 2014
DOI:10.1021/bi500360x
Nitroxyl (HNO), a potential heart failure therapeutic, is known to target cysteine residues to form sulfinamides and/or disulfides. Because HNO-derived modifications may depend on their local environment, we have investigated the reactivity of HNO with cysteine derivatives and C-terminal cysteine-containing peptides at physiological pH and temperature. Our findings indicate that the nature of HNO-derived modifications of C-terminal cysteines is affected by the C-terminal carboxylate. Apart from the lack of sulfinamide formation, these studies have revealed the presence of new products, a sulfohydroxamic acid derivative (RS(O)2NHOH) and a thiosulfonate (RS(O)2SR), presumably produced under our experimental conditions via the intermediacy of a cyclic structure that is hydrolyzed to give a sulfenic acid (RSOH). Moreover, these modifications are formed independent of oxygen.
Co-reporter:Gizem Keceli, Cathy D. Moore, John P. Toscano
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 16) pp:3710-3713
Publication Date(Web):15 August 2014
DOI:10.1016/j.bmcl.2014.07.014
Recent discoveries of important pharmacological properties have drawn attention to the reactivity of HNO (azanone, nitroxyl) with biologically relevant substrates. Apart from its role in thiol oxidation, HNO has been reported to have nitrosative properties, for example, with tryptophan resulting in N-nitrosotryptophan formation. We have investigated the reactivity of HNO with tryptophan and small peptides containing either tryptophan or both a tryptophan and a cysteine residue. Our results point to the more reactive nature of cysteine towards HNO compared with tryptophan.
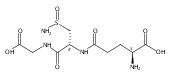
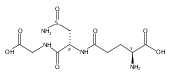
Co-reporter:Meredith R. Cline, Tyler A. Chavez, John P. Toscano
Journal of Inorganic Biochemistry 2013 Volume 118() pp:148-154
Publication Date(Web):January 2013
DOI:10.1016/j.jinorgbio.2012.09.024
Recent research has shown that nitroxyl (HNO) has important and unique biological activity, especially as a potential alternative to current treatments of cardiac failure. HNO is a reactive molecule that undergoes efficient dimerization and subsequent dehydration to form nitrous oxide (N2O), making its detection in solution or biologically relevant preparations difficult. Due to this limitation, HNO has not yet been observed in vivo, though several pathways for its endogenous generation have been postulated. Here, we investigate the oxidation of N-hydroxy-l-arginine (NOHA) by hypochlorous acid (HOCl), which is generated in vivo from hydrogen peroxide and chloride by the heme enzyme, myeloperoxidase. NOHA is an intermediate in the enzymatic production of nitric oxide (NO) by NO synthases, and has been shown previously to be chemically oxidized to either HNO or NO, depending on the oxidant employed. Using membrane inlet mass spectrometry and standard N2O analysis by gas chromatography, we find that NOHA is oxidized by excess HOCl to form HNO-derived N2O. In addition, we also observe the analogous production of HNO from the HOCl oxidation of hydroxylamine, hydroxyurea, and (to a lesser extent) acetohydroxamic acid.Oxidation of N-hydroxy-l-arginine by excess hypochlorous acid (HOCl) is examined by membrane inlet mass spectrometry and gas chromatography. The major products are determined to be HNO and HNO-derived N2O. The analogous production of HNO from HOCl oxidation of hydroxylamine, hydroxyurea, and (to a lesser extent) acetohydroxamic acid is also observed.Highlights► Oxidation of N-hydroxy-l-arginine by excess hypochlorous acid (HOCl) is examined. ► Analytical methods used are membrane inlet MS and nitrous oxide (N2O) analysis. ► Formation of nitroxyl (HNO) and HNO-derived N2O is observed. ► Analogous results are found for the HOCl oxidation of other substrates.
Co-reporter:Gizem Keceli, Cathy D. Moore, Jason W. Labonte, and John P. Toscano
Biochemistry 2013 Volume 52(Issue 42) pp:7387-7396
Publication Date(Web):September 27, 2013
DOI:10.1021/bi401110f
Nitroxyl (HNO), a potential heart failure therapeutic, is known to post-translationally modify cysteine residues. Among reactive nitrogen oxide species, the modification of cysteine residues to sulfinamides [RS(O)NH2] is unique to HNO. We have applied 15N-edited 1H NMR techniques to detect the HNO-induced thiol to sulfinamide modification in several small organic molecules, peptides, and the cysteine protease, papain. Relevant reactions of sulfinamides involve reduction to free thiols in the presence of excess thiol and hydrolysis to form sulfinic acids [RS(O)OH]. We have investigated sulfinamide hydrolysis at physiological pH and temperature. Studies with papain and a related model peptide containing the active site thiol suggest that sulfinamide hydrolysis can be enhanced in a protein environment. These findings are also supported by modeling studies. In addition, analysis of peptide sulfinamides at various pH values suggests that hydrolysis becomes more facile under acidic conditions.
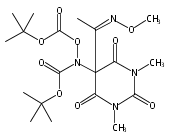
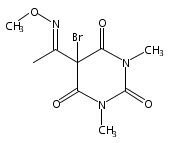
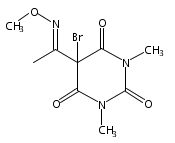
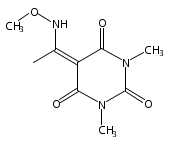
Co-reporter:Daryl A. Guthrie ; Nam Y. Kim ; Maxime A. Siegler ; Cathy D. Moore
Journal of the American Chemical Society 2012 Volume 134(Issue 4) pp:1962-1965
Publication Date(Web):January 9, 2012
DOI:10.1021/ja2103923
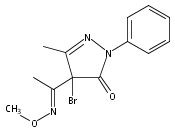
Due to its inherent reactivity, nitroxyl (HNO), must be generated in situ through the use of donor compounds, but very few physiologically useful HNO donors exist. Novel N-substituted hydroxylamines with carbon-based leaving groups have been synthesized, and their structures confirmed by X-ray crystallography. These compounds generate HNO under nonenzymatic, physiological conditions, with the rate and amount of HNO released being dependent mainly on the nature of the leaving group. A barbituric acid and a pyrazolone derivative have been developed as efficient HNO donors with half-lives at pH 7.4, 37 °C of 0.7 and 9.5 min, respectively.
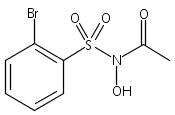
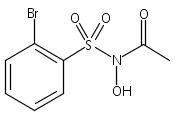
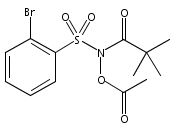
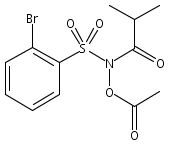
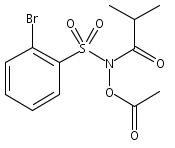
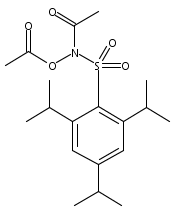
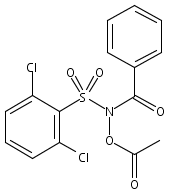
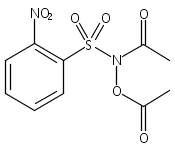
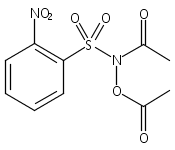
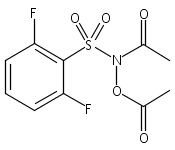
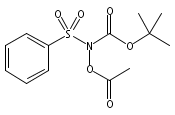
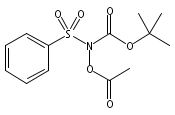
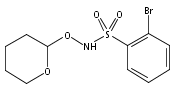
Co-reporter:Art D. Sutton, Morgan Williamson, Hilary Weismiller, and John P. Toscano
Organic Letters 2012 Volume 14(Issue 2) pp:472-475
Publication Date(Web):December 23, 2011
DOI:10.1021/ol203016c
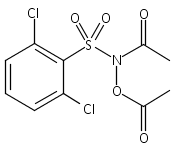
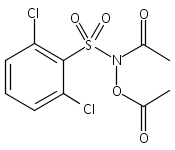
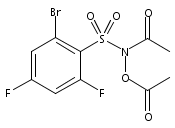
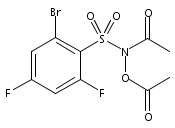
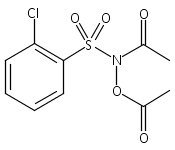
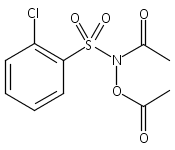
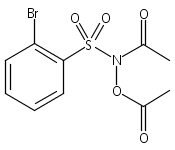
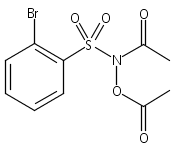
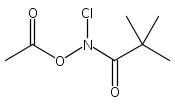
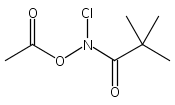
A wide range of N,O-bis-acylated hydroxylamine derivatives with chloro or arenesulfonyl leaving groups, and a related set of N-hydroxy-N-acylsulfonamides, have been synthesized and evaluated for nitroxyl (HNO) production. Mechanistic studies have revealed that the observed aqueous chemistry is more complicated than originally anticipated, and have been used to develop a new series of efficient HNO precursors (4u–4x, 7c–7d) with tunable half-lives.
Co-reporter:Yonglin Liu, Anthony S. Evans and John P. Toscano
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 30) pp:10438-10444
Publication Date(Web):20 Mar 2012
DOI:10.1039/C2CP40327G
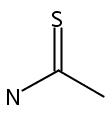
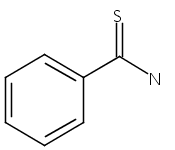
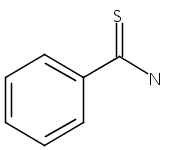
Nanosecond time-resolved infrared (TRIR) spectroscopy has been used to observe singlet thiobenzoylnitrene at 1740 cm−1 upon photolysis of 5-phenyl-1,2,3,4-thiatriazole in acetonitrile and dichloromethane. Consistent with the experimental observations, thiobenzoylnitrene is predicted by B3LYP/6-31G* calculations to have a singlet ground state with an intense IR band at 1752 cm−1. Phenyl isothiocyanate is also produced. Kinetic measurements indicate that it is not formed from singlet thiobenzoylnitrene, but rather directly from the thiatriazole. Unlike benzoylnitrene, singlet thiobenzoylnitrene does not react with acetonitrile or dichloromethane on the nanosecond timescale. However, it does react with dimethyl sulfoxide (DMSO) to produce a sulfoximine detected at 1180 cm−1 (kDMSO = 3 × 105 M−1 s−1). Benzonitrile (observed at 2230 cm−1) is produced from both singlet thiobenzoylnitrene (presumably through a short-lived, unobservable benzonitrile sulfide intermediate) and directly from the thiatriazole. B3LYP/6-31G* calculations also show that the structure of singlet thiobenzoylnitrene is analogous to that of related acylnitrenes, with a significant bonding interaction between the nitrogen and sulfur. Triplet thiobenzoylnitrene, on the other hand, is predicted computationally to have a biradical structure.
Co-reporter:Gizem Keceli and John P. Toscano
Biochemistry 2012 Volume 51(Issue 20) pp:4206-4216
Publication Date(Web):May 9, 2012
DOI:10.1021/bi300015u
Sulfinamide [RS(O)NH2] formation is known to occur upon exposure of cysteine residues to nitroxyl (HNO), which has received recent attention as a potential heart failure therapeutic. Because this modification can alter protein structure and function, we have examined the reactivity of sulfinamides in several systems, including a small organic molecule, peptides, and a protein. Although it has generally been assumed that this thiol to sulfinamide modification is irreversible, we show that sulfinamides can be reduced back to the free thiol in the presence of excess thiol at physiological pH and temperature. We have examined this sulfinamide reduction both in peptides, where a cyclic intermediate analogous to that proposed for asparagine deamidation reactions potentially can contribute, and in a small organic molecule, where the mechanism is restricted to a direct thiolysis. These studies suggest that the contribution from the cyclic intermediate becomes more important in environments with lower dielectric constants. In addition, although sulfinic acid [RS(O)OH] formation is observed upon prolonged incubations in water, reduction of sulfinamides is found to dominate in the presence of thiols. Finally, studies with the cysteine protease, papain, suggest that the reduction of sulfinamide to the free thiol is viable in a protein environment.
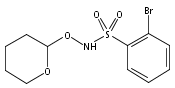
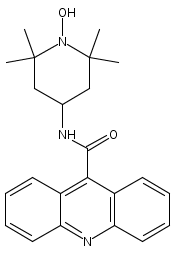
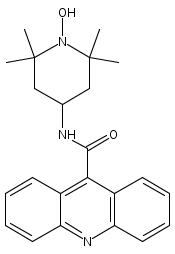
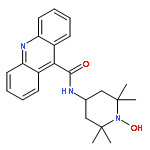
Co-reporter:Meredith R. Cline
Journal of Physical Organic Chemistry 2011 Volume 24( Issue 10) pp:993-998
Publication Date(Web):
DOI:10.1002/poc.1871
Recent research has shown that nitroxyl (HNO) has important and unique biological activity, especially as a potential alternative to current treatments of cardiac failure. HNO is a reactive molecule that spontaneously dimerizes and subsequently dehydrates to form nitrous oxide (N2O), making its detection in solution or biologically relevant preparations difficult. The prefluorescent probe 4-((9-acridinecarbonyl)amino)-2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO-9-AC) has been used to detect HNO in aqueous solution and to differentiate it from nitric oxide (NO). TEMPO-9-AC reacts with HNO via a net hydrogen abstraction to produce the highly fluorescent TEMPO-9-AC-H and NO. The utility of TEMPO-9-AC as a probe for HNO has been shown using the common HNO donors Angeli's salt and Piloty's acid (PA) along with a recently reported HNO donor, 2-bromo-PA. The use of TEMPO-9-AC is complicated by intermolecular fluorescence quenching and competitive HNO trapping by the NO produced, but nonetheless, it can be used to study HNO reactivity in aqueous solution. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:Anthony S. Evans, G. S. M. Sundaram, Jörg Saβmannshausen, John P. Toscano, and Eric M. Tippmann
Organic Letters 2010 Volume 12(Issue 20) pp:4616-4619
Publication Date(Web):September 17, 2010
DOI:10.1021/ol101946u
A chloromethylhydroxamiccarbene was generated photochemically in an attempt to form an intramolecularly stabilized carbene. A rapidly formed intermediate at 1645 cm−1 decayed with an observed rate of 1.99 × 106 s−1. Other intermediates were also observed. These also decayed, albeit much more slowly (kobs = 3.47 × 103 and 1.98 × 104 s−1). Multiple intermediates are apparently a function of both the proximal N,O-dimethylhydroxamic ester and multiple conformers of both the carbene and precursor.
Co-reporter:Brett M. Showalter
Journal of Physical Organic Chemistry 2004 Volume 17(Issue 9) pp:743-748
Publication Date(Web):30 JUL 2004
DOI:10.1002/poc.789
A series of α-lactones were generated from the reaction of phenylchlorocarbene, 4-nitrophenylchlorocarbene, diphenylcarbene, bis(4-nitrophenyl)carbene and bis(4-methoxyphenyl)carbene with carbon dioxide and examined by nanosecond time-resolved infrared (TRIR) spectroscopy. Estimated second-order rate constants for the reaction of these carbenes with carbon dioxide indicate that more nucleophilic carbenes react at faster rates, in agreement with previous low-temperature matrix experiments. Spectral TRIR data confirms that the structure of α-lactones is dependent both on substituents at the α-carbon and on solvent polarity, with electron-donating substituents and polar solvents favoring a zwitterionic ring-opened structure as opposed to the three-membered ring oxiranone form. B3LYP calculations using self-consistent reaction field (SCRF) methods also provide support for these experimental investigations. Copyright © 2004 John Wiley & Sons, Ltd.
Co-reporter:Christopher L. Bianco, Tyler A. Chavez, Victor Sosa, Simran, S. Saund, ... Jon M. Fukuto
Free Radical Biology and Medicine (December 2016) Volume 101() pp:20-31
Publication Date(Web):1 December 2016
DOI:10.1016/j.freeradbiomed.2016.09.020
•Hydropersulfides (RSSH) are readily oxidized to the corresponding perthiyl radical (RSS·).•RSS· does not react with O2 or NO.•Hydropersulfides are superior reductants compared to thiols (RSH).•Perthiyl is less reactive than a thiyl (RS·) species.•The RSSH/RSS· redox couple is biologically accessible and possibly relevant.The recent finding that hydropersulfides (RSSH) are biologically prevalent in mammalian systems has prompted further investigation of their chemical properties in order to provide a basis for understanding their potential functions, if any. Hydropersulfides have been touted as hyper-reactive thiol-like species that possess increased nucleophilicity and reducing capabilities compared to their thiol counterparts. Herein, using persulfide generating model systems, the ability of RSSH species to act as one-electron reductants has been examined. Not unexpectedly, RSSH is relatively easily oxidized, compared to thiols, by weak oxidants to generate the perthiyl radical (RSS·). Somewhat surprisingly, however, RSS· was found to be stable in the presence of both O2 and NO and only appears to dimerize. Thus, the RSSH/RSS· redox couple is readily accessible under biological conditions and since dimerization of RSS· may be a rare event due to low concentrations and/or sequestration within a protein, it is speculated that the general lack of reactivity of individual RSS· species may allow this couple to be utilized as a redox component in biological systems.
Co-reporter:Meredith R. Cline, Chingkuang Tu, David N. Silverman, John P. Toscano
Free Radical Biology and Medicine (15 May 2011) Volume 50(Issue 10) pp:1274-1279
Publication Date(Web):15 May 2011
DOI:10.1016/j.freeradbiomed.2011.02.008
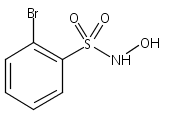
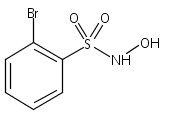
Membrane inlet (or introduction) mass spectrometry (MIMS) was used to detect nitroxyl (HNO) in aqueous solution for the first time. The common HNO donors Angeli's salt (AS) and Piloty's acid (PA), along with a newly developed donor, 2-bromo-N-hydroxybenzenesulfonamide (2-bromo-Piloty's acid, 2BrPA), were examined by this technique. MIMS experiments revealed that under physiological conditions 2BrPA is an essentially pure HNO donor, but AS produces a small amount of nitric oxide (NO). In addition, MIMS experiments also confirmed that PA is susceptible to oxidation and NO production, but that 2BrPA is not as prone to oxidation.
Co-reporter:Yonglin Liu, Anthony S. Evans and John P. Toscano
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 30) pp:NaN10444-10444
Publication Date(Web):2012/03/20
DOI:10.1039/C2CP40327G
Nanosecond time-resolved infrared (TRIR) spectroscopy has been used to observe singlet thiobenzoylnitrene at 1740 cm−1 upon photolysis of 5-phenyl-1,2,3,4-thiatriazole in acetonitrile and dichloromethane. Consistent with the experimental observations, thiobenzoylnitrene is predicted by B3LYP/6-31G* calculations to have a singlet ground state with an intense IR band at 1752 cm−1. Phenyl isothiocyanate is also produced. Kinetic measurements indicate that it is not formed from singlet thiobenzoylnitrene, but rather directly from the thiatriazole. Unlike benzoylnitrene, singlet thiobenzoylnitrene does not react with acetonitrile or dichloromethane on the nanosecond timescale. However, it does react with dimethyl sulfoxide (DMSO) to produce a sulfoximine detected at 1180 cm−1 (kDMSO = 3 × 105 M−1 s−1). Benzonitrile (observed at 2230 cm−1) is produced from both singlet thiobenzoylnitrene (presumably through a short-lived, unobservable benzonitrile sulfide intermediate) and directly from the thiatriazole. B3LYP/6-31G* calculations also show that the structure of singlet thiobenzoylnitrene is analogous to that of related acylnitrenes, with a significant bonding interaction between the nitrogen and sulfur. Triplet thiobenzoylnitrene, on the other hand, is predicted computationally to have a biradical structure.
.jpg)










































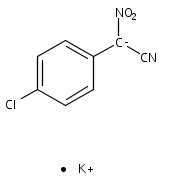

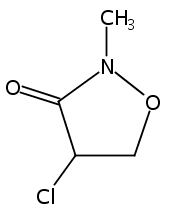


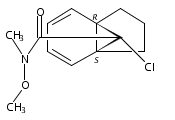
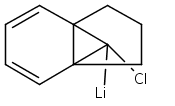

![Acetamide,N-(acetyloxy)-N-[(4-chlorophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/142900/142867-52-5.png)
![Acetamide,N-(acetyloxy)-N-[(4-chlorophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/142900/142867-52-5_b.png)





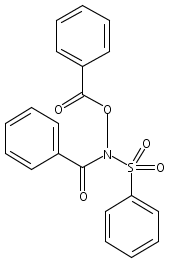
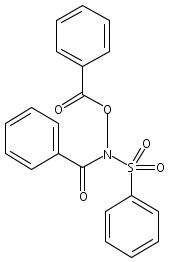
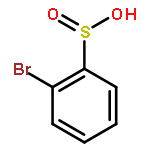
![[(2-methylpropan-2-yl)oxycarbonylamino] Acetate](http://img.cochemist.com/ccimg/99800/99768-83-9.png)
![[(2-methylpropan-2-yl)oxycarbonylamino] Acetate](http://img.cochemist.com/ccimg/99800/99768-83-9_b.png)
![BENZAMIDE, 4-NITRO-N-[(4-NITROBENZOYL)OXY]-](http://img.cochemist.com/ccimg/59200/59101-31-4.png)
![BENZAMIDE, 4-NITRO-N-[(4-NITROBENZOYL)OXY]-](http://img.cochemist.com/ccimg/59200/59101-31-4_b.png)

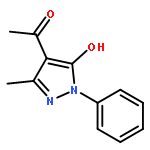
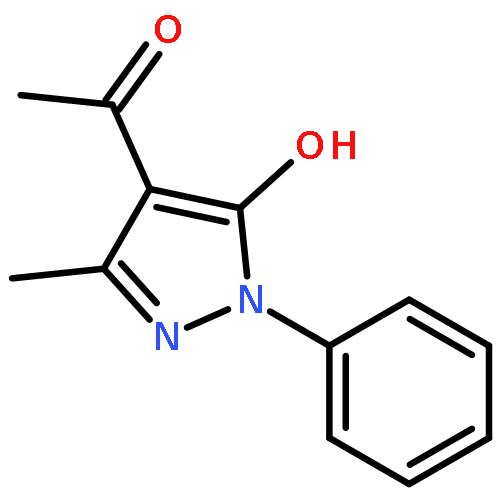
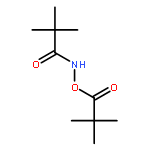
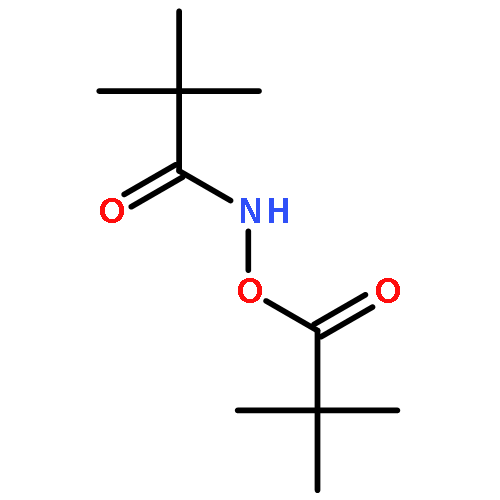
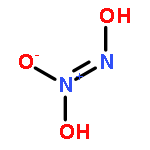
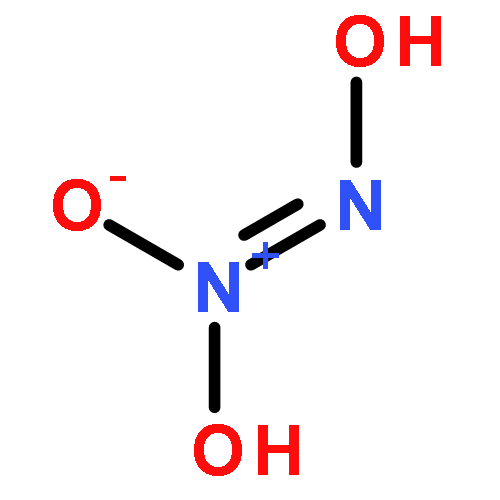
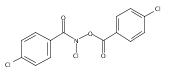
![Benzamide, 4-chloro-N-[(4-chlorobenzoyl)oxy]-](http://img.cochemist.com/ccimg/13200/13173-91-6.png)
![Benzamide, 4-chloro-N-[(4-chlorobenzoyl)oxy]-](http://img.cochemist.com/ccimg/13200/13173-91-6_b.png)

![11-[5-[(5-CYANO-4-METHYL-6-OXO-1-PROPYL-2-PYRROLIDIN-1-YLPYRIDIN-3-YL)METHYLIDENE]-4-OXO-2-SULFANYLIDENE-1,3-THIAZOLIDIN-3-YL]UNDECANOIC ACID](http://img.cochemist.com/ccimg/7100/7064-08-6.png)
![11-[5-[(5-CYANO-4-METHYL-6-OXO-1-PROPYL-2-PYRROLIDIN-1-YLPYRIDIN-3-YL)METHYLIDENE]-4-OXO-2-SULFANYLIDENE-1,3-THIAZOLIDIN-3-YL]UNDECANOIC ACID](http://img.cochemist.com/ccimg/7100/7064-08-6_b.png)

![Carbamic acid, [(ethoxycarbonyl)oxy]-, ethyl ester](http://img.cochemist.com/ccimg/6100/6092-85-9.png)
![Carbamic acid, [(ethoxycarbonyl)oxy]-, ethyl ester](http://img.cochemist.com/ccimg/6100/6092-85-9_b.png)



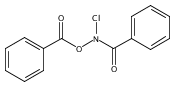
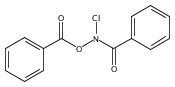
![Benzamide, N-[(4-nitrobenzoyl)oxy]-](http://img.cochemist.com/ccimg/1100/1037-48-5.png)
![Benzamide, N-[(4-nitrobenzoyl)oxy]-](http://img.cochemist.com/ccimg/1100/1037-48-5_b.png)


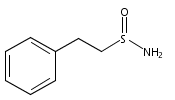
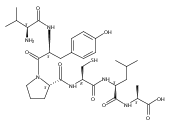
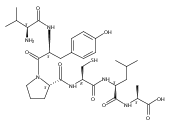
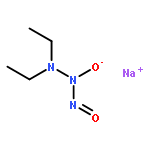
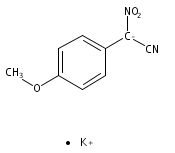
![L-Ornithine,N5-[(hydroxyamino)iminomethyl]-](http://img.cochemist.com/ccimg/53100/53054-07-2.png)
![L-Ornithine,N5-[(hydroxyamino)iminomethyl]-](http://img.cochemist.com/ccimg/53100/53054-07-2_b.png)