Co-reporter:Hai-Xia Wang;Yu-Cheng Ding
RSC Advances (2011-Present) 2017 vol. 7(Issue 78) pp:49626-49632
Publication Date(Web):2017/10/20
DOI:10.1039/C7RA09317A
The mechanism of ring-opening polymerization of oxetane cation series compounds was investigated using the B3LYP and MP2 methods of density functional theory and ab initio methods, at the basis set levels of 6-31G(d,p) and 6-311++G(d,p). The geometrical parameters of the reactant, transition state, intermediate and product of a series of multi-polymer species in the reaction pathway were fully optimized. The structural changes of species in the reaction pathway are explained herein. The computing results show that the polymerization of oxetane is performed by the O atom of oxetane continuously attacking the C atom of the oxetane cation. The energy analysis of the reaction process shows that the acidized oxetane can easily polymerize with other oxetane molecules to form a copolymer, and the activation energy in the initial step is very low. The equilibrium and transition state characteristics of every stationary point in the reaction pathway were determined through vibrational analysis. The corresponding reactant and product of each transition state were verified according to the intrinsic reaction coordinates traced from the transition state of different hierarchical polymers. Finally, the solvent effects of tetrahydrofuran and dichloromethane are discussed herein based on the self-consistent reaction field theory.
Co-reporter:Xiaofei Zhang and Min Pu
RSC Advances 2016 vol. 6(Issue 24) pp:19742-19750
Publication Date(Web):09 Feb 2016
DOI:10.1039/C5RA26808G
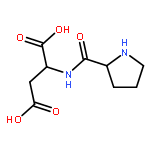
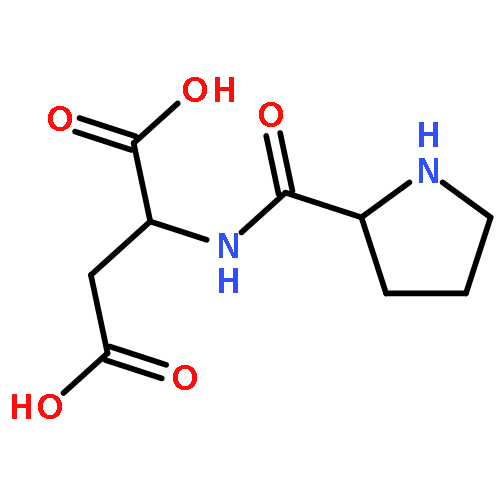
The mechanism of the dipeptide (S)-pro-(S)-asp catalyzed intermolecular aldol reaction with acetone as the donor and three aromatic aldehydes (benzaldehyde, p-methyl benzaldehyde and p-nitrobenzaldehyde) as the acceptors was studied by means of density functional theory (DFT) at the level of B3LYP/6-31G(d,p). The calculated results showed that there were four steps in the reaction path: (i) the nucleophilic attack of an amino group on carbonyl for the formation of intermediate A, which was the rate-determining step due to it having the largest energy barrier of 44.33 kcal mol−1; (ii) the dehydration process to form an s-cis- or s-trans-enamine through an imine-generating step; (iii) the electrophilic addition of aldehyde, which decided the stereoselectivity of the product because of the steric repulsion interactions between the enamine and aldehyde; (iv) the removal of the dipeptide to generate the final products. According to the results analysis, it was found that the dipeptide-catalyzed aldol reaction via an s-trans-enamine was more energetically favorable to obtain the R-product (with an ee value > 99%). The energy variations in the reaction path were verified using CAM-B3LYP and M06-2X methods in the same basis set. The solvation effects were explored based on B3LYP/6-31G(d,p) combined with a polarizable continuum model (PCM), the substituent effects of aromatic aldehydes were also considered. The computed results provided a reference for experiments that DMSO and H2O as the solvents could decrease the energy barriers in the reaction path and the impact of substituent effects might be small. The feasibility of the dipeptide provided a possibility for proteins to act as catalysts which are green and nontoxic.
Co-reporter:Jun-Nan Li, Min Pu, Chi-Cheng Ma, Ye Tian, Jing He, David G. Evans
Journal of Molecular Catalysis A: Chemical 2012 Volume 359() pp:14-20
Publication Date(Web):July 2012
DOI:10.1016/j.molcata.2012.03.015
The mechanisms of acetylene hydrogenation on palladium clusters (Pdn, n = 2–8) are researched by using the B3PW91/GENECP method of density functional theory. The calculation results indicate that there are two possible pathways for the hydrogenation reaction on Pdn cluster from the reactant acetylene to the product ethane. One of the pathways undergoes through two intermediates, the vinyl (Pdn(H)⋯CHCH2) and ethene (Pdn⋯CH2CH2) to form the ethane, and the other goes along vinylidene (Pdn(2H)⋯CCH2), ethylidyne Pdn(2H)⋯CCH3) and ethylidene (Pdn(2H)⋯CHCH3) to ethane. Those intermediates in the two pathways can convert into each other which make the reaction profile complicated. The value of n in Pdn cluster can directly affect the reaction pathway: when n ≤ 4, the acetylene hydrogenation reaction will proceed via the pathway of Pdn(2H)⋯CHCH → Pdn(H)⋯CHCH2 → Pdn⋯CH2CH2 → Pdn(2H)⋯CH2CH2 to form ethane. However, when n > 4, the reaction choose the following pathway: Pdn(2H)⋯CHCH → Pdn(H)⋯CHCH2 → Pdn⋯CHCH3 → Pdn(2H)⋯CHCH3. In addition, the value of their turnover frequency (TOF) for the ethylene formation catalyzed by Pdn cluster is larger than that for ethane, which indicates that the catalytic cycles in the formation of ethylene is efficient.Graphical abstractWhen the value of n in Pdn is equal or lesser than 4 (n ≤ 4), the acetylene hydrogenation reaction will proceed via the pathway of Pdn(2H)⋯CHCH → Pdn(H)⋯CHCH2 → Pdn⋯CH2CH2 → Pdn(2H)⋯CH2CH2 to form ethane. However, when n > 4, the reaction choose the following pathway: Pdn(2H)⋯CHCH → Pdn(H)⋯CHCH2 → Pdn⋯CHCH3 → Pdn(2H)⋯CHCH3.Highlights► Two feasible pathways of the acetylene hydrogenation on Pdn clusters. ► One of the pathways goes along the vinyl and ethene intermediates to form the ethane. ► The other way proceeds via vinylidene, ethylidyne and ethylidene to form ethane. ► The intermediates of the two pathways can convert each other. ► The catalytic cycles in ethylene forming are efficient than that for ethane.
Co-reporter:Jun Nan Li, Min Pu, De Cai Fang, Min Wei, Jing He, David G. Evans
Journal of Molecular Structure 2012 1015() pp: 106-111
Publication Date(Web):
DOI:10.1016/j.molstruc.2012.02.014
Co-reporter:Gao-Jie Hu, Hai-Xia Wang, Liu Ling-Yan, Min Pu, Jing He, David G. Evans
Journal of Physics and Chemistry of Solids 2010 Volume 71(Issue 9) pp:1290-1294
Publication Date(Web):September 2010
DOI:10.1016/j.jpcs.2010.05.009
p-Hydroxybenzoate pillared layered double hydroxides with different Zn/Al mole ratios have been prepared by three different methods: rehydration of calcined LDH precursor, coprecipitation and anion exchange. The products have been characterized by several experimental techniques: PXRD, FT-IR, TG–DTA and UV–vis. PXRD patterns show that the interlayer distance of p-hydroxybenzoic acid (PHBA)-Zn/Al-LDH varies with ratio Zn/Al, from 14.8 to 15.3 Å, indicating that altering Zn/Al ratios can change the arrangement of the intercalated PHBA anions. Not only do infrared spectra display the characteristic absorption of both the PHBA anion and the Zn/Al layer, but also provide further evidence of the interaction between these two parts. Thermal analysis confirms that the intercalation can make PHBA stable up to 434 °C, which is 213 °C higher than that for pure PHBA. By UV–vis it is found that such a product can control blocking of UV radiation in a wider range of wavelength.
Co-reporter:Yue Zhu;De-Cai Fang;Yong-Qiang Ji;Jing He
Structural Chemistry 2010 Volume 21( Issue 4) pp:817-825
Publication Date(Web):2010 August
DOI:10.1007/s11224-010-9616-8
The cis–trans isomerization pathways of 3,3′-azobenzene disulphonate in the S0 and T1 states are studied by DFT method at the B3LYP/6-31G(d,p) level. In the S0 state, the cis–trans isomerization concerns the complex pathway that is characterized by the inversion of one NNC angle combined with rotation around the NC bond, and the three sequential transition states are also found on the potential energy profile. Therefore, the cis–trans isomerization of 3,3′-azobenzene disulphonate can be understood in terms of a pathway involving successive rotation, inversion, and rotation processes. The energy barrier of the S0 state is 22.79 kcal mol−1. In the T1 state, the isomerization mainly concerns the rotational pathway around the NN double bond, and the two isomers are connected through only one transition state. The isomerization of the T1 state is related to a lower energy barrier, 5.02 kcal mol−1, but requires a change in spin-multiplicity.
Co-reporter:Yue Zhu, Min Pu, De-Cai Fang, Jing He, David G. Evans
Journal of Photochemistry and Photobiology A: Chemistry 2010 Volume 211(2–3) pp:89-98
Publication Date(Web):15 April 2010
DOI:10.1016/j.jphotochem.2010.01.014
The isomerization pathways of 4,4′-azobenzene disulfonate in the S0 and T1 states have been studied by using density functional theory (DFT) with B3LYP method at the levels of 6-31G(d,p) and 6-311++G(d,p), respectively. There are two isomerization pathways in the S0 state. One is the inversion of one CNN angle combined with certain degree of rotation around the CN bond, and it is worthy to notice that the potential energy profile includes three sequential transition states. The other pathway is the rotation of CNNC dihedral angle involved inversion of one CNN angle, while the two isomers are connected through the only one transition state. Calculation indicates that the molecular structures at the highest points on the potential energy profiles of two pathways are identical, and the energy barriers are the same, 20.52 kcal/mol. In the T1 state, there exists the rotation pathway (rotation of CNNC dihedral angle) and its energy barrier is 4.11 kcal/mol. In the excited states (T1, S1, T2, and S2), the potential energy profiles of the vertical excitation are obtained by time dependent density functional theory (TD-DFT) at the B3LYP/6-311++G(d,p) level. The photoexcitation at 342 nm results in the reactant molecule populated in the S2 state, but isomerization does not occur directly on the S2 state due to the high energy barrier. It could undergo a rapid relaxation to the minimum of S1 state, and then the isomerization occurs using the inversion or rotation pathway. The results show that the rapid energy redistribution among the various vibrations renders the concentration of such amounts of energy in the inversion coordinate very improbable, while there are two possible photoisomerization pathways by the rotation pathway. The isomerization can easily occur through the S0/S1 conical intersection and the S0–T1–S0 crossing to reach the product. The primary isomerization pathways for 4,4′-azobenzene disulfonate can go through the inversion and rotation forms in the S0 state and the rotation mechanism in the excited state.
Co-reporter:Ling-Yan Liu, Min Pu, Lan Yang, Dian-Qing Li, David G. Evans, Jing He
Materials Chemistry and Physics 2007 Volume 106(2–3) pp:422-427
Publication Date(Web):15 December 2007
DOI:10.1016/j.matchemphys.2007.06.022
Acid orange 7-pillared layered double hydroxide (AO7-LDH) has been prepared by coprecipitation and studied with both experimental characterizations and theoretical calculations. XRD patterns indicate that the product has the typical intercalated structure of LDHs with a gallery height of 1.79 nm, showing that a kind of large anion has been intercalated into the interlayer space of the LDH. Infrared spectrum of AO7-LDH shows that the product has the characteristic absorption bands of both azo and hydrazone forms of AO7 anion, as well as the Mg–O and Al–O vibrations of LDH layers, well convincing the intercalation of AO7 anions successful. TG–DTA curves demonstrate clearly that the thermal stability of AO7 can be enhanced by nearly 130 °C when it is intercalated into the LDH layers. According to the size of AO7 anion (1.25–1.27 nm) obtained by density functional calculations of quantum chemistry at the level of B3LYP/6-31G(d,p), the AO7 anions should be vertical-arranging in the interlayer space, forming a interdigitated structure.
Co-reporter:Min Pu, Bao-Fang Zhang
Materials Letters 2005 Volume 59(Issue 27) pp:3343-3347
Publication Date(Web):November 2005
DOI:10.1016/j.matlet.2005.05.009
The microstructures of lamellae of hydrotalcites with Mg/Al ratio of two are studied using quantum chemical method. A series of molecular cluster models of lamellae are established. The geometries of lamellae are computed by semiempirical molecular orbital methods (MNDO/d and PM3). The optimized cluster models show a hexagonal morphology. The Mg/Al ratio of lamella is found to be an alterable number and its limit is two when the lamella diameter becomes large. The molecular orbital calculations show that the edge and the center of lamellae are respectively easy to attract cations and accept anions, to form intercalated structure.
Co-reporter:Hui-Ying Li, Min Pu, Yong-Qiang Ji, Zhen-Feng Xu, Wen-Lin Feng
Chemical Physics 2004 Volume 307(Issue 1) pp:35-43
Publication Date(Web):6 December 2004
DOI:10.1016/j.chemphys.2004.07.014
The direct dynamics of the hydrogen abstraction reactions of CH2O with CH3/OH are studied using ab initio molecular orbital theory. Both the geometry optimizations of all the stationary points and the vibrational frequency calculations are carried out at the UQCISD/6-311G(d, p) level. The single-point energy is obtained by the multicoefficient Gaussian 3-version 3s (MCG3/3) method. The analysis to the changes of the interatomic distances on the minimum energy paths show that, the breaking of C–H bonds of CH2O and (i) the forming of C–H bond of CH4 in the reaction of CH2O with CH3, (ii) the forming of O–H bond of H2O in the reaction of CH2O with OH, are both concerted. For each reaction, there exists a reactive vibrational normal-mode, and its frequencies change is relevant to the forming and breaking of the above covalent bond. Furthermore, the theoretical forward reaction rate constants in the temperature range 300–3000 K are computed by canonical variational transition state theory with small-curvature tunneling correction (CVT/SCT) method. The computed values of the rate constants are in good agreement with the available experimental data in the measured temperature range. Moreover, the tunneling effects are found to contribute significantly to the rate constants at low temperatures.









![Benzoic acid,2-hydroxy-5-[2-(4-sulfophenyl)diazenyl]-](http://img.cochemist.com/ccimg/21600/21542-82-5.png)
![Benzoic acid,2-hydroxy-5-[2-(4-sulfophenyl)diazenyl]-](http://img.cochemist.com/ccimg/21600/21542-82-5_b.png)















