Co-reporter:James M. Eagan;Masahiro Hori;Dr. Jianbin Wu;Dr. Kyalo Stephen Kanyiva;Dr. Scott A. Snyder
Angewandte Chemie International Edition 2015 Volume 54( Issue 27) pp:7842-7846
Publication Date(Web):
DOI:10.1002/anie.201500925
Abstract
Numerous natural products possess ring systems and functionality for which Hajos–Parrish ketone isomers with a transposed methyl group (termed “iso-Hajos–Parrish ketones”) would be of value. However, such building blocks have not been exploited to the same degree as the more typical Hajos–Parrish hydrindane. An efficient three-step synthesis of such materials was fueled by a simple method for the rapid preparation of highly functionalized cyclopentenones, several of which are new chemical entities that would be challenging to access through other approaches. Furthermore, one iso-Hajos–Parrish ketone was converted into two distinct natural product analogues and one natural product. As one indication of the value of these new building blocks, that latter target was obtained in 10 steps, having previously been accessed in 18 steps using the Hajos–Parrish ketone.
Co-reporter:Stéphane Quideau, Scott A. Snyder
Tetrahedron 2015 Volume 71(Issue 20) pp:2969-2970
Publication Date(Web):20 May 2015
DOI:10.1016/j.tet.2015.02.086
Co-reporter:Stéphane Quideau, Scott A. Snyder
Tetrahedron 2015 Volume 71(Issue 20) pp:2967-2968
Publication Date(Web):20 May 2015
DOI:10.1016/j.tet.2015.02.085
Co-reporter:James M. Eagan;Masahiro Hori;Dr. Jianbin Wu;Dr. Kyalo Stephen Kanyiva;Dr. Scott A. Snyder
Angewandte Chemie 2015 Volume 127( Issue 27) pp:7953-7957
Publication Date(Web):
DOI:10.1002/ange.201500925
Abstract
Numerous natural products possess ring systems and functionality for which Hajos–Parrish ketone isomers with a transposed methyl group (termed “iso-Hajos–Parrish ketones”) would be of value. However, such building blocks have not been exploited to the same degree as the more typical Hajos–Parrish hydrindane. An efficient three-step synthesis of such materials was fueled by a simple method for the rapid preparation of highly functionalized cyclopentenones, several of which are new chemical entities that would be challenging to access through other approaches. Furthermore, one iso-Hajos–Parrish ketone was converted into two distinct natural product analogues and one natural product. As one indication of the value of these new building blocks, that latter target was obtained in 10 steps, having previously been accessed in 18 steps using the Hajos–Parrish ketone.
Co-reporter:Trevor C. Sherwood ; Adam H. Trotta
Journal of the American Chemical Society 2014 Volume 136(Issue 27) pp:9743-9753
Publication Date(Web):June 24, 2014
DOI:10.1021/ja5045852
Although dimeric natural products can often be synthesized in the laboratory by directly merging advanced monomers, these approaches sometimes fail, leading instead to non-natural architectures via incorrect unions. Such a situation arose during our studies of the coccinellid alkaloids, when attempts to directly dimerize Nature’s presumed monomeric precursors in a putative biomimetic sequence afforded only a non-natural analogue through improper regiocontrol. Herein, we outline a unique strategy for dimer formation that obviates these difficulties, one which rapidly constructs the coccinellid dimers psylloborine A and isopsylloborine A through a terminating sequence of two reaction cascades that generate five bonds, five rings, and four stereocenters. In addition, a common synthetic intermediate is identified which allows for the rapid, asymmetric formal or complete total syntheses of eight monomeric members of the class.
Co-reporter:Nathan E. Wright, Adel M. ElSohly, and Scott A. Snyder
Organic Letters 2014 Volume 16(Issue 14) pp:3644-3647
Publication Date(Web):July 2, 2014
DOI:10.1021/ol501284s
Although interest in cyclotriveratrylene and its analogues has been significant, limitations in the ability to adjust its structure fully have hampered studies into their complete range of properties. A unique strategy to synthesize a previously unobtainable cyclotriveratrylene analogue and a procedure which adjusts the inner methylene bridges of that material to a triketone is reported. A second triketone synthesis and computational studies indicate the parameters needed for success.
Co-reporter:Nathan E. Wright;Dr. Scott A. Snyder
Angewandte Chemie 2014 Volume 126( Issue 13) pp:3477-3481
Publication Date(Web):
DOI:10.1002/ange.201311299
Abstract
Explorations into a series of different approaches for 9-membered carbocycle formation have afforded the first reported example of a 9-exo-dig ring closure via a AuIII-promoted reaction between an alkyne and an aryl ring as well as several additional, unique Friedel–Crafts-type cyclizations. Analyses of the factors leading to the success of these transformations are provided, with the application of one of the developed 9-membered ring closures affording an efficient and scalable synthesis of the bioactive resveratrol trimer caraphenol A. That synthesis proceeded with an average yield of 89 % per step (7.8 % overall yield) and has provided access to more than 600 mg of the target molecule.
Co-reporter:Tue H. Jepsen;Stephen B. Thomas;Yunqing Lin;Dr. Christos I. Stathakis;Irene deMiguel;Dr. Scott A. Snyder
Angewandte Chemie International Edition 2014 Volume 53( Issue 26) pp:6747-6751
Publication Date(Web):
DOI:10.1002/anie.201402858
Abstract
Although quinone methides are often postulated as intermediates in the biosynthesis of many polyphenolic natural products, deploying their power in a laboratory setting to achieve similar bond constructions has sometimes proven challenging. Herein, a total synthesis of the resveratrol trimer vaticanol A has been achieved through three instances of quinone methide chemistry. These operations, one of which succeeded only under very specific conditions, expediently generated its [7,5]-carbocyclic core, afforded a unique sequence for dihydrobenzofuran formation, and concurrently generated, in addition to the target molecule, a series of diastereomers reflective of many other isolates.
Co-reporter:Nathan E. Wright;Dr. Scott A. Snyder
Angewandte Chemie International Edition 2014 Volume 53( Issue 13) pp:3409-3413
Publication Date(Web):
DOI:10.1002/anie.201311299
Abstract
Explorations into a series of different approaches for 9-membered carbocycle formation have afforded the first reported example of a 9-exo-dig ring closure via a AuIII-promoted reaction between an alkyne and an aryl ring as well as several additional, unique Friedel–Crafts-type cyclizations. Analyses of the factors leading to the success of these transformations are provided, with the application of one of the developed 9-membered ring closures affording an efficient and scalable synthesis of the bioactive resveratrol trimer caraphenol A. That synthesis proceeded with an average yield of 89 % per step (7.8 % overall yield) and has provided access to more than 600 mg of the target molecule.
Co-reporter:Tue H. Jepsen;Stephen B. Thomas;Yunqing Lin;Dr. Christos I. Stathakis;Irene deMiguel;Dr. Scott A. Snyder
Angewandte Chemie 2014 Volume 126( Issue 26) pp:6865-6869
Publication Date(Web):
DOI:10.1002/ange.201402858
Abstract
Although quinone methides are often postulated as intermediates in the biosynthesis of many polyphenolic natural products, deploying their power in a laboratory setting to achieve similar bond constructions has sometimes proven challenging. Herein, a total synthesis of the resveratrol trimer vaticanol A has been achieved through three instances of quinone methide chemistry. These operations, one of which succeeded only under very specific conditions, expediently generated its [7,5]-carbocyclic core, afforded a unique sequence for dihydrobenzofuran formation, and concurrently generated, in addition to the target molecule, a series of diastereomers reflective of many other isolates.
Co-reporter:Daniel R. Griffith, Lorenzo Botta, Tyler G. St. Denis, and Scott A. Snyder
The Journal of Organic Chemistry 2014 Volume 79(Issue 1) pp:88-105
Publication Date(Web):December 13, 2013
DOI:10.1021/jo4023167
Yunnaneic acids A–D, isolated from the roots of Salvia yunnanensis, are hexameric (A and B) and trimeric (C and D) assemblies of caffeic acid that feature an array of synthetically challenging and structurally interesting domains. In addition to being caffeic acid oligomers, yunnaneic acids A and B are formally dimeric and heterodimeric adducts of yunnaneic acids C and D. Herein we report the first total syntheses of yunnaneic acids C and D featuring the formation of their bicyclo[2.2.2]octene cores in a single step from simple precursors via an oxidative dearomatization/Diels–Alder cascade that may have biogenetic relevance. In addition, exploitation of the key intermediate resulting from this cascade reaction has enabled rapid access to the structurally related caffeic acid metabolite rufescenolide through an unexpected Lewis acid-mediated reduction. Finally, we report the results of extensive model studies toward forming the dimeric yunnaneic acids A and B. These explorations indicate that the innate reactivities of the monomeric fragments do not favor spontaneous formation of the desired dimeric linkages. Consequently, enzymatic involvement may be required for the biosynthesis of these more complex family members.
.jpg)


![1H-INDOLE, 4-BROMO-3-[2-(IODOMETHYL)-5-OXAZOLYL]-1-METHYL-](http://img.cochemist.com/ccimg/761500/761433-94-7.png)
![1H-INDOLE, 4-BROMO-3-[2-(IODOMETHYL)-5-OXAZOLYL]-1-METHYL-](http://img.cochemist.com/ccimg/761500/761433-94-7_b.png)

![Silane, [[2-bromo-6-(2-phenyloxiranyl)phenoxy]methyl]trimethyl-](http://img.cochemist.com/ccimg/761500/761433-44-7.png)
![Silane, [[2-bromo-6-(2-phenyloxiranyl)phenoxy]methyl]trimethyl-](http://img.cochemist.com/ccimg/761500/761433-44-7_b.png)

![Oxirane, 2-[3-bromo-2-[(phenylsulfonyl)methoxy]phenyl]-2-phenyl-](http://img.cochemist.com/ccimg/761500/761433-42-5.png)
![Oxirane, 2-[3-bromo-2-[(phenylsulfonyl)methoxy]phenyl]-2-phenyl-](http://img.cochemist.com/ccimg/761500/761433-42-5_b.png)
![OXIRANE, 2-[3-BROMO-2-[(PHENYLSULFINYL)METHOXY]PHENYL]-2-PHENYL-](http://img.cochemist.com/ccimg/761500/761433-41-4.png)
![OXIRANE, 2-[3-BROMO-2-[(PHENYLSULFINYL)METHOXY]PHENYL]-2-PHENYL-](http://img.cochemist.com/ccimg/761500/761433-41-4_b.png)
![Oxirane, 2-[3-bromo-2-[(phenylthio)methoxy]phenyl]-2-phenyl-](http://img.cochemist.com/ccimg/761500/761433-40-3.png)
![Oxirane, 2-[3-bromo-2-[(phenylthio)methoxy]phenyl]-2-phenyl-](http://img.cochemist.com/ccimg/761500/761433-40-3_b.png)
![METHANONE, [3-BROMO-2-(METHOXYMETHOXY)PHENYL]PHENYL-](http://img.cochemist.com/ccimg/761500/761433-39-0.png)
![METHANONE, [3-BROMO-2-(METHOXYMETHOXY)PHENYL]PHENYL-](http://img.cochemist.com/ccimg/761500/761433-39-0_b.png)
![Silane, [[2-bromo-6-(1-phenylethenyl)phenoxy]methyl]trimethyl-](http://img.cochemist.com/ccimg/761500/761433-38-9.png)
![Silane, [[2-bromo-6-(1-phenylethenyl)phenoxy]methyl]trimethyl-](http://img.cochemist.com/ccimg/761500/761433-38-9_b.png)
![METHANONE, [3-BROMO-2-[(PHENYLTHIO)METHOXY]PHENYL]PHENYL-](http://img.cochemist.com/ccimg/761500/761433-37-8.png)
![METHANONE, [3-BROMO-2-[(PHENYLTHIO)METHOXY]PHENYL]PHENYL-](http://img.cochemist.com/ccimg/761500/761433-37-8_b.png)
![4-[(3-HYDROXYPROPYLAMINO)METHYL]BENZONITRILE](http://img.cochemist.com/ccimg/722000/721958-90-3.png)
![4-[(3-HYDROXYPROPYLAMINO)METHYL]BENZONITRILE](http://img.cochemist.com/ccimg/722000/721958-90-3_b.png)
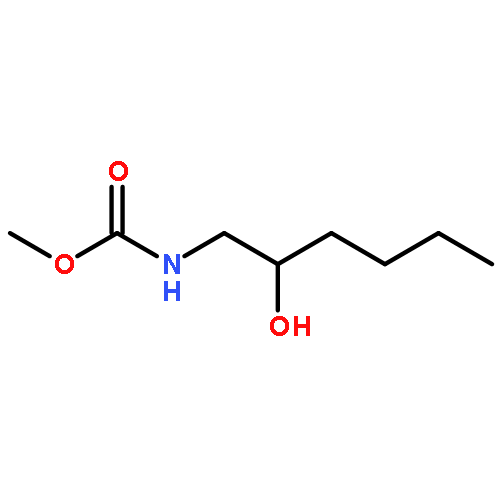
![Carbamic acid, [2-hydroxy-1-(3-nitrophenyl)propyl]-, methyl ester](http://img.cochemist.com/ccimg/439600/439585-37-2.png)
![Carbamic acid, [2-hydroxy-1-(3-nitrophenyl)propyl]-, methyl ester](http://img.cochemist.com/ccimg/439600/439585-37-2_b.png)


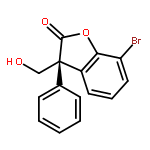

![D-erythro-Pent-1-enitol,1,4-anhydro-2-deoxy-3,5-bis-O-[(1,1-dimethylethyl)dimethylsilyl]-](http://img.cochemist.com/ccimg/173400/173327-56-5.png)
![D-erythro-Pent-1-enitol,1,4-anhydro-2-deoxy-3,5-bis-O-[(1,1-dimethylethyl)dimethylsilyl]-](http://img.cochemist.com/ccimg/173400/173327-56-5_b.png)
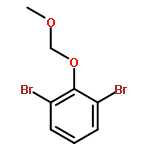
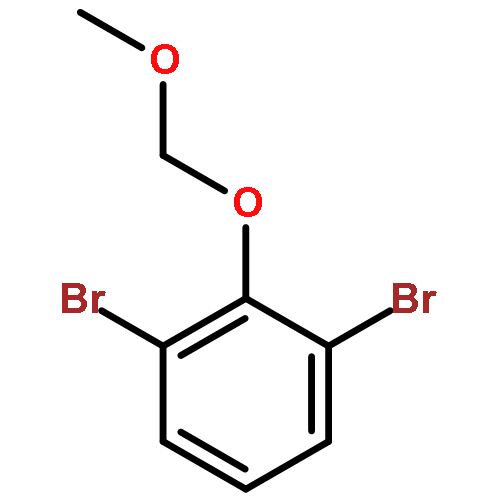


![3-Oxazolidinecarboxylic acid,4-[(4-methoxyphenyl)methyl]-2,2-dimethyl-, phenylmethyl ester, (S)-](http://img.cochemist.com/ccimg/138000/137960-07-7.png)
![3-Oxazolidinecarboxylic acid,4-[(4-methoxyphenyl)methyl]-2,2-dimethyl-, phenylmethyl ester, (S)-](http://img.cochemist.com/ccimg/138000/137960-07-7_b.png)


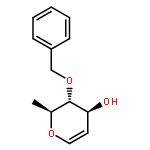













![Phosphonic acid, [(phenylsulfinyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/50800/50746-65-1.png)
![Phosphonic acid, [(phenylsulfinyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/50800/50746-65-1_b.png)

![Ethanol, 2-[[(3-nitrophenyl)methyl]amino]-](http://img.cochemist.com/ccimg/40200/40172-07-4.png)
![Ethanol, 2-[[(3-nitrophenyl)methyl]amino]-](http://img.cochemist.com/ccimg/40200/40172-07-4_b.png)


![1-PHENYLBENZO[E][1]BENZOFURAN](http://img.cochemist.com/ccimg/32800/32724-17-7.png)
![1-PHENYLBENZO[E][1]BENZOFURAN](http://img.cochemist.com/ccimg/32800/32724-17-7_b.png)


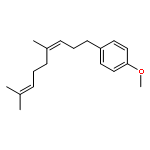
![Benzene, [(3E)-4,8-dimethyl-3,7-nonadienyl]-](http://img.cochemist.com/ccimg/22600/22555-66-4.png)
![Benzene, [(3E)-4,8-dimethyl-3,7-nonadienyl]-](http://img.cochemist.com/ccimg/22600/22555-66-4_b.png)










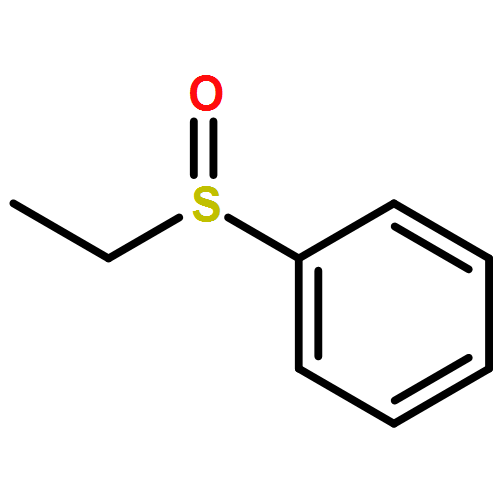
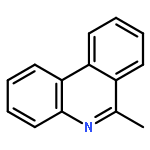
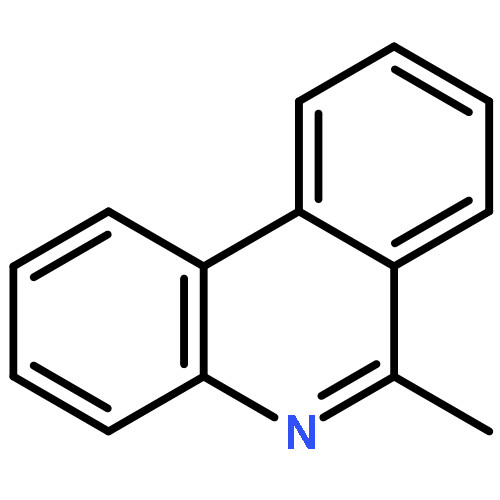


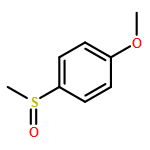


![Benzene, [(phenylmethyl)sulfinyl]-](/data/chemimg/1595300/833-82-9.png)
![Benzene, [(phenylmethyl)sulfinyl]-](/data/chemimg/1595300/833-82-9_b.png)


![Serine, N-[(1,1-dimethylethoxy)carbonyl]-L-valyl-, methyl ester](http://img.cochemist.com/ccimg/793800/793727-88-5.png)
![Serine, N-[(1,1-dimethylethoxy)carbonyl]-L-valyl-, methyl ester](http://img.cochemist.com/ccimg/793800/793727-88-5_b.png)

![BENZENECARBOTHIOAMIDE, N-[2-(1H-INDOL-3-YL)-2-OXOETHYL]-](http://img.cochemist.com/ccimg/793800/793724-26-2.png)
![BENZENECARBOTHIOAMIDE, N-[2-(1H-INDOL-3-YL)-2-OXOETHYL]-](http://img.cochemist.com/ccimg/793800/793724-26-2_b.png)
![1H-Indole, 3-[2-(3-pyridinyl)-5-oxazolyl]-](http://img.cochemist.com/ccimg/793800/793724-25-1.png)
![1H-Indole, 3-[2-(3-pyridinyl)-5-oxazolyl]-](http://img.cochemist.com/ccimg/793800/793724-25-1_b.png)
![3-PYRIDINECARBOXAMIDE, N-[2-(1H-INDOL-3-YL)-2-OXOETHYL]-](http://img.cochemist.com/ccimg/793800/793724-24-0.png)
![3-PYRIDINECARBOXAMIDE, N-[2-(1H-INDOL-3-YL)-2-OXOETHYL]-](http://img.cochemist.com/ccimg/793800/793724-24-0_b.png)
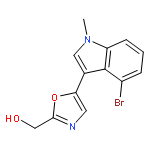
![2-Oxazoleacetonitrile, 5-[4-bromo-1-(methoxymethyl)-1H-indol-3-yl]-](http://img.cochemist.com/ccimg/761000/760979-84-8.png)
![2-Oxazoleacetonitrile, 5-[4-bromo-1-(methoxymethyl)-1H-indol-3-yl]-](http://img.cochemist.com/ccimg/761000/760979-84-8_b.png)
![1H-INDOLE, 3-[2-(AZIDOMETHYL)-5-OXAZOLYL]-4-BROMO-1-(METHOXYMETHYL)-](http://img.cochemist.com/ccimg/761000/760979-83-7.png)
![1H-INDOLE, 3-[2-(AZIDOMETHYL)-5-OXAZOLYL]-4-BROMO-1-(METHOXYMETHYL)-](http://img.cochemist.com/ccimg/761000/760979-83-7_b.png)
![2-methyl-2-propanyl [2-(1h-indol-3-yl)-2-oxoethyl]carbamate](http://img.cochemist.com/ccimg/689300/689232-04-0.png)
![2-methyl-2-propanyl [2-(1h-indol-3-yl)-2-oxoethyl]carbamate](http://img.cochemist.com/ccimg/689300/689232-04-0_b.png)
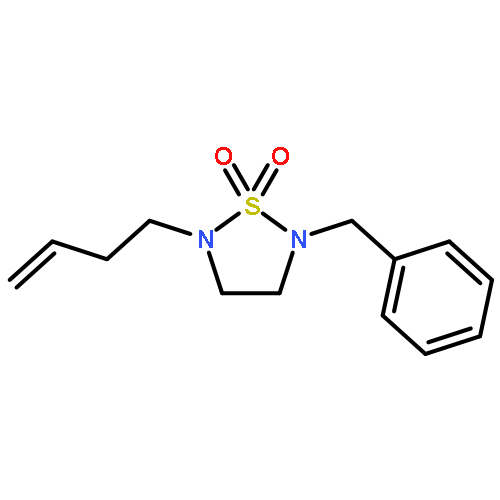
![(3aR)-hexahydro-2H-[1,2,5]Thiadiazolo[2,3-a]pyridine 1,1-dioxide](http://img.cochemist.com/ccimg/503400/503310-76-7.png)
![(3aR)-hexahydro-2H-[1,2,5]Thiadiazolo[2,3-a]pyridine 1,1-dioxide](http://img.cochemist.com/ccimg/503400/503310-76-7_b.png)
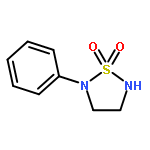
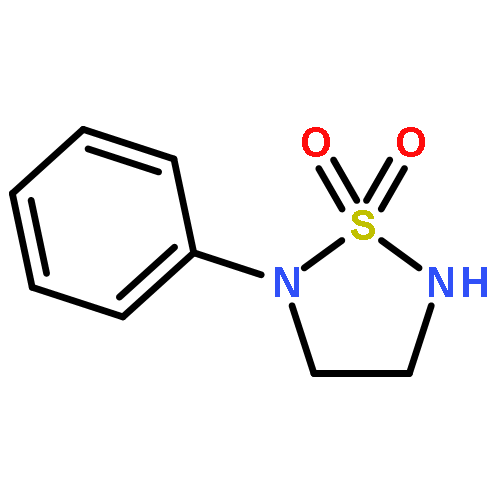
![1,2,5-Thiadiazolidine, 2-[3,5-bis(trifluoromethyl)phenyl]-, 1,1-dioxide](http://img.cochemist.com/ccimg/503400/503310-70-1.png)
![1,2,5-Thiadiazolidine, 2-[3,5-bis(trifluoromethyl)phenyl]-, 1,1-dioxide](http://img.cochemist.com/ccimg/503400/503310-70-1_b.png)
![Carbamic acid, [[(4-cyanophenyl)amino]sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/503400/503310-68-7.png)
![Carbamic acid, [[(4-cyanophenyl)amino]sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/503400/503310-68-7_b.png)
![Carbamic acid, [[(4-methoxyphenyl)amino]sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/503400/503310-67-6.png)
![Carbamic acid, [[(4-methoxyphenyl)amino]sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/503400/503310-67-6_b.png)
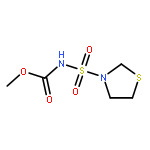



![Carbamic acid, [(dicyclohexylamino)sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/503400/503310-64-3.png)
![Carbamic acid, [(dicyclohexylamino)sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/503400/503310-64-3_b.png)
![Carbamic acid, [[methyl(phenylmethyl)amino]sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/503400/503310-63-2.png)
![Carbamic acid, [[methyl(phenylmethyl)amino]sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/503400/503310-63-2_b.png)
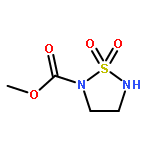
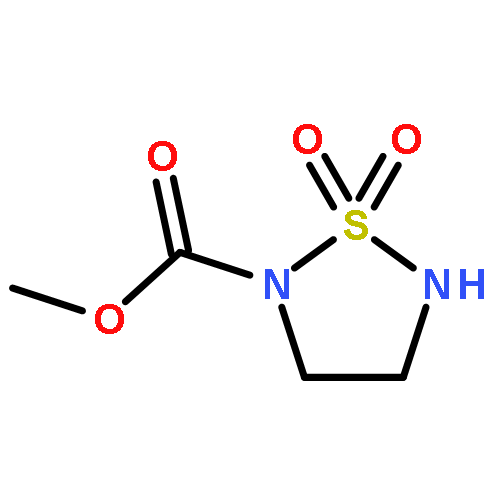
![Ethanol, 2-[[3,5-bis(trifluoromethyl)phenyl]amino]-](http://img.cochemist.com/ccimg/503400/503310-51-8.png)
![Ethanol, 2-[[3,5-bis(trifluoromethyl)phenyl]amino]-](http://img.cochemist.com/ccimg/503400/503310-51-8_b.png)
![CARBAMIC ACID, [2-HYDROXY-1-(2-METHOXYPHENYL)ETHYL]-, 2-PROPENYL ESTER](http://img.cochemist.com/ccimg/439600/439585-43-0.png)
![CARBAMIC ACID, [2-HYDROXY-1-(2-METHOXYPHENYL)ETHYL]-, 2-PROPENYL ESTER](http://img.cochemist.com/ccimg/439600/439585-43-0_b.png)
![CARBAMIC ACID, [2-HYDROXY-1-(2,4,6-TRIMETHYLPHENYL)ETHYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/439600/439585-35-0.png)
![CARBAMIC ACID, [2-HYDROXY-1-(2,4,6-TRIMETHYLPHENYL)ETHYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/439600/439585-35-0_b.png)
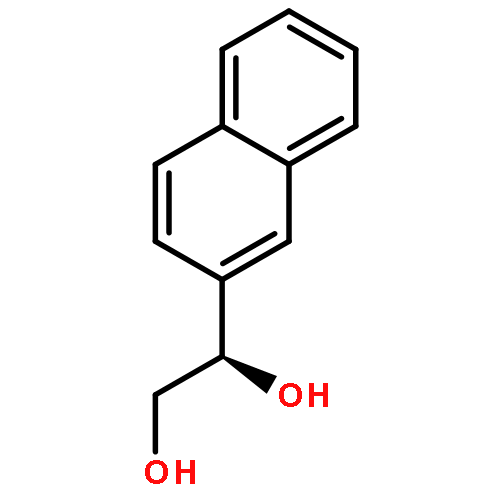
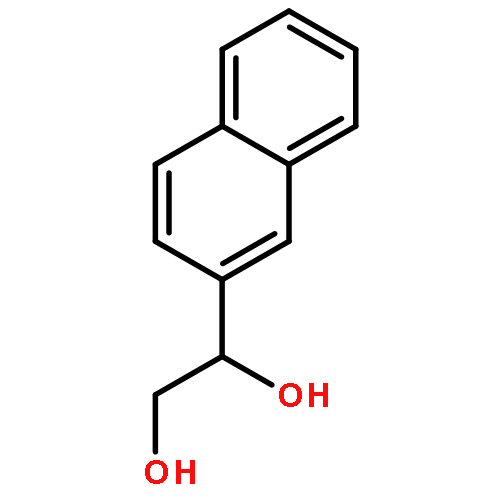
![Carbamic acid, [2-hydroxy-1-(2-naphthalenyl)ethyl]-, methyl ester](http://img.cochemist.com/ccimg/439600/439585-33-8.png)
![Carbamic acid, [2-hydroxy-1-(2-naphthalenyl)ethyl]-, methyl ester](http://img.cochemist.com/ccimg/439600/439585-33-8_b.png)

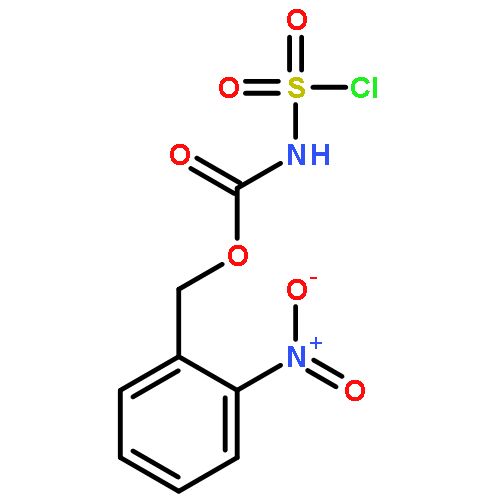
![1,2-Ethanediol, 1-[3,5-bis(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/439600/439584-69-7.png)
![1,2-Ethanediol, 1-[3,5-bis(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/439600/439584-69-7_b.png)
![1,2-Ethanediol, 1-[4-(acetyloxy)phenyl]-](http://img.cochemist.com/ccimg/439600/439584-65-3.png)
![1,2-Ethanediol, 1-[4-(acetyloxy)phenyl]-](http://img.cochemist.com/ccimg/439600/439584-65-3_b.png)

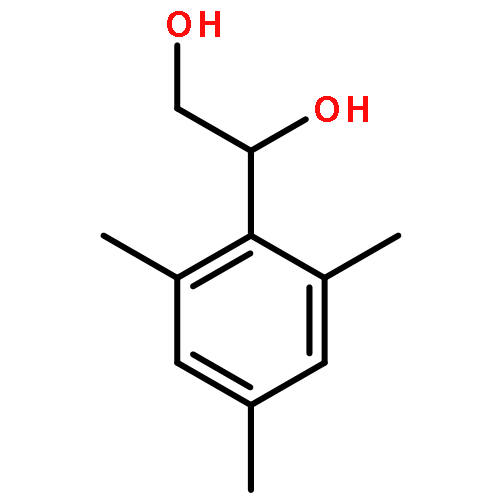
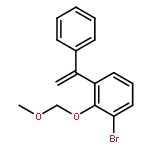

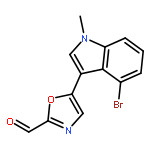

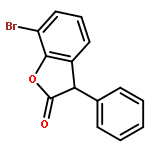

![1,2-Ethanediol, 1-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/175700/175605-64-8.png)
![1,2-Ethanediol, 1-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/175700/175605-64-8_b.png)
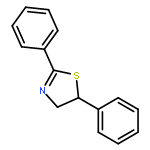
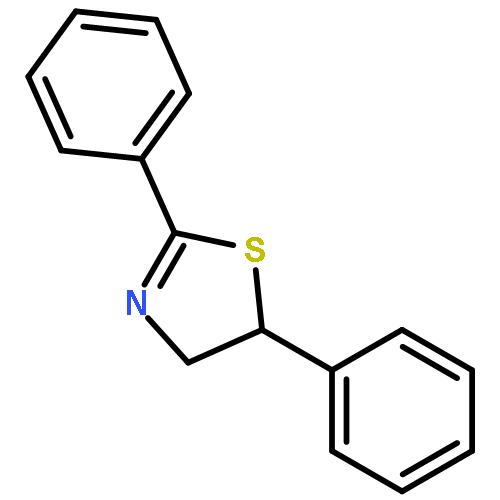


![Ethanol, 2-[(3,4,5-trimethoxyphenyl)amino]-](http://img.cochemist.com/ccimg/121500/121495-51-0.png)
![Ethanol, 2-[(3,4,5-trimethoxyphenyl)amino]-](http://img.cochemist.com/ccimg/121500/121495-51-0_b.png)
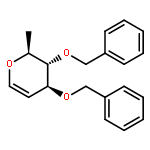



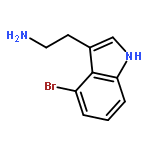
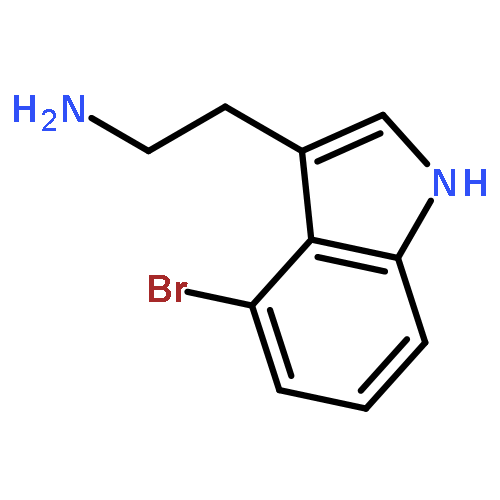

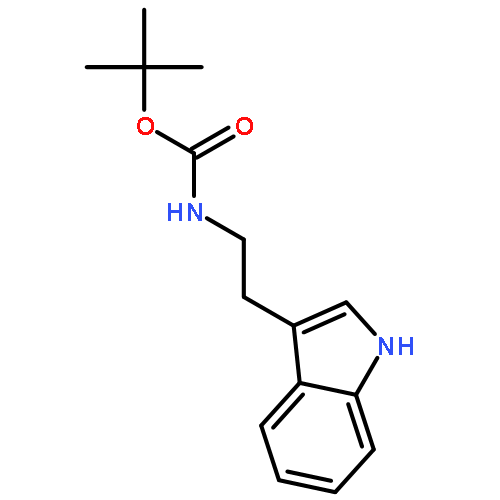
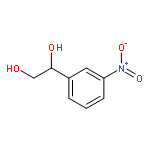
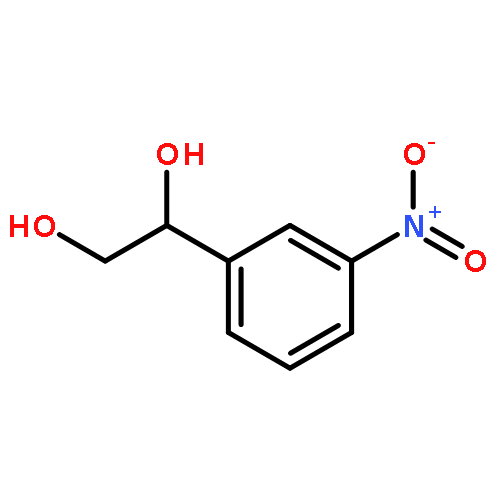
![Carbamic acid, [(cyclohexylamino)sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/90300/90222-26-7.png)
![Carbamic acid, [(cyclohexylamino)sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/90300/90222-26-7_b.png)
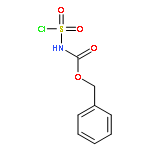



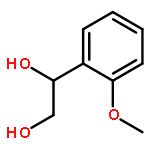





![BENZAMIDE, N-[2-(1H-INDOL-3-YL)-2-OXOETHYL]-](http://img.cochemist.com/ccimg/73100/73053-94-8.png)
![BENZAMIDE, N-[2-(1H-INDOL-3-YL)-2-OXOETHYL]-](http://img.cochemist.com/ccimg/73100/73053-94-8_b.png)









![N-[2-(1H-indol-3-yl)ethyl]-Benzamide](http://img.cochemist.com/ccimg/4800/4753-09-7.png)
![N-[2-(1H-indol-3-yl)ethyl]-Benzamide](http://img.cochemist.com/ccimg/4800/4753-09-7_b.png)


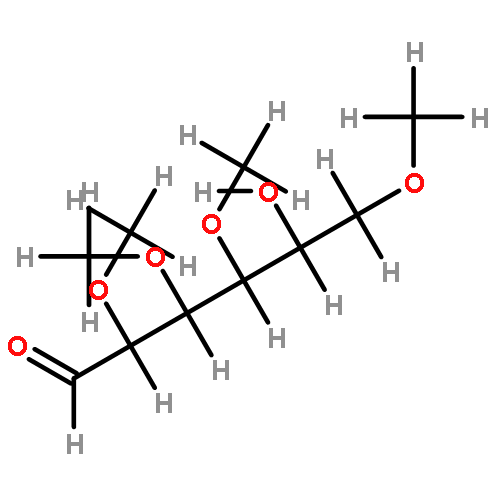
![2-[(4-Nitrophenyl)amino]ethanol](http://img.cochemist.com/ccimg/2000/1965-54-4.png)
![2-[(4-Nitrophenyl)amino]ethanol](http://img.cochemist.com/ccimg/2000/1965-54-4_b.png)
![1H-INDOLE, 4-BROMO-3-[2-(IODOMETHYL)-5-OXAZOLYL]-1-(METHOXYMETHYL)-](http://img.cochemist.com/ccimg/761000/760980-34-5.png)
![1H-INDOLE, 4-BROMO-3-[2-(IODOMETHYL)-5-OXAZOLYL]-1-(METHOXYMETHYL)-](http://img.cochemist.com/ccimg/761000/760980-34-5_b.png)
![2-OXAZOLEMETHANOL, 5-[4-BROMO-1-(METHOXYMETHYL)-1H-INDOL-3-YL]-](http://img.cochemist.com/ccimg/761000/760979-81-5.png)
![2-OXAZOLEMETHANOL, 5-[4-BROMO-1-(METHOXYMETHYL)-1H-INDOL-3-YL]-](http://img.cochemist.com/ccimg/761000/760979-81-5_b.png)
![L-Tyrosine,3-[(3S)-3-[2-[(1S)-1-amino-2-methylpropyl]-4-[(phenylmethoxy)methyl]-5-oxazolyl]-7-bromo-2,3-dihydro-1-(methoxymethyl)-2-oxo-1H-indol-3-yl]-O-(methoxymethyl)-N-[(phenylmethoxy)carbonyl]-](http://img.cochemist.com/ccimg/496800/496789-55-0.png)
![L-Tyrosine,3-[(3S)-3-[2-[(1S)-1-amino-2-methylpropyl]-4-[(phenylmethoxy)methyl]-5-oxazolyl]-7-bromo-2,3-dihydro-1-(methoxymethyl)-2-oxo-1H-indol-3-yl]-O-(methoxymethyl)-N-[(phenylmethoxy)carbonyl]-](http://img.cochemist.com/ccimg/496800/496789-55-0_b.png)
![Carbamic acid, [(1S)-1-[4-(hydroxymethyl)-2-oxazolyl]-2-methylpropyl]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/496800/496789-53-8.png)
![Carbamic acid, [(1S)-1-[4-(hydroxymethyl)-2-oxazolyl]-2-methylpropyl]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/496800/496789-53-8_b.png)

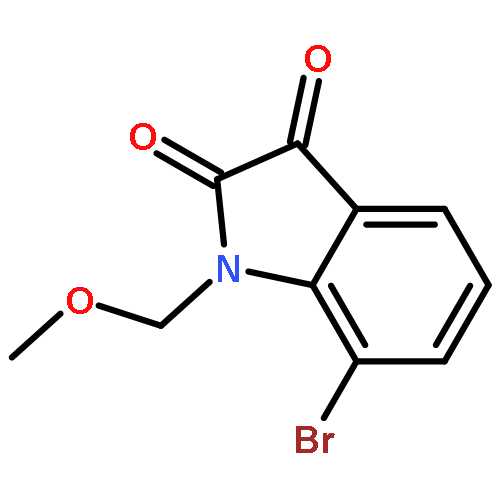
![Carbamic acid,[(1S)-2-methyl-1-[4-[(phenylmethoxy)methyl]-2-oxazolyl]propyl]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/496800/496789-39-0.png)
![Carbamic acid,[(1S)-2-methyl-1-[4-[(phenylmethoxy)methyl]-2-oxazolyl]propyl]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/496800/496789-39-0_b.png)