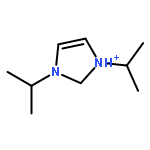
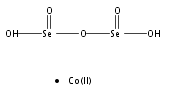Co-reporter:Douglas H. Fabini, Ting Ann Siaw, Constantinos C. Stoumpos, Geneva Laurita, Daniel Olds, Katharine Page, Jerry G. Hu, Mercouri G. Kanatzidis, Songi Han, and Ram Seshadri
Journal of the American Chemical Society November 22, 2017 Volume 139(Issue 46) pp:16875-16875
Publication Date(Web):November 2, 2017
DOI:10.1021/jacs.7b09536
The role of organic molecular cations in the high-performance perovskite photovoltaic absorbers, methylammonium lead iodide (MAPbI3) and formamidinium lead iodide (FAPbI3), has been an enigmatic subject of great interest. Beyond aiding in the ease of processing of thin films for photovoltaic devices, there have been suggestions that many of the remarkable properties of the halide perovskites can be attributed to the dipolar nature and the dynamic behavior of these cations. Here, we establish the dynamics of the molecular cations in FAPbI3 between 4 K and 340 K and the nature of their interaction with the surrounding inorganic cage using a combination of solid state nuclear magnetic resonance and dielectric spectroscopies, neutron scattering, calorimetry, and ab initio calculations. Detailed comparisons with the reported temperature dependence of the dynamics of MAPbI3 are then carried out which reveal the molecular ions in the two different compounds to exhibit very similar rotation rates (≈8 ps) at room temperature, despite differences in other temperature regimes. For FA, rotation about the N···N axis, which reorients the molecular dipole, is the dominant motion in all phases, with an activation barrier of ≈21 meV in the ambient phase, compared to ≈110 meV for the analogous dipole reorientation of MA. Geometrical frustration of the molecule–cage interaction in FAPbI3 produces a disordered γ-phase and subsequent glassy freezing at yet lower temperatures. Hydrogen bonds suggested by atom–atom distances from neutron total scattering experiments imply a substantial role for the molecules in directing structure and dictating properties. The temperature dependence of reorientation of the dipolar molecular cations systematically described here can clarify various hypotheses including those of large-polaron charge transport and fugitive electron spin polarization that have been invoked in the context of these unusual materials.
Co-reporter:Leo K. Lamontagne, Geneva Laurita, Michael Knight, Huma Yusuf, Jerry Hu, Ram Seshadri, and Katharine Page
Inorganic Chemistry May 1, 2017 Volume 56(Issue 9) pp:5158-5158
Publication Date(Web):April 13, 2017
DOI:10.1021/acs.inorgchem.7b00307
The cubic semiconducting compounds APd3O4 (A = Ca, Sr) can be hole-doped by Na substitution on the A site and driven toward more conducting states. This process has been followed here by a number of experimental techniques to understand the evolution of electronic properties. While an insulator-to-metal transition is observed in Ca1–xNaxPd3O4 for x ≥ 0.15, bulk metallic behavior is not observed for Sr1–xNaxPd3O4 up to x = 0.20. Given the very similar crystal and (calculated) electronic structures of the two materials, the distinct behavior is a matter of interest. We present evidence of local disorder in the A = Sr materials through the analysis of the neutron pair distribution function, which is potentially at the heart of the distinct behavior. Solid-state 23Na nuclear magnetic resonance studies additionally suggest a percolative insulator-to-metal transition mechanism, wherein presumably small regions with a signal resembling metallic NaPd3O4 form almost immediately upon Na substitution, and this signal grows monotonically with substitution. Some signatures of increased local disorder and a propensity for Na clustering are seen in the A = Sr compounds.
Co-reporter:Jean-Luc Brédas (Associate Editor), Kristin Persson (Associate Editor), and Ram Seshadri (Associate Editor)
Chemistry of Materials March 28, 2017 Volume 29(Issue 6) pp:2399-2399
Publication Date(Web):March 28, 2017
DOI:10.1021/acs.chemmater.7b00990
Co-reporter:Clayton Cozzan;Geneva Laurita;Jerry G. Hu;Kent J. Griffith;Clare P. Grey;Ram Seshadri
Inorganic Chemistry February 20, 2017 Volume 56(Issue 4) pp:2153-2158
Publication Date(Web):February 6, 2017
DOI:10.1021/acs.inorgchem.6b02780
SiAlON ceramics, solid solutions based on the Si3N4 structure, are important, lightweight structural materials with intrinsically high strength, high hardness, and high thermal and chemical stability. Described by the chemical formula β-Si6–zAlzOzN8–z, from a compositional viewpoint, these materials can be regarded as solid solutions between Si3N4 and Al3O3N. A key aspect of the structural evolution with increasing Al and O (z in the formula) is to understand how these elements are distributed on the β-Si3N4 framework. The average and local structural evolution of highly phase-pure samples of β-Si6–zAlzOzN8–z with z = 0.050, 0.075, and 0.125 are studied here, using a combination of X-ray diffraction, NMR studies, and density functional theory calculations. Synchrotron X-ray diffraction establishes sample purity and indicates subtle changes in the average structure with increasing Al content in these compounds. Solid-state magic-angle-spinning 27Al NMR experiments, coupled with detailed ab initio calculations of NMR spectra of Al in different AlOqN4–q tetrahedra (0 ≤ q ≤ 4), reveal a tendency of Al and O to cluster in these materials. Independently, the calculations suggest an energetic preference for Al–O bond formation, instead of a random distribution, in the β-SiAlON system.
Co-reporter:Megan M. Butala, Martin Mayo, Vicky V. T. Doan-Nguyen, Margaret A. Lumley, Claudia Göbel, Kamila M. Wiaderek, Olaf J. Borkiewicz, Karena W. Chapman, Peter J. Chupas, Mahalingam Balasubramanian, Geneva Laurita, Sylvia Britto, Andrew J. Morris, Clare P. Grey, and Ram Seshadri
Chemistry of Materials April 11, 2017 Volume 29(Issue 7) pp:3070-3070
Publication Date(Web):March 27, 2017
DOI:10.1021/acs.chemmater.7b00070
In the pursuit of high-capacity electrochemical energy storage, a promising domain of research involves conversion reaction schemes, wherein electrode materials are fully transformed during charge and discharge. There are, however, numerous difficulties in realizing theoretical capacity and high rate capability in many conversion schemes. Here we employ operando studies to understand the conversion material FeS2, focusing on the local structure evolution of this relatively reversible material. X-ray absorption spectroscopy, pair distribution function analysis, and first-principles calculations of intermediate structures shed light on the mechanism of charge storage in the Li–FeS2 system, with some general principles emerging for charge storage in chalcogenide materials. Focusing on second and later charge/discharge cycles, we find small, disordered domains that locally resemble Fe and Li2S at the end of the first discharge. Upon charge, this is converted to a Li–Fe–S composition whose local structure reveals tetrahedrally coordinated Fe. With continued charge, this ternary composition displays insertion–extraction behavior at higher potentials and lower Li content. The finding of hybrid modes of charge storage, rather than simple conversion, points to the important role of intermediates that appear to store charge by mechanisms that more closely resemble intercalation.
Co-reporter:Hayden A. Evans, John G. Labram, Sara R. Smock, Guang Wu, Michael L. ChabinycRam Seshadri, Fred Wudl
Inorganic Chemistry 2017 Volume 56(Issue 1) pp:395-401
Publication Date(Web):December 14, 2016
DOI:10.1021/acs.inorgchem.6b02287
Two new compounds containing tetrathiafulvalene (TTF) cations with extended and discrete anions based on Bi and I are reported. The compound (TTF)BiI4 comprises [BiI2I4/2]− chains of edge-shared octahedra that are interspersed with stacks of TTF+•. The compound (TTF)4BiI6 has mixed-valence stacks of TTF and TTF+• and discrete molecules of TTF+• separated by discrete [BiI6]−3 anions. The optical and electrical transport properties of these compounds are reported. Due to the mixed-valence stacks of TTF, (TTF)4BiI6 is the significantly better electrical conductor than (TTF)BiI4, despite the discrete nature of the inorganic moiety.
Co-reporter:Douglas H. Fabini, John G. Labram, Anna J. Lehner, Jonathon S. Bechtel, Hayden A. Evans, Anton Van der Ven, Fred Wudl, Michael L. Chabinyc, and Ram Seshadri
Inorganic Chemistry 2017 Volume 56(Issue 1) pp:11-25
Publication Date(Web):September 15, 2016
DOI:10.1021/acs.inorgchem.6b01539
Main-group halide perovskites have generated much excitement of late because of their remarkable optoelectronic properties, ease of preparation, and abundant constituent elements, but these curious and promising materials differ in important respects from traditional semiconductors. The distinguishing chemical, structural, and electronic features of these materials present the key to understanding the origins of the optoelectronic performance of the well-studied hybrid organic–inorganic lead halides and provide a starting point for the design and preparation of new functional materials. Here we review and discuss these distinguishing features, among them a defect-tolerant electronic structure, proximal lattice instabilities, labile defect migration, and, in the case of hybrid perovskites, disordered molecular cations. Additionally, we discuss the preparation and characterization of some alternatives to the lead halide perovskites, including lead-free bismuth halides and hybrid materials with optically and electronically active organic constituents.
Co-reporter:Clayton Cozzan;Geneva Laurita;Michael W. Gaultois;Marcus Cohen;Alexander A. Mikhailovsky;Mahalingam Balasubramanian;Ram Seshadri
Journal of Materials Chemistry C 2017 vol. 5(Issue 38) pp:10039-10046
Publication Date(Web):2017/10/05
DOI:10.1039/C7TC03039H
Inorganic phosphor materials play a crucial role in the creation of white light from blue and near-UV solid-state light-emitting diodes. Understanding the intricacies of the phosphor structure is key for setting the stage for improved, more efficient functionality. Average structure and coordination environment analysis of the robust and efficient green-emitting phosphor, β-SiAlON:Eu2+ (β-Si6−zAlzOzN8−zEu0.009), is combined here with a range of property measurements to elucidate the role of Al content (z) in luminescence properties, including the red shift of emission and the thermal quenching of luminescence as a function of increasing Al content z. Average structure techniques reveal changes in polyhedral distortion with increasing z for the 9-coordinate Eu site in β-SiAlON:Eu2+. X-ray absorption near edge structure (XANES) is used to confirm that the majority of the activator Eu is in the Eu2+ state, exhibiting the symmetry-allowed and efficient 4f75d0 → 4f65d1 transitions. Room temperature and temperature-dependent luminescence indicate a curious increase in thermal stability with increasing z over a small range due to an increasing barrier for thermal ionization, which is correlated to an increase in the quantum yield of the phosphor.
Co-reporter:Geneva Laurita;Douglas H. Fabini;Constantinos C. Stoumpos;Mercouri G. Kanatzidis;Ram Seshadri
Chemical Science (2010-Present) 2017 vol. 8(Issue 8) pp:5628-5635
Publication Date(Web):2017/07/24
DOI:10.1039/C7SC01429E
Hybrid halide perovskites combine ease of preparation and relatively abundant constituent elements with fascinating photophysical properties. Descriptions of the chemical and structural drivers of the remarkable properties have often focused on the potential role of the dynamic order/disorder of the molecular A-site cations. We reveal here a key aspect of the inorganic framework that potentially impacts the electronic, thermal, and dielectric properties. The temperature evolution of the X-ray pair distribution functions of hybrid perovskites ABX3 [A+ = CH3NH3 (MA) or CH(NH2)2 (FA); B2+ = Sn or Pb; X− = Br, or I] in their cubic phases above 300 K reveals temperature-activated displacement (off-centering) of the divalent group 14 cations from their nominal, centered sites. This symmetry-lowering distortion phenomenon, previously dubbed emphanisis in the context of compounds such as PbTe, is attributed to Sn2+ and Pb2+ lone pair stereochemistry. Of the materials studied here, the largest displacements from the center of the octahedral sites are found in tin iodides, a more moderate effect is found in lead bromides, and the weakest effect is seen in lead iodides. The A-site cation appears to play a role as well, with the larger FA resulting in greater off-centering for both Sn2+ and Pb2+. Dynamic off-centering, which is concealed within the framework of traditional Bragg crystallography, is proposed to play a key role in the remarkable defect-tolerant nature of transport in these semiconductors via its effect on the polarizability of the lattice. The results suggest a novel chemical design principle for future materials discovery.
Co-reporter:Molleigh B. Preefer;Dr. Bernd Oschmann; Dr. Craig J. Hawker; Dr. Ram Seshadri; Dr. Fred Wudl
Angewandte Chemie 2017 Volume 129(Issue 47) pp:15314-15318
Publication Date(Web):2017/11/20
DOI:10.1002/ange.201708746
AbstractWe demonstrate a novel crosslinked disulfide system as a cathode material for Li-S cells that is designed with the two criteria of having only a single point of S−S scission and maximizing the ratio of S−S to the electrochemically inactive framework. The material therefore maximizes theoretical capacity while inhibiting the formation of polysulfide intermediates that lead to parasitic shuttle. The material we report contains a 1:1 ratio of S:C with a theoretical capacity of 609 mAh g−1. The cell gains capacity through 100 cycles and has 98 % capacity retention thereafter through 200 cycles, demonstrating stable, long-term cycling. Raman spectroscopy confirms the proposed mechanism of disulfide bonds breaking to form a S−Li thiolate species upon discharge and reforming upon charge. Coulombic efficiencies near 100 % for every cycle, suggesting the suppression of polysulfide shuttle through the molecular design.
Co-reporter:Joshua D. Bocarsly, Emily E. Levin, Christina A. C. Garcia, Kai Schwennicke, Stephen D. Wilson, and Ram Seshadri
Chemistry of Materials 2017 Volume 29(Issue 4) pp:
Publication Date(Web):February 13, 2017
DOI:10.1021/acs.chemmater.6b04729
Alternating cycles of isothermal magnetization and adiabatic demagnetization applied to a magnetocaloric material can drive refrigeration in very much the same manner as cycles of gas compression and expansion. The material property of interest in finding candidate magnetocaloric materials is ΔSM, their gravimetric entropy change upon application of a magnetic field under isothermal conditions. There is, however, no general method for screening materials for such an entropy change without actually performing the relevant, time- and effort-intensive magnetic measurements. Here we propose a simple computational proxy based on performing nonmagnetic and magnetic density functional theory calculations on magnetic materials. This proxy, which we term the magnetic deformation ΣM, is a measure of how much the unit cell deforms comparing with the relaxed structures with and without the inclusion of spin polarization. ΣM appears to correlate very well with experimentally measured magnetic entropy change values. The proxy has been tested against 33 ferromagnetic materials with known ΔSM, including nine materials newly measured for this study. It has then been used to screen 134 ferromagnetic materials for which the magnetic entropy has not yet been reported, identifying 30 compounds as being promising for further study. As a demonstration of the effectiveness of our approach, we have prepared one of these compounds and measured its isothermal entropy change. MnCoP, with a TC of 575 K, shows a maximal ΔSM of −6.0 J kg–1 K–1 for an H = 5 T applied field.
Co-reporter:Anne-Marie Zieschang, Joshua D. Bocarsly, Michael Dürrschnabel, Leopoldo Molina-Luna, Hans-Joachim Kleebe, Ram SeshadriBarbara Albert
Chemistry of Materials 2017 Volume 29(Issue 2) pp:
Publication Date(Web):December 27, 2016
DOI:10.1021/acs.chemmater.6b04088
ε-Fe3N shows interesting magnetism but is difficult to obtain as a pure and single-phase sample. We report a new preparation method using the reduction of iron(II) bromide with elemental sodium in liquid ammonia at −78 °C, followed by annealing at 573 K. Nanostructured and monophasic oxygen-free iron nitride, ε-Fe3N, is produced according to X-ray diffraction and transmission electron microscopy experiments. The magnetic properties between 2 and 625 K were characterized using a vibrating sample magnetometer, revealing soft ferromagnetic behavior with a low-temperature average moment of 1.5 μB/Fe and a Curie temperature of 500 K. TC is lower than that of bulk ε-Fe3N (575 K) [ Chem. Phys. Lett 2010, 493, 299], which corresponds well with the small particle size within the agglomerates (15.4 (±4.1) nm according to TEM, 15.8(1) according to XRD). Samples were analyzed before and after partial oxidation (Fe3N–FexOy core–shell nanoparticles with a 2–3 nm thick shell) by X-ray diffraction, transmission electron microscopy, electron energy-loss spectroscopy, and magnetic measurements. Both the pristine Fe3N nanoparticles and the oxidized core–shell particles showed shifting and broadening of the magnetic hysteresis loops upon cooling in a magnetic field.
Co-reporter:Molleigh B. Preefer;Dr. Bernd Oschmann; Dr. Craig J. Hawker; Dr. Ram Seshadri; Dr. Fred Wudl
Angewandte Chemie International Edition 2017 Volume 56(Issue 47) pp:15118-15122
Publication Date(Web):2017/11/20
DOI:10.1002/anie.201708746
AbstractWe demonstrate a novel crosslinked disulfide system as a cathode material for Li-S cells that is designed with the two criteria of having only a single point of S−S scission and maximizing the ratio of S−S to the electrochemically inactive framework. The material therefore maximizes theoretical capacity while inhibiting the formation of polysulfide intermediates that lead to parasitic shuttle. The material we report contains a 1:1 ratio of S:C with a theoretical capacity of 609 mAh g−1. The cell gains capacity through 100 cycles and has 98 % capacity retention thereafter through 200 cycles, demonstrating stable, long-term cycling. Raman spectroscopy confirms the proposed mechanism of disulfide bonds breaking to form a S−Li thiolate species upon discharge and reforming upon charge. Coulombic efficiencies near 100 % for every cycle, suggesting the suppression of polysulfide shuttle through the molecular design.
Co-reporter:Hayden A. Evans, Emily C. Schueller, Sara R. Smock, Guang Wu, Ram Seshadri, Fred Wudl
Inorganica Chimica Acta 2017 Volume 468(Volume 468) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.ica.2017.04.060
Noble metal halide compounds are an exciting class of materials which display unique crystal chemistry with temperature and pressure. Here we report two new compounds, single-valence formamidinium platinum iodide [(FA)2PtIVI6] and mixed-valence methylammonium gold iodide [(MA)2AuIAuIIII6]. Structural changes between 300 K and 100 K have been monitored. The compounds crystallize in two different perovskite-related structure classes, with the added complexity of a disordered, “tumbling”, organic cation at 300 K. This tumbling is significantly reduced at 100 K. (FA)2PtIVI6 undergoes in-phase octahedral tilting similar to related vacancy-ordered perovskite materials. At 100 K, (MA)2AuIAuIIII6 undergoes a transition which facilitates stronger Au–I covalent bonding as made evident by a contraction of the unit cell and distances between Au and I atoms in the ab plane.Download high-res image (129KB)Download full-size image
Co-reporter:Douglas H. Fabini, Geneva Laurita, Jonathon S. Bechtel, Constantinos C. Stoumpos, Hayden A. Evans, Athanassios G. Kontos, Yannis S. Raptis, Polycarpos Falaras, Anton Van der Ven, Mercouri G. Kanatzidis, and Ram Seshadri
Journal of the American Chemical Society 2016 Volume 138(Issue 36) pp:11820-11832
Publication Date(Web):September 1, 2016
DOI:10.1021/jacs.6b06287
Co-reporter:Leo K. Lamontagne, Geneva Laurita, Michael W. Gaultois, Michael Knight, Leila Ghadbeigi, Taylor D. Sparks, Markus E. Gruner, Rossitza Pentcheva, Craig M. Brown, and Ram Seshadri
Chemistry of Materials 2016 Volume 28(Issue 10) pp:3367
Publication Date(Web):May 9, 2016
DOI:10.1021/acs.chemmater.6b00447
PbPdO2 is a band semiconductor with a band gap arising from the filled d8 nature of square-planar Pd2+. We establish that hole doping through Li substitution for Pd in PbPdO2 results in a p-type metallic oxide with a positive temperature coefficient of resistance for substitution amounts as small as 2 mol % Li for Pd. Furthermore, PbPd1–xLixO2 demonstrates a high Seebeck coefficient and is therefore an oxide thermoelectric material with high thermopower despite the metallic conductivity. Up to 4 mol % Li is found to substitute for Pd as verified by Rietveld refinement of neutron diffraction data. At this maximal Li substitution, the resistivity is driven below the Mott metallic maximum to 3.5 × 10–3 Ω cm with a Seebeck coefficient of 115 μV/K at room temperature, which increases to 175 μV/K at 600 K. These electrical properties are almost identical to those of the well-known p-type oxide thermoelectric NaxCoO2. Nonmagnetic Li-substituted PbPdO2 does not possess a correlated, magnetic state with high-spin degeneracy as found in some complex cobalt oxides. This suggests that there are other avenues to achieving high Seebeck coefficients with metallic conductivities in oxide thermoelectrics. The electrical properties coupled with the moderately low lattice thermal conductivities allow for a zT of 0.12 at 600 K, the maximal temperature measured here. The trend suggests yet higher values at elevated temperatures. First-principles calculations of the electronic structure and electrical transport provide insight into the observed properties.
Co-reporter:Hayden A. Evans, Anna J. Lehner, John G. Labram, Douglas H. Fabini, Omar Barreda, Sara R. Smock, Guang Wu, Michael L. Chabinyc, Ram Seshadri, and Fred Wudl
Chemistry of Materials 2016 Volume 28(Issue 11) pp:3607
Publication Date(Web):May 27, 2016
DOI:10.1021/acs.chemmater.6b00633
Co-reporter:Megan M. Butala, Katherine R. Danks, Margaret A. Lumley, Shiliang Zhou, Brent C. Melot, and Ram Seshadri
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 10) pp:6496
Publication Date(Web):February 16, 2016
DOI:10.1021/acsami.5b12840
Complex manganese oxides have been extensively studied as intercalation Li-ion battery electrodes. The simple oxide MnO has been proposed as a conversion anode material with a theoretical capacity of 756 mAh g–1 for full reduction to the metal. We report the reaction of MnO with Li using in situ X-ray diffraction and find no sign of crystalline products upon either discharge or charge. However, the absence of reflections, paired with electrochemical impedance spectroscopy, suggests disordered discharge products. We also examine composite electrodes with porous particles of MnO as the active component, with pores generated through the reductive heating of Mn3O4. We compare the behavior of these with more dense MnO powders, including studies of the electrode morphologies pre- and postcyling. We find differences in the first discharge relevant to the utility of such mesostructuring in conversion reaction materials. Specifically, we find this type of mesostructure, which gives advantage in intercalation and pseudocapacitive storage, does not yield the same benefits for conversion reaction systems.Keywords: conversion electrode; in situ XRD; lithium-ion battery; manganese oxide; mesoporosity; transition metal oxide
Co-reporter:Douglas H. Fabini; Tom Hogan; Hayden A. Evans; Constantinos C. Stoumpos; Mercouri G. Kanatzidis;Ram Seshadri
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 3) pp:376-381
Publication Date(Web):January 14, 2016
DOI:10.1021/acs.jpclett.5b02821
Hybrid main group halide perovskites hold great technological promise in optoelectronic applications and present rich and complex evolution of structure and dynamics. Here we present low-temperature dielectric measurements and calorimetry of APbI3 [A = CH3NH3+, HC(NH2)2+] that suggest glassy behavior on cooling. In both compounds, the dielectric loss displays frequency-dependent peaks below 100 K characteristic of a glassy slowing of relaxation dynamics, with HC(NH2)2PbI3 exhibiting greater glass fragility. Consistent with quenched disorder, the low-temperature heat capacity of both perovskites deviates substantially from the ∼T3 acoustic phonon contribution predicted by the Debye model. We suggest that static disorder of the A-site molecular cation, potentially coupled to local distortions of the Pb–I sublattice, is responsible for these phenomena. The distinct low-temperature dynamics observed in these two perovskites suggest qualitative differences in the interaction between the molecular cation and the surrounding inorganic framework, with potential implications for defect screening and device performance at ambient temperatures.
Co-reporter:Dr. Sheri Madhu;Hayden A. Evans;Dr. Vicky V. T. Doan-Nguyen;Dr. John G. Labram;Dr. Guang Wu;Dr. Michael L. Chabinyc;Dr. Ram Seshadri;Dr. Fred Wudl
Angewandte Chemie International Edition 2016 Volume 55( Issue 28) pp:
Publication Date(Web):
DOI:10.1002/anie.201604535
Co-reporter:Dr. Sheri Madhu;Hayden A. Evans;Dr. Vicky V. T. Doan-Nguyen;Dr. John G. Labram;Dr. Guang Wu;Dr. Michael L. Chabinyc;Dr. Ram Seshadri;Dr. Fred Wudl
Angewandte Chemie 2016 Volume 128( Issue 28) pp:
Publication Date(Web):
DOI:10.1002/ange.201604535
Co-reporter:Dr. Sheri Madhu;Hayden A. Evans;Dr. Vicky V. T. Doan-Nguyen;Dr. John G. Labram;Dr. Guang Wu;Dr. Michael L. Chabinyc;Dr. Ram Seshadri;Dr. Fred Wudl
Angewandte Chemie International Edition 2016 Volume 55( Issue 28) pp:8032-8035
Publication Date(Web):
DOI:10.1002/anie.201601585
Abstract
We report the preparation and X-ray crystallographic characterization of the first crystalline homoatomic polymer chain, which is part of a semiconducting pyrroloperylene–iodine complex. The crystal structure contains infinite polyiodide I∞δ−. Interestingly, the structure of iodine within the insoluble, blue starch–iodine complex has long remained elusive, but has been speculated as having infinite chains of iodine. Close similarities in the low-wavenumber Raman spectra of the title compound and starch–iodine point to such infinite polyiodide chains in the latter as well.
Co-reporter:Dr. Sheri Madhu;Hayden A. Evans;Dr. Vicky V. T. Doan-Nguyen;Dr. John G. Labram;Dr. Guang Wu;Dr. Michael L. Chabinyc;Dr. Ram Seshadri;Dr. Fred Wudl
Angewandte Chemie 2016 Volume 128( Issue 28) pp:8164-8167
Publication Date(Web):
DOI:10.1002/ange.201601585
Abstract
We report the preparation and X-ray crystallographic characterization of the first crystalline homoatomic polymer chain, which is part of a semiconducting pyrroloperylene–iodine complex. The crystal structure contains infinite polyiodide I∞δ−. Interestingly, the structure of iodine within the insoluble, blue starch–iodine complex has long remained elusive, but has been speculated as having infinite chains of iodine. Close similarities in the low-wavenumber Raman spectra of the title compound and starch–iodine point to such infinite polyiodide chains in the latter as well.
Co-reporter:Kimberly A. See, Stephan Hug, Katharina Schwinghammer, Margaret A. Lumley, Yonghao Zheng, Jaya M. Nolt, Galen D. Stucky, Fred Wudl, Bettina V. Lotsch, and Ram Seshadri
Chemistry of Materials 2015 Volume 27(Issue 11) pp:3821
Publication Date(Web):May 15, 2015
DOI:10.1021/acs.chemmater.5b00772
Redox active electrode materials derived from organic precursors are of interest for use as alternative cathodes in Li batteries due to the potential for their sustainable production from renewable resources. Here, a series of organic networks that either contain triazine units or are derived from triazine-containing precursors are evaluated as cathodes versus Li metal anodes as possible active materials in Li batteries. The role of the molecular structure on the electrochemical performance is studied by comparing several materials prepared across a range of conditions allowing control over functionality and long-range order. Well-defined structures in which the triazine unit persists in the final material exhibit very low capacities at voltages relevant for cathode materials (<10 mA·h g–1). Relatively high, reversible capacity (around 150 mA·h g–1) is in fact displayed by amorphous materials with little evidence of triazine functionality. This result directly contradicts previous suggestions that the triazine unit is responsible for charge storage in this family of materials. While the gently sloping discharge and charge profiles suggest a capacitive-type mechanism—further confirmed by the trend of increasing capacity with increasing surface area—electron paramagnetic resonance (EPR) spectroscopy studies show that the materials exhibiting higher capacities also display substantial EPR signals, potentially implicating unpaired spins in a charge storage mechanism that could involve charge transfer.
Co-reporter:Kristin A. Denault, Jakoah Brgoch, Simon D. Kloß, Michael W. Gaultois, Joan Siewenie, Katharine Page, and Ram Seshadri
ACS Applied Materials & Interfaces 2015 Volume 7(Issue 13) pp:7264
Publication Date(Web):March 27, 2015
DOI:10.1021/acsami.5b00445
The average and local structure of the oxides Ba2SiO4, BaAl2O4, SrAl2O4, and Y2SiO5 are examined to evaluate crystal rigidity in light of recent studies suggesting that highly connected and rigid structures yield the best phosphor hosts. Simultaneous momentum-space refinements of synchrotron X-ray and neutron scattering yield accurate average crystal structures, with reliable atomic displacement parameters. The Debye temperature ΘD, which has proven to be a useful proxy for structural rigidity, is extracted from the experimental atomic displacement parameters and compared with predictions from density functional theory calculations and experimental low-temperature heat capacity measurements. The role of static disorder on the measured displacement parameters, and the resulting Debye temperatures, are also analyzed using pair distribution function of total neutron scattering, as refined over varying distance ranges of the pair distribution function. The interplay between optimal bonding in the structure, structural rigidity, and correlated motion in these structures is examined, and the different contributions are delineated.Keywords: correlated atomic motion; Debye temperature; heat capacity; pair distribution function; phosphors; structural rigidity
Co-reporter:Alexander Birkel, Nicholas A. DeCino, Clayton Cozzan, Alexander A. Mikhailovsky, Byung-Chul Hong, Ram Seshadri
Solid State Sciences 2015 Volume 48() pp:82-89
Publication Date(Web):October 2015
DOI:10.1016/j.solidstatesciences.2015.07.005
•Single-phase Ba3MgSi2O8 was prepared using sol–gel route with microwave heating and doped with the ions Eu2+, Tb3+, and Mn2+.•Sol–gel route provides better phase purity than conventional methods.•Full color emission (cool white) achieved and thermally robust emission observed.We present a rapid and energy-efficient microwave-assisted approach to prepare a single-phase full-color phosphor based on Ba3MgSi2O8. The samples were prepared using a citric acid based sol–gel preparation pathway with a microwave-assisted heating step, which reduces the time required for the final heat treatment to less than 30 min. Thermogravimetric analysis was utilized to optimize the solution-based preparation prior to microwave heating. The structural properties of the obtained luminescent materials have been thoroughly investigated by means of X-ray powder diffraction and Rietveld analyses. To study the optical behavior, the excitation and emission spectra were recorded. Full-color emission is achieved using Eu2+ (blue), Tb3+ (green), and Mn2+ (red) as the activator ions. The thermally robust emission was investigated using temperature-dependent luminescence spectroscopy. The energy-transfer processes within the samples were studied using time-dependent spectroscopy, and the quantum yield of this true color phosphor as a function of the composition was determined.
Co-reporter:Kimberly A. See ; Michal Leskes ; John M. Griffin ; Sylvia Britto ; Peter D. Matthews ; Alexandra Emly ; Anton Van der Ven ; Dominic S. Wright ; Andrew J. Morris ; Clare P. Grey ;Ram Seshadri
Journal of the American Chemical Society 2014 Volume 136(Issue 46) pp:16368-16377
Publication Date(Web):November 10, 2014
DOI:10.1021/ja508982p
The high theoretical gravimetric capacity of the Li–S battery system makes it an attractive candidate for numerous energy storage applications. In practice, cell performance is plagued by low practical capacity and poor cycling. In an effort to explore the mechanism of the discharge with the goal of better understanding performance, we examine the Li–S phase diagram using computational techniques and complement this with an in situ 7Li NMR study of the cell during discharge. Both the computational and experimental studies are consistent with the suggestion that the only solid product formed in the cell is Li2S, formed soon after cell discharge is initiated. In situ NMR spectroscopy also allows the direct observation of soluble Li+-species during cell discharge; species that are known to be highly detrimental to capacity retention. We suggest that during the first discharge plateau, S is reduced to soluble polysulfide species concurrently with the formation of a solid component (Li2S) which forms near the beginning of the first plateau, in the cell configuration studied here. The NMR data suggest that the second plateau is defined by the reduction of the residual soluble species to solid product (Li2S). A ternary diagram is presented to rationalize the phases observed with NMR during the discharge pathway and provide thermodynamic underpinnings for the shape of the discharge profile as a function of cell composition.
Co-reporter:Kristin A. Denault, Jakoah Brgoch, Michael W. Gaultois, Alexander Mikhailovsky, Ralf Petry, Holger Winkler, Steven P. DenBaars, and Ram Seshadri
Chemistry of Materials 2014 Volume 26(Issue 7) pp:2275
Publication Date(Web):March 19, 2014
DOI:10.1021/cm500116u
The orthosilicate phosphors SrxBa2–xSiO4:Eu2+ have now been known for over four decades and have found extensive recent use in solid-state white lighting. It is well-recognized in the literature and in practice that intermediate compositions in the solid-solutions between the orthosilicates Sr2SiO4 and Ba2SiO4 yield the best phosphor hosts when the thermal stability of luminescence is considered. We employ a combination of synchrotron X-ray diffraction, total scattering measurements, density functional theory calculations, and low-temperature heat capacity measurements, in conjunction with detailed temperature- and time-resolved studies of luminescence properties to understand the origins of the improved luminescence properties. We observe that in the intermediate compositions, the two cation sites in the crystal structure are optimally bonded as determined from bond valence sum calculations. Optimal bonding results in a more rigid lattice, as established by the intermediate compositions possessing the highest Debye temperature, which are determined experimentally from low-temperature heat capacity measurements. Greater rigidity in turn results in the highest luminescence efficiency for intermediate compositions at elevated temperatures.
Co-reporter:Steffi Rades, Stephan Kraemer, Ram Seshadri, and Barbara Albert
Chemistry of Materials 2014 Volume 26(Issue 4) pp:1549
Publication Date(Web):January 24, 2014
DOI:10.1021/cm403167a
A nanoscale boride, α-FeB, with grains of variable size and crystallinity was synthesized by precipitation from solution followed by heat treatment (450 °C, 550 °C, 750 °C, 1050 °C). Analysis of transmission electron micrographs, electron diffraction, and magnetic measurements suggests superparamagnetism at room temperature for the smaller, more disordered particles of FeB, while the larger, more crystalline particles of α-FeB, with a particle size of approximately 20 nm, display open magnetic hysteresis loops and blocking. In contrast to the soft ferromagnetism of bulk β-FeB, which was synthesized by conventional solid state reaction at 1500 °C, the sample of α-FeB annealed at 1050 °C is a harder ferromagnet, possibly due to stacking faults that pin the magnetic domains; these stacking faults are apparent in the high resolution transmission electron micrographs. The changes in magnetic behavior are visible from the varying blocking temperatures (63 K, 94 K, 150 K, and >320 K from the smallest to the largest particles) and correlate with the transformations from amorphous to α-FeB and from α-FeB to β-FeB.
Co-reporter:Kimberly A. See, Young-Si Jun, Jeffrey A. Gerbec, Johannes K. Sprafke, Fred Wudl, Galen D. Stucky, and Ram Seshadri
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 14) pp:10908
Publication Date(Web):February 13, 2014
DOI:10.1021/am405025n
The Li–S system offers a tantalizing battery for electric vehicles and renewable energy storage due to its high theoretical capacity of 1675 mAh g–1 and its employment of abundant and available materials. One major challenge in this system stems from the formation of soluble polysulfides during the reduction of S8, the active cathode material, during discharge. The ability to deploy this system hinges on the ability to control the behavior of these polysulfides by containing them in the cathode and allowing for further redox. Here, we exploit the high surface areas and good electrical conductivity of mesoporous carbons (MC) to achieve high sulfur utilization while functionalizing the MC with sulfur (S–MC) in order to modify the surface chemistry and attract polysulfides to the carbon material. S–MC materials show enhanced capacity and cyclability trending as a function of sulfur functionality, specifically a 50% enhancement in discharge capacity is observed at high cycles (60–100 cycles). Impedance spectroscopy suggests that the S-MC materials exhibit a lower charge-transfer resistance compared with MC materials which allows for more efficient electrochemistry with species in solution at the cathode. Isothermal titration calorimetry shows that the change in surface chemistry from unfunctionalized to S-functionalized carbons results in an increased affinity of the polysulfide intermediates for the S–MC materials, which is the likely cause for enhanced cyclability.Keywords: enhance polysulfude affinity; isothermal titration calorimetry; lithium−sulfur cell; mesoporous carbons; sulfur cathode; sulfur-functionalized porous carbons
Co-reporter:Lauren M. Misch, Jakoah Brgoch, Alexander Birkel, Thomas E. Mates, Galen D. Stucky, and Ram Seshadri
Inorganic Chemistry 2014 Volume 53(Issue 5) pp:2628-2634
Publication Date(Web):February 11, 2014
DOI:10.1021/ic4030124
We present a rapid microwave-assisted solid-state approach to prepare complex platinum-group metal oxides with the formula La2BaMO5 (M = Pd, Pt). While conventional furnace-based preparations of these compounds take several days and often require oxidizing conditions, the microwave-assisted pathway enables the target compounds to be obtained with high phase purity in about 20 min of reaction time in air without the multiple regrindings that are required of conventional solid-state synthesis. These complex oxides are stable in various atmospheres up to 1000 °C unlike the simple noble metal oxides, which are reduced even at room temperature. Density functional theory-based calculations have been employed to establish the stability of these complex oxides and to understand the electronic structure origins of the stability, most notably the influence of electropositive cations. It is shown that the presence of electropositive ions in the oxide crystal structure “softens” the oxygen anion and results in more covalent (Pd/Pt)–O interactions.
Co-reporter:Claudia Lermer, Megan M. Butala, Bethany R. Lettiere, and Ram Seshadri
Crystal Growth & Design 2014 Volume 14(Issue 9) pp:4526-4530
Publication Date(Web):August 8, 2014
DOI:10.1021/cg500625h
Vapor-phase leaching of Zn and O from complex oxides was performed with the goal of creating mesoporous metal oxides with connected porosity. At elevated temperatures, complex Zn–M–O oxides (M = Nb, Mo, W) can be reduced to yield textured product materials, including reduced M–O oxides, nitrides, and the metal. The nature of the product varies with temperature, time, reducing atmosphere, and the identity of the metal M. M = Nb results in the formation of porous NbO2 without the need for extraneous templates or pore formers. The crystal chemistry of the starting Nb compound is found to influence the nature of the texturing or porosity of the final product. The evolution of morphology is also impacted by the starting Zn:Nb ratio. In the case of the Mo and W compounds, reductive leaching yields the metal or metal nitride (for M = Mo). Morphology change is also observed, and varies for each product phase. An additional interesting aspect of the process is that the reductive leaching occurs in stages, allowing intermediate Zn–M–O compositions with reduced M to be stabilized. The evolution of morphology also appears to be dependent on the initial and final crystal structures.
Co-reporter:Jakoah Brgoch ; Anna J. Lehner ; Michael Chabinyc ;Ram Seshadri
The Journal of Physical Chemistry C 2014 Volume 118(Issue 48) pp:27721-27727
Publication Date(Web):November 7, 2014
DOI:10.1021/jp508880y
Lead halide perovskites have attracted great interest because of rapid improvements in the efficiency of photovoltaics based on these materials. To predict new related functional materials, a good understanding of the correlations between crystal chemistry, electronic structure, and optoelectronic properties is required. Describing the electronic structure of these materials using density functional theory provides a choice of exchange-correlation functionals, including hybrid functionals, and inclusion of spin-orbit coupling, which is critical for the correct description of band gap and absolute band positions (ionization energy). Here, various computational schemes that employ different choices of exchange-correlation and hybrid functionals, and include or exclude spin-orbit coupling were implemented to examine these effects. Using PbI2 as an initial structural model, it is found that standard exchange correlation functionals (PBE) in conjunction with spin-orbit coupling suffice to locate ionization energies efficiently through the use of slab calculations. Band gaps require the use of hybrid functionals carried out on single unit cells and spin-orbit coupling. Polymorphs of alkali metal lead halides, APbI3 (A = Rb, Cs) are examined in the cubic perovskite structure and the reduced dimensional NH4CdCl3/Sn2S3 structure with quasi-two-dimensional connectivity. The somewhat elevated Born effective charges computed for these structures suggest that while the Pb2+ 6s lone-pairs are stereochemically inert, the presence of proximal instabilities could have implications for the functional properties of these materials.
Co-reporter:Kimberly A. See;Jeffrey A. Gerbec;Young-Si Jun;Fred Wudl;Galen D. Stucky;Ram Seshadri
Advanced Energy Materials 2013 Volume 3( Issue 8) pp:1056-1061
Publication Date(Web):
DOI:10.1002/aenm.201300160
Abstract
Conversion reaction cells afford the ability to explore new energy storage paradigms that transcend the dogma of small, low-charge cations essential to intercalative processes. Here we report the use of earth-abundant and green calcium and sulfur in unprecedented conversion reaction Ca–S primary cells. Using S-infiltrated mesoporous carbon (abbreviated S@meso-C) cathodes, we achieve discharge capacities as high as 600 mAh g−1 (S basis) within the geometry Ca|Ca(ClO4)2/CH3CN|S@meso-C, at a discharge rate of C/3.5. The electrolyte system in the Ca–S battery is of paramount importance as the solid electrolyte interface (SEI) formed on the Ca anode limits the capacity and stability of the cell. We determine that 0.5 M Ca(ClO4)2 in CH3CN forms an SEI that advantageously breaks down under anodic bias to allow oxidation of the anode. This same SEI, however, exhibits high impedance which increases over time at open circuit limiting the shelf life of the cell.
Co-reporter:Michael W. Gaultois, Taylor D. Sparks, Christopher K. H. Borg, Ram Seshadri, William D. Bonificio, and David R. Clarke
Chemistry of Materials 2013 Volume 25(Issue 15) pp:2911
Publication Date(Web):May 6, 2013
DOI:10.1021/cm400893e
In this review, we describe the creation of a large database of thermoelectric materials prepared by abstracting information from over 100 publications. The database has over 18 000 data points from multiple classes of compounds, whose relevant properties have been measured at several temperatures. Appropriate visualization of the data immediately allows certain insights to be gained with regard to the property space of plausible thermoelectric materials. Of particular note is that any candidate material needs to display an electrical resistivity value that is close to 1 mΩ cm at 300 K, that is, samples should be significantly more conductive than the Mott minimum metallic conductivity. The Herfindahl–Hirschman index, a commonly accepted measure of market concentration, has been calculated from geological data (known elemental reserves) and geopolitical data (elemental production) for much of the periodic table. The visualization strategy employed here allows rapid sorting of thermoelectric compositions with respect to important issues of elemental scarcity and supply risk.Keywords: datamining; elemental abundance; Herfindahl−Hirschman Index; thermoelectrics;
Co-reporter:Nathan C. George, Andrew J. Pell, Géraldine Dantelle, Katharine Page, Anna Llobet, M. Balasubramanian, Guido Pintacuda, Bradley F. Chmelka, and Ram Seshadri
Chemistry of Materials 2013 Volume 25(Issue 20) pp:3979
Publication Date(Web):October 9, 2013
DOI:10.1021/cm401598n
The oxide garnet Y3Al5O12 (YAG), when substituted with a few percent of the activator ion Ce3+ to replace Y3+, is a luminescent material that is nearly ideal for phosphor-converted solid-state white lighting. The local environments of the small number of substituted Ce3+ ions are known to critically influence the optical properties of the phosphor. Using a combination of powerful experimental methods, the nature of these local environments is determined and is correlated with the macroscopic luminescent properties of Ce-substituted YAG. The rigidity of the garnet structure is established and is shown to play a key role in the high quantum yield and in the resistance toward thermal quenching of luminescence. Local structural probes reveal compression of the Ce3+ local environments by the rigid YAG structure, which gives rise to the unusually large crystal-field splitting, and hence yellow emission. Effective design rules for finding new phosphor materials inferred from the results establish that efficient phosphors require rigid, highly three-dimensionally connected host structures with simple compositions that manifest a low number of phonon modes, and low activator ion concentrations to avoid quenching.Keywords: electron and nuclear magnetic resonance; inorganic phosphors; structure−property relations; white solid-state lighting; X-ray absorption; X-ray and neutron scattering;
Co-reporter:Maosheng Miao, Jakoah Brgoch, Aditi Krishnapriyan, Abby Goldman, Joshua A. Kurzman, and Ram Seshadri
Inorganic Chemistry 2013 Volume 52(Issue 14) pp:8183-8189
Publication Date(Web):July 3, 2013
DOI:10.1021/ic400947p
The “lone” 6s electron pair often plays a key role in determining the structure and physical properties of compounds containing sixth-row elements in their lower oxidation states: Tl+, Pb2+, and Bi3+ with the [Xe]4f145d106s2 electronic configuration. The lone pairs on these ions are associated with reduced structural symmetries, including ferroelectric instabilities and other important phenomena. Here we consider the isoelectronic auride Au– ion with the [Xe]4f145d106s2 electronic configuration. Ab initio density functional theory methods are employed to probe the effect of the 6s lone pair in alkali-metal aurides (KAu, RbAu, and CsAu) with the CsCl structure. The dielectric constants, Born effective charges, and structural instabilities suggest that the 6s lone pair on the Au– anion is stereochemically inert to minor mechanical and electrical perturbation. Pressures greater than 14 GPa, however, lead to reorganization of the electronic structure of CsAu and activate lone-pair involvement and Au–Au interactions in bonding, resulting in a transformation from the cubic CsCl structure type to an orthorhombic Cmcm structure featuring zigzag Au–Au chains.
Co-reporter:Nathan C. George ; Alexander Birkel ; Jakoah Brgoch ; Byung-Chul Hong ; Alexander A. Mikhailovsky ; Katharine Page ; Anna Llobet ;Ram Seshadri
Inorganic Chemistry 2013 Volume 52(Issue 23) pp:13730-13741
Publication Date(Web):November 15, 2013
DOI:10.1021/ic402318k
Structural intricacies of the orange-red nitride phosphor system La3–xCexSi6N11 (0 < x ≤ 3) have been elucidated using a combination of state-of-the art tools, in order to understand the origins of the exceptional optical properties of this important solid-state lighting material. In addition, the optical properties of the end-member (x = 3) compound, Ce3Si6N11, are described for the first time. A combination of synchrotron powder X-ray diffraction and neutron scattering is employed to establish site preferences and the rigid nature of the structure, which is characterized by a high Debye temperature. The high Debye temperature is also corroborated from ab initio electronic structure calculations. Solid-state 29Si nuclear magnetic resonance, including paramagnetic shifts of 29Si spectra, are employed in conjunction with low-temperature electron spin resonance studies to probes of the local environments of Ce ions. Detailed wavelength-, time-, and temperature-dependent luminescence properties of the solid solution are presented. Temperature-dependent quantum yield measurements demonstrate the remarkable thermal robustness of luminescence of La2.82Ce0.18Si6N11, which shows little sign of thermal quenching, even at temperatures as high as 500 K. This robustness is attributed to the highly rigid lattice. Luminescence decay measurements indicate very short decay times (close to 40 ns). The fast decay is suggested to prevent strong self-quenching of luminescence, allowing even the end-member compound Ce3Si6N11 to display bright luminescence.
Co-reporter:Joshua A. Kurzman, Lauren M. Misch and Ram Seshadri
Dalton Transactions 2013 vol. 42(Issue 41) pp:14653-14667
Publication Date(Web):06 Sep 2013
DOI:10.1039/C3DT51818C
The platinum group metals (PGMs) are widely employed as catalysts, especially for the mitigation of automotive exhaust pollutants. The low natural abundance of PGMs and increasing demand from the expanding automotive sector necessitates strategies to improve the efficiency of PGM use. Conventional catalysts typically consist of PGM nanoparticles dispersed on high surface area oxide supports. However, high PGM loadings must be used to counter sintering, ablation, and deactivation of the catalyst such that sufficient activity is maintained over the operating lifetime. An appealing strategy for reducing metal loading is the substitution of PGM ions into oxide hosts: the use of single atoms (ions) as catalytic active sites represents a highly atom-efficient alternative to the use of nanoparticles. This review addresses the crystal chemistry and reactivity of oxide compounds of precious metals that are, or could be relevant to developing an understanding of the role of precious metal ions in heterogeneous catalysis. We review the chemical conditions that facilitate stabilization of the notoriously oxophobic precious metals in oxide environments, and survey complex oxide hosts that have proven to be amenable to reversible redox cycling of PGMs.
Co-reporter:Alexander Birkel, Nicholas A. DeCino, Nathan C. George, Katherine A. Hazelton, Byung-Chul Hong, Ram Seshadri
Solid State Sciences 2013 Volume 19() pp:51-57
Publication Date(Web):May 2013
DOI:10.1016/j.solidstatesciences.2013.02.003
We present a rapid microwave-assisted approach for the preparation of Eu2+-doped orthosilicate phosphors. The preparation method relies on a citrate based sol–gel reaction with subsequent combustion in a domestic microwave oven, in contrast to more conventional solid-state methods. This sol–gel pathway yields phase pure, high quality orthosilicates, in less than 25 min of final heating time. In addition, superior morphology control is achieved employing the sol–gel method compared to solid-state preparations. In order to understand the formation process of the final products, thermogravimetric analyses and temperature-dependent X-ray diffraction data were acquired and compared to the conventional solid-state preparation. The morphology and elemental composition of the obtained luminescent materials were investigated using scanning electron microscopy and energy-dispersive X-ray spectroscopy. The optical properties were elucidated by measuring room-temperature emission and excitation spectra, and the application and efficiency of the obtained phosphors in LED devices was studied.
Co-reporter:Phillip T. Barton;Dr. Y. Daniel Premch;Dr. Philip A. Chater; Ram Seshadri; Matthew J. Rosseinsky
Chemistry - A European Journal 2013 Volume 19( Issue 43) pp:14521-14531
Publication Date(Web):
DOI:10.1002/chem.201301451
Abstract
NiO:Li is an early exemplar for which hole-doping of a correlated insulator gives rise to rich and varied magnetic behavior. It is also an important system from the viewpoint of p-type transparent conducting oxides, and is representative of a large class of materials that have been used in lithium ion batteries, since the end-member compound, LiNiO2, belongs to the class of layered cathode materials. Despite the deceptive structural and compositional simplicity of this system, a complete understanding of its complex magnetic properties has remained elusive. Here a comprehensive investigation of the solid solution LixNi2−xO2, examining samples of precise stoichiometry using a combination of high-resolution synchrotron X-ray powder diffraction and SQUID magnetometry, is provided. The focus is on the interesting region between 0.40<x<1.00 in which the magnetic ordering temperature changes drastically with composition. The magnetism evolves from strong G-type antiferromagnetism of x=0.40 with TN=327 K to robust uncompensated magnetic order at TN=240 K when x is close to 0.7, and to glassy A-type antiferromagnetism of x=1.00 at TN=9 K. This study demonstrates this magnetic behavior is linked to the Li–Ni chemical order that develops from short- to long-range. The interfaces between ordered domains give rise to magnetic exchange bias, which manifests as a shift in the magnetization-field loop for samples with nanoscale coherence lengths (0.54<x<0.66).
Co-reporter:Jakoah Brgoch ; Steven P. DenBaars ;Ram Seshadri
The Journal of Physical Chemistry C 2013 Volume 117(Issue 35) pp:17955-17959
Publication Date(Web):July 15, 2013
DOI:10.1021/jp405858e
Efficient phosphor performance, meaning a high photoluminescence quantum yield (Φ), requires the Ce3+ ion to be hosted in a rigid crystal structure. This is particularly important when the phosphor is operated at slightly elevated temperatures, as is becoming conventional in high brightness, phosphor-converted white solid-state light-emitting diodes. We find the Debye temperature (ΘD) of the undoped host crystal structure, which is readily calculated using ab initio methods within the quasi-harmonic approximation, is a useful proxy for structural rigidity. ΘD is found here to correlate well with the experimentally measured Φ for a number of Ce3+ phosphors. In addition to being rigid, the host lattice must possess a large enough band gap (Eg) that the Ce3+ 4f to 5d transition can occur without interference from a host energy channel. As a sorting diagram for efficient hosts with high Φ, we propose using Eg of the host, also readily calculated with high reliability using hybrid functionals, as one of the axes and ΘD as the other. Such a diagram is used to group the phosphor hosts that we have tested so far. It is interesting to note that a large Eg and high ΘD are often contraindicated, suggesting a challenge in the search for new host structures. While the strategy is applied here for oxides hosts with Ce3+ as the activator ion, it is transferable to other classes of host compounds, and to Eu2+ based phosphors as well.
Co-reporter:Alexander Birkel, Kristin A. Denault, Nathan C. George, Courtney E. Doll, Bathylle Héry, Alexander A. Mikhailovsky, Christina S. Birkel, Byung-Chul Hong, and Ram Seshadri
Chemistry of Materials 2012 Volume 24(Issue 6) pp:1198
Publication Date(Web):February 8, 2012
DOI:10.1021/cm3000238
Ce3+-substituted aluminum garnet compounds of yttrium (Y3Al5O12) and lutetium (Lu3Al5O12)—both important compounds in the generation of (In,Ga)N-based solid state white lighting—have been prepared using a simple microwave heating technique involving the use of a microwave susceptor to provide the initial heat source. Carbon used as the susceptor additionally creates a reducing atmosphere around the sample that helps stabilize the desired luminescent compound. High quality, phase-pure materials are prepared within a fraction of the time and using a fraction of the energy required in a conventional ceramic preparation; the microwave technique allows for a reduction of about 95% in preparation time, making it possible to obtain phase pure, Ce3+-substituted garnet compounds in under 20 min of reaction time. It is estimated that the overall reduction in energy compared with ceramic routes as practised in the lab is close to 99%. Conventionally prepared material is compared with material prepared using microwave heating in terms of structure, morphology, and optical properties, including quantum yield and thermal quenching of luminescence. Finally, the microwave-prepared compounds have been incorporated into light-emitting diode “caps” to test their performance characteristics in a real device, in terms of their photon efficiency and color coordinates.Keywords: inorganic materials; microwave preparation; rare-earth phosphors;
Co-reporter:Mao-Sheng Miao, Joshua A. Kurzman, Nisha Mammen, Shobhana Narasimhan, and Ram Seshadri
Inorganic Chemistry 2012 Volume 51(Issue 14) pp:7569-7578
Publication Date(Web):July 5, 2012
DOI:10.1021/ic3002674
First-principles electronic structure calculations are presented on a variety of Au compounds and species—encompassing a wide range of formal oxidation states, coordination geometries, and chemical environments—in order to understand the potentially systematic behavior in the nature and energetics of d states that are implicated in catalytic activity. In particular, we monitor the position of the d-band center, which has been suggested to signal catalytic activity for reactions such as CO oxidation. We find a surprising absence of any kind of correlation between the formal oxidation state of Au and the position of the d-band center. Instead, we find that the center of the d band displays a nearly linear dependence on the degree of its filling, and this is a general relationship for Au irrespective of the chemistry or geometry of the particular Au compound. Across the compounds examined we find that even small calculated changes in the d-band filling result in a relatively large effect on the position of the d-band center. The results presented here have some important implications for the question of the catalytic activity of Au and indicate that the formal oxidation state is not a determining factor.
Co-reporter:Alexander Birkel, Lucy E. Darago, Alasdair Morrison, Laurianne Lory, Nathan C. George, Alexander A. Mikhailovsky, Christina S. Birkel, Ram Seshadri
Solid State Sciences 2012 Volume 14(Issue 6) pp:739-745
Publication Date(Web):June 2012
DOI:10.1016/j.solidstatesciences.2012.03.014
A rapid and energy efficient microwave assisted solid state preparative route for europium-doped Åkermanite (Ca2MgSi2O7) has been developed. This method reduces the reaction time and energy needed by more than 90%, compared to the preparation carried out in a conventional furnace. The obtained samples are phase pure as has been determined using synchrotron X-ray powder diffraction data and Rietveld analyses. Scanning electron microscopy was employed to investigate the morphology of the microwave prepared compounds whilst energy dispersive X-ray spectroscopy (EDX) was used to verify the elemental composition of the specimens. A systematic investigation of the influence of the utilized microwave setup is presented. Finally, the microwave prepared materials were subject to temperature dependent photoluminescence measurements in order to investigate the thermal quenching of the luminescence.Graphical abstractHighlights► Eu-doped Ca2MgSi2O7 has been prepared by a microwave assisted synthesis. ► The influence of some important experimental parameters has been examined. ► Phase pure materials have been obtained in less than one hour.
Co-reporter:Lauren M. Misch, Joshua A. Kurzman, Alan R. Derk, Young-Il Kim, Ram Seshadri, Horia Metiu, Eric W. McFarland, and Galen D. Stucky
Chemistry of Materials 2011 Volume 23(Issue 24) pp:5432
Publication Date(Web):November 16, 2011
DOI:10.1021/cm202709y
Substituted metal oxides containing ionic species have been attracting a great deal of attention because of their potential ability to reduce the usage of precious metals in heterogeneous catalysts. We investigate Pd-substituted CeO2 for C–H bond activation reactions including the partial oxidation and dry reforming of CH4. This catalyst has been previously studied for CO oxidation, NOx reduction, and the water-gas shift reaction. Pd-substituted CeO2, Ce1–xPdxO2−δ, was prepared as a powder with high surface area and a hollow sphere morphology using ultrasonic spray pyrolysis. The catalysts were extensively characterized using synchrotron X-ray diffraction and other techniques, confirming phase pure samples up to 10 mol % Pd substitution. Ce0.95Pd0.05O2−δ was found to be active for partial oxidation of CH4 around 500 °C and higher. Our studies, including postcatalytic synchrotron diffraction, suggest that the single-phase Ce1–xPdxO2−δ material is not the active species and that catalysis occurs instead over the reduced two-phase Pd0/CeO2. This observation has been further confirmed by verifying the activity of the reduced Pd0/CeO2 catalysts for ethylene hydrogenation, a reaction that is known to require Pd0.Keywords: catalyst; hydrocarbon; nanoparticle;
Co-reporter:Joshua A. Kurzman, Jun Li, Thomas D. Schladt, César R. Parra, Xiaoying Ouyang, Ryan Davis, Jeffrey T. Miller, Susannah L. Scott, and Ram Seshadri
Inorganic Chemistry 2011 Volume 50(Issue 17) pp:8073-8084
Publication Date(Web):July 29, 2011
DOI:10.1021/ic200455a
Complex oxides—containing at least two different cations on crystallographically distinct sites—have recently been shown to display redox cycling of platinum group metals (PGMs), such as Pd; for example, Pd-substituted complex oxides can reversibly extrude metallic Pd under reducing conditions and then reincorporate Pd2+ ions into the lattice under oxidizing conditions. The title compounds, YMn0.5Fe0.5–xPdxO3−δ (0 ≤ x ≤ 0.07) crystallizing in the noncentrosymmetric YMnO3 structure, were prepared using a sol–gel process at 800 °C, and the structures were refined from high-resolution synchrotron X-ray powder diffraction data. Their redox cycling behavior was monitored using synchrotron X-ray diffraction and EXAFS studies. In contrast to the previously studied complex oxide host compounds, YMn0.5Fe0.5–xPdxO3−δ is only modestly tolerant to cycling: repeated redox cycling leads to the formation of PdO, which, on the time-scale of the oxidation cycles, does not reincorporate in the complex oxide lattice. Both oxidized and reduced samples were tested for the oxidation of CO to CO2 under CO-lean conditions. YMn0.5Fe0.5–xPdxO3−δ performs essentially as well as previously studied YFe1–xPdxO3−δ. The CO oxidation light-off characteristics of the hexagonal hosts are very similar to finely dispersed PdO. Despite evidence that Pd is almost fully dispersed as divalent ions in the host lattice, which is presumably accompanied by the concurrent creation of oxygen vacancies (2 Pd2+:1 VO2–), the as-prepared hexagonal materials do not display any significant improvement in catalytic activity as a function of Pd substitution level. This suggests that the corner-connected trigonal bipyramids that characterize this structural family do not enable the transport of oxygen through the bulk of the lattice. The study casts light on factors in the solid-state chemistry of precious metal-substituted complex oxides that influence the efficacy of redox cycling of the precious metal, and catalytic performance.
Co-reporter:Joshua A. Kurzman ; Xiaoying Ouyang ; Won Bin Im ; Jun Li ; Jerry Hu ; Susannah L. Scott ;Ram Seshadri
Inorganic Chemistry 2010 Volume 49(Issue 10) pp:4670-4680
Publication Date(Web):April 15, 2010
DOI:10.1021/ic100486g
La4LiAuO8 and La2BaPdO5, two previously known oxides, are presented as model compounds for examining the role of isolated and immobilized Au3+ and Pd2+ ions in heterogeneous catalysis. Structural characterization, stability, surface composition, and electronic structure of these compounds are presented. These are examined in studies ranging from synchrotron X-ray scattering, including pair distribution function (PDF) and maximum entropy method (MEM) analysis, to density functional calculations of the electronic structures. The exceptional stability displayed by these compounds as verified by thermogravimetric analysis can be attributed to the presence of covalent Au−O and Pd−O interactions revealed in MEM studies, which suggests a criterion for stabilizing these highly oxophobic transition metals in oxide environments. Catalytic testing of the two compounds as heterogeneous catalysts in the oxidation of CO to CO2 are presented. La2BaPdO5 appears to be an effective catalyst for CO oxidation, despite the low surface area of the oxide being used. This is the first time that a fully ordered (rather than doped) Pd2+ oxide had been used to catalyze CO oxidation. La4LiAuO8 on the other hand, is much less effective at catalyzing CO oxidation. Differences in the reactivities of the two compounds are discussed with respect to differences in their density functional electronic structures.
Co-reporter:Yifeng Shi, Bingkun Guo, Serena A. Corr, Qihui Shi, Yong-Sheng Hu, Kevin R. Heier, Liquan Chen, Ram Seshadri and Galen D. Stucky
Nano Letters 2009 Volume 9(Issue 12) pp:4215-4220
Publication Date(Web):September 23, 2009
DOI:10.1021/nl902423a
Highly ordered mesoporous crystalline MoO2 materials with bicontinuous Ia3d mesostructure were synthesized by using phosphomolybdic acid as a precursor and mesoporous silica KIT-6 as a hard template in a 10% H2 atmosphere via nanocasting strategy. The prepared mesoporous MoO2 material shows a typical metallic conductivity with a low resistivity (∼0.01Ω cm at 300 K), which makes it different from all previously reported mesoporous metal oxides materials. Primary test found that mesoporous MoO2 material exhibits a reversible electrochemical lithium storage capacity as high as 750 mA h g−1 at C/20 after 30 cycles, rendering it as a promising anode material for lithium ion batteries.
Co-reporter:Won Bin Im, Katharine Page, Steven P. DenBaars and Ram Seshadri
Journal of Materials Chemistry A 2009 vol. 19(Issue 46) pp:8761-8766
Publication Date(Web):15 Oct 2009
DOI:10.1039/B912793C
The compound LaSr2AlO5 was recently introduced as a competitive Ce3+ host material for blue-pumped yellow phosphors for use in white light emitting diodes. A crucial feature of the crystal structure of LaSr2AlO5 is that La, which is the host site for Ce3+, is located in the 8h positions of the I4/mcm crystal structure, a site equally shared with Sr. While the average crystal structure of LaSr2AlO5 as revealed by Rietveld analysis of laboratory and synchrotron X-ray diffraction data suggests nothing untoward, maximum entropy method analysis of the synchrotron X-ray data reveals the existence of conspicuous non-sphericity of the electron density. Pair distribution function analysis of the data suggests that despite their occupying the same crystallographic site, La and Sr possess distinct coordination environments, and the environment around La is more compact and regular than the environment suggested by the Rietveld refinement of the average structure. The absorption and emission from Ce3+ centers is controlled by the local coordination and symmetry, and the use of powerful new tools in unraveling details of these strengthens the rational search for new phosphors for solid state white lighting.
Co-reporter:Won Bin Im, Natalie N. Fellows, Steven P. DenBaars and Ram Seshadri
Journal of Materials Chemistry A 2009 vol. 19(Issue 9) pp:1325-1330
Publication Date(Web):26 Jan 2009
DOI:10.1039/B818313A
Solid solutions of two isotypic compounds from the end members LaSr2AlO5 and Sr3SiO5 are hosts for Ce3+ activator ions, allowing for careful tuning of the various parameters associated with efficient solid state (blue + yellow =) white lighting. By incorporating this phosphor with an encapsulant on InGaN light-emitting diodes (λmax = 430 nm), we obtain white light with a color rendering index (Ra) between 67 and 70 and color temperatures between 6550 and 7345 K, with relatively high efficacies, suggesting these solid solutions are promising for applications in solid state white lighting.
Co-reporter:Jun Li, Udayshankar G. Singh, Thomas D. Schladt, Judith K. Stalick, Susannah L. Scott and Ram Seshadri
Chemistry of Materials 2008 Volume 20(Issue 20) pp:6567
Publication Date(Web):September 25, 2008
DOI:10.1021/cm801534a
Metastable YFeO3 with the hexagonal YAlO3 structure was obtained by a sol−gel process at 700 °C, using metal nitrate precursors with pH control and the appropriate citric acid to nitrate ratio. Under similar conditions, YFe1−xPdxO3−δ (0 < x ≤ 0.1) compositions were also prepared. The substitution of Fe by Pd stabilizes the YAlO3 structure at higher temperatures. The crystal structures of YFe1−xPdxO3−δ (0 ≤ x ≤ 0.1) were refined by Rietveld analysis of X-ray and neutron powder diffraction data. The parent hexagonal YFeO3 (x = 0) crystallizes in the space group P63/mmc with a = 3.5099(3) Å and c = 11.759(2) Å. The redox-driven mobility of Pd to integrate into the oxide host as ions and to dissociate from it as fcc-Pd nanoparticles was monitored by a combination of X-ray diffraction and X-ray photoelectron spectroscopy. Pd nanoparticles in the reduced samples were detected by scanning backscattered electron microscopy and transmission electron microscopy. The Pd2+-containing materials showed significant low-temperature (near 100 °C) catalytic activity for CO oxidation, comparable to that of highly dispersed PdO/Al2O3, despite their relatively low surface areas.
Co-reporter:Katharine Page, Jun Li, Robert Savinelli, Holly N. Szumila, Jinping Zhang, Judith K. Stalick, Thomas Proffen, Susannah L. Scott, Ram Seshadri
Solid State Sciences 2008 Volume 10(Issue 11) pp:1499-1510
Publication Date(Web):November 2008
DOI:10.1016/j.solidstatesciences.2008.03.018
As possible substitute materials for platinum group metal heterogeneous catalysts, high surface area carbides of the early transition metals Mo and W are of great interest. Here we report nanostructured, high surface area Mo2C and WC prepared by decomposing and carburizing ammonium paramolybdate [(NH4)6Mo7O24·4H2O] and ammonium paratungstate [(NH4)10W12O41·5H2O] in flowing 50%CH4/50%H2. Surface areas as high as 52 m2/g for Mo2C and 24 m2/g for WC were obtained, with both structures crystallizing in structures appropriate for catalytic activity. We have studied these materials using a combination of neutron diffraction Rietveld refinement, X-ray photoelectron spectroscopy, surface area measurements, and scanning transmission electron microscopy. In addition, we have used pair-distribution function (PDF) analysis of the neutron total scattering data as a means of establishing the presence of graphitic carbon in the as-prepared materials.
Co-reporter:Ombretta Masala, Darin Hoffman, Nalini Sundaram, Katharine Page, Thomas Proffen, Gavin Lawes, Ram Seshadri
Solid State Sciences 2006 Volume 8(Issue 9) pp:1015-1022
Publication Date(Web):September 2006
DOI:10.1016/j.solidstatesciences.2006.04.014
Hard/soft CoFe2O4/ZnFe2O4 and soft/hard ZnFe2O4/CoFe2O4 core/shell nanoparticles were prepared by combining high-temperature thermolysis of metal oxide precursors with seed-mediated growth. Magnetic properties of the core/shell nanoparticles were compared to those of individual CoFe2O4 and ZnFe2O4 nanoparticles of similar size prepared by the same method. The structure of the core/shell materials was established using a combination of X-ray and neutron powder diffraction, and transmission electron microscopy. Further evidence for core/shell structure was obtained from magnetic measurements using a SQUID magnetometer. Magnetization measurements as a function of temperature reveal that the core/shell nanoparticles display a single blocking temperature suggesting that the spins of the hard CoFe2O4 and the soft ZnFe2O4 are strongly coupled and respond jointly to changes of temperature and magnetic field. The blocking temperature increases according to the relative amount of hard magnetic material (CoFe2O4) in the nanoparticles in the range of 46–150 K. Magnetic measurements on the nanoparticles as pressed powders and as dispersions in paraffin wax indicate that interparticle interactions significantly influence magnetization and coercivity of the particles, and these must be taken into account before the magnetization behavior of the core/shell structures can be interpreted in terms of coupling between the soft and hard magnetic materials.
Co-reporter:E. S. Toberer;R. Seshadri
Advanced Materials 2005 Volume 17(Issue 18) pp:
Publication Date(Web):3 AUG 2005
DOI:10.1002/adma.200500668
An unusual route to hierarchically porous MnO monoliths is demonstrated. The monoliths are obtained by successive stages of liquid- and vapor-phase leaching. The accompanying Figure shows 10–30 nm sized pores created by the conversion of zinc manganese oxide to manganese oxide. The leaching methodology is general and can be extended to the preparation of a number of porous inorganics.
Co-reporter:Lauren P. Snedeker, Aditi S. Risbud, Ombretta Masala, Jin Ping Zhang, Ram Seshadri
Solid State Sciences 2005 Volume 7(Issue 12) pp:1500-1505
Publication Date(Web):December 2005
DOI:10.1016/j.solidstatesciences.2005.08.020
We describe the all-organic phase conversion of bulk commercial ZnO in the wurtzite modification to sub-30 nm ZnO that we find to be partially in the zinc blende [F4¯3m, a=4.568(3) Åa=4.568(3) Å] modification. The conversion involves refluxing ZnO in 2,4-pentanedione (acetylacetone) at 413 K to form the zinc 2,4-pentanedionate, which is decomposed by heating at 573 K in an appropriate high-temperature solvent such as dibenzylether to form nanophase ZnO. This nanophase, partially zinc blende ZnO can also be obtained in a single step by heating commercial zinc 2,4-pentanedionate in refluxing dibenzylether. Thermodiffractometry suggests that the conversion of zinc blende ZnO to wurtzite ZnO commences near 650 K.
Co-reporter:Ram Seshadri
Current Opinion in Solid State and Materials Science 2005 Volume 9(1–2) pp:1-7
Publication Date(Web):February–April 2005
DOI:10.1016/j.cossms.2006.03.002
The current experimental situation on the occurrence or absence of ferromagnetism in diluted magnetic semiconductors based on wurtzite zinc oxide hosts is presented, focusing mainly on the many recent experiments which have been performed on bulk systems. Numerous reports have suggested that partial (typically less than 10 at.%) substitution of Zn2+ in ZnO by magnetic transition metal (tM) ions, particularly Mn2+ and Co2+, can result in samples with ferromagnetic Curie temperatures above the room temperature. Reports which cast doubt on the very existence of any kind of long range magnetic order in clean samples of Zn1−xtMxO at low transition metal (tM) concentrations are also appearing with increasing frequency. The confusing situation clearly calls for a critical, even subjective position to be taken on this topic. The experimental situation on bulk samples strongly favors the view that in cases when ferromagnetism is found, it is not intrinsic to Zn1−xtMxO.
Co-reporter:K Ramesha, Ram Seshadri
Solid State Sciences 2004 Volume 6(Issue 8) pp:841-845
Publication Date(Web):August 2004
DOI:10.1016/j.solidstatesciences.2004.04.018
We present a simple and convenient route for the preparation of spinel CuCr2Se4 based on the solvothermal reaction of Cu2+ and Cr3+ salts with H2Se in toluene at 330 °C. H2Se required for the reaction is generated in situ by the aromatization of sitosterol by Se. Scanning electron microscopy shows particles in the 25–200 nm range agglomerating into spheroidal assemblies. The particles display the expected ferromagnetic behavior of bulk spinel CuCr2Se4. At 5 K, the saturation magnetization is found to be 2.3 μBper Cr atom, reduced from the expected spin-only value. Results of LMTO calculations on this ferromagnetic compound which is nearly half-metallic are also presented.Graphic
Co-reporter:Ujjal K Gautam, Ram Seshadri
Materials Research Bulletin 2004 Volume 39(4–5) pp:669-676
Publication Date(Web):2 April 2004
DOI:10.1016/j.materresbull.2003.12.003
A new solvothermal route for the preparation of nanocrystals of PbS and PbSe, involving the reaction of lead stearate with sulfur or selenium and tetralin (tetrahydronaphthalene) in toluene solvent is described. Tetralin in the presence of S/Se gives H2S/H2Se and gets aromatized to naphthalene. The nanocrystals have been characterized by powder X-ray diffraction and electron microscopy. Use of surfactant Triton X-100 (polyoxyethylene(10)isooctylphenyl ether) resulted in both nanorods and nanoparticles of PbSe. Capping by citric acid and malonic acid reduce the particle sizes to less than 10 nm.
Co-reporter:M. Dinamani, P.Vishnu Kamath, Ram Seshadri
Solid State Sciences 2003 Volume 5(Issue 5) pp:805-810
Publication Date(Web):May 2003
DOI:10.1016/S1293-2558(03)00085-2
We report the use of electrogeneration of acid to electrochemically precipitate crystalline strontium sulfate coatings from EDTA stabilized solutions onto stainless steel anodes. This is the first report of the electrodeposition of strontium sulfate. A study of the deposited SrSO4 by microscopy and diffraction suggests that by changing conditions of current and time, control over crystallite habit and crystallite orientation with respect to the substrate can be achieved.Graphic
Co-reporter:Udayshankar G. Singh, Jun Li, Joseph W. Bennett, Andrew M. Rappe, Ram Seshadri, Susannah L. Scott
Journal of Catalysis (25 July 2007) Volume 249(Issue 2) pp:349-358
Publication Date(Web):25 July 2007
DOI:10.1016/j.jcat.2007.04.023
Perovskite BaCeO3 materials with low levels of substitution of Pd(II) on the Ce site and a corresponding number of oxygen vacancies were prepared by a high-temperature synthesis method. Although their surface areas are low (ca. 1.0 m2 g−1), their low-temperature (<200 °C) activity for CO oxidation is comparable to that of highly dispersed PdO/Al2O3. When the doped perovskites are reduced extensively in H2, causing extrusion of Pd(0) from the lattice, their catalytic activity declines dramatically. Consequently, activity is attributed to the presence of cationic Pd(II) in the perovskite lattice. Density functional theory was used to investigate the atomic and electronic character of the structures containing oxygen vacancies. Both experimental and theoretical evidence support a catalytic mechanism involving labilization of lattice and surface oxygen by cationic Pd(II).
Co-reporter:Geneva Laurita, Douglas H. Fabini, Constantinos C. Stoumpos, Mercouri G. Kanatzidis and Ram Seshadri
Chemical Science (2010-Present) 2017 - vol. 8(Issue 8) pp:NaN5635-5635
Publication Date(Web):2017/06/16
DOI:10.1039/C7SC01429E
Hybrid halide perovskites combine ease of preparation and relatively abundant constituent elements with fascinating photophysical properties. Descriptions of the chemical and structural drivers of the remarkable properties have often focused on the potential role of the dynamic order/disorder of the molecular A-site cations. We reveal here a key aspect of the inorganic framework that potentially impacts the electronic, thermal, and dielectric properties. The temperature evolution of the X-ray pair distribution functions of hybrid perovskites ABX3 [A+ = CH3NH3 (MA) or CH(NH2)2 (FA); B2+ = Sn or Pb; X− = Br, or I] in their cubic phases above 300 K reveals temperature-activated displacement (off-centering) of the divalent group 14 cations from their nominal, centered sites. This symmetry-lowering distortion phenomenon, previously dubbed emphanisis in the context of compounds such as PbTe, is attributed to Sn2+ and Pb2+ lone pair stereochemistry. Of the materials studied here, the largest displacements from the center of the octahedral sites are found in tin iodides, a more moderate effect is found in lead bromides, and the weakest effect is seen in lead iodides. The A-site cation appears to play a role as well, with the larger FA resulting in greater off-centering for both Sn2+ and Pb2+. Dynamic off-centering, which is concealed within the framework of traditional Bragg crystallography, is proposed to play a key role in the remarkable defect-tolerant nature of transport in these semiconductors via its effect on the polarizability of the lattice. The results suggest a novel chemical design principle for future materials discovery.
Co-reporter:Joshua A. Kurzman, Lauren M. Misch and Ram Seshadri
Dalton Transactions 2013 - vol. 42(Issue 41) pp:NaN14667-14667
Publication Date(Web):2013/09/06
DOI:10.1039/C3DT51818C
The platinum group metals (PGMs) are widely employed as catalysts, especially for the mitigation of automotive exhaust pollutants. The low natural abundance of PGMs and increasing demand from the expanding automotive sector necessitates strategies to improve the efficiency of PGM use. Conventional catalysts typically consist of PGM nanoparticles dispersed on high surface area oxide supports. However, high PGM loadings must be used to counter sintering, ablation, and deactivation of the catalyst such that sufficient activity is maintained over the operating lifetime. An appealing strategy for reducing metal loading is the substitution of PGM ions into oxide hosts: the use of single atoms (ions) as catalytic active sites represents a highly atom-efficient alternative to the use of nanoparticles. This review addresses the crystal chemistry and reactivity of oxide compounds of precious metals that are, or could be relevant to developing an understanding of the role of precious metal ions in heterogeneous catalysis. We review the chemical conditions that facilitate stabilization of the notoriously oxophobic precious metals in oxide environments, and survey complex oxide hosts that have proven to be amenable to reversible redox cycling of PGMs.
Co-reporter:Won Bin Im, Katharine Page, Steven P. DenBaars and Ram Seshadri
Journal of Materials Chemistry A 2009 - vol. 19(Issue 46) pp:NaN8766-8766
Publication Date(Web):2009/10/15
DOI:10.1039/B912793C
The compound LaSr2AlO5 was recently introduced as a competitive Ce3+ host material for blue-pumped yellow phosphors for use in white light emitting diodes. A crucial feature of the crystal structure of LaSr2AlO5 is that La, which is the host site for Ce3+, is located in the 8h positions of the I4/mcm crystal structure, a site equally shared with Sr. While the average crystal structure of LaSr2AlO5 as revealed by Rietveld analysis of laboratory and synchrotron X-ray diffraction data suggests nothing untoward, maximum entropy method analysis of the synchrotron X-ray data reveals the existence of conspicuous non-sphericity of the electron density. Pair distribution function analysis of the data suggests that despite their occupying the same crystallographic site, La and Sr possess distinct coordination environments, and the environment around La is more compact and regular than the environment suggested by the Rietveld refinement of the average structure. The absorption and emission from Ce3+ centers is controlled by the local coordination and symmetry, and the use of powerful new tools in unraveling details of these strengthens the rational search for new phosphors for solid state white lighting.
Co-reporter:Won Bin Im, Natalie N. Fellows, Steven P. DenBaars and Ram Seshadri
Journal of Materials Chemistry A 2009 - vol. 19(Issue 9) pp:NaN1330-1330
Publication Date(Web):2009/01/26
DOI:10.1039/B818313A
Solid solutions of two isotypic compounds from the end members LaSr2AlO5 and Sr3SiO5 are hosts for Ce3+ activator ions, allowing for careful tuning of the various parameters associated with efficient solid state (blue + yellow =) white lighting. By incorporating this phosphor with an encapsulant on InGaN light-emitting diodes (λmax = 430 nm), we obtain white light with a color rendering index (Ra) between 67 and 70 and color temperatures between 6550 and 7345 K, with relatively high efficacies, suggesting these solid solutions are promising for applications in solid state white lighting.
.jpg)
















![[2,2'-Bipyridine]-5,5'-dicarbonitrile](/data/chemimg/1593000/1802-29-5.png)
![[2,2'-Bipyridine]-5,5'-dicarbonitrile](/data/chemimg/1593000/1802-29-5_b.png)