Co-reporter:Lei Yu, Minhao Huang, Tianfeng Xu, Linjiang Tong, Xiao-e Yan, Zhang Zhang, Yong Xu, Caihong Yun, Hua Xie, Ke Ding, Xiaoyun Lu
European Journal of Medicinal Chemistry 2017 Volume 126(Volume 126) pp:
Publication Date(Web):27 January 2017
DOI:10.1016/j.ejmech.2016.12.006
•Optimization of pyrido[2,3-d]pyrimidin-7-ones with improved pharmacokinetic properties.•Compound 9s potently suppressed EGFRL858R/T790M kinase and H1975 cells.•Compound 9s exhibited moderate plasma exposure and an oral bioavailability value of 16%.Structural optimization of pyrido[2,3-d]pyrimidin-7-ones was conducted to yield a series of new selective EGFRT790M inhibitors with improved pharmacokinetic properties. One of the most promising compound 9s potently suppressed EGFRL858R/T790M kinase and inhibited the proliferation of H1975 cells with IC50 values of 2.0 nM and 40 nM, respectively. The compound dose-dependently induced reduction of the phosphorylation of EGFR and downstream activation of ERK in NCIH1975 cells. It also exhibited moderate plasma exposure after oral administration and an oral bioavailability value of 16%. Compound 9s may serve as a promising lead compound for further drug discovery overcoming the acquired resistance of non-small cell lung cancer (NSCLC) patients.Structural optimization of pyrido[2,3-d]pyrimidin-7-ones yielded new selective EGFRT790M inhibitors. Compound 9s exhibited good pharmacokinetic properties with F value of 16%, and inhibited EGFRL858R/T790M kinase and H1975 cells with IC50 values of 2.0 and 40 nM, respectively.Download high-res image (191KB)Download full-size image
Co-reporter:Zhen Wang; Huan Bian; Sergio G. Bartual; Wenting Du; Jinfeng Luo; Hu Zhao; Shasha Zhang; Cheng Mo; Yang Zhou; Yong Xu; Zhengchao Tu; Xiaomei Ren; Xiaoyun Lu; Rolf A. Brekken; Libo Yao; Alex N. Bullock; Jin Su
Journal of Medicinal Chemistry 2016 Volume 59(Issue 12) pp:5911-5916
Publication Date(Web):May 24, 2016
DOI:10.1021/acs.jmedchem.6b00140
The structure-based design of 1, 2, 3, 4-tetrahydroisoquinoline derivatives as selective DDR1 inhibitors is reported. One of the representative compounds, 6j, binds to DDR1 with a Kd value of 4.7 nM and suppresses its kinase activity with an IC50 value of 9.4 nM, but it is significantly less potent for a panel of 400 nonmutated kinases. 6j also demonstrated reasonable pharmacokinetic properties and a promising oral therapeutic effect in a bleomycin-induced mouse pulmonary fibrosis model.
Co-reporter:Xiaokai Li, Jiayi Shen, Li Tan, Zhang Zhang, Donglin Gao, Jinfeng Luo, Huimin Cheng, Xiaoping Zhou, Jie Ma, Ke Ding, Xiaoyun Lu
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 12) pp:2760-2763
Publication Date(Web):15 June 2016
DOI:10.1016/j.bmcl.2016.04.076
B-RafV600E was an effective target for the treatment of human cancers. Based on a pan-Raf inhibitor TAK-632, a series of N-(4-aminopyridin-2-yl)amide derivatives were designed as novel B-RafV600E inhibitors. Detailed structure–activity studies of the compounds revealed that most of the compounds displayed potent enzymatic activity against B-RafV600E, and good selectivity over B-RafWT. One of the most promising compound 4l exhibited potent inhibitory activity with an IC50 value of 38 nM for B-rafV600E, and displayed antiproliferative activities against colo205 and HT29 cells with IC50 values of 0.136 and 0.094 μM, respectively. It also displayed good selectivity on both enzymatic and cellular assays over B-RafWT. These inhibitors may serve as lead compounds for further developing novel B-RafV600E inhibitors as anticancer drugs.
Co-reporter:Yi Zou, Jianhu Xiao, Zhengchao Tu, Yingyi Zhang, Kun Yao, Minghao Luo, Ke Ding, Yihua Zhang, Yisheng Lai
Bioorganic & Medicinal Chemistry Letters 2016 26(13) pp: 3052-3059
Publication Date(Web):1 July 2016
DOI:10.1016/j.bmcl.2016.05.014
A series of novel 4,5,6-trisubstituted pyrimidines were designed as potent covalent Bruton’s tyrosine kinase (BTK) inhibitors based on the structure of ibrutinib by using a ring-opening strategy. Among these derivatives, compound I1 exhibited the most potent inhibitory activity with an IC50 value of 0.07 μM. The preliminary structure–activity relationship was discussed and the primary amino group at the C-4 position of pyrimidine was crucial for maintaining BTK activity. Furthermore, molecular dynamics simulations and binding free energy calculations were performed for three inhibitor-BTK complexes to determine the probable binding model, which provided a comprehensive guide for further structural modification and optimization.
Co-reporter:Yupeng Li; Xiaoyun Lu; Xiaomei Ren
Journal of Medicinal Chemistry 2015 Volume 58(Issue 8) pp:3287-3301
Publication Date(Web):January 8, 2015
DOI:10.1021/jm5012319
Discoidin domain receptors (DDRs) are members of the transmembrane receptor tyrosine kinase (RTK) superfamily which are distinguished from others by the presence of a discoidin motif in the extracellular domain and their utilization of collagens as internal ligands. Two types of DDRs, DDR1 and DDR2, have been identified with distinct expression profiles and ligand specificities. These DDRs play important roles in the regulation of fundamental cellular process, such as proliferation, survival, differentiation, adhesion, and matrix remodeling. They have also been closely linked to a number of human diseases, including various fibrotic disorders, atherosclerosis, and cancer. As a consequence, DDRs have been considered as novel potential molecular targets for drug discovery and increasing efforts are being devoted to the identification of new small molecule inhibitors targeting the receptors. In this review, we offer a contemporary overview on the discovery of DDRs inhibitors and their potential medical application for the treatment of cancer and inflammation related disorders.
Co-reporter:Yingjun Li, Huimin Cheng, Zhang Zhang, Xiaoxi Zhuang, Jinfeng Luo, Huoyou Long, Yang Zhou, Yong Xu, Rana Taghipouran, Dan Li, Adam Patterson, Jeff Smaill, Zhengchao Tu, Donghai Wu, Xiaomei Ren, and Ke Ding
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 5) pp:543
Publication Date(Web):March 18, 2015
DOI:10.1021/acsmedchemlett.5b00039
A series of N-(3-ethynyl-2,4-difluorophenyl)sulfonamides were identified as new selective Raf inhibitors. The compounds potently inhibit B-RafV600E with low nanomolar IC50 values and exhibit excellent target specificity in a selectivity profiling investigation against 468 kinases. They strongly suppress proliferation of a panel of human cancer cell lines and patient-derived melanoma cells with B-RafV600E mutation while being significantly less potent to the cells with B-RafWT. The compounds also display favorable pharmacokinetic properties with a preferred example (3s) demonstrating significant in vivo antitumor efficacy in a xenograft mouse model of B-RafV600E mutated Colo205 human colorectal cancer cells, supporting it as a promising lead compound for further anticancer drug discovery.Keywords: B-Raf; colon cancer; kinase inhibitor; melanoma; targeted therapy
Co-reporter:Shilin Xu, Liufeng Mao, Ping Ding, Xiaoxi Zhuang, Yang Zhou, Lei Yu, Yingxue Liu, Tao Nie, Tingting Xu, Yong Xu, Jinsong Liu, Jeff Smaill, Xiaomei Ren, Donghai Wu, Ke Ding
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 13) pp:3751-3760
Publication Date(Web):1 July 2015
DOI:10.1016/j.bmc.2015.03.082
The estrogen-related receptor γ (ERRγ) is a potential molecular target for the development of small molecules to stimulate the adipose browning process, which may represent a novel attractive strategy to treat obesity related disorders. The receptor possesses a very small ligand binding cavity and therefore identification of small molecule ERRγ modulators is a considerable challenge. We have successfully designed and synthesized a series of 1-benzyl-4-phenyl-1H-1,2,3-triazoles and demonstrated that they improve the transcriptional functions of ERRγ, potently elevating both the mRNA levels and the protein levels of ERRγ downstream targets. One of the most promising compounds, 4-(1-(4-iso-propylbenzyl)-1H-1,2,3-triazol-4-yl)benzene-1,2-diol (2e) was further shown to directly bind with the ERRγ ligand binding domain (ERRγ-LBD) in an isothermal calorimetric (ITC) assay and to thermally stabilize ERRγ-LBD protein by increasing its melting temperature (Tm) as demonstrated by circular dichroism (CD) spectroscopy. Furthermore, 2e potently stimulates the adipocyte browning process and induces mitochondrial biogenesis both in vitro and in vivo, suggesting the considerable therapeutic potential of this compound for the treatment of obesity and related disorders.
Co-reporter:Jian Tang, Bangxing Wang, Tian Wu, Junting Wan, Zhengchao Tu, Moses Njire, Baojie Wan, Scott G. Franzblauc, Tianyu Zhang, Xiaoyun Lu, and Ke Ding
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 7) pp:814
Publication Date(Web):June 11, 2015
DOI:10.1021/acsmedchemlett.5b00176
A series of pyrazolo[1,5-a]pyridine-3-carboxamide derivatives were designed and synthesized as new anti-Mycobacterium tuberculosis (Mtb) agents. The compounds exhibit promising in vitro potency with nanomolar MIC values against the drug susceptive H37Rv strain and a panel of clinically isolated multidrug-resistant Mtb (MDR-TB) strains. One of the representative compounds (5k) significantly reduces the bacterial burden in an autoluminescent H37Ra infected mouse model, suggesting its promising potential to be a lead compound for future antitubercular drug discovery.Keywords: Antitubercular agent; H37Rv; pyrazolo[1,5-a]pyridine; structure−activity relationship; tuberculosis
Co-reporter:Shingpan Chan, Kun Han, Rong Qu, Linjiang Tong, Yingjun Li, Zhang Zhang, Huimin Cheng, Xiaoyun Lu, Adam Patterson, Jeff Smaill, Xiaomei Ren, Jian Ding, Hua Xie, Ke Ding
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 19) pp:4277-4281
Publication Date(Web):1 October 2015
DOI:10.1016/j.bmcl.2015.07.089
IGF1R amplification was recently implied to be related to the secondary acquired resistance against the 2nd or 3rd generation EGFR inhibitor therapies. We have successfully identified a series of 2,4-diarylamino-pyrimidines as new IGF1R/EGFRL858R/T790M co-targeting agents. One of the most promising compounds 8g potently inhibits both kinases with low nanomolar IC50 values, but is significantly less potent in inhibiting the wild type EGFR. The compound also displays a good kinase selectivity profile against a panel of 468 kinases. Moreover, 8g strongly suppresses the proliferation of CO-1686-resistant H1975-IGF1R cancer cells, suggesting its promising potential as a new lead compound for future anticancer drug discovery.
Co-reporter:Xiaoyun Lu, Zhang Zhang, Xiaomei Ren, Xiaofeng Pan, Deping Wang, Xiaoxi Zhuang, Jingfeng Luo, Rongmin Yu, Ke Ding
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 17) pp:3458-3463
Publication Date(Web):1 September 2015
DOI:10.1016/j.bmcl.2015.07.006
A series of pyrimidine alkynyl derivatives were designed and synthesized as new Bcr-Abl inhibitors by hybriding the structural moieties from GNF-7, ponatinib and nilotinib. One of the most potent compounds 4e strongly suppresses Bcr-AblWT and Bcr-AblT315I kinase with IC50 values of 5.0 and 9.0 nM, and inhibits the proliferation of K562 and murine Ba/F3 cells ectopically expressing Bcr-AblT315I cells with IC50 values of 2 and 50 nM, respectively. It also displays good pharmacokinetics properties with an oral bioavailability of 35.3% and T1/2 value of 48.7 h, and demonstrates significantly suppression on tumor growth in xenografted mice of K562 and Ba/F3 cells expressing Bcr-AblT315I. These inhibitors may serve as lead compounds for further developing new anticancer drugs overcoming the clinically acquired resistance against current Bcr-Abl inhibitors.A series of pyrimidine alkynyl derivatives were designed and synthesized as new Bcr-Abl inhibitors by hybriding the structural moieties from GNF-7, ponatinib and nilotinib. Compound 4e is one of the most potent compounds.
Co-reporter:Huimin Cheng ; Yu Chang ; Lianwen Zhang ; Jinfeng Luo ; Zhengchao Tu ; Xiaoyun Lu ; Qingwen Zhang ; Jibu Lu ; Xiaomei Ren
Journal of Medicinal Chemistry 2014 Volume 57(Issue 6) pp:2692-2703
Publication Date(Web):March 3, 2014
DOI:10.1021/jm500007h
Epidermal growth factor receptor (EGFR) amplification has been demonstrated to be critical for the inherent and/or acquired resistance against current B-RafV600E inhibitor therapy for melanoma and colorectal cancer patients. We describe the discovery and structure–activity relationship study of a series of 1H-pyrazolo[3,4-b]pyridine-5-carboxamide analogues as novel dual inhibitors of EGFR and B-RafV600E mutant. One of the most promising compounds, 6a, potently inhibited both of the kinases with IC50 values of 8.0 and 51 nM, respectively. The compound also strongly suppressed the proliferation of a panel of intrinsic and acquired resistant melanoma and/or colorectal cancer cells harboring overexpressed EGFR with submicromolar IC50 values. Further mechanism investigation revealed that 6a could sustainably inhibit the activation of the MAPK path way in the resistant SK-MEL-28 PR30 melanoma cancer cells and WiDr colorectal cancer cells with EGFR amplification. Our results support the hypothesis that the EGFR/B-RafV600E dual inhibition might be a tractable strategy to overcome the intrinsic and acquired resistance of melanoma and/or colorectal cancers against the current B-RafV600E inhibitor therapy.
Co-reporter:Xiaoyun Lu
Journal of Medicinal Chemistry 2014 Volume 57(Issue 4) pp:1167-1169
Publication Date(Web):February 5, 2014
DOI:10.1021/jm500178r
The study by Huang et al. is an excellent example of rational structure-based and lipophilic-efficiency optimization of crizotinib (Xalkori) aimed at novel ALK inhibitors capable of overcoming clinically acquired resistance against the current drug in NSCLC patients. One of the most promising new compounds, 8e, displayed subnanomolar potency against ALKWT and a panel of the crizotinib-resistant mutants and demonstrated robust in vivo antitumor efficacy. The super suppressing potency of this compound on ROS1 kinase may also indicate its great potential to overcome the resistance associated with the most recently identified ROS1 mutation.
Co-reporter:F Yang, R Deng, X-J Qian, S-H Chang, X-Q Wu, J Qin, G-K Feng, K Ding and X-F Zhu
Cell Death & Disease 2014 5(3) pp:e1114
Publication Date(Web):2014-03-01
DOI:10.1038/cddis.2014.43
The serine/threonine kinase AKT is generally accepted as a promising anticancer therapeutic target. However, the relief of feedback inhibition and enhancement of other survival pathways often attenuate the anticancer effects of AKT inhibitors. These compensatory mechanisms are very complicated and remain poorly understood. In the present study, we found a novel 2-pyrimidyl-5-amidothiazole compound, DC120, as an ATP competitive AKT kinase inhibitor that suppressed proliferation and induced apoptosis in liver cancer cells both in vitro and in vivo. DC120 blocked the phosphorylation of downstream molecules in the AKT signal pathway in dose- and time-dependent manners both in vitro and in vivo. However, unexpectedly, DC120 activated mammalian target of rapamycin complex 1 (mTORC1) pathway that was suggested by increased phosphorylation of 70KD ribosomal protein S6 kinase (P70S6K) and eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1). The activated mTORC1 signal was because of increase of intracellular Ca2+ via Ca2+/calmodulin (CaM)/ signaling to human vacuolar protein sorting 34 (hVps34) upon AKT inhibition. Meanwhile, DC120 attenuated the inhibitory effect of AKT on CRAF by decreasing phosphorylation of CRAF at Ser259 and thus activated the mitogen-activated protein kinase (MAPK) pathway. The activation of the mTORC1 and MAPK pathways by DC120 was not mutually dependent, and the combination of DC120 with mTORC1 inhibitor and/or MEK inhibitor induced significant apoptosis and growth inhibition both in vitro and in vivo. Taken together, the combination of AKT, mTORC1 and/or MEK inhibitors would be a promising therapeutic strategy for liver cancer treatment.
Co-reporter:Wenna Wang, Dexin Kong, Huimin Cheng, Li Tan, Zhang Zhang, Xiaoxi Zhuang, Huoyou Long, Yang Zhou, Yong Xu, Xiaohong Yang, Ke Ding
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 17) pp:4250-4253
Publication Date(Web):1 September 2014
DOI:10.1016/j.bmcl.2014.07.035
Emerging drug resistance and other drawbacks limit tubulin inhibitors’ therapeutic applications and developing novel tubulin inhibitors still attracts intensive efforts. We describe the discovery and structure–activity relationship study of a series of benzimidazole-2-urea derivatives as novel β tubulin inhibitors. The representative compound 6o potently suppressed the proliferation of a panel of human cancer cells (NCI-H460, Colo205, K562, A431, HepG2, Hela, MDA-MB-435S) with IC50 values of 0.040, 0.050, 0.006, 0.026, 1.774, 0.452 and 0.052 μM, respectively. Compound 6o obviously inhibited NCI-H460 spindles formation and induced cell cycle arrest at G2/M phase at 0.10 μM. Computational study suggested that 6o interacts with β tubulin in a novel binding mode. Our results suggested that benzimidazole-2-urea derivatives might be promising tubulin inhibitors with novel binding mode for further development.
Co-reporter:Shilin Xu ; Tianfeng Xu ; Lianwen Zhang ; Zhang Zhang ; Jinfeng Luo ; Yingxue Liu ; Xiaoyun Lu ; Zhengchao Tu ; Xiaomei Ren
Journal of Medicinal Chemistry 2013 Volume 56(Issue 21) pp:8803-8813
Publication Date(Web):October 14, 2013
DOI:10.1021/jm4012388
Structural optimization of a series of 2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidinyl compounds, potential new irreversible EGFR inhibitors, was performed to improve pharmacokinetic properties of the compounds. This led to compound 2v with improved aqueous solubility and good pharmacokinetic properties which at the nanomolar level potently inhibits gefitinib-resistant EGFRL858R/T790M kinase and displays strong antiproliferative activity against H1975 nonsmall cell lung cancer cells. The new inhibitor also shows promising antitumor efficacy in a murine EGFRL858R/T790M-driven H1975 xenograft model without effect on body weight. These studies provide new lead compounds for further development of drugs for treatment of gefitinib-resistant nonsmall cell lung cancer patients.
Co-reporter:Wei Zhou ; Xiaofeng Liu ; Zhengchao Tu ; Lianwen Zhang ; Xin Ku ; Fang Bai ; Zhenjiang Zhao ; Yufang Xu ; Ke Ding ;Honglin Li
Journal of Medicinal Chemistry 2013 Volume 56(Issue 20) pp:7821-7837
Publication Date(Web):September 22, 2013
DOI:10.1021/jm401045n
The EGFR T790M variant is an important mutation, resulting in approximately 50% of the clinically acquired resistance to approved EGFR inhibitors. Starting with a previously reported pyrimidine-based EGFR inhibitor, a novel pteridin-7(8H)-one scaffold with a high 3D similarity was found and transformed into irreversible inhibitors of the EGFR T790M mutant. The most potent compounds, 3q and 3x, exhibited excellent enzyme inhibitory activities, with subnanomolar IC50 values for both the wild-type and T790M/L858R double mutant EGFRs, as well as potent cellular antiproliferative activities against both gefitinib-sensitive and -resistant cancer cell lines. The in vivo antitumor efficacy study demonstrated that compound 3x significantly inhibited tumor growth and induced tumor stasis in an EGFR-T790M/L858R-driven human nonsmall-cell lung cancer xenograft mouse model. This work demonstrated the utility of this sophisticated computational design strategy for fast 3D scaffold hopping with competitive bioactivities to meet an important clinical need.
Co-reporter:Chun Han ; Zhangjian Huang ; Chao Zheng ; Ledong Wan ; Lianwen Zhang ; Sixun Peng ; Ke Ding ; Hongbin Ji ; Jide Tian ;Yihua Zhang
Journal of Medicinal Chemistry 2013 Volume 56(Issue 11) pp:4738-4748
Publication Date(Web):May 13, 2013
DOI:10.1021/jm400463q
A series of hybrids (12a–k) from (phenylsulfonyl)furoxan and anilinopyrimidine were synthesized and biologically evaluated as epidermal growth factor receptor (EGFR) inhibitors for intervention of non-small-cell lung cancer (NSCLC). Compound 12k exhibited strong and selective EGFR L858R/T790M inhibitory activity (IC50 = 0.047 μM) and displayed antiproliferative effects on EGFR mutation NSCLC cell lines HCC827 (del E746_A750) and H1975 (L858R/T790M) with IC50 values of 0.007 and 0.029 μM, respectively. Additionally, 12k released high levels of NO in H1975 cells but not in normal human cells, and its activity was diminished by pretreatment with a NO scavenger. Furthermore, 12k induced apoptosis of H1975 and HCC827 cells more strongly than WZ4002 (1), inhibited EGFR downstream signaling in H1975 cells, and suppressed the nuclear factor-κB activation in H1975 cells, while 1 had no significant effects under the same conditions. Finally, 12k substantially inhibited tumor growth in an H1975 xenograft mouse model. Overall, 12k might be a promising candidate for the treatment of NSCLC.
Co-reporter:Xiaomei Ren ; Xiaofen Pan ; Zhang Zhang ; Deping Wang ; Xiaoyun Lu ; Yupeng Li ; Donghai Wen ; Huoyou Long ; Jinfeng Luo ; Yubing Feng ; Xiaoxi Zhuang ; Fengxiang Zhang ; Jianqi Liu ; Fang Leng ; Xingfen Lang ; Yang Bai ; Miaoqin She ; Zhengchao Tu ; Jingxuan Pan
Journal of Medicinal Chemistry 2013 Volume 56(Issue 3) pp:879-894
Publication Date(Web):January 9, 2013
DOI:10.1021/jm301581y
Bcr-AblT315I mutation-induced imatinib resistance remains a major challenge for clinical management of chronic myelogenous leukemia (CML). Herein, we report GZD824 (10a) as a novel orally bioavailable inhibitor against a broad spectrum of Bcr-Abl mutants including T315I. It tightly bound to Bcr-AblWT and Bcr-AblT315I with Kd values of 0.32 and 0.71 nM, respectively, and strongly inhibited the kinase functions with nanomolar IC50 values. The compound potently suppressed proliferation of Bcr-Abl-positive K562 and Ku812 human CML cells with IC50 values of 0.2 and 0.13 nM, respectively. It also displayed good oral bioavailability (48.7%), a reasonable half-life (10.6 h), and promising in vivo antitumor efficacy. It induced tumor regression in mouse xenograft tumor models driven by Bcr-AblWT or the mutants and significantly improved the survival of mice bearing an allograft leukemia model with Ba/F3 cells harboring Bcr-AblT315I. GZD824 represents a promising lead candidate for development of Bcr-Abl inhibitors to overcome acquired imatinib resistance.
Co-reporter:Mingshan Gao ; Lei Duan ; Jinfeng Luo ; Lianwen Zhang ; Xiaoyun Lu ; Yan Zhang ; Zhang Zhang ; Zhengchao Tu ; Yong Xu ; Xiaomei Ren
Journal of Medicinal Chemistry 2013 Volume 56(Issue 8) pp:3281-3295
Publication Date(Web):March 22, 2013
DOI:10.1021/jm301824k
Discoidin domain receptor 1 (DDR1) is an emerging potential molecular target for new anticancer drug discovery. We have discovered a series of 3-(2-(pyrazolo[1,5-a]pyrimidin-6-yl) ethynyl)benzamides that are selective and orally bioavailable DDR1 inhibitors. The two most promising compounds (7rh and 7rj) inhibited the enzymatic activity of DDR1, with IC50 values of 6.8 and 7.0 nM, respectively, but were significantly less potent in suppressing the kinase activities of DDR2, Bcr-Abl, and c-Kit. Further study revealed that 7rh bound with DDR1 with a Kd value of 0.6 nM, while it was significantly less potent to the other 455 kinases tested. The S(35) and S(10) selectivity scores of 7rh were 0.035 and 0.008, respectively. The compounds also potently inhibited the proliferation of cancer cells expressing high levels of DDR1 and strongly suppressed cancer cell invasion, adhesion, and tumorigenicity. Preliminary pharmacokinetic studies suggested that they possessed good PK profiles, with oral bioavailabilities of 67.4% and 56.2%, respectively.
Co-reporter:Shilin Xu ; Xiaoxi Zhuang ; Xiaofen Pan ; Zhang Zhang ; Lei Duan ; Yingxue Liu ; Lianwen Zhang ; Xiaomei Ren
Journal of Medicinal Chemistry 2013 Volume 56(Issue 11) pp:4631-4640
Publication Date(Web):May 8, 2013
DOI:10.1021/jm4003928
Estrogen-related receptor α is a potential candidate target for therapeutic treatment of breast cancer. We describe the discovery and structure–activity relationship study of a series of 1-phenyl-4-benzoyl-1H-1,2,3-triazoles as novel suppressors of ERRα transcriptional functions. The most promising compound, 2-aminophenyl-(1-(3-isopropylphenyl)-1H-1,2,3-triazol-4-yl)methanone (14n), potently suppressed the transcriptional functions of ERRα with IC50 = 0.021 μM in a cell-based reporter gene assay and also decreased both the mRNA levels and the protein levels of ERRα and the downstream targets. This compound inhibited the proliferation and migration of breast cancer cells with high level of ERRα. Preliminary pharmacokinetic studies suggested that it possessed a good pharmacokinetic profile with an oral bioavailability of 71.8%. The compounds may serve as novel small molecule probes for further validation of ERRα as a molecular target for anticancer drug development.
Co-reporter:Chun Han, Zhangjian Huang, Chao Zheng, Ledong Wan, Yisheng Lai, Sixun Peng, Ke Ding, Hongbin Ji, Yihua Zhang
European Journal of Medicinal Chemistry 2013 Volume 66() pp:82-90
Publication Date(Web):August 2013
DOI:10.1016/j.ejmech.2013.05.026
•NO donating anilinopyrimidines were synthesized and evaluated as EGFR inhibitors.•Compounds 10f–h exhibited potent inhibitory activity against EGFR L858R/T790M.•Compounds 10f–h displayed antiproliferative activity as potent as WZ4002.•10h produced high levels of NO in H1975 cells but not in normal human cells.•10h inhibited the EGFR activation and downstream signaling in H1975 cells.To search for potent nitric oxide (NO) donating epidermal growth factor receptor (EGFR) inhibitors, a series of phenylsulfonylfuroxan-based anilinopyrimidines 10a–h were synthesized and biologically evaluated. Compounds 10f–h exhibited potent inhibitory activity against EGFR L858R/T790M and were as potent as WZ4002 in inhibition of H1975 cells harboring EGFR L858R/T790M. Additionally, 10h produced high levels of NO in H1975 cells but not in normal human cells, and its antiproliferative activity was diminished by hemoglobin, an NO scavenger. Furthermore, 10h inhibited EGFR activation and downstream signaling in H1975 cells. These results suggest that the strong antiproliferative activity of 10h could be attributed to the synergic effects of high levels of NO production and inhibition of EGFR and downstream signaling in the cancer cells.Compound 10h exhibited potent inhibitory activity against EGFR mutant L858R/T790M and antiproliferative effects on both gefitinib-resistant H1975 (harboring EGFR L858R/T790M) and gefitinib-sensitive HCC827 (bearing EGFR del E746_A750) cells.
Co-reporter:Tianfeng Xu;Lianwen Zhang;Shilin Xu;Dr. Chao-Yie Yang;Jinfeng Luo;Fang Ding;Xiaoyun Lu;Yingxue Liu;Zhengchao Tu;Shiliang Li; Duanqing Pei;Qian Cai; Honglin Li;Xiaomei Ren; Shaomeng Wang; Ke Ding
Angewandte Chemie International Edition 2013 Volume 52( Issue 32) pp:8387-8390
Publication Date(Web):
DOI:10.1002/anie.201302313
Co-reporter:Tianfeng Xu;Lianwen Zhang;Shilin Xu;Dr. Chao-Yie Yang;Jinfeng Luo;Fang Ding;Xiaoyun Lu;Yingxue Liu;Zhengchao Tu;Shiliang Li; Duanqing Pei;Qian Cai; Honglin Li;Xiaomei Ren; Shaomeng Wang; Ke Ding
Angewandte Chemie 2013 Volume 125( Issue 32) pp:8545-8548
Publication Date(Web):
DOI:10.1002/ange.201302313
Co-reporter:Jiajie Yan, Fengtao Zhou, Dongguang Qin, Tong Cai, Ke Ding, and Qian Cai
Organic Letters 2012 Volume 14(Issue 5) pp:1262-1265
Publication Date(Web):February 15, 2012
DOI:10.1021/ol300114w
A simple and efficient approach for the synthesis of [1,2,3]triazolo[1,5-a]quinoxalin-4(5H)-ones is described. The methodology is based on a tandem reaction of 1-(2-haloaryl)propiolamides with sodium azide through a [3 + 2] azide–alkyne cycloaddition and intramolecular Ullmann-type C–N coupling process.
Co-reporter:Huimin Cheng ; Junting Wan ; Meng-I Lin ; Yingxue Liu ; Xiaoyun Lu ; Jinsong Liu ; Yong Xu ; Jianxin Chen ; Zhengchao Tu ; Yih-Shyun E. Cheng
Journal of Medicinal Chemistry 2012 Volume 55(Issue 5) pp:2144-2153
Publication Date(Web):February 14, 2012
DOI:10.1021/jm2013503
The influenza virus nucleoprotein (NP) is an emerging target for anti-influenza drug development. Nucleozin (1) and its closely related derivatives had been identified as NP inhibitors displaying anti-influenza activity. Utilizing 1 as a lead molecule, we successfully designed and synthesized a series of 1H-1,2,3-triazole-4-carboxamide derivatives as new anti-influenza A agents. One of the most potent compounds, 3b, inhibited the replication of various H3N2 and H1N1 influenza A virus strains with IC50 values ranging from 0.5 to 4.6 μM. Compound 3b also strongly inhibited the replication of H5N1 (RG14), amantidine-resistant A/WSN/33 (H1N1), and oseltamivir-resistant A/WSN/1933 (H1N1, 274Y) virus strains with IC50 values in sub-μM ranges. Further computational studies and mechanism investigation suggested that 3b might directly target influenza virus A nucleoprotein to inhibit its nuclear accumulation.
Co-reporter:Shaohua Chang ; Lianwen Zhang ; Shilin Xu ; Jinfeng Luo ; Xiaoyun Lu ; Zhang Zhang ; Tianfeng Xu ; Yingxue Liu ; Zhengchao Tu ; Yong Xu ; Xiaomei Ren ; Meiyu Geng ; Jian Ding ; Duanqing Pei
Journal of Medicinal Chemistry 2012 Volume 55(Issue 6) pp:2711-2723
Publication Date(Web):February 16, 2012
DOI:10.1021/jm201591k
The EGFRT790M mutant contributes approximately 50% to clinically acquired resistance against gefitinib or erlotinib. However, almost all the single agent clinical trials of the second generation irreversible EGFR inhibitors appear inadequate to overcome the EGFRT790M-related resistance. We have designed and synthesized a series of 2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidinyl derivatives as novel EGFR inhibitors. The most potent compounds, 2q and 2s, inhibited the enzymatic activities of wild-type and mutated EGFRs, with IC50 values in subnanomolar ranges, including the T790M mutants. The kinase inhibitory efficiencies of the compounds were further validated by Western blot analysis of the activation of EGFR and downstream signaling in cancer cells harboring different mutants of EGFR. The compounds also strongly inhibited the proliferation of H1975 non small cell lung cancer cells bearing EGFRL858R/T790M, while being significantly less toxic to normal cells. Moreover, 2s displayed promising anticancer efficacy in a human NSCLC (H1975) xenograft nude mouse model.
Co-reporter:Yupeng Li ; Mengjie Shen ; Zhang Zhang ; Jinfeng Luo ; Xiaofen Pan ; Xiaoyun Lu ; Huoyou Long ; Donghai Wen ; Fengxiang Zhang ; Fang Leng ; Yingjun Li ; Zhengchao Tu ; Xiaomei Ren
Journal of Medicinal Chemistry 2012 Volume 55(Issue 22) pp:10033-10046
Publication Date(Web):October 22, 2012
DOI:10.1021/jm301188x
A series of 3-(1H-1,2,3-triazol-1-yl)benzamide derivatives were designed and synthesized as new Bcr-Abl inhibitors by using combinational strategies of bioisosteric replacement, scaffold hopping, and conformational constraint. The compounds displayed significant inhibition against a broad spectrum of Bcr-Abl mutants including the gatekeeper T315I and p-loop mutations, which are associated with disease progression in CML. The most potent compounds 6q and 6qo strongly inhibited the kinase activities of Bcr-AblWT and Bcr-AblT315I with IC50 values of 0.60, 0.36 and 1.12, 0.98 nM, respectively. They also potently suppressed the proliferation of K562, KU812 human CML cells, and a panel of murine Ba/F3 cells ectopically expressing either Bcr-AblWT or any of a panel of other Bcr-Abl mutants that have been shown to contribute to clinical acquired resistance, including Bcr-AblT315I, with IC50 values in low nanomolar ranges. These compounds may serve as lead compounds for further development of new Bcr-Abl inhibitors capable of overcoming clinical acquired resistance against imatinib.
Co-reporter:Shaohua Chang, Zhang Zhang, Xiaoxi Zhuang, Jinfeng Luo, Xianwen Cao, Honglin Li, Zhengchao Tu, Xiaoyun Lu, Xiaomei Ren, Ke Ding
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 2) pp:1208-1212
Publication Date(Web):15 January 2012
DOI:10.1016/j.bmcl.2011.11.080
A new series of 2-substituted thiazole carboxamides were identified as potent pan inhibitors against all three isoforms of Akt (Akt1, Akt2 and Akt3) by systematic optimization of weak screening hit N-(1-amino-3-phenylpropan-2-yl)-2-phenylthiazole-5-carboxamide (1). One of the most potent compounds, 5m, inhibited the kinase activities of Akt1, Akt2 and Akt3 with IC50 values of 25, 196 and 24 nM, respectively. The compound also potently inhibited the phosphorylation of downstream MDM2 and GSK3β proteins, and displayed strongly antiproliferative activity in prostate cancer cells. The inhibitors might serve as lead compounds for further development of novel effective anticancer agents.
Co-reporter:Shilin Xu, Lianwen Zhang, Shaohua Chang, Jinfeng Luo, Xiaoyun Lu, Zhengchao Tu, Yingxue Liu, Zhang zhang, Yong Xu, Xiaomei Ren and Ke Ding
MedChemComm 2012 vol. 3(Issue 9) pp:1155-1159
Publication Date(Web):29 Jun 2012
DOI:10.1039/C2MD20078C
A series of 5,8-dioxo-pyrimido[4,5-e][1,4]diazepine derivatives were designed and synthesized as new inhibitors against wild-type EGFR and a panel of mutants, including the clinical resistance related T790M mutants. One of the most potent compounds 2l inhibited all forms of EGFR evaluated with low nM IC50 values. It also strongly suppressed the proliferation of H1975 and HCC827 non-small cell lung cancer cells with IC50 values of 69 and 71 nM, respectively.
Co-reporter:Jin Zhang;Jing Zhou;Xiaomei Ren;Yanyan Diao;Honglin Li
Investigational New Drugs 2012 Volume 30( Issue 2) pp:490-507
Publication Date(Web):2012 April
DOI:10.1007/s10637-010-9577-1
Receptor tyrosine kinases (RTKs) modulate a variety of cellular events, including cell proliferation, differentiation, mobility and apoptosis. In addition, RTKs have been validated as targets for cancer therapies. Microtubules are another class of proven targets for many clinical anticancer drugs. Here, we report that 1-(4-chloro-3-(trifluoromethyl) phenyl)-3-(2-cyano-4-hydroxyphenyl)urea (D181) functions as both a receptor tyrosine kinase inhibitor and a tubulin polymerization enhancer. D181 displayed potent inhibitory activities against a panel of RTKs, including Flt3, VEGFR, cKit, FGFR1 and PDGFRβ. D181 also enhanced tubulin polymerization and modified the secondary structure of tubulin proteins to disrupt their dynamic instability. Because of synergistic cooperation, D181 strongly inhibited the proliferation of various cancer cell lines, induced LoVo cell cycle arrest in the G1 and M phases and suppressed tumor growth in nude mice bearing human LoVo and HT29 xenografts. Our studies have provided a new, promising lead compound and novel clues for multi-target anticancer drug design and development.
Co-reporter:Qian Cai, Fengtao Zhou, Tianfeng Xu, Liangbing Fu, and Ke Ding
Organic Letters 2011 Volume 13(Issue 2) pp:340-343
Publication Date(Web):December 17, 2010
DOI:10.1021/ol102826f
A novel copper-catalyzed tandem reaction of 1-(2-iodoaryl)-2-yn-1-ones with isocyanides is described. The reaction is through a formal [3 + 2] cycloaddition/coupling tandem process and leads to efficient formation of 4-oxo-indeno[1,2-b]pyrroles.
Co-reporter:Lijie Peng ; Xuefei Gao ; Lei Duan ; Xiaomei Ren ; Donghai Wu
Journal of Medicinal Chemistry 2011 Volume 54(Issue 21) pp:7729-7733
Publication Date(Web):September 29, 2011
DOI:10.1021/jm200976s
The nuclear estrogen-related receptor α (ERRα) plays a central role in the regulation of expression of the genes involved in mitochondrial biogenesis and oxidative metabolism. We have successfully identified a series of pyrido[1,2-a]pyrimidin-4-ones as new agonists enhancing the transcriptional functions of ERRα. The compounds potently elevated the mRNA levels and the protein levels of ERRα downstream targets. Consequently, the compounds improved the glucose and fatty acid uptake in C2C12 muscle cells.
Co-reporter:Deping Wang, Zhang Zhang, Xiaoyun Lu, Yubing Feng, Kun Luo, Jirong Gan, Liu Yingxue, Junting Wan, Xiang Li, Fengxiang Zhang, Zhengchao Tu, Qian Cai, Xiaomei Ren, Ke Ding
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 7) pp:1965-1968
Publication Date(Web):1 April 2011
DOI:10.1016/j.bmcl.2011.02.029
A series of 2,4-disubstituted thiazole derivatives were designed and synthesized as new Bcr/Abl inhibitors by hybriding the structural moieties from FDA approved imatinib, nilotinib and dasatinib. The new inhibitors strongly suppressed the activity of Bcr/Abl kinase and potently inhibited the proliferation of K562 and KU812 leukemia cancer cells. Compound 4i displayed comparable potency with that of nilotinib in both biochemical kinase assay and cancer cell growth inhibition assay. These inhibitors might serve as lead compounds for further developing new anticancer drugs.
Co-reporter:Xiujie Liu, Xiaoli Huang, Wanhua Lin, Dongye Wang, Yanyan Diao, Honglin Li, Xiaoyan Hui, Yu Wang, Aimin Xu, Donghai Wu, Ding Ke
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 10) pp:2949-2952
Publication Date(Web):15 May 2011
DOI:10.1016/j.bmcl.2011.03.063
a-FABP is indespensible in inflammation and may serve as a new potential drug target for inflammation related diseases. We have successfully designed and synthesized a series of aromatic substituted pyrazoles as new human a-FABP inhibitors. The compounds strongly bound to the hydrophobic binding pocket of a-FABP, while showed significantly lower binding affinities to the closely related homologue protein h-FABP. The most potent and selective compound 5g bound to a-FABP with an apparent Ki value below 1.0 nM, while did not inhibit h-FABP at 50 μM and thus represents one of the most potent and selective a-FABP inhibitors to date. The strong binding capacity of these inhibitors was further validated by their effective blockade of inflammatory responses as determined by the production of pro-inflammatory cytokines upon LPS stimulation. Compound 5g may serve as a lead compound for developing new effective therapeutic agent for prevention and treatment of atherosclerosis, type 2 diabetes and other inflammatory and metabolic related diseases.
Co-reporter:Mingshan Gao;Xiujie Liu;Xianyang Wang;Qian Cai
Chinese Journal of Chemistry 2011 Volume 29( Issue 6) pp:1199-1204
Publication Date(Web):
DOI:10.1002/cjoc.201190223
Abstract
A general synthesis of 1-aryl-1-H-indazoles from o-halogenated aryl aldehydes or ketones and aryl hydrazines was described. This protocol included an intermolecular condensation and a ligand-free copper-catalyzed intramolecular Ullmann-type coupling reaction. This method was applied to a wide range of substrates to produce the indazole products in good yields.
Co-reporter:Lijie Peng, Lei Duan, Xiaofeng Liu, Mengjie Shen, Yingjun Li, Jiajie Yan, Honglin Li, Ke Ding
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 15) pp:4457-4461
Publication Date(Web):1 August 2011
DOI:10.1016/j.bmcl.2011.06.009
A series of α-glutamic acid scaffold based 4-(benzamido)-4-(1,3,4-oxadiazol-2-yl) butanoic acids were designed and synthesized as new ADAMTS inhibitors. The compounds dose-dependently inhibited the enzymatic activities of ADAMTS-4 and ADAMTS-5. One of the most active compound 2h potently inhibited ADAMTS-4 and ADAMTS-5 with IC50 values of 1.2 and 0.8 μM, respectively. These inhibitors may serve as new lead compounds for further development of therapeutics to treat osteoarthritis.
Co-reporter:Fengtao Zhou;Dr. Ke Ding;Dr. Qian Cai
Chemistry - A European Journal 2011 Volume 17( Issue 44) pp:12268-12271
Publication Date(Web):
DOI:10.1002/chem.201102459
Co-reporter:Qian Cai, Zhengqiu Li, Jiajia Wei, Liangbin Fu, Chengyong Ha, Duanqing Pei and Ke Ding
Organic Letters 2010 Volume 12(Issue 7) pp:1500-1503
Publication Date(Web):March 2, 2010
DOI:10.1021/ol1002225
A new and efficient method for the synthesis of aza-fused polycyclic quinolines (e.g., benzimidazo[1,2-a]quinolines) is described. This protocol includes an intermolecular condensation followed by a copper-catalyzed intramolecular C−N coupling reaction. The method is applied to a wide range of 2-iodo, 2-bromo, and 2-chloro aryl aldehyde substrates to yield the aza-fused polycyclic quinolines in good yields.
Co-reporter:Xiaomei Ren, Lei Duan, Qiang He, Zhang Zhang, Yi Zhou, Donghai Wu, Jingxuan Pan, Duanqing Pei, and Ke Ding
ACS Medicinal Chemistry Letters 2010 Volume 1(Issue 9) pp:454
Publication Date(Web):September 7, 2010
DOI:10.1021/ml100146z
Inhibition of the signal transducer and activator of transcription 3 (STAT3) signaling pathway has been considered a novel therapeutic strategy to treat human cancers with constitutively active STAT3. In this study, we report the identification of niclosamide, an FDA-approved anthelmintic drug, as a new small-molecule inhibitor of the STAT3 signaling pathway. This compound potently inhibited the activation and transcriptional function of STAT3 and consequently induced cell growth inhibition, apoptosis, and cell cycle arrest of cancer cells with constitutively active STAT3. Our study provides a new promising lead compound with a salicylic amide scaffold for the development of STAT3 pathway inhibitors as novel molecularly targeted anticancer drugs.Keywords (keywords): apoptosis; cell cycle arrest; inhibitor; niclosamide; STAT3 signaling pathway
Co-reporter:Qian Cai, Zhengqiu Li, Jiajia Wei, Chengyong Ha, Duanqing Pei and Ke Ding
Chemical Communications 2009 (Issue 48) pp:7581-7583
Publication Date(Web):04 Nov 2009
DOI:10.1039/B918345K
A straightforward synthesis of indole-2-carboxylic esters was developed through a ligand-free copper-catalysed condensation/coupling/deformylation cascade process from 2-halo aryl aldehydes or ketones with ethyl isocyanoacetate. The reactions proceeded well for most of the 2-iodo-, bromo-, and chloro-subtrates under room temperature or mild conditions.
Co-reporter:Deping Wang and Ke Ding
Chemical Communications 2009 (Issue 14) pp:1891-1893
Publication Date(Web):20 Feb 2009
DOI:10.1039/B821212K
2-Pyridinyl β-ketones were identified as new efficient ligands for CuI-catalysed N-arylation of aliphatic amines at room temperature with great selectivity and substrate scope tolerance.
Co-reporter:Deping Wang;Qian Cai
Advanced Synthesis & Catalysis 2009 Volume 351( Issue 11-12) pp:1722-1726
Publication Date(Web):
DOI:10.1002/adsc.200900327
Abstract
The copper(I) bromide/1-(5,6,7,8-tetrahydroquinolin-8-yl)-2-methylpropan-1-one (CuBr-L3) combination catalyzed the cross-coupling reactions between aryl or heteroaryl halides and aqueous ammonia with high yields to produce primary aromatic or heteroaromatic amines at room temperature or under mild conditions.
Co-reporter:Jing Zhou, Lei Duan, Huaming Chen, Xiaomei Ren, Zhang Zhang, Fengtao Zhou, Jinsong Liu, Duanqing Pei, Ke Ding
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 17) pp:5091-5094
Publication Date(Web):1 September 2009
DOI:10.1016/j.bmcl.2009.07.044
2-Piperazinyl naphthoquinones (2) and 2-piperidinyl naphthoquinones (3) were designed and synthesized as new cytotoxic and apoptosis inducing agents by utilizing the anti-parasite drug atovaquone as lead compound. Several compounds displayed significantly improved cytotoxic activities against a panel of cancer cell lines than that of atovaquone. These compounds also induced apoptosis through activating pro-apoptotic caspases 9 and 3.2-Piperazinyl and 2-piperidinyl analogues of anti-parasite atovaquone were designed and synthesized as potent cytotoxic and apoptosis inducing agents.
Co-reporter:Qian Cai, Zhengqiu Li, Jiajia Wei, Chengyong Ha, Duanqing Pei and Ke Ding
Chemical Communications 2009(Issue 48) pp:NaN7583-7583
Publication Date(Web):2009/11/04
DOI:10.1039/B918345K
A straightforward synthesis of indole-2-carboxylic esters was developed through a ligand-free copper-catalysed condensation/coupling/deformylation cascade process from 2-halo aryl aldehydes or ketones with ethyl isocyanoacetate. The reactions proceeded well for most of the 2-iodo-, bromo-, and chloro-subtrates under room temperature or mild conditions.
Co-reporter:Deping Wang and Ke Ding
Chemical Communications 2009(Issue 14) pp:NaN1893-1893
Publication Date(Web):2009/02/20
DOI:10.1039/B821212K
2-Pyridinyl β-ketones were identified as new efficient ligands for CuI-catalysed N-arylation of aliphatic amines at room temperature with great selectivity and substrate scope tolerance.

-](http://img.cochemist.com/ccimg/341100/341001-38-5.png)
-](http://img.cochemist.com/ccimg/341100/341001-38-5_b.png)


![1-[2-(dimethylamino)ethyl]-1h-pyrazol-4-amine](http://img.cochemist.com/ccimg/1153000/1152939-98-4.png)
![1-[2-(dimethylamino)ethyl]-1h-pyrazol-4-amine](http://img.cochemist.com/ccimg/1153000/1152939-98-4_b.png)
![7H-Pyrrolo[2,3-d]pyrimidine, 4-(4-nitrophenoxy)-](http://img.cochemist.com/ccimg/924700/924655-26-5.png)
![7H-Pyrrolo[2,3-d]pyrimidine, 4-(4-nitrophenoxy)-](http://img.cochemist.com/ccimg/924700/924655-26-5_b.png)








![D-glycero-L-gulo-Nonitol,4,8-anhydro-3,5-dideoxy-9-C-[[(2S)-2-hydroxy-2-[(2R,5R,6R)-tetrahydro-2-methoxy-5,6-dimethyl-4-methylene-2H-pyran-2-yl]acetyl]amino]-5,5-dimethyl-6-O-methyl-7,9-O-methylene-,(9S)-](http://img.cochemist.com/ccimg/115200/115185-92-7.png)
![D-glycero-L-gulo-Nonitol,4,8-anhydro-3,5-dideoxy-9-C-[[(2S)-2-hydroxy-2-[(2R,5R,6R)-tetrahydro-2-methoxy-5,6-dimethyl-4-methylene-2H-pyran-2-yl]acetyl]amino]-5,5-dimethyl-6-O-methyl-7,9-O-methylene-,(9S)-](http://img.cochemist.com/ccimg/115200/115185-92-7_b.png)




![Ethanone, 1-[4-chloro-2-(methylthio)-5-pyrimidinyl]-](http://img.cochemist.com/ccimg/66200/66116-82-3.png)
![Ethanone, 1-[4-chloro-2-(methylthio)-5-pyrimidinyl]-](http://img.cochemist.com/ccimg/66200/66116-82-3_b.png)
![2-CHLORO-N-[1-(2-FLUOROBENZOYL)PIPERIDIN-4-YL]ACETAMIDE](http://img.cochemist.com/ccimg/56100/56090-54-1.png)
![2-CHLORO-N-[1-(2-FLUOROBENZOYL)PIPERIDIN-4-YL]ACETAMIDE](http://img.cochemist.com/ccimg/56100/56090-54-1_b.png)
![5-BROMO-3-METHOXY-1H-PYRAZOLO[3,4-B]PYRIDINE](http://img.cochemist.com/ccimg/1363400/1363381-82-1.png)
![5-BROMO-3-METHOXY-1H-PYRAZOLO[3,4-B]PYRIDINE](http://img.cochemist.com/ccimg/1363400/1363381-82-1_b.png)


![(S)-tert-Butyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate](http://img.cochemist.com/ccimg/1268600/1268524-70-4.png)
![(S)-tert-Butyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate](http://img.cochemist.com/ccimg/1268600/1268524-70-4_b.png)


![Methyl 1H-pyrazolo[3,4-b]pyridine-5-carboxylate](http://img.cochemist.com/ccimg/1196200/1196156-42-9.png)
![Methyl 1H-pyrazolo[3,4-b]pyridine-5-carboxylate](http://img.cochemist.com/ccimg/1196200/1196156-42-9_b.png)
![Ethyl 5-((tert-butoxycarbonyl)amino)pyrazolo[1,5-a]pyridine-3-carboxylate](http://img.cochemist.com/ccimg/1101200/1101120-33-5.png)
![Ethyl 5-((tert-butoxycarbonyl)amino)pyrazolo[1,5-a]pyridine-3-carboxylate](http://img.cochemist.com/ccimg/1101200/1101120-33-5_b.png)
![3H-Pyrazolo[3,4-b]pyridin-3-one, 5-bromo-1,2-dihydro-](/data/chemimg/349300/1086064-44-9.png)
![3H-Pyrazolo[3,4-b]pyridin-3-one, 5-bromo-1,2-dihydro-](/data/chemimg/349300/1086064-44-9_b.png)


![3-iodo-4-methyl-N-[3-(4-methylimidazol-1-yl)-5-(trifluoromethyl)phenyl]benzamide](http://img.cochemist.com/ccimg/927000/926922-18-1.png)
![3-iodo-4-methyl-N-[3-(4-methylimidazol-1-yl)-5-(trifluoromethyl)phenyl]benzamide](http://img.cochemist.com/ccimg/927000/926922-18-1_b.png)
![Propanedioic acid, [1-[(4-ethylphenyl)amino]ethylidene]-, diethyl ester](http://img.cochemist.com/ccimg/886600/886588-89-2.png)
![Propanedioic acid, [1-[(4-ethylphenyl)amino]ethylidene]-, diethyl ester](http://img.cochemist.com/ccimg/886600/886588-89-2_b.png)
![ethyl 5-chloropyrazolo[1,5-a]pyridine-3-carboxylate](http://img.cochemist.com/ccimg/886400/886364-13-2.png)
![ethyl 5-chloropyrazolo[1,5-a]pyridine-3-carboxylate](http://img.cochemist.com/ccimg/886400/886364-13-2_b.png)
![1H-Pyrazolo[3,4-b]pyridine, 5-bromo-3-methyl-](/data/chemimg/906800/885223-65-4.png)
![1H-Pyrazolo[3,4-b]pyridine, 5-bromo-3-methyl-](/data/chemimg/906800/885223-65-4_b.png)
![1H-PYRAZOLO[3,4-B]PYRIDINE, 5-BROMO-3-PHENYL-](http://img.cochemist.com/ccimg/882100/882029-94-9.png)
![1H-PYRAZOLO[3,4-B]PYRIDINE, 5-BROMO-3-PHENYL-](http://img.cochemist.com/ccimg/882100/882029-94-9_b.png)
![1H-Pyrazolo[3,4-b]pyridine,5-bromo-3-iodo-1-[[2-(trimethylsilyl)ethoxy]methyl]-](http://img.cochemist.com/ccimg/875800/875781-19-4.png)
![1H-Pyrazolo[3,4-b]pyridine,5-bromo-3-iodo-1-[[2-(trimethylsilyl)ethoxy]methyl]-](http://img.cochemist.com/ccimg/875800/875781-19-4_b.png)
![1H-PYRROLO[2,3-D]PYRIMIDINE, 4-(2-FLUORO-4-NITROPHENOXY)-](http://img.cochemist.com/ccimg/875800/875764-99-1.png)
![1H-PYRROLO[2,3-D]PYRIMIDINE, 4-(2-FLUORO-4-NITROPHENOXY)-](http://img.cochemist.com/ccimg/875800/875764-99-1_b.png)
![Furo[2,3-d]pyrimidine, 4-chloro-5-phenyl-](http://img.cochemist.com/ccimg/873400/873306-25-3.png)
![Furo[2,3-d]pyrimidine, 4-chloro-5-phenyl-](http://img.cochemist.com/ccimg/873400/873306-25-3_b.png)

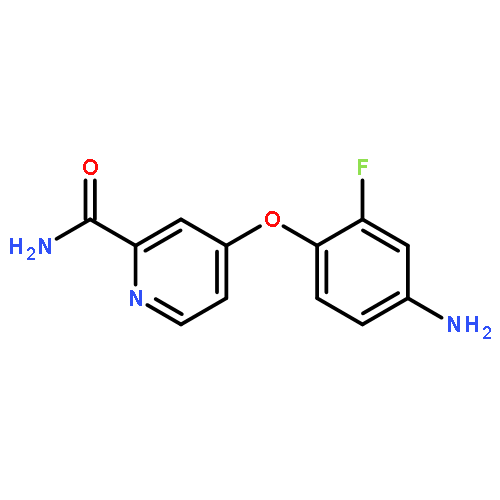
![Benzenamine, 4-[(2-chloro-4-pyridinyl)oxy]-3-fluoro-](http://img.cochemist.com/ccimg/868800/868733-61-3.png)
![Benzenamine, 4-[(2-chloro-4-pyridinyl)oxy]-3-fluoro-](http://img.cochemist.com/ccimg/868800/868733-61-3_b.png)
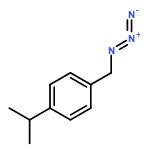
![Benzenamine, 3-[(4-methyl-1-piperazinyl)methyl]-5-(trifluoromethyl)-](http://img.cochemist.com/ccimg/853300/853296-94-3.png)
![Benzenamine, 3-[(4-methyl-1-piperazinyl)methyl]-5-(trifluoromethyl)-](http://img.cochemist.com/ccimg/853300/853296-94-3_b.png)

![[1,1'-BIPHENYL]-4-METHANAMINE, 4'-(TRIFLUOROMETHOXY)-](http://img.cochemist.com/ccimg/473000/472964-25-3.png)
![[1,1'-BIPHENYL]-4-METHANAMINE, 4'-(TRIFLUOROMETHOXY)-](http://img.cochemist.com/ccimg/473000/472964-25-3_b.png)
![Benzenamine, 4-[(6,7-dimethoxy-4-quinolinyl)oxy]-3-fluoro-](http://img.cochemist.com/ccimg/347200/347161-74-4.png)
![Benzenamine, 4-[(6,7-dimethoxy-4-quinolinyl)oxy]-3-fluoro-](http://img.cochemist.com/ccimg/347200/347161-74-4_b.png)
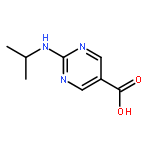
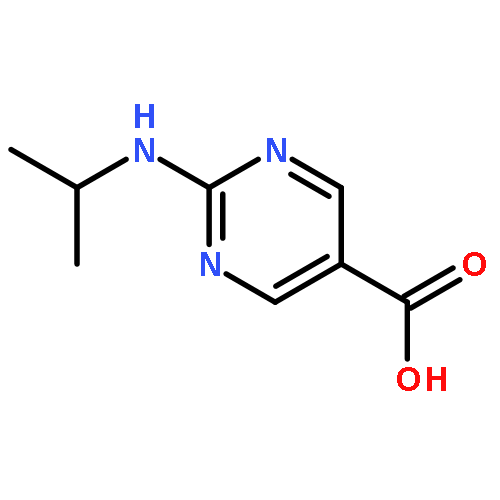
amino]benzoic acid](http://img.cochemist.com/ccimg/342400/342398-30-5.png)
amino]benzoic acid](http://img.cochemist.com/ccimg/342400/342398-30-5_b.png)
![3-[(1-BENZYL-1H-BENZIMIDAZOL-2-YL)THIO]-PROPANOIC ACID](http://img.cochemist.com/ccimg/303200/303132-83-4.png)
![3-[(1-BENZYL-1H-BENZIMIDAZOL-2-YL)THIO]-PROPANOIC ACID](http://img.cochemist.com/ccimg/303200/303132-83-4_b.png)




![Benzoic acid, 2-[[bis(phenylmethyl)amino]carbonyl]-](http://img.cochemist.com/ccimg/211000/210984-34-2.png)
![Benzoic acid, 2-[[bis(phenylmethyl)amino]carbonyl]-](http://img.cochemist.com/ccimg/211000/210984-34-2_b.png)
![Benzoic acid, 4-[2-[(trifluoroacetyl)amino]ethyl]-, methyl ester](http://img.cochemist.com/ccimg/181600/181514-22-7.png)
![Benzoic acid, 4-[2-[(trifluoroacetyl)amino]ethyl]-, methyl ester](http://img.cochemist.com/ccimg/181600/181514-22-7_b.png)












![Pyrazolo[1,5-a]pyridine-3-carboxylic acid, 2,7-dimethyl-](http://img.cochemist.com/ccimg/143900/143803-85-4.png)
![Pyrazolo[1,5-a]pyridine-3-carboxylic acid, 2,7-dimethyl-](http://img.cochemist.com/ccimg/143900/143803-85-4_b.png)


![Furo[2,3-d]pyrimidin-4(1H)-one, 5-phenyl-](http://img.cochemist.com/ccimg/141800/141764-36-5.png)
![Furo[2,3-d]pyrimidin-4(1H)-one, 5-phenyl-](http://img.cochemist.com/ccimg/141800/141764-36-5_b.png)





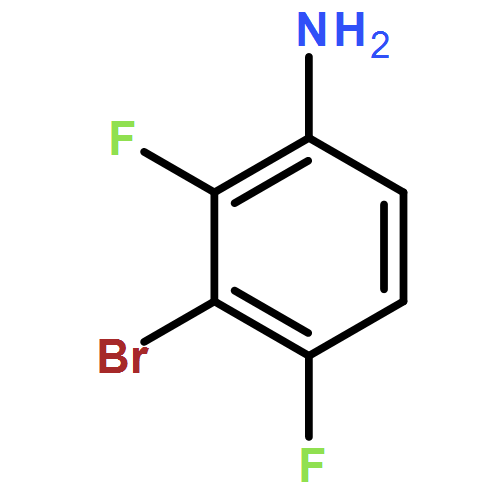


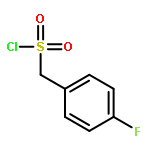

![2-Methylpyrazolo[1,5-a]pyridine-3-carboxylic acid](http://img.cochemist.com/ccimg/80600/80537-08-2.png)
![2-Methylpyrazolo[1,5-a]pyridine-3-carboxylic acid](http://img.cochemist.com/ccimg/80600/80537-08-2_b.png)
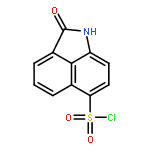
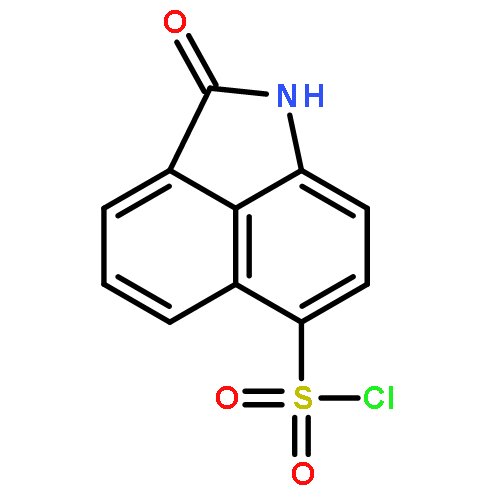
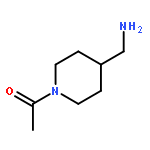
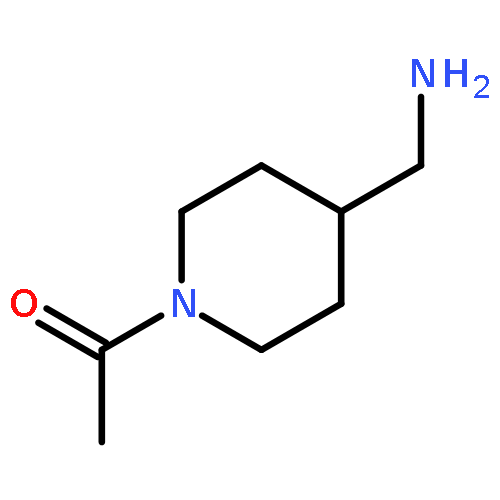
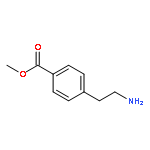
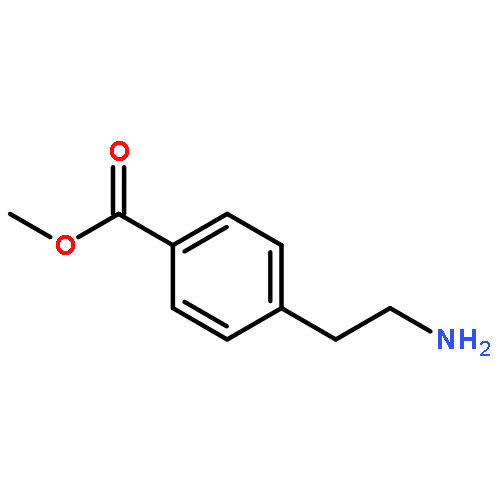
![Benz[cd]indol-2(1H)-one, 1-ethyl-6-nitro-](http://img.cochemist.com/ccimg/64700/64663-16-7.png)
![Benz[cd]indol-2(1H)-one, 1-ethyl-6-nitro-](http://img.cochemist.com/ccimg/64700/64663-16-7_b.png)
![BENZ[CD]INDOLE-6-SULFONAMIDE, 1-ETHYL-1,2-DIHYDRO-2-OXO-N-PHENYL-](http://img.cochemist.com/ccimg/62200/62104-14-7.png)
![BENZ[CD]INDOLE-6-SULFONAMIDE, 1-ETHYL-1,2-DIHYDRO-2-OXO-N-PHENYL-](http://img.cochemist.com/ccimg/62200/62104-14-7_b.png)
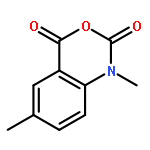
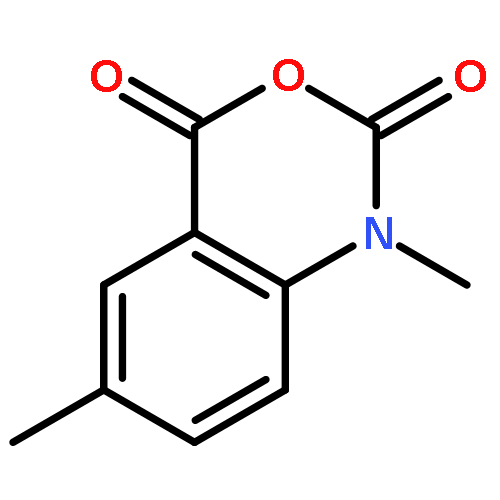
![2-{[5-(2-PYRAZINYL)-1,3,4-OXADIAZOL-2-YL]SULFANYL}-1-INDANONE](http://img.cochemist.com/ccimg/58000/57999-46-9.png)
![2-{[5-(2-PYRAZINYL)-1,3,4-OXADIAZOL-2-YL]SULFANYL}-1-INDANONE](http://img.cochemist.com/ccimg/58000/57999-46-9_b.png)
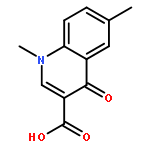
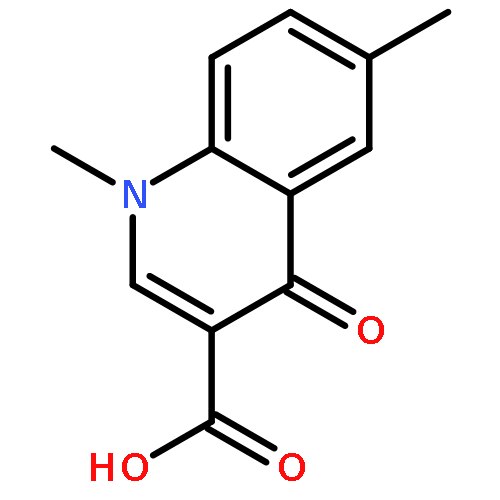
![6-Amino-1-ethyl-1H-benzo[cd]indol-2-one](http://img.cochemist.com/ccimg/51400/51331-93-2.png)
![6-Amino-1-ethyl-1H-benzo[cd]indol-2-one](http://img.cochemist.com/ccimg/51400/51331-93-2_b.png)
![2-[(4,5-Diphenyl-1H-imidazol-2-yl)thio]-propanoic acid](http://img.cochemist.com/ccimg/50700/50677-46-8.png)
![2-[(4,5-Diphenyl-1H-imidazol-2-yl)thio]-propanoic acid](http://img.cochemist.com/ccimg/50700/50677-46-8_b.png)
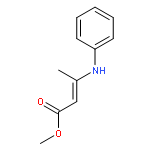
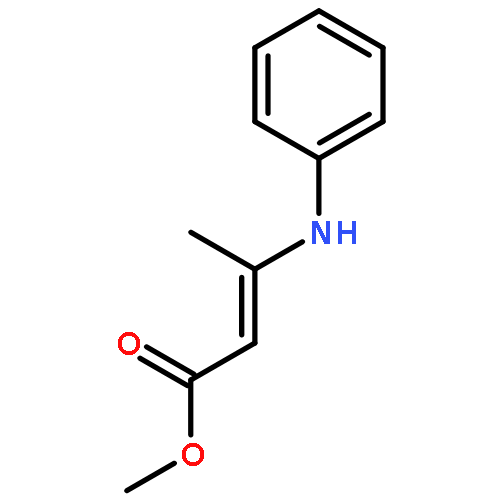
![Benz[cd]indol-2(1H)-one, 1-propyl-](http://img.cochemist.com/ccimg/39300/39273-38-6.png)
![Benz[cd]indol-2(1H)-one, 1-propyl-](http://img.cochemist.com/ccimg/39300/39273-38-6_b.png)
![Pyrazolo[1,5-a]pyridine-3-carboxylic acid, 2,7-dimethyl-, ethyl ester](http://img.cochemist.com/ccimg/34800/34760-55-9.png)
![Pyrazolo[1,5-a]pyridine-3-carboxylic acid, 2,7-dimethyl-, ethyl ester](http://img.cochemist.com/ccimg/34800/34760-55-9_b.png)
![1-Ethyl-2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonyl chloride](http://img.cochemist.com/ccimg/32200/32103-15-4.png)
![1-Ethyl-2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonyl chloride](http://img.cochemist.com/ccimg/32200/32103-15-4_b.png)
![ETHYL 2-METHYLPYRAZOLO[1,5-A]PYRIDINE-3-CARBOXYLATE](http://img.cochemist.com/ccimg/30900/30843-10-8.png)
![ETHYL 2-METHYLPYRAZOLO[1,5-A]PYRIDINE-3-CARBOXYLATE](http://img.cochemist.com/ccimg/30900/30843-10-8_b.png)
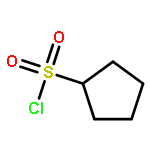
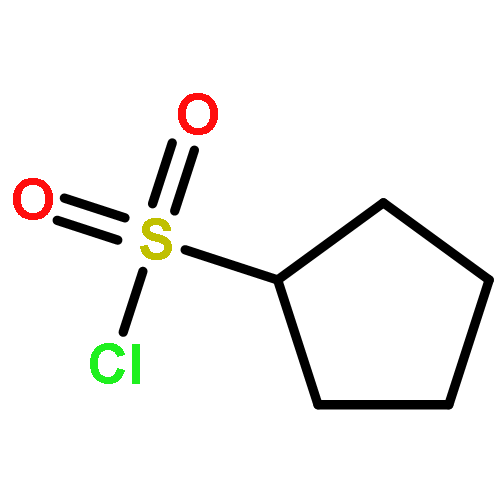











![Benz[cd]indol-2(1H)-one,1-ethyl-](http://img.cochemist.com/ccimg/1900/1830-56-4.png)
![Benz[cd]indol-2(1H)-one,1-ethyl-](http://img.cochemist.com/ccimg/1900/1830-56-4_b.png)
![Benz[cd]indol-2(1H)-one,1-methyl-](http://img.cochemist.com/ccimg/1800/1710-20-9.png)
![Benz[cd]indol-2(1H)-one,1-methyl-](http://img.cochemist.com/ccimg/1800/1710-20-9_b.png)
![N-(3-(5-(4-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide](http://img.cochemist.com/ccimg/918600/918504-65-1.png)
![N-(3-(5-(4-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide](http://img.cochemist.com/ccimg/918600/918504-65-1_b.png)