Co-reporter:Henning Henschel, Jan-Peter Klöckner, Ian A. Nicholls, Marc H. Prosenc
Journal of Molecular Structure 2012 Volume 1007() pp:45-51
Publication Date(Web):11 January 2012
DOI:10.1016/j.molstruc.2011.09.050
Four different complexes of the cobalt(II) acetate–pyridine–water system were obtained as dominant species by crystallization from a series of dichloromethane and toluene solutions. The complexes were characterized by terms of X-ray crystal structure determination. Factors in solution properties leading to crystallization of certain complexes are discussed. Furthermore, trends in terms of structure and binding energies in a systematic series of mononuclear cobalt(II) complexes were studied using density functional calculations.Highlights► A consistent series of Co(II) complexes with water and pyridine was characterized. ► DFT calculations reveal trends within this series. ► A dinuclear Co(II) complex with hitherto unknown coordination environment was found.
Co-reporter:Marc Heinrich Prosenc, Hauke Reddmann, Hanns-Dieter Amberger
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2012 Volume 87() pp:126-134
Publication Date(Web):15 February 2012
DOI:10.1016/j.saa.2011.11.025
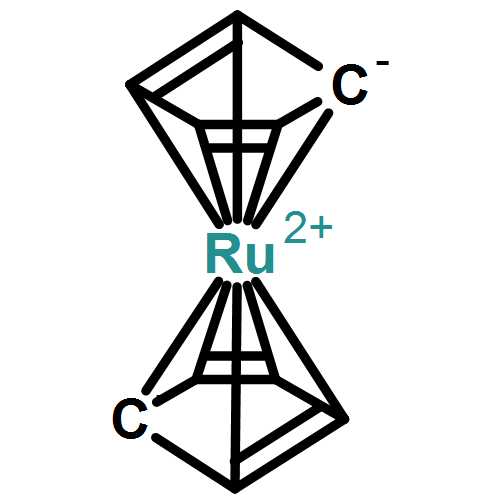
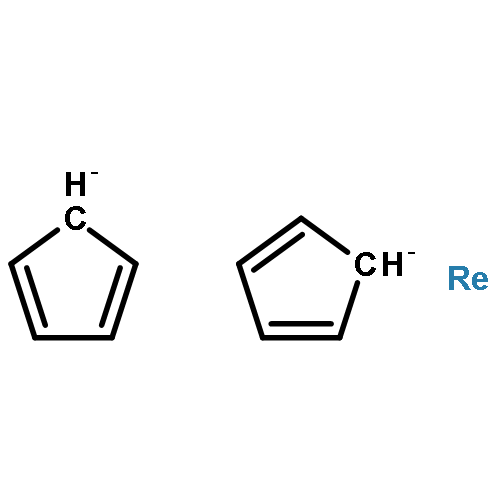
Previous empirical assignments of the normal modes of Ru(η5-C5H5)2 were checked against the results of a calculation applying density functional theory (DFT). After some reassignments, following those recently suggested for Fe(η5-C5H5)2 (after theoretical model calculations), a satisfactory agreement was observed.Recently communicated polarized Raman spectra of an oriented Ru(η5-C5Me5)2 single crystal were used here for the identification of the irreducible representations of a number of Raman active normal modes (assuming molecular D5h symmetry) which agree well with the results of the DFT calculation. The energies of IR active fundamental vibrations, extracted from recently communicated FIR/MIR spectra (pellets), were correlated with comparable energies of IR allowed irreducible representations of the DFT calculation and assigned. Both the skeletal and the intra-ligand normal modes could be correlated with the idealized standard motions (νis) of the model sandwich complex Ru(C5C5)2, and previous assignments had to be revised. Neglecting the νCH vibrations (which are off by ca. 50 cm−1) an r.m.s. deviation of 9.8 cm−1 (for 47 assignments) of the remaining normal modes could be achieved.Graphical abstractHighlights► DFT calculations on normal modes of an organometallic sandwich complex. ► Polarized single crystal Raman spectra of an organometallic sandwich complex. ► Excellent agreement of experimental spectra and DFT predictions.
Co-reporter:Hanns-Dieter Amberger, Marc Heinrich Prosenc, Hauke Reddmann
Journal of Organometallic Chemistry 2012 698() pp: 42-48
Publication Date(Web):
DOI:10.1016/j.jorganchem.2011.10.023
Co-reporter:Nils Pagels;Dr. Ole Albrecht;Dr. Detlef Görlitz;Dr. Andrey Y. Rogachev;Dr. Marc H. Prosenc;Dr. Jürgen Heck
Chemistry - A European Journal 2011 Volume 17( Issue 15) pp:4166-4176
Publication Date(Web):
DOI:10.1002/chem.201003462
Abstract
The paramagnetic dinuclear complexes 1,8-bis(cobaltocenyl)naphthalene (2) and 1,8-bis[(pentamethyl-η5-cyclopentadienyl)(η5-cyclopentadiendiyl)cobalt(II)]naphthalene (4) were synthesized. The molecular structures were characterized by X-ray structure analysis and consisted of two cobaltocenes linked through a distorted naphthalene clamp. Electronic interactions between the two cobalt atoms were observed by cyclic voltammetric studies. Superconducting quantum interference device (SQUID) measurements of the pure compounds and diluted in their diamagnetic iron derivatives, as well as variable-temperature NMR spectroscopy experiments in solution are presented. Magnetic measurements revealed an antiferromagnetic coupling of the electrons in complexes 2 and 4. From NMR spectroscopy experiments, Curie behavior in the temperature range from −60 to +60 °C can be deduced. The electronic structure and magnetic behavior is supported by results of broken-symmetry DFT and multireference calculations along with UV/Vis spectroscopic data, which revealed an intramolecular through space π–π interaction between the cobaltocene units.
Co-reporter:Henning Henschel, Andreas M. Schneider and Marc H. Prosenc
Chemistry of Materials 2010 Volume 22(Issue 17) pp:5105
Publication Date(Web):August 10, 2010
DOI:10.1021/cm100401f
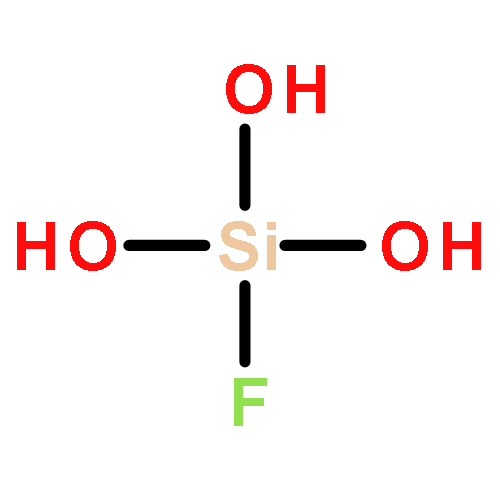
The initial step of the sol−gel process, that is, the condensation of two molecules of silicic acid has been studied by means of density functional theory. The chosen system represents the reagents under basic reaction conditions. Calculations were performed in the gas phase as well as by employing a solvent model for aqueous solution. For both systems, a reaction intermediate with one pentacoordinated silicon center was found as the most stable structure. The influence of intramolecular hydrogen bonds on the stability of the intermediate structure is discussed, and different pathways for the subsequent condensation step are investigated. Furthermore, the effect of fluoride substitution on the reaction path is investigated.
Co-reporter:Sabrina Trtica, Marc Heinrich Prosenc, Michael Schmidt and Jürgen Heck, Ole Albrecht and Detlef Görlitz, Frank Reuter and Eva Rentschler
Inorganic Chemistry 2010 Volume 49(Issue 4) pp:1667-1673
Publication Date(Web):January 19, 2010
DOI:10.1021/ic902058n
The disubstitution of 1,8-diiodonaphthalene (1) with cyclopentadienyl nucleophiles reveals 1,8-(dicyclopentadienyl)-naphthalene, which rapidly undergoes Diels−Alder reaction forming 1,8-(3a′,4′,7′,7a′-tetrahydro-4′,7′-methanoindene-7a′,8′-diyl)-naphthalene (2). A subsequent retro-Diels−Alder reaction in the presence of sodium hydride yields the disodium salt of 1,8-(dicyclopentadiendiyl)-naphthalene 3. The disodium salt 3 was the starting material to obtain the paramagnetic bisnickelocene derivative 4, which structure was obtained by X-ray structure analysis, revealing two nickelocenes kept together in a stacked fashion by a 1,8-naphthalene clamp. An electronic interaction between the two nickel atoms is found as a result of cyclic voltammetry, indicating five different oxidation states +4, +3, +2, +1, and 0. The magnetic properties of 4 in solution were studied by variable temperature paramagnetic 1H NMR spectroscopy and Evans method and revealed Curie behavior between 213 and 293 K. The magnetic susceptibility of a powdered sample of 4 was measured, and an antiferromagnetic interaction with an exchange coupling of J12 = −31.49 cm−1 is found. In accord with experimental data, broken symmetry density functional theory (DFT) calculations revealed four antiferromagnetically coupled electrons resulting in an open shell singlet ground state.
Co-reporter:Timo Paul Rieckborn;Elvira Huber;Emine Karakoc
European Journal of Inorganic Chemistry 2010 Volume 2010( Issue 30) pp:4757-4761
Publication Date(Web):
DOI:10.1002/ejic.201000879
Abstract
The decomposition of formic acid catalyzed by a cationic hydridoplatinum tetradentate phosphane complex was studied by NMR spectroscopy, isotope labeling and DFT calculations. In the initial step dihydrogen is formed by the reaction of the formic acid with the hydrido ligand yielding a platinum formate cation. The formato ligand is then transformed to CO2 by a β-H transfer of the formate C–H bond to the Pt center revealing the initial cationic Pt–H complex. The reaction sequence was illuminated by deuterium and C-13 isotope labeling revealing the cationic Pt–D complex as only Pt complex product if a deuterated formate ligand was used. The transfer of the hydrido ligand to an ensuing coordination site at the Pt center is assisted by the labile bonding character of the phosphane ligand. The activation energy was calculated to be +110 kJ/mol above the initial formate complex. The decomposition reaction was observed to be completely reversible yielding formic acid from CO2 and H2 at a pressure of around 40 bar. Thus, this reaction provides an excellent example for dihydrogen storage and release on demand using a Pt complex as catalyst.

![Phosphine, tris[2-(dicyclohexylphosphino)ethyl]-](http://img.cochemist.com/ccimg/146400/146337-60-2.png)
![Phosphine, tris[2-(dicyclohexylphosphino)ethyl]-](http://img.cochemist.com/ccimg/146400/146337-60-2_b.png)


![4-[2-[4-(DIMETHYLAMINO)PHENYL]ETHYNYL]-N,N-DIMETHYLANILINE](http://img.cochemist.com/ccimg/62100/62063-67-6.png)
![4-[2-[4-(DIMETHYLAMINO)PHENYL]ETHYNYL]-N,N-DIMETHYLANILINE](http://img.cochemist.com/ccimg/62100/62063-67-6_b.png)
![PHENOL, 2-[[(2-AMINOPHENYL)IMINO]METHYL]-4-BROMO-](http://img.cochemist.com/ccimg/51900/51800-88-5.png)
![PHENOL, 2-[[(2-AMINOPHENYL)IMINO]METHYL]-4-BROMO-](http://img.cochemist.com/ccimg/51900/51800-88-5_b.png)
![Tris[2-(diphenylphosphino)ethyl]phosphine](http://img.cochemist.com/ccimg/23600/23582-03-8.png)
![Tris[2-(diphenylphosphino)ethyl]phosphine](http://img.cochemist.com/ccimg/23600/23582-03-8_b.png)