Co-reporter:Dr. Luca Rocchigiani;Dr. Julio Fernez-Cestau;Gabriele Agonigi;Dr. Isabelle Chambrier; Dr. Peter H. M. Budzelaar; Dr. Manfred Bochmann
Angewandte Chemie 2017 Volume 129(Issue 44) pp:14049-14053
Publication Date(Web):2017/10/23
DOI:10.1002/ange.201708640
AbstractThe synthesis and characterization of hitherto hypothetical AuIII π-alkyne complexes is reported. Bonding and stability depend strongly on the trans effect and steric factors. Bonding characteristics shed light on the reasons for the very different stabilities between the classical alkyne complexes of PtII and their drastically more reactive AuIII congeners. Lack of back-bonding facilitates alkyne slippage, which is energetically less costly for gold than for platinum and explains the propensity of gold to facilitate C−C bond formation. Cycloaddition followed by aryl migration and reductive deprotonation is presented as a new reaction sequence in gold chemistry.
Co-reporter:Morwen Williams;Adam I. Green;Julio Fernandez-Cestau;David L. Hughes;Maria A. O'Connell;Mark Searcey;Benoît Bertrand
Dalton Transactions 2017 vol. 46(Issue 39) pp:13397-13408
Publication Date(Web):2017/10/10
DOI:10.1039/C7DT02804K
A series of cyclometallated gold(III) complexes supported by pyrazine-based (C^Npz^C)-type pincer ligands were synthesized via two different pathways. Nucleophilic attack on the isocyanide complex [(C^Npz^C)Au(CNC6H3Me2-2,6)]SbF6 (2) gave [(C^Npz^C)Au(ACC)]SbF6 complexes with aniline (4·SbF6), adamantylamine (5), glycine ethyl ester (6), alanine methyl ester (7), valine methyl ester (8), phenylglycine methyl ester (9) and methionine methyl ester (10) substituents (ACC = acyclic carbene). The pathway via isocyanide insertion into gold–amide bonds was also investigated; e.g. the reaction of xylyl isocyanide with (C^Npz^C)AuNHPh followed by protonation with HBF4·OEt2 gave the acyclic carbene complex 4·BF4. To the best of our knowledge compounds 6–10 represent the first examples of gold(III) acyclic carbene complexes bearing amino acid functions. The compounds provide a versatile platform for the study of the anti-proliferative properties of gold(III) complexes. Tests against human adenoma-type lung cancer cells identified 5, 6, 7 and 10 as particularly promising and demonstrate the synthetic flexibility of acyclic carbene complexes and the potential of that class of compounds for anticancer applications. Compared to cisplatin, amino ester-containing ACC complexes showed improved selectivity for MCF-7 breast cancer cells over that for healthy fibroblasts.
Co-reporter:Alexander S. Romanov, Manfred Bochmann
Journal of Organometallic Chemistry 2017 Volume 847(Volume 847) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.jorganchem.2017.02.045
•Facile synthesis of silver complexes of cyclic (alkyl)(amino) carbene (CAAC) ligands is described.•Surprisingly, ethyl-substituted CAAC leds to the formation of a chloride-bridged dimer.•Remarkably for silver carbene complexes, the new compounds are stable to light and show no evidence of ligand dissociation.•The complexes show blue photoluminescence, which is blue-shifted by 20 nm compared to copper analogues.Silver complexes of cyclic (alkyl)(amino)carbenes (CAACs), (RL)nAgX (n = 1, X = Cl, Br, I; n = 2, X = OTf; R = Me2, Et2, or adamantyl) are accessible in high yields by reacting free carbenes with silver salts. The smaller carbene ligand Me2L leads to the formation of a mixture of neutral (Me2L)AgCl and cationic [(Me2L)2Ag]+ products. The transmetallation of (AdL)AgCl with copper and gold halides gives the corresponding copper and gold compounds (AdL)MCl (M = Cu and Au) in a clean and quantitative reaction. Whereas (Me2L)AgCl is monomeric in the solid state, (Et2L)AgCl crystallizes as a Cl-bridged dimer. None of the compounds show metal-metal interactions. The complexes show blue photoluminescence, which consists of a fluorescence component with a lifetime of several nanoseconds, as well as a long-lived emission in the microsecond regime.Silver complexes with cyclic(alkyl)(amino) carbene ligands can be mononuclear or binuclear in the solid state. Complexes with sterically hindered CAAC ligands show blue photoluminescence.Download high-res image (198KB)Download full-size image
Co-reporter:L. Rocchigiani;J. Fernandez-Cestau;P. H. M. Budzelaar;M. Bochmann
Chemical Communications 2017 vol. 53(Issue 31) pp:4358-4361
Publication Date(Web):2017/04/13
DOI:10.1039/C7CC01628J
Selective Au–C bond cleavage and arene–C–H activation in (C^N^C)Au(III) pincer complexes are reversible, leading to a solvent-dependent proton shuttling process. The ether-free cleavage products are non-fluxional and show weak gold(III)–arene interactions commensurate with intermediates postulated for CMD-type arene activation.
Co-reporter:Alexander S. Romanov;Richard H. Friend;Johannes M. Richter;Le Yang;Dawei Di;Dan Credgington;Saul Jones;Mikko Linnolahti;Mojtaba Abdi Jalebi;Tudor H. Thomas;Jasmine P. H. Rivett
Science 2017 Volume 356(Issue 6334) pp:
Publication Date(Web):
DOI:10.1126/science.aah4345
Adding a twist for enhanced performance
The efficiency of organic light-emitting diodes (OLEDs) is fundamentally governed by the ratio of emissive singlet to dark triplet excitons that are formed from spin-polarized electron and hole currents within the material. Typically, this has set an upper limit of 25% internal quantum efficiency for OLEDs. Di et al. manipulated the ratio of spin states through a modification of process chemistry. They introduced a rotation of the molecular structure, which inverted the spin-state energetics and enhanced OLED performance.
Science, this issue p. 159
Co-reporter:Alexander S. Romanov, Dawei Di, Le Yang, Julio Fernandez-Cestau, Ciaran R. Becker, Charlotte E. James, Bonan Zhu, Mikko Linnolahti, Dan Credgington and Manfred Bochmann
Chemical Communications 2016 vol. 52(Issue 38) pp:6379-6382
Publication Date(Web):12 Apr 2016
DOI:10.1039/C6CC02349E
Linear two-coordinate copper complexes of cyclic (alkyl)(amino)carbenes (CAAC)CuX (X = halide) show photoluminescence with solid-state quantum yields of up to 96%; in contrast to previously reported Cu photoemitters the emission is independent of temperature over the range T = 4–300 K and occurs very efficiently by prompt rather than delayed fluorescence, with lifetimes in the sub-nanosecond range.
Co-reporter:Dragoş-Adrian Roşca and Manfred Bochmann
Organometallics 2016 Volume 35(Issue 1) pp:27-31
Publication Date(Web):December 17, 2015
DOI:10.1021/acs.organomet.5b00846
The Au(II) C∧N∧C pincer complex [(C∧N∧C)Au]2 is stable under thermal conditions but disproportionates on irradiation in solution to give an Au(I)4Au(III)4 mixed-valence aggregate with a 20-membered macrocyclic structure, consisting of four linear Au(I) C–Au–N building blocks, each of which is decorated with a square planar (C∧N∧C)Au(III) substituent. In the crystal, the rings are stacked to form solvent-filled channels with an internal diameter of 8.3 Å and a cross-channel AuI···AuI distance of 7.7 Å.
Co-reporter:Julio Fernandez-Cestau, Benoît Bertrand, Maria Blaya, Garth A. Jones, Thomas J. Penfold and Manfred Bochmann
Chemical Communications 2015 vol. 51(Issue 93) pp:16629-16632
Publication Date(Web):25 Sep 2015
DOI:10.1039/C5CC07523H
The first examples of pyrazine-based gold(III) pincer complexes are reported; their intense photoemissions can be modified by protonation, N-alkylation or metal ions, without the need for altering the ligand framework. Emissions shift from red (77 K) to blue (298 K) due to thermally activated delayed fluorescence (TADF).
Co-reporter:Isabelle Chambrier; Chiranjib Banerjee; Sonia Remiro-Buenamañana; Yimin Chao; Andrew N. Cammidge
Inorganic Chemistry 2015 Volume 54(Issue 15) pp:7368-7380
Publication Date(Web):July 14, 2015
DOI:10.1021/acs.inorgchem.5b00892
Cadmium selenide quantum dots of 2.2–2.3 nm diameter were prepared by phosphorus-free methods using oleic acid as stabilizing surface ligand. Ligand exchange monitored quantitatively by 1H NMR spectroscopy gave an estimate of 30–38 monodentate ligands per nanocrystal, with a ligand density of 1.8–2.3 nm–2. The extent of ligand exchange with macrocycles carrying one or more functional groups was investigated, with the aim of producing nanocrystal–macrocycle conjugates with a limited number of coligands. Metal-free porphyrins are able to sequester the Cd2+ ions from the Cd(oleate)2 outer layer of the nanocrystals. Zinc porphyrin complexes carrying one carboxylate function displace oleate efficiently to give porphyrin/CdSe composites with porphyrins stacked upright on the crystal surface. Porphyrins with four potential ligating sites are able to bind to the crystal surface only if the donors are at the end of sufficiently long and flexible tethers. High-dilution methods allowed the synthesis and isolation of well-defined composites of composition [CdSe{porphyrin}2], where porphyrin = 5,10,15,20-tetrakis{3-(carboxy-n-alkyloxy)phenyl}porphyrinato zinc (n = 5 or 10) and 5,10,15,20-tetrakis{3-(11-undecenyloxythiol)phenyl}porphyrinato zinc. Comparison of the composition data obtained by 1H NMR spectroscopy with luminescence quenching behavior suggests a dependence of quenching efficiency on the tether length. Luminescence quenching was also observed for porphyrins that, according to 1H NMR results, do not undergo surface ligand exchange.
Co-reporter:Dragoş-Adrian Roşca, Joseph A. Wright and Manfred Bochmann
Dalton Transactions 2015 vol. 44(Issue 48) pp:20785-20807
Publication Date(Web):13 Nov 2015
DOI:10.1039/C5DT03930D
Gold, the archetypal “noble metal”, used to be considered of little interest in catalysis. It is now clear that this was a misconception, and a multitude of gold-catalysed transformations has been reported. However, one consequence of the long-held view of gold as inert metal is that its organometallic chemistry contains many “unknowns”, and catalytic cycles devised to explain gold's reactivity draw largely on analogies with other transition metals. How realistic are such mechanistic assumptions? In the last few years a number of key compound classes have been discovered that can provide some answers. This Perspective attempts to summarise these developments, with particular emphasis on recently discovered gold(III) complexes with bonds to hydrogen, oxygen, alkenes and CO ligands.
Co-reporter:Alexander S. Romanov and Manfred Bochmann
Organometallics 2015 Volume 34(Issue 11) pp:2439-2454
Publication Date(Web):January 29, 2015
DOI:10.1021/om501211p
The chemistry of Au(I) complexes with two types of cyclic (alkyl)(amino)carbene (CAAC) ligands has been explored, using the sterically less demanding dimethyl derivative Me2CAAC and the 2-adamantyl ligand AdCAAC. The conversion of (AdCAAC)AuCl into (AdCAAC)AuOH by treatment with KOH is significantly accelerated by the addition of tBuOH. (AdCAAC)AuOH is a convenient starting material for the high-yield syntheses of (AdCAAC)AuX complexes by acid/base and C–H activation reactions (X = OAryl, CF3CO2, N(Tf)2, C2Ph, C6F5, C6HF4, C6H2F3, CH2C(O)C6H4OMe, CH(Ph)C(O)Ph, CH2SO2Ph), while the cationic complexes [(AdCAAC)AuL]+ (L = CO, CNtBu) and (AdCAAC)AuCN were obtained by chloride substitution from (AdCAAC)AuCl. The reactivity toward variously substituted fluoroarenes suggests that (AdCAAC)AuOH is able to react with C–H bonds with pKa values lower than about 31.5. This, together with the spectroscopic data, confirm the somewhat stronger electron-donor properties of CAAC ligands in comparison to imidazolylidene-type N-heterocyclic carbenes (NHCs). In spite of this, the oxidation of Me2CAAC and AdCAAC gold compounds is much less facile. Oxidations proceed with C–Au cleavage by halogens unless light is strictly excluded. The oxidation of (AdCAAC)AuCl with PhICl2 in the dark gives near-quantitative yields of (AdCAAC)AuCl3, while [Au(Me2CAAC)2]Cl leads to trans-[AuCl2(Me2CAAC)2]Cl. In contrast to the chemistry of imidazolylidene-type gold NHC complexes, oxidation products containing Au–Br or Au–I bonds could not be obtained; whereas the reaction with CsBr3 cleaves the Au–C bond to give mixtures of [AdCAAC-Br]+[AuBr2]− and [(AdCAAC-Br)]+ [AuBr4]−, the oxidation of (AdCAAC)AuI with I2 leads to the adduct (AdCAAC)AuI·I2. Irrespective of the steric demands of the CAAC ligands, their gold complexes proved more resistant to oxidation and more prone to halogen cleavage of the Au–C bonds than gold(I) complexes of imidazole-based NHC ligands.
Co-reporter:Dragoş-Adrian Roşca, Julio Fernandez-Cestau, David L. Hughes, and Manfred Bochmann
Organometallics 2015 Volume 34(Issue 11) pp:2098-2101
Publication Date(Web):December 18, 2014
DOI:10.1021/om501165z
Dioxygen reacts with the gold(I) hydride (IPr)AuH under insertion to give the hydroperoxide (IPr)AuOOH, a long-postulated reaction in gold catalysis and the first demonstration of O2 activation by Au–H in a well-defined system. Subsequent condensation gave the peroxide (IPr)Au–OO–Au(IPr) (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene). The reaction kinetics are reported, as well as the reactivity of Au(I) hydrides with radical scavengers.
Co-reporter:James Morris;Julio Fernandez-Cestau;Dragoş-Adrian Roşca;Joseph A. Wright
Science Advances 2015 Volume 1(Issue 9) pp:
Publication Date(Web):
DOI:10.1126/sciadv.1500761
The first isolable gold(III)-CO complex reveals unexpected differences in CO bonding to gold and platinum catalysts.
Co-reporter:Lingfang Wang, Valentin Poirier, Fabio Ghiotto, Manfred Bochmann, Roderick D. Cannon, Jean-François Carpentier, and Yann Sarazin
Macromolecules 2014 Volume 47(Issue 8) pp:2574-2584
Publication Date(Web):April 1, 2014
DOI:10.1021/ma500124k
A kinetic analysis of the metal-catalyzed immortal ring-opening polymerization (ROP) of cyclic esters is presented, based on a first-principles approach without making assumptions regarding the active species. The kinetics of all immortal ROP reactions performed with a metal catalyst and an exogenous chain transfer agent are characterized by the initiation, propagation and exchange rate constants (ki, kp, and ke, respectively). Curve fitting to this kinetic scheme in the initial stage of the polymerization allows the extraction of ki and kp from a single experiment. This has been illustrated for the ROP of l-lactide using tin(II) complexes of the type {LOi}Sn(X) ({LOi} = aminophenolate ancillary ligand, X = N(SiMe3)2 or methyl (S,S)-lactate), Sn(OiPr)2 or Sn(N(SiMe3)2)2 as precatalysts paired with excess iPrOH as a coactivator. Nonlinear regressions (R2 > 0.999) illustrate the three possible scenarios, ki < kp, ki = kp, and ki > kp. The kinetic model can be extended to any metal (pre)catalyst for the immortal ROP of any cyclic ester, as exemplified using trimethylene carbonate as a monomer or employing a germylene precatalyst. A kinetic treatment for the late phase of immortal ROP reactions is introduced, which also gives direct access to kp. In agreement with the ROP kinetic data for {LOi}Sn(N(SiMe3)2), Sn(OiPr)2, Sn(N(SiMe3)2)2, and the new Sn(OiPr)(N(SiMe3)2 recorded in the presence of various quantities of iPrOH, synthetic and 119Sn{1H} NMR data provide evidence for reversible production of tin(II) bis(alkoxide) when a small excess (1–3 equiv) of alcohol is used with tin(II) precatalysts. It is also shown that, regardless of the identity of the precatalyst, Sn(OiPr)2 and Sn(O-polymeryl)2 are, respectively, the actual initiating and propagating species when immortal ROP reactions are performed in the presence of a larger excess of alcohol (7 equiv or more vs Sn).
Co-reporter:Thomas Dann, Dragoş-Adrian Roşca, Joseph A. Wright, Gregory G. Wildgoose and Manfred Bochmann
Chemical Communications 2013 vol. 49(Issue 86) pp:10169-10171
Publication Date(Web):17 Sep 2013
DOI:10.1039/C3CC45984E
The bond energy of the unsupported Au–Au bond in the Au(II) dimer [(C∧N∧C)Au]2 and the difference between AuIII–OH and AuIII–H bond enthalpies have been determined experimentally by electrochemical methods, with Au–OH and Au–H complexes showing unexpected differences in their reduction pathways, supported by DFT modelling.
Co-reporter:Fabio Ghiotto, Chrysoula Pateraki, John R. Severn, Nic Friederichs and Manfred Bochmann
Dalton Transactions 2013 vol. 42(Issue 25) pp:9040-9048
Publication Date(Web):05 Mar 2013
DOI:10.1039/C3DT00107E
The influence of methylaluminoxane (MAO) catalyst activators of different concentrations and preparative histories on the performance of 1-hexene polymerisations was investigated by kinetic methods, using rac-Me2Si(2-Me-Benz[e]Ind)2ZrCl2 as the standard catalyst precursor. Fast sampling and analysis allow the time dependence of monomer conversion and the growth of the number-average polymer molecular weight to be determined at a sufficiently short timescale to make this a feasible method for routine catalyst evaluation. Differences in productivity, polymer molecular weight and active species count are shown to be primarily a linear function of the trimethylaluminium concentration. The results in toluene and heptane as solvents are compared; the data show that the inferior performance in heptane is due to a substantially lower active species concentration.
Co-reporter:Lingfang Wang;Roderick D. Cannon;Jean-François Carpentier;Thierry Roisnel;Yann Sarazin
European Journal of Inorganic Chemistry 2013 Volume 2013( Issue 34) pp:
Publication Date(Web):
DOI:10.1002/ejic.201300964
Abstract
A general kinetic model to describe the initiation and monomer depletion phases of the metal-catalysed living ring-opening polymerisation (ROP) of cyclic esters is presented. The model allows the description of ROP reactions in terms of rates of initiation (ki) and propagation (kp) and is in principle applicable to all metal-mediated ROP reactions. The model was validated by curve-fitting of data obtained for the living ROP of L-lactide catalysed by a variety of tin(II) catalysts. The catalyst was chosen from tin(II) diisopropoxide or from heteroleptic complexes of the type (LOx)Sn(OR), in which (LOx) is an amino or aminoether phenolate ancillary ligand and OR is isopropoxide or O-tert-butyl lactate. All tested catalysts promote the controlled, living polymerisation of L-lactide. No initiation phase was discerned for any of the considered catalysts in polymerisations performed at 60 °C, whereas at 25 °C initiation is an order of magnitude slower than propagation and, therefore, the inclusion of ki is required for an accurate kinetic description. Tin diisopropoxide is polymeric in the solid state, but it is dimeric in toluene solution. [Sn(OiPr)2]2 is an excellent example of an aggregated catalyst precursor and is catalytically more active than the heteroleptic (LOx)Sn(OR). As dissociation of the inactive dimer into a catalytically active monomeric complex is required, half-order dependence on [metal] is to be expected and was indeed found. A method to estimate the related monomer–dimer equilibrium constant KD under polymerisation conditions is also provided.
Co-reporter:Dr. Nicky Savjani;Drago&x15f;-Adrian Ro&x15f;ca;Dr. Mark Schormann ;Dr. Manfred Bochmann
Angewandte Chemie International Edition 2013 Volume 52( Issue 3) pp:874-877
Publication Date(Web):
DOI:10.1002/anie.201208356
Co-reporter:Fabio Ghiotto, Chrysoula Pateraki, Jukka Tanskanen, John R. Severn, Nicole Luehmann, André Kusmin, Jörg Stellbrink, Mikko Linnolahti, and Manfred Bochmann
Organometallics 2013 Volume 32(Issue 11) pp:3354-3362
Publication Date(Web):May 28, 2013
DOI:10.1021/om4002878
The composition of methylalumoxane (MAO) and its interaction with trimethylaluminum (TMA) have been investigated by a combination of chemical, spectroscopic, neutron scattering, and computational methods. The interactions of MAO with donor molecules such as THF, pyridine, and PPh3 as a means of quantifying the content of “free” and “bound” TMA have been evaluated, as well as the ability of MAO to produce [Me2AlL2]+ cations, a measure of the electrophilic component likely to be involved in the activation of single-site catalysts. THF, pyridine, and diphenylphosphinopropane (dppp) give the corresponding TMA–donor ligand complexes accompanied by the formation of [Me2AlL2]+ cations. The results suggest that MAO contains not only Lewis acid sites but also structures capable of acting as sources of [AlMe2]+ cations. Another unique, but still unresolved, structural aspect of MAO is the nature of “bound” and “free” TMA. The addition of the donors OPPh3, PMe3, and PCy3 leads to the precipitation of polymeric MAO and shows that about one-fourth of the total TMA content is bound to the MAO polymers. This conclusion was independently confirmed by pulsed field gradient spin echo (PFG-SE) NMR measurements, which show fast and slow diffusion processes resulting from free and MAO-bound TMA, respectively. The hydrodynamic radius Rh of polymeric MAO in toluene solutions was found to be 12 ± 0.3 Å, leading to an estimate for the average size of MAO polymers of about 50–60 Al atoms. Small-angle neutron scattering (SANS) resulted in the radius RS = 12.0 ± 0.3 Å for the MAO polymer, in excellent agreement with PFG-SE NMR experiments, a molecular weight of 1800 ± 100, and about 30 Al atoms per MAO polymer. The MAO structures capable of releasing [AlMe2]+ on reaction with a base were studied by quantum chemical calculations on the MAO models (OAlMe)n(TMA)m for up to n = 8 and m = 5. Both −O–AlMe2–O– and −O–AlMe2–μ-Me– four-membered rings are about equally likely to lead to dissociation of [AlMe2]+ cations. The resulting MAO anions rearrange, with structures containing separated Al2O2 4-rings being particularly favorable. The results support the notion that catalyst activation by MAO can occur by both Lewis acidic cluster sites and [AlMe2]+ cation formation.
Co-reporter:Dr. Nicky Savjani;Drago&x15f;-Adrian Ro&x15f;ca;Dr. Mark Schormann ;Dr. Manfred Bochmann
Angewandte Chemie 2013 Volume 125( Issue 3) pp:908-911
Publication Date(Web):
DOI:10.1002/ange.201208356
Co-reporter:Dragoş-Adrian Roşca, Dan A. Smith and Manfred Bochmann
Chemical Communications 2012 vol. 48(Issue 58) pp:7247-7249
Publication Date(Web):23 May 2012
DOI:10.1039/C2CC33104G
The gold(III) hydroxide κ3-(C∧N∧C)*Au(OH) reacts with C–H and N–H compounds and arylboronic acids to produce a range of perfluoroaryls, N-heterocyclic and alkynyl compounds in high yields; some of which show unexpectedly strong modulation of their photoluminescence from yellow to blue [(C∧N∧C)* = 2,6-(C6H3But)2pyridine].
Co-reporter:Chiranjib Banerjee, David L. Hughes, Manfred Bochmann and Thomas Nann
Dalton Transactions 2012 vol. 41(Issue 24) pp:7244-7248
Publication Date(Web):26 Apr 2012
DOI:10.1039/C2DT30283G
Indium phosphide nanofibres were grown from a single-molecule precursor, [(PhCH2)2InP(SiMe3)2]2, using hot injection techniques by a solution–liquid–solid (SLS) process, under “surfactant-free” conditions and without the use of protic additives. The fibres are 85–95 nm in diameter and grow from In metal droplets of 100 nm diameter. The length of the nanofibres is a function of the precursor injection temperature (rather than the growth temperature) and can be varied from 6000 nm at 210 °C to 1000 nm at 310 °C. The indium metal tip can be readily removed under mild, non-etching conditions by treatment with thiophenol–P(SiMe3)3 mixtures.
Co-reporter:Nicky Savjani, Sean P. Bew, David L. Hughes, Simon J. Lancaster, and Manfred Bochmann
Organometallics 2012 Volume 31(Issue 7) pp:2534-2537
Publication Date(Web):December 2, 2011
DOI:10.1021/om201035v
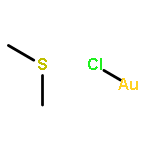
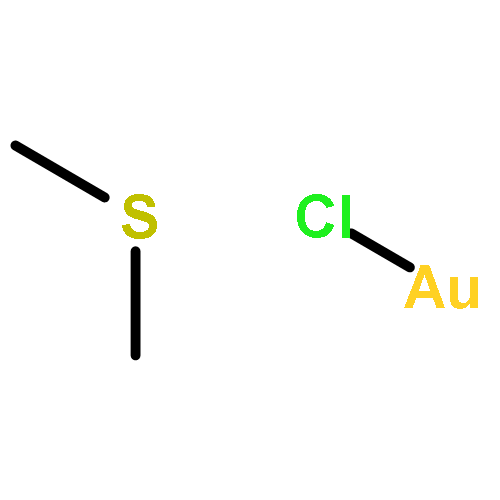
[(Me2S)Au]+ cations, generated from (Me2S)AuCl and AgSbF6 in dichloromethane at 0–20 °C, serve as sources of solvated Au+ (alongside unreactive [Au(SMe2)2]+), which reacts with the methyl-substituted arenes C6Me6–nHn (n = 0–2) with C–H bond cleavage to give the sulfonium salts [C6Me5–nHnCH2SMe2]+. There was no evidence for arene π coordination to Au+ or for the formation of σ-bonded Au–benzyl species. Surprisingly, the reaction of Au+ with CH2Ar2 leads to C–C bond cleavage (Ar = 2,4,6-C6H2Me3). The reactions are highly selective for benzylic C–H and C–C bonds, whereas metalation of the arene ring is not observed.
Co-reporter:Dan A. Smith, Dragoş-Adrian Roşca, and Manfred Bochmann
Organometallics 2012 Volume 31(Issue 17) pp:5998-6000
Publication Date(Web):August 8, 2012
DOI:10.1021/om300666j
Treatment of gold(III) pincer complexes (C∧N∧C)*AuX with trifluoroacetic acid (X = C6F5, thiophenyl, Me, Et) or of (C∧N∧C)*AuOAcF with AgOAcF/arylboronic acids leads to the selective cleavage of a C–Au bond under mild conditions to give the bidentate complexes (HC-C∧N)*Au(X)(OAcF) [(C∧N∧C)* = 2,6-(C6H3But)2pyridine]. Alkylation of (C∧N∧C)*Au(OAcF) with AlR3 (R = Me, Et) proved to be a high-yielding route to gold(III) alkyls. Au–C cleavage significantly influences reactivity, e.g., with boronic acids. The photoemission of the cleavage product (HC-C∧N)*Au(C6H4F)(OAcF) is about an order of magnitude more intense than that of its tridentate parent compound.
Co-reporter:Nicky Savjani, Luke A. Wilkinson, David L. Hughes, Mark Schormann, and Manfred Bochmann
Organometallics 2012 Volume 31(Issue 21) pp:7600-7609
Publication Date(Web):October 31, 2012
DOI:10.1021/om300911h
Mixtures of silver salts AgX (X = NO3, CF3CO2, CF3SO3) with M[Au(C6F5)2] (M = NBu4, PPh4) gave respectively the ionic mixed-metal clusters [M2{(C6F5)4Au2Ag2X2}]n (1, X = NO3; a, M = NBu4; b, M = PPh4) and [M{(C6F5)4Au2Ag2X}]n, (2a,b, X = CF3CO2; 3a,b, X = CF3SO3). The degree of aggregation n of these cluster compounds depends strongly on the method of isolation (solvent evaporation or precipitation); for example, recrystallization of 1a gave a crystalline salt of the tetraanion [(C6F5)4Au2Ag2X2]24– as well as the polymer [(NBu4)2{(C6F5)4Au2Ag2(NO3)2}]n. The aurophilic Au···Au interactions strongly influence the photoemission wavelength. The anion X has remarkably little effect on the luminescence color but strongly influences the conformation of the polyanionic chains, leading to a variety of solid-state structures, from well-defined dimers (1a1) to linear (1b) and curved (1a2, 2a) polymeric chain aggregates.
Co-reporter:Nicky Savjani, Mark Schormann, Manfred Bochmann
Polyhedron 2012 Volume 38(Issue 1) pp:137-140
Publication Date(Web):11 May 2012
DOI:10.1016/j.poly.2012.02.026
The reaction of the potassium diketiminate K[RNC(CF3)CH(CF3)CNR)], where R = 3,5-C6H3Me2, with PPh3AuCl afford the complex [RNC(CF3)CH(CF3)CNR)AuPPh3]. Unlike gold(I) diketiminates without backbone-CF3 substituents, the complex is thermally stable in the solid state and in solution. The crystal structure confirms that, unlike previous examples of Au(I) ketiminates, this complex possesses a three-coordinate metal centre with a distorted Y-shaped coordination geometry.Graphical abstractA new thermally stable gold(I) diketiminato complex has been prepared which shows an unusual, asymmetrically chelated three-coordinate metal centre.Highlights► Gold(I) diketiminato complex synthesis. ► High thermal stability in solution. ► First example of unsymmetrical N,N-chelate formation to Au(I). ► Large structural effects of subtle electronic ligand modification.
Co-reporter:Drago&x15f;-Adrian Ro&x15f;ca;Dr. Dan A. Smith;Dr. David L. Hughes ; Manfred Bochmann
Angewandte Chemie International Edition 2012 Volume 51( Issue 42) pp:10643-10646
Publication Date(Web):
DOI:10.1002/anie.201206468
Co-reporter:Drago&x15f;-Adrian Ro&x15f;ca;Dr. Dan A. Smith;Dr. David L. Hughes ; Manfred Bochmann
Angewandte Chemie 2012 Volume 124( Issue 42) pp:10795-10798
Publication Date(Web):
DOI:10.1002/ange.201206468
Co-reporter:Nicky Savjani, Simon J. Lancaster, Sean Bew, David L. Hughes and Manfred Bochmann
Dalton Transactions 2011 vol. 40(Issue 5) pp:1079-1090
Publication Date(Web):16 Dec 2010
DOI:10.1039/C0DT01134G
Compounds of the new tetrafluorophthalimido anion, [C6F4(CO)2N]−, are readily accessible by treatment of tetrafluorophthalimide with either LiNPri2 or mixtures of NEt3 and Me3ECl (E = Si or Sn), to give C6F4(CO)2N-X (X = Li 3, SiMe34, and SnMe35). The reaction of the trimethylsilyl derivative 4 with AgF leads cleanly to the ion pair complex [Ag(NCMe)2][Ag(N(CO)2C6F4)2] (6·2MeCN), which contains a linear [Ag{N(CO)2C6F4}2]− anion and a tetracoordinate Ag+ cation. Compound 6 reacts with iodine to give the N-iodo compound C6F4(CO)2NI 7, which crystallises as an acetonitrile adduct. Treatment of 6 with LAuCl affords LAu{N(CO)2C6F4} (L = Ph3P 8a, Cy3P 8b, or THT 9), whereas the reaction with AuCl in acetonitrile affords the heterobinuclear compound [Ag(MeCN)2][Au{N(CO)2C6F4}2]·MeCN (10·3MeCN). The tetrafluorophthalimido ligand is not readily displaced by donor ligands; however, the addition of B(C6F5)3(Et2O) to a diethyl ether solution of 8a leads to the salt [Au(PPh3)2][N{COB(C6F5)3}2C6F4)] 11. The analogous reaction of (THT)Au{N(CO)2C6F4} with B(C6F5)3 in toluene in the presence of excess norbornene (nb) gives [Au(nb)3][N{COB(C6F5)3}2C6F4)] 12. Compounds 11 and 12 contain a new non-coordinating phthalimido-bridged diborate anion with O-bonded boron atoms. The crystal structures of compounds 2–11 are reported.
Co-reporter:Nora Carrera, Nicky Savjani, Jason Simpson, David L. Hughes and Manfred Bochmann
Dalton Transactions 2011 vol. 40(Issue 5) pp:1016-1019
Publication Date(Web):09 Dec 2010
DOI:10.1039/C0DT01422B
While most metallic elements across the Periodic Table form stable chelating β-diketiminato complexes, examples of Au(I) are conspicuous by their absence. We report here the reaction of K[HC(F3CCNR)2] with AuCl(PPh3) which provides a rare example of a thermally stable gold(I) diketiminato complex, (Ph3P)Au[RNC(CF3)CH(CF3)CNR] [R = 3,5-C6H3(CF3)2]. The complex is highly fluxional in solution but in the solid state adopts a U-conformation. By contrast, the analogous reaction of K[HC(F3CCNR)2] with CuBr(PPh3)3 gives the rigid 18-electron chelate complex (Ph3P)2Cu[κ2-HC{(CF3)CNR}2].
Co-reporter:Manfred Bochmann
Accounts of Chemical Research 2010 Volume 43(Issue 9) pp:1267
Publication Date(Web):July 12, 2010
DOI:10.1021/ar100044s
Ion−ion interactions are a crucial but often overlooked aspect of many polymerization reactions. The precise nature of cation−anion binding is as yet poorly understood, and little is known of the extent of ionic interactions in the typically nonaqueous, low-polarity reaction media of most polymerizations. Nevertheless, adequate control of cation−anion interactions can greatly enhance the productivity and efficiency of chemical processes and can provide low-energy alternatives to current methods. This is illustrated here with the carbocationic polymerization of isoalkenes. Carbocationic polymerizations involve, as the name implies, carbocations as propagating species. Of the various types of substrates that can be polymerized cationically, the copolymerization of isobutene to isobutene−isoprene rubber stands out as the only large-scale, industrially important implementation of this reaction type. The products, elastomers with controlled degrees of unsaturation for subsequent cross-linking, have excellent gas barrier and mechanical dampening properties that make them indispensable components in polymer composites. For such applications, the polymer molecular weight has to be high, ∼5 × 105 g/mol, with 1−2 mol % isoprene. Cationic polymerizations are however notoriously difficult to control. As a means of suppressing chain transfer, the process is carried out at temperatures as low as −100 °C, with aluminum chloride initiators in chloromethane. Current industrial production of isobutene−isoprene butyl rubber is thus highly energy intensive and produces aluminum and chloride effluent. This Account summarizes how highly electrophilic organometallics coupled with new types of very weakly coordinating counteranions can provide the basis for a more environmentally friendly, lower energy alternative. Because any copolymerization of two monomers, here primarily isobutene and isoprene, leads to two different propagating species, each of which is characterized by different chain growth and chain termination kinetics, variation of the associated counteranions can give rather unexpected results. With judicious choice of the initiator and the counteranion, new chemistry can be injected into such processes and can open avenues to new families of polymer materials. Mechanistic investigations of the initiation process with zirconocene hydrides illustrate the complexity of this first step. Replacing aluminum with zinc initiators not only provides a nontoxic alternative but also generates a system in which the polymer molecular weight is much less affected by temperature and comonomer concentration, which can lead to a range of products, from oligomeric lubricant precursors to C═C-rich high-molecular-weight elastomers. The key in all these cases is the construction of either preformed or in situ-generated complex anions that are resistant to electrophilic or redox degradation and are capable of stabilizing tightly associated carbocations. Such initiator systems allow much more benign operating temperatures, reduce the need for chlorocarbon solvents, and can operate at concentrations as low as 5 × 10−5 M. Along the way are provided the first examples of structurally characterized sec-alkyl carbocations and carbocation salts of organometallic zincates.
Co-reporter:Lewis M. Broomfield, David Boschert, Joseph A. Wright, David L. Hughes, Manfred Bochmann
Journal of Organometallic Chemistry 2009 694(25) pp: 4084-4089
Publication Date(Web):
DOI:10.1016/j.jorganchem.2009.08.033
Co-reporter:Antonio Guerrero;Kevin Kulbaba
Macromolecular Chemistry and Physics 2008 Volume 209( Issue 16) pp:1714-1720
Publication Date(Web):
DOI:10.1002/macp.200800150
Co-reporter:Carlos Alonso-Moreno, Simon J. Lancaster, Joseph A. Wright, David L. Hughes, Cristiano Zuccaccia, Andrea Correa, Alceo Macchioni, Luigi Cavallo and Manfred Bochmann
Organometallics 2008 Volume 27(Issue 21) pp:5474-5487
Publication Date(Web):September 30, 2008
DOI:10.1021/om800486p
The mixed-alkyl metallocene complexes (IPCF)M(Me)(CH2SiMe3) (M = Zr, Hf; IPCF = Me2C(C5H4)(C13H8)) were synthesized by the reaction of (IPCF)M(Me)Cl (M = Zr, Hf) with Me3SiCH2MgCl. The crystal structures of (IPCF)Zr(CH2SiMe3)2, (IPCF)HfMe2, and (IPCF)Zr(Me)Cl were determined by X-ray diffraction. The kinetics of site epimerization of the ion pairs (IPCF)M(CH2SiMe3)(μ-Me)B(C6F5)3 and [(IPCF)MCH2SiMe3+···B(C6F5)4−] (M = Zr, Hf) were studied by variable-temperature NMR spectroscopy, while the solution ground-state structures of the ion pairs [LZrCH2SiMe3+···B(C6F5)4−] (L = SBI, IPCF; SBI = rac-Me2Si(Ind)2) were probed experimentally by 19F,1H HOESY NMR spectroscopy and theoretically by DFT and molecular dynamics calculations. They reveal differences in the strength of anion interactions between the SBI and IPCF systems which may be significant for their catalytic activity. The tetraarylborate salts are stabilized by agostic interactions to ligand Si−Me moieties, with Hf > Zr. The exchange rates of both the MeB(C6F5)3− and the B(C6F5)4− compounds increase with increasing ion pair concentration. This acceleration is also seen on addition of excess [Ph3C][B(C6F5)4]. Pulsed-field gradient spin−echo (PGSE) NMR measurements indicated that both [(IPCF)ZrCH2SiMe3+···B(C6F5)4−] and [(SBI)ZrCH2SiMe3+···B(C6F5)4−] were present mainly as ion quadruples in toluene-d8/1,2-F2C6H4 (8/2 in volume) at millimolar concentrations and, notably, their aggregation increased to a similar extent with the addition of an excess of [Ph3C][B(C6F5)4]. The results demonstrate the formation of mixed-ion aggregates of the type {[(L)MR+···X−][CPh3+···X−]n}. However, whereas the site epimerization rates kex of the system (SBI)ZrMe(CH2SiMe3)/[Ph3C][B(C6F5)4] continue to increase linearly with the total ion concentration, for (IPCF)ZrMe(CH2SiMe3)/[Ph3C][B(C6F5)4] mixtures kex reaches a plateau at ca. 400 s−1 (at 20 °C). Measurement of site epimerization rates as a function of ion pair concentration [(A+)x(B+)1−xX−] therefore provides evidence for the existence of a rate-limiting barrier in the IPCF system, while it is absent in others.
Co-reporter:Konstantin P. Bryliakov, Evgenii P. Talsi, Alexander Z. Voskoboynikov, Simon J. Lancaster and Manfred Bochmann
Organometallics 2008 Volume 27(Issue 23) pp:6333-6342
Publication Date(Web):October 30, 2008
DOI:10.1021/om800664p
The formation of cationic species relevant to olefin polymerization based on (SBI)HfCl2, Me2C(C5H4)(Flu)HfCl2, Ph2C(C5H4)(Flu)HfCl2, and L′HfCl2 activated by MAO, AlMe3/CPh3[B(C6F5)4], and AlBui3/CPh3[B(C6F5)4] (SBI = rac-Me2Si(Ind)2; L′ = C2H4(Flu)(5,6-C3H6-2-MeInd)) was studied by 1H, 13C, and 19F NMR spectroscopy. Thermally stable heterobinuclear intermediates of the type [LHf(μ-Me)2AlMe2]+[MeMAO]− and [LHf(μ-Me)2AlMe2]+[B(C6F5)4]− were identified when using MAO and AlMe3/CPh3[B(C6F5)4] as activators, respectively. The stability of these species explains the low productivity of hafnocene catalysts in the presence of AlMe3-containing activators, compared to zirconocenes. By contrast, in the ternary systems LHfCl2/AlBui3/CPh3[B(C6F5)4] hydride species were detected that must be responsible for the formation of the highly active sites in olefin polymerization. The ionic hydrido species differ significantly in stability. The formation of the mixed-alkyl complex L′Hf(Me)CH2SiMe3 proceeds with surprisingly high diastereoselectivity; the sterically more hindered isomer is produced preferentially. It reacts with CPh3[B(C6F5)4] to afford the ion pair [L′Hf-CH2SiMe3]+[B(C6F5)4]− as two diastereomers that exist in dynamic equilibrium. The rates of site epimerization of this ion pair indicate only small energy differences between the two isomers.
Co-reporter:Musa Said, Mark Thornton-Pett, David L. Hughes, Manfred Bochmann
Inorganica Chimica Acta 2007 Volume 360(Issue 4) pp:1354-1363
Publication Date(Web):1 March 2007
DOI:10.1016/j.ica.2006.03.013
Sterically highly hindered phosphiniminato complexes MCl3(NCP) were prepared from MCl4 and Li[NPC] in toluene [M = Zr or Hf; NPC = 4-ButC6H4C(SiMe3)P(Ph)2NC6H2Me3-2,4,6]. Reaction with methyl lithium readily affords the corresponding zirconium and hafnium trimethyl complexes. The structures of representative zirconium and hafnium complexes MX3(NPC) (X = Cl, M = Zr, Hf; X = Me, M = Hf) were determined by X-ray diffraction. In all cases the NPC ligand acts as C–N chelate, with an additional bonding contribution from the ipso-carbon atom of the C-bound aryl substituent, which results in a η1:η2-coordination mode. The reaction of the hafnium trimethyl complex with CPh3+ salts of perfluoroarylborate anions results either in the diastereoselective formation of the binuclear cation [{(NPC)HfMe2}2(μ-Me)]+ or in the formation of the mononuclear cation [(NPC)HfMe2]+, depending on the molar ratio of reagents.Bulky phosphiniminates MX3(NCP) (X = Cl or Me) bind to M = Zr and Hf not only as N–C chelates but involve arene π-bonding, to give an η1:η2-coordination mode. The reaction of HfMe3(NCP) with CPh3+ gives selectively either the binuclear cation [{(NPC)HfMe2}2(μ-Me)]+ or mononuclear [(NPC)HfMe2]+, depending on the reagent ratio.
Co-reporter:Yann Sarazin;Simon J. Coles;David L. Hughes;Michael B. Hursthouse
European Journal of Inorganic Chemistry 2006 Volume 2006(Issue 16) pp:
Publication Date(Web):28 JUN 2006
DOI:10.1002/ejic.200600397
Ph3SnN(SiMe3)2 (1) was prepared in good yields by reaction of [{NaN(SiMe3)2}2·THF] (2) with Ph3SnF. Treatment of 1 with [H(OEt2)2][H2N{B(C6F5)3}2] (4) in dichloromethane afforded the stannylium cation [Ph3Sn(OEt2)][H2N{B(C6F5)3}2] (5), which was characterised by 1H, 13C{1H}, 11B, 19F and 119Sn NMR spectroscopy. The reaction of Sn(NMe2)4 with [Ph2MeNH][B(C6F5)4] (3) gave the amidotin(IV) compound [Sn(NMe2)3(HNMe2)2][B(C6F5)4] (6) which proved very stable towards ligand substitution and resisted treatment with Et2O, THF, TMEDA and pyrazine. Two new Brønsted acid salts [H(NMe2H)2][B(C6F5)4] (7) and [(C4H4N2)H·OEt2][H2N{B(C6F5)3}2] (8) were synthesised. The reaction of 7 with Sn(NMe2)4 in Et2O allowed the preparation of 6 in a much improved yield (83 %). The treatment of 7 with Me3SnN(SiMe3)2 in Et2O yielded [Me3Sn(HNMe2)2][B(C6F5)4] (9) nearly quantitatively. Compounds 1, 2, 6, 8 and 9 were characterised by single-crystal X-ray diffraction analyses; 6 is the first example of a structurally characterised amidotin(IV)cation.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Co-reporter:Musa Said, David L. Hughes, Manfred Bochmann
Inorganica Chimica Acta 2006 Volume 359(Issue 11) pp:3467-3473
Publication Date(Web):1 August 2006
DOI:10.1016/j.ica.2005.10.041
The reaction of the lithium iminophenolate LiOC6H3-2-But-6-CHNAr, where Ar = 3-C6H4P{C6H3(CF3)2-3,5}2, [Li(O–NP)] with ZrCl4 affords ZrCl2(O–NP)2. Whereas previously reported zirconium bis(salicylaldiminato) complexes are typified by the trans orientation of the O-atoms, the molecular structure ZrCl2(O–NP)2 is unusual in adopting an all-cis arrangement of N, O and Cl donors. The crystal structure of the parent ligand, HOC6H3-2-But-6-CHNC6H4PPh2-3, is reported for comparison. Upon activation with methylaluminoxane the complex is highly active for the polymerisation of ethylene to oligomers and low-molecular weight polymers. Notably, the catalytic activity is unaffected by the presence of phosphine donors.Zirconium complexes with phosphine-substituted salicyliminato ligands have been prepared, ZrCl2(O–NP)2 [N–OP = OC6H3-2-But-6-CHNAr, Ar = 3-C6H4P{C6H3(CF3)2-3,5}2. Unlike previously reported examples of this type which have O-donors in mutually trans positions, ZrCl2(O–NP)2 adopts the unusual all-cis configuration. The catalytic activity in ethylene polymerizations is unaffected by the presence of phosphine donors.
Co-reporter:Yann Sarazin;Gerhard Fink;Klaus Hauschild
Macromolecular Rapid Communications 2005 Volume 26(Issue 15) pp:1208-1213
Publication Date(Web):29 JUL 2005
DOI:10.1002/marc.200500241
Summary: The metallocenes rac-C2H4(Ind)2ZrCl2 (1), rac-Me2Si(Ind)2ZrCl2 (2), and rac-Me2Si(2-Me-benz[e]Ind)2ZrCl2 (3) efficiently copolymerize propene and 5-vinyl-2-norbornene (VNB). 1 and 2 give a high VNB content and high productivities, whereas 3 gives moderate incorporation. Surprisingly, precatalysts 1 and 2, which have very closely related structures, showed very different reactivities toward VNB, with 1 having a greater affinity for VNB than for propene. The copolymers are quantitatively converted into polyolefins with polar functionalities.
Co-reporter:Yann Sarazin;Gerhard Fink;Klaus Hauschild;Gerhard Fink;Yann Sarazin;Klaus Hauschild
Macromolecular Rapid Communications 2005 Volume 26(Issue 15) pp:
Publication Date(Web):3 AUG 2005
DOI:10.1002/marc.200590030
Co-reporter:Robyn K. J. Bott, Max Hammond, Peter N. Horton, Simon J. Lancaster, Manfred Bochmann and Peter Scott
Dalton Transactions 2005 (Issue 22) pp:3611-3613
Publication Date(Web):14 Oct 2005
DOI:10.1039/B509807F
Octahedral titanium and zirconium complexes based on salicyloxazoline ligands with sterically demanding ortho-substituents provide a new family of extremely active ethene polymerization catalysts [up to 108 g PE (mol bar h)−1] which are in some cases “single site”.
Co-reporter:Mark D. Hannant, Mark Schormann, David L. Hughes, Manfred Bochmann
Inorganica Chimica Acta 2005 Volume 358(Issue 5) pp:1683-1691
Publication Date(Web):15 March 2005
DOI:10.1016/j.ica.2004.11.012
Protonation of (MeCNC6H3Pr2i-2,6)2 (PriDAD) with [H(OEt2)2][H2N{B(C6F5)3}2] affords the immonium salt [PriDADH][H2N{B(C6F5)3}2] (1) which shows intra- and inter-ionic NH⋯F hydrogen bonding in the solid state. Protolysis of ZnEt2 with [PriDADH][H2N{B(C6F5)3}2] gives [(PriDAD)ZnEt][H2N{B(C6F5)3}2] as a reactive intermediate. Repeated recrystallizations allowed the isolation of the dimeric zinc hydroxo complex [{(PriDAD)Zn(μ-OH)}2][H2N{B(C6F5)3}2]2 (2) which shows asymmetric OH-bridges. The analogous reaction of [Ph*DADH][H2N{B(C6F5)3}2] [Ph*DAD = (MeCNC6H3Ph2-2,6)2 with ZnMe2 or M{N(SiMe3)2gives the three-coordinate Zn and Cd compounds [(Ph*DAD)ZnMe][H2N{B(C6F5)3}2] (5) and [(Ph*DAD)M–N(SiMe3)2][H2N{B(C6F5)3}2] (6, M = Zn; 7, M = Cd). The crystal structures of 1, 2 and 6 are reported.Protonation of diazadienes (DAD) with [H(OEt2)2][H2N{B(C6F5)3}2] gives salts of the extremely weakly coordinating amido-diborate anion, [DADH][H2N{B(C6F5)3}2], which show inter-ionic NH⋯F hydrogen bonding in the solid state. The compounds react with zink alkyls and amides to give three- and four-coordinate complexes such as [{(PriDAD)Zn(μ-OH)}2][H2N{B(C6F5)3}2]2 and [(Ph*DAD)ZnR][H2N{B(C6F5)3}2] [R = Me, N(SiMe3)2; Ph*DAD = (MeCNC6H3Ph2-2,6)2]. Here, too, inter-ionic hydrogen bonding to organic fluorine atoms is an important structural element.
Co-reporter:Timothy J. Woodman, Yann Sarazin, Shaun Garratt, Gerhard Fink, Manfred Bochmann
Journal of Molecular Catalysis A: Chemical 2005 Volume 235(1–2) pp:88-97
Publication Date(Web):1 July 2005
DOI:10.1016/j.molcata.2005.03.017
Homoleptic allyl complexes of divalent metals, MII[1,3-C3H3(SiMe3)2]2 (1, M = Cr; 2, M = Fe; 3, M = Ni; 4, M = Co) activated with methylaluminoxane (MAO) have been tested as catalysts for the polymerization of norbornene. Whereas the iron and cobalt systems were poorly active, both the nickel and chromium complexes were very productive and gave high molecular weight poly(norbornene)s, although the polymers generated by Cr and Ni differed significantly in the degree of stereoregularity. CrII[1,3-C3H3(SiMe3)2]2 polymerized ethylene in the absence of any cocatalyst but, surprisingly, was deactivated by MAO, whereas 1/B(C6F5)3 showed moderate activity at 20 °C under 1 bar of ethylene. NMR experiments suggest that B(C6F5)3 acts as a one-electron oxidant to generate the [CrIII(allyl)2]+ cation. Although ethylene/norbornene copolymerizations were possible with 1/B(C6F5)3, the activity was much lower than in ethylene homopolymerizations, and only limited amounts of norbornene could be incorporated. The catalytic behavior of this Cr(II) precursor contrasts sharply with that of the Cr(IV) alkyl complex Cr(CH2SiMe3)4 (5)/MAO which polymerizes ethylene but reacts with norbornene to give oligomers, mostly trimers–pentamers. On the other hand, (5)/MAO is highly active for ethylene/norbornene copolymerizations and gives high molecular weight copolymers. Significantly, catalyst activity increases with increasing norbornene concentration. The copolymers show block-structure, and incorporated norbornene is present at the least as di-norbornene units, even at incorporation levels as low as 10 mol%. At higher norbornene concentrations, NNN sequences prevail.The catalytic behavior of Cr(II) and Cr(IV) catalyst precursors for the polymerization of ethylene and norbornene are compared. Cr(allyl)2/MAO catalysts polymerize norbornene but not ethylene, whereas Cr(II) undergoes one-electron transfer with B(C6F5)3 to generate [CrIII(allyl)2]+ which readily polymerizes ethylene. By contrast, the Cr(IV) complex Cr(CH2SiMe3)4/MAO oligomerizes norbornene but is highly active for ethylene/norbornene copolymerizations to high molecular weight copolymers with up to 60 mol% norbornene content. Significantly, activity increases with norbornene concentration.
Co-reporter:Fuquan Song, Roderick D. Cannon and Manfred Bochmann
Chemical Communications 2004 (Issue 5) pp:542-543
Publication Date(Web):05 Feb 2004
DOI:10.1039/B314845A
Whereas the rates of propene polymerisation catalysed by zirconocene ion pairs are strongly anion-dependent, hexene polymerisations are not; the findings demonstrate the existence of very different kinetic regimes for two closely related reactions.
Co-reporter:Musa Said, David L. Hughes and Manfred Bochmann
Dalton Transactions 2004 (Issue 3) pp:359-360
Publication Date(Web):07 Jan 2004
DOI:10.1039/B315711C
Treatment of the N–P ligand ArPN(SiMe3)2 with TiCl4 affords the imido-bridged binuclear titanium complex [TiCl2(THF)(μ-NArP)]2
(ArP
=
m-C6H4PR2) which reacts with Ni(0) or Pd(II) to give heterotrinuclear compounds, while activation with methylaluminoxane generates a new type of imido-based ethene polymerisation catalyst that is tolerant of –PR2 functional groups.
Co-reporter:Manfred Bochmann
Journal of Organometallic Chemistry 2004 Volume 689(Issue 24) pp:3982-3998
Publication Date(Web):29 November 2004
DOI:10.1016/j.jorganchem.2004.07.006
The nature of the counteranion is an essential component of metallocene polymerisation catalysts. Detailed mechanistic investigations show how the anion is able to determine the activity and, in many cases, also the stereoselectivity of the catalyst. This review summarises recent advances in mechanistic understanding of well defined metallocene catalysts based on ion pairs [L2ZrR+ ⋯X−] and describes recent insights in ion mobility and kinetics of alkene polymerisation processes. The interplay of ligand structure and nature of the counteranions demonstrates a fascinating versatility and subtlety that continually challenge our ability to rationalise and predict catalyst performance.For over 40 years metallocene complexes of titanium, zirconium and hafnium have been studied as catalysts in olefin polymerisations and are now of major industrial importance. This review summarises recent advances in mechanistic understanding of well defined metallocene catalysts based on ion pairs [L2ZrR+ ⋯ X−]. The interplay of ligand structure and nature of the counteranions demonstrates a fascinating subtlety that continually challenges our ability to rationalise and predict catalyst performance.
Co-reporter:Antonio Rodriguez-Delgado;Mark D. Hannant;Simon J. Lancaster
Macromolecular Chemistry and Physics 2004 Volume 205(Issue 3) pp:
Publication Date(Web):9 FEB 2004
DOI:10.1002/macp.200300143
Summary: The influence of perfluoroborate anions on catalyst activity and stereospecificity has been determined for propene polymerisations at 20–60 °C. For C2-symmetric ansa-zirconocenes, activities were strongly ligand-dependent, with systems activated by TIBA/Ph3C+X−, where TIBA = AliBu3, X = {Z[B(C6F5)3]2}, and Z = CN or NH2, giving significantly higher activities than methylaluminoxane under comparable conditions. For high-activity catalysts at 60 °C, {H2N[B(C6F5)3]2}− proved superior to {CN[B(C6F5)3]2}−, presumably due to the higher thermal stability of the amidoborate. The influence of counteranions on PP stereospecificity for these catalysts is negligible. On the other hand, the conformationally flexible catalyst, (2-PhInd)2ZrCl2/TIBA/Ph3C+X−, shows not only a strong anion dependence of its activity, but also significant variation in poly(propylene) isotacticity, mainly due to the formation of small fractions of highly isotactic (mmmm > 70%) PP. This effect was particularly pronounced for {CN[B(C6F5)3]2}− and the dianion, {Ni[CNB(C6F5)3]4}2−.
Co-reporter:Elaine Farrow, Yann Sarazin, David L. Hughes, Manfred Bochmann
Journal of Organometallic Chemistry 2004 Volume 689(Issue 24) pp:4624-4629
Publication Date(Web):29 November 2004
DOI:10.1016/j.jorganchem.2004.07.012
The reaction of Cp2Zr(OPri)2 with [H(OEt2)2][H2N{B(C6F5)3}2] in dichloromethane at room temperature gives [Cp2Zr(OPri)(HOPri)]+[H2N{B(C6F5)3}2]− · Et2O in high yield. The crystal structure is reported. The complex contains a short Zr-alkoxide and a longer Zr-alcohol bond; the OH group of the coordinated isopropanol is hydrogen-bonded to a diethyl ether molecule. The complex initiates the polymerisation of propylene oxide, most probably via a cationic mechanism.The reaction of Cp2Zr(OPri)2 with [H(OEt2)2][H2N{B(C6F5)3}2] in gives [Cp2Zr(OPri)(HOPri)]+[H2N{B(C6F5)3}2]− · Et2O in high yield. The complex initiates the polymerisation of propylene oxide, most probably via a cationic mechanism.
Co-reporter:Shaun Garratt Dr.;Antonio Guerrero;David L. Hughes Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 16) pp:
Publication Date(Web):15 MAR 2004
DOI:10.1002/anie.200353787
Better rubber: The initiator system [Zn(C6F5)2]/tBuCl for isobutene/isoprene copolymerizations (see scheme) gave products with high molecular weights, significantly increased isoprene incorporation, and minimal cross-linking.
Co-reporter:Fuquan Song;Mark D. Hannant;Roderick D. Cannon
Macromolecular Symposia 2004 Volume 213(Issue 1) pp:173-186
Publication Date(Web):29 JUN 2004
DOI:10.1002/masy.200450917
The olefin polymerisation activity of metallocene catalysts strongly depends on the counteranion provided by the activator system. The relative activities of a number of new diborate anions [Z(BAr3)2]− have been quantified (Z = CN, NH2, N(CN)2; Ar = C6F5 or o-C6F4C6F5). The kinetic parameters for the initiation, propagation and termination steps of propene polymerisations catalysed by (SBI)ZrCl2 have been determined using quenched-flow kinetic and batch techniques [SBI = rac-Me2Si(1-Ind)2]. Comparison of two activator systems, (i) CPh3[B(C6F5)4] / triisobutylaluminium (TIBA) and (ii) methylaluminoxane (MAO) shows, surprisingly, that the concentration of species actively involved in chain growth at any one time is comparable for both systems, although the MAO-activated catalyst is about 20 times less active than the borate system. It is concluded that the counteranion remains sufficiently strongly bound to the metal centre throughout the chain growth sequence to modulate the energetics of monomer insertion. A model suggesting that the monomer binding follows an associative interchange (Ia) mechanism is proposed.
Co-reporter:Shaun Garratt Dr.;Antonio Guerrero;David L. Hughes Dr.
Angewandte Chemie 2004 Volume 116(Issue 16) pp:
Publication Date(Web):15 MAR 2004
DOI:10.1002/ange.200453787
Metallorganische Zink-Lewis-Säuren wie [Zn(C6F5)2] sind hocheffiziente Initiatoren in Isopren-Isobuten-Copolymerisationen (siehe Schema). Gegenüber klassischen Katalysatorsystemen ergeben sich Copolymere mit überraschend hohem Isoprengehalt bei hohen Molekulargewichten und geringer Vernetzung.
Co-reporter:Mark D. Hannant, Mark Schormann and Manfred Bochmann
Dalton Transactions 2002 (Issue 22) pp:4071-4073
Publication Date(Web):16 Oct 2002
DOI:10.1039/B208165M
Reaction between DADZnR2 and either B(C6F5)3 or ZnR2 and [DADH][B(C6F5)4] affords three-coordinate alkyl and amide zinc cations which are active for the ring opening polymerisation of epoxides and ε-caprolactone [DAD =
(MeCNC6H3Pri2-2,6)2].
Co-reporter:Mark Schormann;Timothy J. Woodman
Israel Journal of Chemistry 2002 Volume 42(Issue 4) pp:283-293
Publication Date(Web):8 MAR 2010
DOI:10.1560/JL3X-DXE1-CD6R-FCB0
The synthesis of new lanthanide allyl complexes of enhanced stability and solubility in saturated hydrocarbons based on silyl-substituted allyl ligands is reported. Thus the potassium salt K(CH2CHCHSiMe3) (1) reacts with YCl3 in tetrahydrofuran to give the tris-allyl complex Y(CH2CHCHSiMe3)3 (2), while K(CH2CHCHSiMe2tBu) (3) affords Y(CH2CHCHSiMe2tBu)3(THF)1.5 (4). Slow re-crystallization of 4 from light petroleum in the presence of tert-butylcyanide led to multiple insertion to give the sec-amido complex Y{NHC(tBu)(CH)3SiMe2tBu}2{η2-NHC(tBu)CH=CHCH2SiMe2tBu)CH(CHCHSiMe2tBu)CtBuNH}(THF)·(CH3CH(Me)(CH2)2CH3) (5), which was crystallographically characterized. The reaction of ScCl3(THF)3 with two equivalents of Li{1,3-C3H3(SiMe3)2} in tetrahydrofuran gives the bis-allyl complex {1,3-C3H3(SiMe3)2}2Sc(μ-Cl)2Li(THF)2 (6), while the analogous reaction of K{1,3-C3H3(SiMe3)2} (7) with either LaCl3 or YCl3 in tetrahydrofuran affords the bis-allyl complexes MCl{1,3-C3H3(SiMe3)2}2(THF)x (8, M = La, x = 1; 9, M = Y, x = 0). An attempt to prepare the similar neodymium complex gave the mono-allyl complex NdI2{1,3-C3H3(SiMe3)2}(THF)1.25 (10). The reactions of 8 and 9 with triisobutyl aluminum in benzene-d6 show allyl exchange between lanthanide and aluminum. Complexes 8, 9, and 10 have been tested with a variety of activator systems as catalysts for the polymerization of 1,3-butadiene.
Co-reporter:Fuquan Song;Daniela Pappalardo;Anthony F. Johnson;Bernhard Rieger
Journal of Polymer Science Part A: Polymer Chemistry 2002 Volume 40(Issue 10) pp:1484-1497
Publication Date(Web):26 MAR 2002
DOI:10.1002/pola.10230
The copolymerization of propene with 7-methyl-1,6-octadiene (MOD) catalyzed by Cp*TiMe3/B(C6F5)3 (A) and rac-C2H4(Ind)2ZrCl2/methylaluminoxane (B) in toluene under 1 bar propene gave copolymers with unsaturated side chains. Under these conditions, catalyst A produced copolymers with an atactic backbone structure of type 1, with 3.5–19.6 mol % MOD incorporation and weight-average molecular weight = 0.7–2.7 × 105. Using catalyst B, copolymers 2 with 0.4–3.8 mol % MOD incorporation were prepared. The comonomer incorporation was a linear function of the feed ratio. The titanium catalyst A had a significantly higher affinity for MOD than the sterically more hindered zirconocene B. Postpolymerization modification of the side-chain CC bond allowed the facile introduction of a wide variety of functional groups. Epoxidation and especially ozonolysis of the CC bond, to give CHO and COOH functionalized copolymers, proved to be very facile routes to functionalized polypropenes. According to monitoring by NMR, most of these transformations proceed in an essentially quantitative conversion. As an example of potential applications of such polymers, polypropenes with covalently attached dyes were prepared that are suitable for blending. © 2002 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 40: 1484–1497, 2002
Co-reporter:Timothy J. Woodman, Mark Thornton-Pett and Manfred Bochmann
Chemical Communications 2001(Issue 4) pp:NaN330-330
Publication Date(Web):2001/02/01
DOI:10.1039/B009016F
Warming mixtures of
(CpR)Zr(η3-C4H7)(η
4-C4H6) and
B(C6F5)3 leads to complete transfer of all
three C6F5 substituents of a
B(C6F5)3 molecule to give borole-bridged
triple-decker complexes with a Zr2C4B core, a
zwitterionic structure and an unusually strong Zr–F donor
interaction.
Co-reporter:Chiranjib Banerjee, David L. Hughes, Manfred Bochmann and Thomas Nann
Dalton Transactions 2012 - vol. 41(Issue 24) pp:NaN7248-7248
Publication Date(Web):2012/04/26
DOI:10.1039/C2DT30283G
Indium phosphide nanofibres were grown from a single-molecule precursor, [(PhCH2)2InP(SiMe3)2]2, using hot injection techniques by a solution–liquid–solid (SLS) process, under “surfactant-free” conditions and without the use of protic additives. The fibres are 85–95 nm in diameter and grow from In metal droplets of 100 nm diameter. The length of the nanofibres is a function of the precursor injection temperature (rather than the growth temperature) and can be varied from 6000 nm at 210 °C to 1000 nm at 310 °C. The indium metal tip can be readily removed under mild, non-etching conditions by treatment with thiophenol–P(SiMe3)3 mixtures.
Co-reporter:Dragoş-Adrian Roşca, Joseph A. Wright and Manfred Bochmann
Dalton Transactions 2015 - vol. 44(Issue 48) pp:NaN20807-20807
Publication Date(Web):2015/11/13
DOI:10.1039/C5DT03930D
Gold, the archetypal “noble metal”, used to be considered of little interest in catalysis. It is now clear that this was a misconception, and a multitude of gold-catalysed transformations has been reported. However, one consequence of the long-held view of gold as inert metal is that its organometallic chemistry contains many “unknowns”, and catalytic cycles devised to explain gold's reactivity draw largely on analogies with other transition metals. How realistic are such mechanistic assumptions? In the last few years a number of key compound classes have been discovered that can provide some answers. This Perspective attempts to summarise these developments, with particular emphasis on recently discovered gold(III) complexes with bonds to hydrogen, oxygen, alkenes and CO ligands.
Co-reporter:Alexander S. Romanov, Dawei Di, Le Yang, Julio Fernandez-Cestau, Ciaran R. Becker, Charlotte E. James, Bonan Zhu, Mikko Linnolahti, Dan Credgington and Manfred Bochmann
Chemical Communications 2016 - vol. 52(Issue 38) pp:NaN6382-6382
Publication Date(Web):2016/04/12
DOI:10.1039/C6CC02349E
Linear two-coordinate copper complexes of cyclic (alkyl)(amino)carbenes (CAAC)CuX (X = halide) show photoluminescence with solid-state quantum yields of up to 96%; in contrast to previously reported Cu photoemitters the emission is independent of temperature over the range T = 4–300 K and occurs very efficiently by prompt rather than delayed fluorescence, with lifetimes in the sub-nanosecond range.
Co-reporter:Nicky Savjani, Simon J. Lancaster, Sean Bew, David L. Hughes and Manfred Bochmann
Dalton Transactions 2011 - vol. 40(Issue 5) pp:NaN1090-1090
Publication Date(Web):2010/12/16
DOI:10.1039/C0DT01134G
Compounds of the new tetrafluorophthalimido anion, [C6F4(CO)2N]−, are readily accessible by treatment of tetrafluorophthalimide with either LiNPri2 or mixtures of NEt3 and Me3ECl (E = Si or Sn), to give C6F4(CO)2N-X (X = Li 3, SiMe34, and SnMe35). The reaction of the trimethylsilyl derivative 4 with AgF leads cleanly to the ion pair complex [Ag(NCMe)2][Ag(N(CO)2C6F4)2] (6·2MeCN), which contains a linear [Ag{N(CO)2C6F4}2]− anion and a tetracoordinate Ag+ cation. Compound 6 reacts with iodine to give the N-iodo compound C6F4(CO)2NI 7, which crystallises as an acetonitrile adduct. Treatment of 6 with LAuCl affords LAu{N(CO)2C6F4} (L = Ph3P 8a, Cy3P 8b, or THT 9), whereas the reaction with AuCl in acetonitrile affords the heterobinuclear compound [Ag(MeCN)2][Au{N(CO)2C6F4}2]·MeCN (10·3MeCN). The tetrafluorophthalimido ligand is not readily displaced by donor ligands; however, the addition of B(C6F5)3(Et2O) to a diethyl ether solution of 8a leads to the salt [Au(PPh3)2][N{COB(C6F5)3}2C6F4)] 11. The analogous reaction of (THT)Au{N(CO)2C6F4} with B(C6F5)3 in toluene in the presence of excess norbornene (nb) gives [Au(nb)3][N{COB(C6F5)3}2C6F4)] 12. Compounds 11 and 12 contain a new non-coordinating phthalimido-bridged diborate anion with O-bonded boron atoms. The crystal structures of compounds 2–11 are reported.
Co-reporter:Nora Carrera, Nicky Savjani, Jason Simpson, David L. Hughes and Manfred Bochmann
Dalton Transactions 2011 - vol. 40(Issue 5) pp:NaN1019-1019
Publication Date(Web):2010/12/09
DOI:10.1039/C0DT01422B
While most metallic elements across the Periodic Table form stable chelating β-diketiminato complexes, examples of Au(I) are conspicuous by their absence. We report here the reaction of K[HC(F3CCNR)2] with AuCl(PPh3) which provides a rare example of a thermally stable gold(I) diketiminato complex, (Ph3P)Au[RNC(CF3)CH(CF3)CNR] [R = 3,5-C6H3(CF3)2]. The complex is highly fluxional in solution but in the solid state adopts a U-conformation. By contrast, the analogous reaction of K[HC(F3CCNR)2] with CuBr(PPh3)3 gives the rigid 18-electron chelate complex (Ph3P)2Cu[κ2-HC{(CF3)CNR}2].
Co-reporter:L. Rocchigiani, J. Fernandez-Cestau, P. H. M. Budzelaar and M. Bochmann
Chemical Communications 2017 - vol. 53(Issue 31) pp:NaN4361-4361
Publication Date(Web):2017/04/03
DOI:10.1039/C7CC01628J
Selective Au–C bond cleavage and arene–C–H activation in (C^N^C)Au(III) pincer complexes are reversible, leading to a solvent-dependent proton shuttling process. The ether-free cleavage products are non-fluxional and show weak gold(III)–arene interactions commensurate with intermediates postulated for CMD-type arene activation.
Co-reporter:Julio Fernandez-Cestau, Benoît Bertrand, Maria Blaya, Garth A. Jones, Thomas J. Penfold and Manfred Bochmann
Chemical Communications 2015 - vol. 51(Issue 93) pp:NaN16632-16632
Publication Date(Web):2015/09/25
DOI:10.1039/C5CC07523H
The first examples of pyrazine-based gold(III) pincer complexes are reported; their intense photoemissions can be modified by protonation, N-alkylation or metal ions, without the need for altering the ligand framework. Emissions shift from red (77 K) to blue (298 K) due to thermally activated delayed fluorescence (TADF).
Co-reporter:Dragoş-Adrian Roşca, Dan A. Smith and Manfred Bochmann
Chemical Communications 2012 - vol. 48(Issue 58) pp:NaN7249-7249
Publication Date(Web):2012/05/23
DOI:10.1039/C2CC33104G
The gold(III) hydroxide κ3-(C∧N∧C)*Au(OH) reacts with C–H and N–H compounds and arylboronic acids to produce a range of perfluoroaryls, N-heterocyclic and alkynyl compounds in high yields; some of which show unexpectedly strong modulation of their photoluminescence from yellow to blue [(C∧N∧C)* = 2,6-(C6H3But)2pyridine].
Co-reporter:Fabio Ghiotto, Chrysoula Pateraki, John R. Severn, Nic Friederichs and Manfred Bochmann
Dalton Transactions 2013 - vol. 42(Issue 25) pp:NaN9048-9048
Publication Date(Web):2013/03/05
DOI:10.1039/C3DT00107E
The influence of methylaluminoxane (MAO) catalyst activators of different concentrations and preparative histories on the performance of 1-hexene polymerisations was investigated by kinetic methods, using rac-Me2Si(2-Me-Benz[e]Ind)2ZrCl2 as the standard catalyst precursor. Fast sampling and analysis allow the time dependence of monomer conversion and the growth of the number-average polymer molecular weight to be determined at a sufficiently short timescale to make this a feasible method for routine catalyst evaluation. Differences in productivity, polymer molecular weight and active species count are shown to be primarily a linear function of the trimethylaluminium concentration. The results in toluene and heptane as solvents are compared; the data show that the inferior performance in heptane is due to a substantially lower active species concentration.
Co-reporter:Thomas Dann, Dragoş-Adrian Roşca, Joseph A. Wright, Gregory G. Wildgoose and Manfred Bochmann
Chemical Communications 2013 - vol. 49(Issue 86) pp:NaN10171-10171
Publication Date(Web):2013/09/17
DOI:10.1039/C3CC45984E
The bond energy of the unsupported Au–Au bond in the Au(II) dimer [(C∧N∧C)Au]2 and the difference between AuIII–OH and AuIII–H bond enthalpies have been determined experimentally by electrochemical methods, with Au–OH and Au–H complexes showing unexpected differences in their reduction pathways, supported by DFT modelling.

![Ditelluride, bis[2,4,6-tris(1,1-dimethylethyl)phenyl]](http://img.cochemist.com/ccimg/99400/99354-18-4.png)
![Ditelluride, bis[2,4,6-tris(1,1-dimethylethyl)phenyl]](http://img.cochemist.com/ccimg/99400/99354-18-4_b.png)










![Poly(oxy[1,1'-biphenyl]-4,4'-diylcarbonyl[1,1'-biphenyl]-4,4'-diyl)](http://img.cochemist.com/ccimg/122500/122483-17-4.png)
![Poly(oxy[1,1'-biphenyl]-4,4'-diylcarbonyl[1,1'-biphenyl]-4,4'-diyl)](http://img.cochemist.com/ccimg/122500/122483-17-4_b.png)




![9H-Fluorene, 9-[1-(1,3-cyclopentadienyl)-1-methylethyl]-](http://img.cochemist.com/ccimg/135700/135600-73-6.png)
![9H-Fluorene, 9-[1-(1,3-cyclopentadienyl)-1-methylethyl]-](http://img.cochemist.com/ccimg/135700/135600-73-6_b.png)


![Poly[1,4-phenylene(1,2-dioxo-1,2-ethanediyl)-1,4-phenylene-1,2-ethyne
diyl-1,4-phenylene-1,2-ethynediyl]](http://img.cochemist.com/ccimg/122500/122483-15-2.png)
![Poly[1,4-phenylene(1,2-dioxo-1,2-ethanediyl)-1,4-phenylene-1,2-ethyne
diyl-1,4-phenylene-1,2-ethynediyl]](http://img.cochemist.com/ccimg/122500/122483-15-2_b.png)












![21H,23H-porphine, 5,10,15,20-tetrakis[4-(decyloxy)phenyl]-](http://img.cochemist.com/ccimg/94900/94846-70-5.png)
![21H,23H-porphine, 5,10,15,20-tetrakis[4-(decyloxy)phenyl]-](http://img.cochemist.com/ccimg/94900/94846-70-5_b.png)






![Benzenamine,N-[[[4-(1,1-dimethylethyl)phenyl]methyl]diphenylphosphoranylidene]-2,4,6-trimethyl-](http://img.cochemist.com/ccimg/388200/388123-78-2.png)
![Benzenamine,N-[[[4-(1,1-dimethylethyl)phenyl]methyl]diphenylphosphoranylidene]-2,4,6-trimethyl-](http://img.cochemist.com/ccimg/388200/388123-78-2_b.png)








![Diselenide, bis[2,4,6-tris(1,1-dimethylethyl)phenyl]](http://img.cochemist.com/ccimg/20900/20875-32-5.png)
![Diselenide, bis[2,4,6-tris(1,1-dimethylethyl)phenyl]](http://img.cochemist.com/ccimg/20900/20875-32-5_b.png)




![Phenol, 2-[(cyclohexylimino)methyl]-6-(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/215100/215033-70-8.png)
![Phenol, 2-[(cyclohexylimino)methyl]-6-(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/215100/215033-70-8_b.png)












![Zirconium, dichloro[h10-2,4-cyclopentadien-1-ylidene(1-methylethylidene)-9H-fluoren-9-ylidene]-](http://img.cochemist.com/ccimg/130700/130638-44-7.png)
![Zirconium, dichloro[h10-2,4-cyclopentadien-1-ylidene(1-methylethylidene)-9H-fluoren-9-ylidene]-](http://img.cochemist.com/ccimg/130700/130638-44-7_b.png)


























![Phenol, 2-(1,1-dimethylethyl)-6-[[(2,4,6-trimethylphenyl)imino]methyl]-](http://img.cochemist.com/ccimg/500600/500556-51-4.png)
![Phenol, 2-(1,1-dimethylethyl)-6-[[(2,4,6-trimethylphenyl)imino]methyl]-](http://img.cochemist.com/ccimg/500600/500556-51-4_b.png)


![1,3,5-TRI(PROPAN-2-YL)-2-[[2,4,6-TRI(PROPAN-2-YL)PHENYL]DISELANYL]BENZENE](http://img.cochemist.com/ccimg/71600/71518-96-2.png)
![1,3,5-TRI(PROPAN-2-YL)-2-[[2,4,6-TRI(PROPAN-2-YL)PHENYL]DISELANYL]BENZENE](http://img.cochemist.com/ccimg/71600/71518-96-2_b.png)
![Benzene, [(1Z)-2-iodo-1-propen-1-yl]-](/data/chemimg/1181600/29461-68-5.png)
![Benzene, [(1Z)-2-iodo-1-propen-1-yl]-](/data/chemimg/1181600/29461-68-5_b.png)
![1H-Imidazole, 1-[2,6-bis(1-methylethyl)phenyl]-](http://img.cochemist.com/ccimg/25400/25364-47-0.png)
![1H-Imidazole, 1-[2,6-bis(1-methylethyl)phenyl]-](http://img.cochemist.com/ccimg/25400/25364-47-0_b.png)
![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4.png)
![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4_b.png)








![Zirconium,[(dimethylsilylene)bis[(1,2,3,3a,7a-h)-1H-inden-1-ylidene]]dimethyl-](http://img.cochemist.com/ccimg/146900/146814-57-5.png)
![Zirconium,[(dimethylsilylene)bis[(1,2,3,3a,7a-h)-1H-inden-1-ylidene]]dimethyl-](http://img.cochemist.com/ccimg/146900/146814-57-5_b.png)





![N-[(E)-1-carbamoyl-2-(4-chlorophenyl)ethenyl]furan-2-carboxamide](http://img.cochemist.com/ccimg/6100/6088-88-6.png)
![N-[(E)-1-carbamoyl-2-(4-chlorophenyl)ethenyl]furan-2-carboxamide](http://img.cochemist.com/ccimg/6100/6088-88-6_b.png)




![Bis[1,3-bis(trimethylsilyl)cyclopentadienyl]zirconium dichloride](http://img.cochemist.com/ccimg/61000/60938-62-7.png)
![Bis[1,3-bis(trimethylsilyl)cyclopentadienyl]zirconium dichloride](http://img.cochemist.com/ccimg/61000/60938-62-7_b.png)


















![Ditelluride, bis[2,4,6-tris(1-methylethyl)phenyl]](http://img.cochemist.com/ccimg/132000/131922-65-1.png)
![Ditelluride, bis[2,4,6-tris(1-methylethyl)phenyl]](http://img.cochemist.com/ccimg/132000/131922-65-1_b.png)






![Zinc, [5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/478800/14074-80-7.png)
![Zinc, [5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/478800/14074-80-7_b.png)



![Phenol, 2-(1,1-dimethylethyl)-6-[[(pentafluorophenyl)imino]methyl]-](http://img.cochemist.com/ccimg/352100/352033-68-2.png)
![Phenol, 2-(1,1-dimethylethyl)-6-[[(pentafluorophenyl)imino]methyl]-](http://img.cochemist.com/ccimg/352100/352033-68-2_b.png)





![rac-Ethylenebis(4,5,6,7-tetrahydro-1-indenyl)]zirconium dichloride](http://img.cochemist.com/ccimg/100200/100163-29-9.png)
![rac-Ethylenebis(4,5,6,7-tetrahydro-1-indenyl)]zirconium dichloride](http://img.cochemist.com/ccimg/100200/100163-29-9_b.png)







![Disulfide, bis[2,4,6-tris(1-methylethyl)phenyl]](http://img.cochemist.com/ccimg/20900/20875-34-7.png)
![Disulfide, bis[2,4,6-tris(1-methylethyl)phenyl]](http://img.cochemist.com/ccimg/20900/20875-34-7_b.png)






![Tris[N,N-bis(trimethylsilyl)amide]yttrium (III)](http://img.cochemist.com/ccimg/41900/41836-28-6.png)
![Tris[N,N-bis(trimethylsilyl)amide]yttrium (III)](http://img.cochemist.com/ccimg/41900/41836-28-6_b.png)











![Potassium, [1,3-bis(trimethylsilyl)-2-propenyl]-](http://img.cochemist.com/ccimg/221100/221047-77-4.png)
![Potassium, [1,3-bis(trimethylsilyl)-2-propenyl]-](http://img.cochemist.com/ccimg/221100/221047-77-4_b.png)






![Lanthanum Tris[bis(trimethylsilyl)amide]](http://img.cochemist.com/ccimg/35800/35788-99-9.png)
![Lanthanum Tris[bis(trimethylsilyl)amide]](http://img.cochemist.com/ccimg/35800/35788-99-9_b.png)