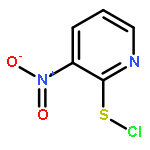
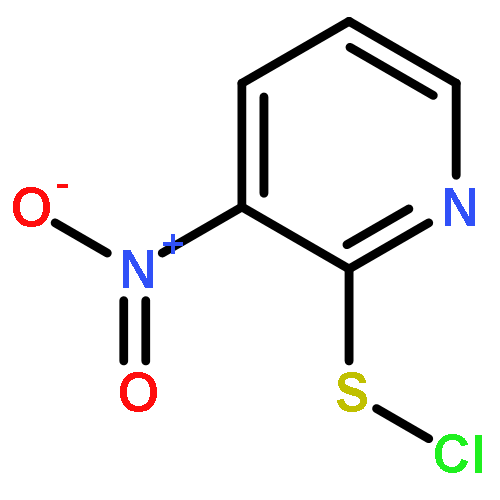
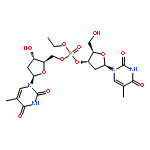
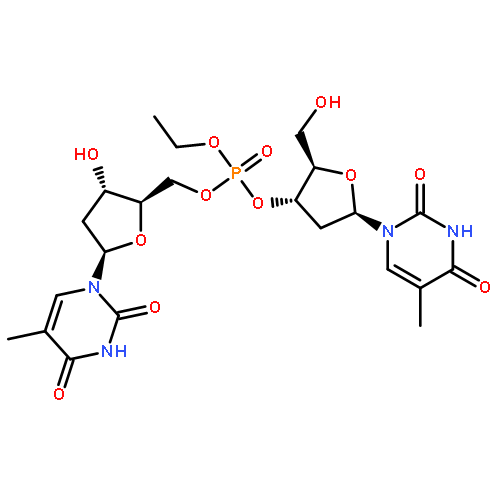
Co-reporter:Shun-ichi Wada, Anna Takesada, Yurie Nagamura, Eri Sogabe, Rieko Ohki, Junsuke Hayashi, Hidehito Urata
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 24(Issue 24) pp:
Publication Date(Web):15 December 2017
DOI:10.1016/j.bmcl.2017.11.018
The conjugation of Aib-containing amphipathic helical peptide with cyclo(-Arg-Gly-Asp-d-Phe-Cys-) (cRGDfC) at the C-terminus of the helix peptide (PI) has been reported to be useful for constructing a carrier for targeted siRNA delivery into cells. In order to explore structure–activity relationships for the development of potential carriers for siRNA delivery, we synthesized conjugates of Aib-containing amphipathic helical peptide with cRGDfC at the N-terminus (PII) and both the N- and C-termini (PIII) of the helical peptide. Furthermore, to examine the influence of PI helical chain length on siRNA delivery, truncated peptides containing 16 (PIV), 12 (PV), and 8 (PVI) amino acid residues at the N-terminus of the helical chain were synthesized. PII and PIII, as well as PI, could deliver anti-luciferase siRNA into cells to induce the knockdown of luciferase stably expressed in cells. In contrast, all of the truncated peptides were unlikely to transport siRNA into cells.Carriers for siRNA: PI: acetyl-KLULKLULKULKAULKLUGC(cRGDfC)-NH2; PII: acetyl-C(cRGDfC)KLULKLULKULKAULKLUG-NH2; PIII: acetyl-C(cRGDfC)KLULKLULKULKAULKLUGC(cRGDfC)-NH2.Download high-res image (47KB)Download full-size image
Co-reporter:Shun-ichi Wada, Masashi Iwata, Yuka Ozaki, Takashi Ozaki, Junsuke Hayashi, Hidehito Urata
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 18) pp:4478-4485
Publication Date(Web):15 September 2016
DOI:10.1016/j.bmc.2016.07.040
To achieve the targeted delivery of siRNA, five conjugates of Aib-containing amphipathic helical peptides with mono-, di-, and trivalent cRGDfC [cyclo(-Arg-Gly-Asp-d-Phe-Cys-)], which is known to bind to αVβ3 integrin, at several positions of the amphipathic helical peptide were designed and synthesized. Among the five conjugates, the monovalent cRGDfC conjugating at position 20 of the amino acid sequence of the helical peptide through the formation of a disulfide bond (PI) and the divalent cRGDfC conjugating at positions 2 and 14 of the amino acid sequence of the helical peptide through the formation of disulfide bonds (PIII) significantly enhanced the delivery of fluorescence-labeled siRNA into A549 cells as the peptide/siRNA complex formed by electrostatic interaction. The cellular uptake of the PI/siRNA complex was mediated by both endocytic and non-endocytic pathways, whereas that of the PIII/siRNA complex was enabled by endocytosis. Furthermore, the cellular uptake of the PI/siRNA complex might involve specific interactions of the RGD group with the αVβ3 integrin receptor. Next, the RNAi effect of the peptide/siRNA complex on luciferase expression in A549-Luc cells was examined. Luciferase expression was significantly decreased in the presence of the complex at the concentration of 1.0 μM PI/10 nM siRNA. In contrast, the PIII/siRNA complex did not show the RNAi effect under the same conditions. However, extending the incubation time led to the suppression of the luciferase expression in the presence of the PIII/siRNA complex. Considering that the cellular uptake of the PIII/siRNA complex is mediated by the endocytic pathway, the release of siRNA from the endosome into the cytosol might require a long time. We present herein a useful and unique tool for the delivery of siRNA.
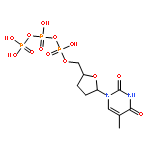
Co-reporter:Shun-ichi Wada, Tomoe Urase, Yuka Hasegawa, Kenta Ban, Aya Sudani, Yui Kawai, Junsuke Hayashi, Hidehito Urata
Bioorganic & Medicinal Chemistry 2014 22(24) pp: 6776-6780
Publication Date(Web):
DOI:10.1016/j.bmc.2014.10.040
Co-reporter:Shun-ichi Wada, Yuki Hashimoto, Yui Kawai, Kaori Miyata, Hirokazu Tsuda, Osamu Nakagawa, Hidehito Urata
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 24) pp:7669-7673
Publication Date(Web):15 December 2013
DOI:10.1016/j.bmc.2013.10.029
A number of cell-penetrating peptides (CPPs) have been characterized and their usefulness as delivery tools has been clarified. As one of the CPPs, model amphipathic peptide (MAP) was developed by integrating both hydrophobic and hydrophilic amino acids in its sequence. In our previous work, we designed MAP(Aib) by replacing five alanine (Ala) residues on the hydrophobic face of the helix in the MAP sequence with α-aminoisobutyric acid (Aib) residues, and the replacement resulted in higher helix propensity, stronger resistance to protease, and higher cell membrane permeability than MAP. As a next step, we examined the efficiency of oligonucleotide (ODN) delivery into cells by MAP(Aib) in comparison with that by MAP. The electrostatically formed MAP(Aib)/ODN complex was more easily taken up by cells than the MAP/ODN complex, and the ODN delivery by MAP(Aib) was via an endocytic pathway. We demonstrated that the incorporation of Aib residues into CPPs enhances the delivery of hydrophilic molecules, such as ODN, into cells.
Co-reporter:Shun-ichi Wada, Yasunari Hitora, Saori Yokoe, Osamu Nakagawa, Hidehito Urata
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 10) pp:3219-3222
Publication Date(Web):15 May 2012
DOI:10.1016/j.bmc.2012.03.054
Using the membrane-modifying peptide, trichorovin-XIIa (TV-XIIa), which is an 11-residual peptaibol isolated from the fungus Trichoderma viride, we synthesized covalent conjugates of 20-mer oligonucleotide with TV-XIIa to examine the potential use of TV-XIIa in cellular delivery. The results indicated that the conjugates were progressively taken up by human lung carcinoma A549 cells. Next, the antisense effects of the conjugates on p53 protein expression were examined. The p53 expression was significantly decreased by ca. 20–50% in the presence of the conjugates upon treatment with the transfection solution at the concentration of 5 μM.
Co-reporter:Shun-ichi Wada, Hirokazu Tsuda, Terumi Okada, Hidehito Urata
Bioorganic & Medicinal Chemistry Letters 2011 21(19) pp: 5688-5691
Publication Date(Web):
DOI:10.1016/j.bmcl.2011.08.030
Co-reporter:Shun-ichi Wada;Teppei Hitomi;Harukuni Tokuda;Reiko Tanaka
Chemistry & Biodiversity 2010 Volume 7( Issue 9) pp:2303-2308
Publication Date(Web):
DOI:10.1002/cbdv.201000147
Abstract
Cancer chemoprevention, the prevention of cancer by ingestion of chemical agents that reduce the risk of carcinogenesis, is one of the potent ways to reduce morbidity and mortality. We have been searching for cancer chemopreventive agents from the leaves and barks of coniferous trees that have been treated as waste in the forestry industry. We have previously reported the isolation of spiro-biflavonoids, named as abiesinols, and a neolignan from the MeOH extract of the bark of Abies sachalinensis. These compounds were tested for their inhibitory effects on the activation of (±)-(E)-methyl-2-[(E)-hydroxyimino]-5-nitro-6-methoxyhex-3-enamide (NOR 1), a nitric oxide (NO) donor, as a primary screening test for anti-tumor initiators. All compounds tested exhibited potent inhibitory effects on NOR 1 activation. Furthermore, abiesinol A, bearing a spiro-biflavonoid skeleton, showed remarkable anti-tumor-initiating activity in the in vivo two-stage mouse skin carcinogenesis test using peroxynitrite (ONOO−; PN) as the initiator and 12-O-tetradecanoylphorbol-13-acetate (TPA) as the promoter.
Co-reporter:Shun-ichi Wada, Yumiko Yasui, Harukuni Tokuda, Reiko Tanaka
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 17) pp:6414-6421
Publication Date(Web):1 September 2009
DOI:10.1016/j.bmc.2009.07.016
We have previously reported the isolation of nine phenolic compounds including three new flavonostilbenes, jezonocinols A, B, and C, from the MeOH extract of the bark of Picea jezoensis var. jezoensis. Further investigation of the MeOH extract led to the isolation of three new stilbene-type compounds and one new 1,4-benzodioxane-type compound, together with seven known phenolic compounds. These compounds were tested for their inhibitory effects on the activation of (±)-(E)-methyl-2-[(E)-hydroxy-imino]-5-nitro-6-methoxy-3-hexemide (NOR 1), a nitric oxide (NO) donor, as a primary screening test for anti-tumor initiators. All compounds tested exhibited potent inhibitory effects on NOR 1 activation. Furthermore, jezonocinol B, the most potent inhibitor of NOR 1 activation, showed remarkable anti-tumor-initiating activity in the in vivo two-stage mouse skin carcinogenesis test using peroxynitrite (ONOO−; PN) as the initiator and 12-O-tetradecanoylphorbol-13-acetate (TPA) as the promoter.
Co-reporter:Shun-ichi Wada;Teppei Hitomi ;Reiko Tanaka
Helvetica Chimica Acta 2009 Volume 92( Issue 8) pp:1610-1620
Publication Date(Web):
DOI:10.1002/hlca.200900032
Abstract
Eight spiro-biflavonoids named abiesinols A–H, along with three neolignans, quercetin, and protocatechuic acid were isolated from the MeOH extract of the bark of Abies sachalinensis. The structures of these phenolic compounds were characterized by spectroscopic methods including NMR and MS. The absolute configurations of the abiesinols were determined by Mosher's method, CD, and NOESY data.
Co-reporter:Shun-ichi Wada, Yumiko Yasui, Teppei Hitomi and Reiko Tanaka
Journal of Natural Products 2007 Volume 70(Issue 10) pp:1605-1610
Publication Date(Web):October 2, 2007
DOI:10.1021/np070104o
The MeOH extract of the bark of Picea jezoensis var. jezoensis was found to show scavenging activity for the DPPH radical. Bioassay-guided fractionation led to the isolation of two new flavonostilbenes named jezonocinols A (1) and B (2) and one nor-flavonostilbene named jezonocinol C (3) together with six known phenolic compounds. Their structures were elucidated by spectroscopic methods, and the absolute configurations of jezonocinols A (1) and B (2) were determined by Mosher’s method, CD, and NOESY data.
Co-reporter:Reiko Tanaka
Chemistry & Biodiversity 2007 Volume 4(Issue 5) pp:991-997
Publication Date(Web):18 MAY 2007
DOI:10.1002/cbdv.200790090
Based on the membrane-modifying peptaibol trichocellin-A-I (1) from Trichoderma viride, we designed a vehicle for the cellular delivery of antisense oligodeoxynucleotides by attaching a (Lys)10 stretch to the C-terminus of 1. The resulting transporter peptide 2, prepared by solid-phase synthesis using Fmoc protocol in combination with amino acid fluorides, was found to be mainly α-helical in solution, in contrast to its precursors 1 and 3. The uptake of the complex formed between carrier 2 and a fluorescence-tagged oligonucleotide, i.e., 4, was studied at different charge ratios by confocal laser-scanning microscopy, using two different eukaryotic cell lines: mouse embryonal fibroblast (NIH3T3) and human lung carcinoma (A549) cells. Peptide 2 readily translocated 4 into the cytoplasms of NIH3T3 cells. However, the peptide/oligonucleotide complex was accumulated around the plasma membrane of the A549 cells.
Co-reporter:Reiko Tanaka
Chemistry & Biodiversity 2006 Volume 3(Issue 4) pp:473-479
Publication Date(Web):19 APR 2006
DOI:10.1002/cbdv.200690050
One new pimarane-type diterpenoid (1) and two new taraxerane-based triterpenoids (2 and 3) were isolated from the bark of Macaranga tanarius, along with seven known compounds. Their structures were identified by spectroscopic methods including NMR and MS analyses. Compounds 1–5 were tested for their in vitro inhibition of DNA topoisomerase II, as well as for their cytotoxicities against human lung carcinoma A549 cells (Table 3). The triterpenoids 2–5 showed strong activities in both assays, but the diterpenoid 1 was only moderately active.
Co-reporter:Shun-ichi Wada;Reiko Tanaka
Chemistry & Biodiversity 2005 Volume 2(Issue 5) pp:689-694
Publication Date(Web):18 MAY 2005
DOI:10.1002/cbdv.200590045
DNA Topoisomerases (Topos) II are target enzymes for anticancer chemotherapeutic drug development. Bioassay-guided fractionation of the CHCl3 extract of the bark of Bischofia javanica led to the isolation of betulinic acid (1) and its derivatives, betulonic acid (2), 3β-O-(Z)-coumaroylbetulinic acid (3), and 3β-O-(E)-coumaroylbetulinic acid (4). These compounds were found to be catalytic inhibitors of Topo II activities with IC50 values ranging from 0.38 to 58 μM. The acylation of the OH group at C(3) of betulinic acid exhibited stronger Topo II inhibitory activity.