Co-reporter:Chenhuan Dong, Zhifu Xie, Yanyan Yu, Jia Li, Junhua Liu, Jingya Li, Lihong Hu
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 12) pp:2688-2696
Publication Date(Web):15 June 2016
DOI:10.1016/j.bmc.2016.04.034
As a follow-up discovery of AMPK activators from natural products, 20S-dammar-24-en-2α,3β,12β,20-tetrol (GP, 1), a dammarane-type triterpenoid, was found to have some favorable metabolic effects on dyslipidemia in Golden Syrian hamsters, and activate AMPKα2β1γ1 by around 2.4 fold with an EC50 of 5.1 μM on molecular level. In order to enhance its potency at AMPK and structure–activity relationship study, GP derivatives were designed, synthesized, and evaluated in pharmacological AMPK activation assays. Structure–activity relationship analysis showed that amine at the 24-position (groups I–IV) effectively and significantly increased the potency and efficacy. GP derivatives 12 and 17–19 exhibited better potency (EC50: 0.3, 0.8, 0.8, and 1.0 μM) and efficacy (fold: 3.2, 2.7, 3.0, and 2.8) in the activation of AMPK heterotrimer α2β1γ1 than positive control (AMP, EC50: 1.6 μM, fold: 3.2). Furthermore, the most potent compounds 12 and 17 obviously inhibited glucose output through increasing the phosphorylation of AMPK, without affecting mitochondrial membrane potential or producing cytotoxicity.
Co-reporter:Junhua Liu, Dakai Chen, Peng Liu, Mengna He, Jia Li, Jingya Li, Lihong Hu
European Journal of Medicinal Chemistry 2014 Volume 79() pp:340-349
Publication Date(Web):22 May 2014
DOI:10.1016/j.ejmech.2014.04.010
•SAR study of PPD has been reported.•Some derivatives showed potent AMPK activation.•12 and 13 exhibited potent inhibition of lipid synthesis.Adenosine 5′-monophosphate-activated protein kinase (AMPK) has been demonstrated as a promising drug target due to its regulatory function in glucose and lipid metabolism. 20(S)-protopanoxadiol (PPD) was firstly identified from high throughput screening as a small molecule activator of AMPK subtype α2β1γ1. In order to enhance its potency on AMPK, a series of PPD derivatives were synthesized and evaluated. Structure–activity relationship study showed that the amine derivatives at the 24-position (groups I–VI) can improve the potency (EC50: 0.7–2.3 μM) and efficacy (fold: 2.5–3.8). Among them, compounds 12 and 13 exhibited the best potency (EC50: 1.2 and 0.7 μM) and efficacy (fold: 3.7 and 3.8). Further study suggested the mechanism of AMPK activation may functioned at the allosteric position, resulting the inhibition of the lipid synthesis in HepG2 cell model.
Co-reporter:Ben-Ren Liao, Hai-Bing He, Ling-Ling Yang, Li-Xin Gao, Liang Chang, Jie Tang, Jing-Ya Li, Jia Li, Fan Yang
European Journal of Medicinal Chemistry 2014 Volume 83() pp:15-25
Publication Date(Web):18 August 2014
DOI:10.1016/j.ejmech.2014.06.011
•Non-phosphorus-based fructose-1,6-bisphosphatase (FBPase) inhibitors, 2,5-diphenyl-1,3,4-oxadiazoles, were explored.•Compounds bearing a linear alkyl with terminal hydrogen-bonding acceptor showed potent inhibition at molecular level.•Compound 22 and 27b inhibit the glucose production in primary rat hepatocytes with an IC50 value of 167.3 μM and 112.5 μM.•Both inhibition and binding mode to the enzyme were investigated by enzymatic kinetics and in silico experiments.With the aim of discovering a novel class of non-phosphorus-based fructose-1,6-bisphosphatase (FBPase) inhibitors, a series of 2,5-diphenyl-1,3,4-oxadiazoles were synthesized based on the hit compound (1) resulting from a high-throughput screening (HTS). Structure–activity relationship (SAR) studies led to the identification of several compounds with comparable inhibitory activities to AMP, the natural allosteric inhibitor of FBPase. Notably, compound 22 and 27b, bearing a terminal carboxyl or 1H-tetrazole, demonstrated remarkable inhibition to gluconeogenesis (GNG). In addition, both inhibition and binding mode to the enzyme were investigated by enzymatic kinetics and in silico experiments for representative compounds 16 and 22.Novel non-phosphorus-based FBPase inhibitors were explored, and some compounds showed comparable inhibitory activities to AMP and remarkable inhibition to gluconeogenesis. Both inhibition and binding mode to the enzyme were investigated.
Co-reporter:Hai-Bing He, Li-Xin Gao, Qi-Feng Deng, Wei-Ping Ma, Chun-Lan Tang, Wen-Wei Qiu, Jie Tang, Jing-Ya Li, Jia Li, Fan Yang
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 23) pp:7237-7242
Publication Date(Web):1 December 2012
DOI:10.1016/j.bmcl.2012.09.040
Protein tyrosine phosphatase 1B (PTP1B) is a major negative regulator of both insulin and leptin signals. For years, inhibiting of PTP1B has been considered to be a potential therapeutics for treating Type 2 diabetes and obesity. Recently, we recognized lithocholic acid (LCA) as a natural inhibitor against PTP1B (IC50 = 12.74 μM) by a vertical screen for the first time. Further SAR research was carried out by synthesizing and evaluating a series of compounds bearing two methyls at C-4 position and a fused heterocycle to ring A. Among them, compound 14b achieved a PTP1B inhibitory activity about eightfold than LCA and a 14-fold selectivity over the homogenous enzyme TCPTP.The synthesis and biological evaluation of a series of heterocyclic ring-substituted 4,4-dimethyl lithocholic acid derivatives are reported for the first time. The active compound 14b displayed potent inhibitory activity on protein tyrosine phosphatase 1B with IC50 value of approximately 1.62 μM.
Co-reporter:Zhe Cheng, An-Feng Chen, Fang Wu, Li Sheng, Han-Kun Zhang, Min Gu, Yuan-Yuan Li, Li-Na Zhang, Li-Hong Hu, Jing-Ya Li, Jia Li
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 16) pp:5915-5924
Publication Date(Web):15 August 2010
DOI:10.1016/j.bmc.2010.06.085
The clinical use of the natural alkaloid berberine (BBR) as an antidiabetic reagent is limited by its poor bioavailability. Our previous work demonstrated that dihydroberberine (dhBBR) has enhanced bioavailability and in vivo efficacy compared with berberine. Here we synthesized the 8,8-dimethyldihydroberberine (Di-Me) with improved stability, and bioavailability over dhBBR. Similar to BBR and dhBBR, Di-Me inhibited mitochondria respiration, increased AMP:ATP ratio, activated AMPK and stimulated glucose uptake in L6 myotubes. In diet-induced obese (DIO) mice, Di-Me counteracted the increased adiposity, tissue triglyceride accumulation and insulin resistance, and improved glucose tolerance at a dosage of 15 mg/kg. Administered to db/db mice with a dosage of 50 mg/kg, Di-Me effectively reduced random fed and fasting blood glucose, improved glucose tolerance, alleviated insulin resistance and reduced plasma triglycerides, with better efficacy than dhBBR at the same dosage. Our work highlights the importance of dihydroberberine analogs as potential therapeutic reagents for type 2 diabetes treatment.8,8-Disubstituted dihydroberberine hydrochlorides were synthesized and evaluated. Di-Me shows enhanced chemical stability, bioavailability and oral efficacy on obese and diabetic mouse models.

![b-D-Glucopyranoside, (3b,12b)-20-[(6-O-b-D-glucopyranosyl-b-D-glucopyranosyl)oxy]-12-hydroxydammar-24-en-3-yl2-O-[6-O-(carboxyacetyl)-b-D-glucopyranosyl]-](http://img.cochemist.com/ccimg/88200/88140-34-5.png)
![b-D-Glucopyranoside, (3b,12b)-20-[(6-O-b-D-glucopyranosyl-b-D-glucopyranosyl)oxy]-12-hydroxydammar-24-en-3-yl2-O-[6-O-(carboxyacetyl)-b-D-glucopyranosyl]-](http://img.cochemist.com/ccimg/88200/88140-34-5_b.png)





















![Methyl (1R,2S,3R,5R,6S,10S,16R,17R)-2,6-dimethyl-20-oxo-8-azahexacyclo[11.5.1.11,5.02,10.03,8.016,19]icos-13(19)-ene-17-carboxylate](http://img.cochemist.com/ccimg/881400/881388-87-0.png)
![Methyl (1R,2S,3R,5R,6S,10S,16R,17R)-2,6-dimethyl-20-oxo-8-azahexacyclo[11.5.1.11,5.02,10.03,8.016,19]icos-13(19)-ene-17-carboxylate](http://img.cochemist.com/ccimg/881400/881388-87-0_b.png)
![Methyl (1S,2S,3R,5R,6S,10S)-2,6-dimethyl-21-oxo-14-oxa-8-azahexacyclo[11.6.1.11,5.02,10.03,8.017,20]henicosa-13(20),17-diene-18-carboxylate](http://img.cochemist.com/ccimg/874300/874201-05-5.png)
![Methyl (1S,2S,3R,5R,6S,10S)-2,6-dimethyl-21-oxo-14-oxa-8-azahexacyclo[11.6.1.11,5.02,10.03,8.017,20]henicosa-13(20),17-diene-18-carboxylate](http://img.cochemist.com/ccimg/874300/874201-05-5_b.png)
![Methyl (1R,2S,3R,5R,6S,10S)-2,6-dimethyl-20-oxo-8-azahexacyclo[11.5.1.11,5.02,10.03,8.016,19]icosa-13(19),16-diene-17-carboxylate](http://img.cochemist.com/ccimg/857700/857672-34-5.png)
![Methyl (1R,2S,3R,5R,6S,10S)-2,6-dimethyl-20-oxo-8-azahexacyclo[11.5.1.11,5.02,10.03,8.016,19]icosa-13(19),16-diene-17-carboxylate](http://img.cochemist.com/ccimg/857700/857672-34-5_b.png)






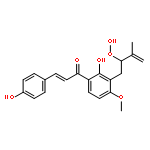
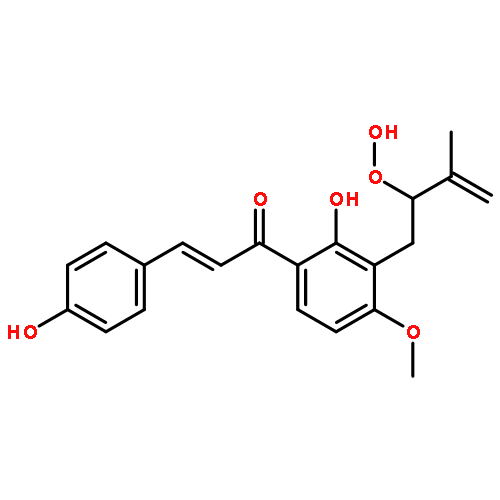


![Benzaldehyde, 4-[(6-hydroxyhexyl)oxy]-](/data/chemimg/996000/96735-91-0.png)
![Benzaldehyde, 4-[(6-hydroxyhexyl)oxy]-](/data/chemimg/996000/96735-91-0_b.png)
![(2E)-1-{3-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-2,4-dihydroxyphenyl}-3-(4-hydroxyphenyl)prop-2-en-1-one](http://img.cochemist.com/ccimg/63000/62949-76-2.png)
![(2E)-1-{3-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-2,4-dihydroxyphenyl}-3-(4-hydroxyphenyl)prop-2-en-1-one](http://img.cochemist.com/ccimg/63000/62949-76-2_b.png)
![(E)-1-[2-HYDROXY-4-METHOXY-3-(3-METHYLBUT-2-ENYL)PHENYL]-3-(4-HYDROXYPHENYL)PROP-2-EN-1-ONE](http://img.cochemist.com/ccimg/56000/55912-03-3.png)
![(E)-1-[2-HYDROXY-4-METHOXY-3-(3-METHYLBUT-2-ENYL)PHENYL]-3-(4-HYDROXYPHENYL)PROP-2-EN-1-ONE](http://img.cochemist.com/ccimg/56000/55912-03-3_b.png)



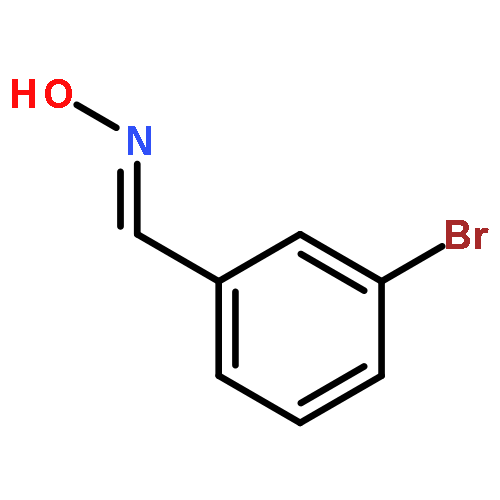


![(9R)-8,8-dimethyl-2-oxo-9,10-dihydro-2H,8H-pyrano[2,3-f]chromen-9-yl (2Z)-2-methylbut-2-enoate](http://img.cochemist.com/ccimg/19500/19427-82-8.png)
![(9R)-8,8-dimethyl-2-oxo-9,10-dihydro-2H,8H-pyrano[2,3-f]chromen-9-yl (2Z)-2-methylbut-2-enoate](http://img.cochemist.com/ccimg/19500/19427-82-8_b.png)
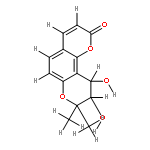
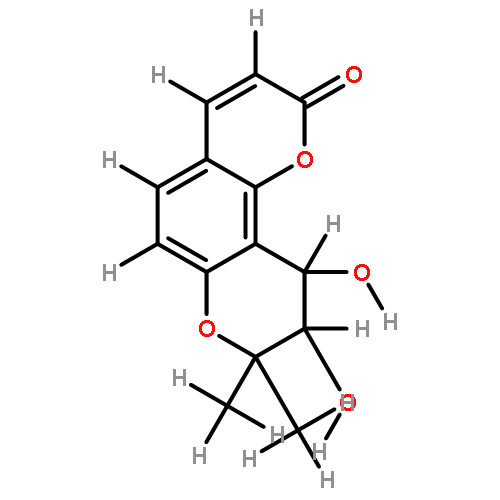
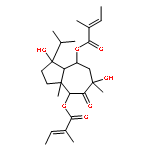
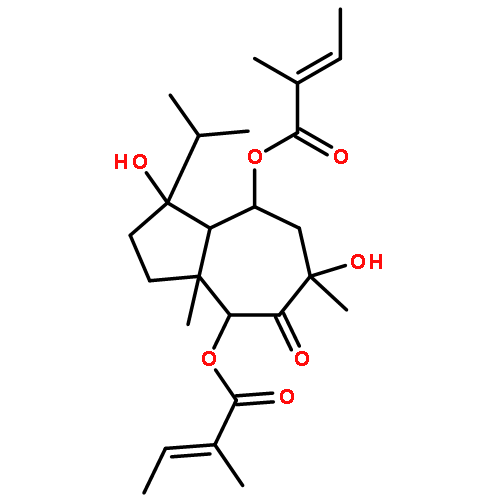
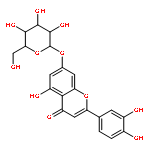

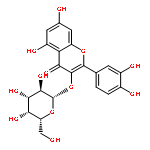
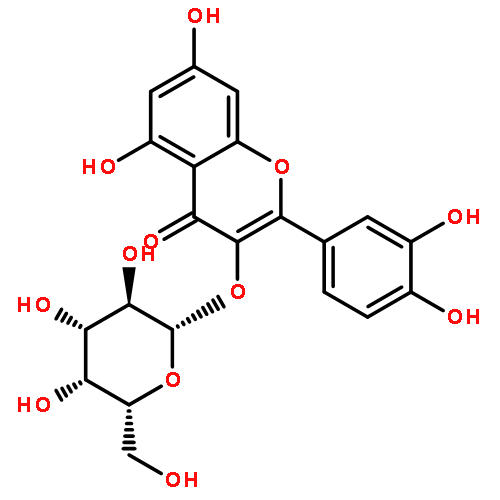
![2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2R,3S,4R,5S,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydropyran-2-yl]oxy-chromen-4-one](http://img.cochemist.com/ccimg/500/482-35-9.png)
![2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2R,3S,4R,5S,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydropyran-2-yl]oxy-chromen-4-one](http://img.cochemist.com/ccimg/500/482-35-9_b.png)