Co-reporter:Katharina Bilotti, Erin E. Kennedy, Chuxuan Li, Sarah Delaney
DNA Repair 2017 Volume 59(Volume 59) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.dnarep.2017.08.010
•Packaged DNA presents a complex substrate for DNA glycosylases.•We found that hOGG1 is able to excise lesions near the ends of nucleosomal DNA.•Transient DNA unwrapping promotes excision of otherwise occluded lesions.•Solution accessibility of DNA bases is not fully predictive of hOGG1 activity.•Acetylation of H2B increases hOGG1 product formation at the studied lesion site.If unrepaired, damage to genomic DNA can cause mutations and/or be cytotoxic. Single base lesions are repaired via the base excision repair (BER) pathway. The first step in BER is the recognition and removal of the nucleobase lesion by a glycosylase enzyme. For example, human oxoguanine glycosylase 1 (hOGG1) is responsible for removal of the prototypic oxidatively damaged nucleobase, 8-oxo-7,8-dihydroguanine (8-oxoG). To date, most studies of glycosylases have used free duplex DNA substrates. However, cellular DNA is packaged as repeating nucleosome units, with 145 base pair segments of DNA wrapped around histone protein octamers. Previous studies revealed inhibition of hOGG1 at the nucleosome dyad axis and in the absence of chromatin remodelers. In this study, we reveal that even in the absence of chromatin remodelers or external cofactors, hOGG1 can initiate BER at positions off the dyad axis and that this activity is facilitated by spontaneous and transient unwrapping of DNA from the histones. Additionally, we find that solution accessibility as determined by hydroxyl radical footprinting is not fully predictive of glycosylase activity and that histone tails can suppress hOGG1 activity. We therefore suggest that local nuances in the nucleosome environment and histone-DNA interactions can impact glycosylase activity.
Co-reporter:Ji Huang and Sarah Delaney
The Journal of Physical Chemistry B 2016 Volume 120(Issue 18) pp:4195-4203
Publication Date(Web):April 26, 2016
DOI:10.1021/acs.jpcb.6b00927
Expansion of a trinucleotide repeat (TNR) sequence is the molecular signature of several neurological disorders. The formation of noncanonical structures by the TNR sequence is proposed to contribute to the expansion mechanism. Furthermore, it is known that the propensity for expansion increases with repeat length. In this work, we use calorimetry to describe the thermodynamic parameters (ΔH, TΔS, and ΔG) of the noncanonical stem-loop hairpins formed by the TNR sequences (CAG)n and (CTG)n, as well as the canonical (CAG)n/(CTG)n duplexes, for n = 6–14. Using a thermodynamic cycle, we calculated the same thermodynamic parameters describing the process of converting from noncanonical stem-loop hairpins to a canonical duplex. In addition to these thermodynamic analyses, we used spectroscopic techniques to determine the rate at which the noncanonical structures convert to duplex and the activation enthalpy ΔH⧧ describing this process. We report that the thermodynamic parameters of unfolding the stem-loop (CTG)n and (CAG)n hairpins, along with the thermodynamic and kinetic properties of hairpin to duplex conversion, do not proportionally correspond to the increase in length, but rather show a unique pattern that depends on whether the sequence has an even or odd number of repeats.
Co-reporter:Ji Huang, Craig J. Yennie, and Sarah Delaney
Chemical Research in Toxicology 2015 Volume 28(Issue 12) pp:2325
Publication Date(Web):November 16, 2015
DOI:10.1021/acs.chemrestox.5b00330
Defining the biological consequences of oxidative DNA damage remains an important and ongoing area of investigation. At the foundation of understanding the repercussions of such damage is a molecular-level description of the action of DNA-processing enzymes, such as polymerases. In this work, we focus on a secondary, or hyperoxidized, oxidative lesion of dG that is formed by oxidation of the primary oxidative lesion, 2′-deoxy-8-oxo-7,8-dihydroguanosine (8-oxodG). In particular, we examine incorporation into DNA of the diastereomers of the hyperoxidized guanosine triphosphate lesion spiroiminodihydantoin-2′-deoxynucleoside-5′-triphosphate (dSpTP). Using kinetic parameters, we describe the ability of the Klenow fragment of Escherichia coli DNA polymerase I lacking 3′ → 5′ exonuclease activity (KF–) to utilize (S)-dSpTP and (R)-dSpTP as building blocks during replication. We find that both diastereomers act as covert lesions, similar to a Trojan horse: KF– incorporates the lesion dNTP opposite dC, which is a nonmutagenic event; however, during the subsequent replication, it is known that dSp is nearly 100% mutagenic. Nevertheless, using kpol/Kd to define the nucleotide incorporation specificity, we find that the extent of oxidation of the dGTP-derived lesion correlates with its ability to be incorporated into DNA. KF– has the highest specificity for incorporation of dGTP opposite dC. The selection factors for incorporating 8-oxodGTP, (S)-dSpTP, and (R)-dSpTP are 1700-, 64000-, and 850000-fold lower, respectively. Thus, KF– is rigorous in its discrimination against incorporation of the hyperoxidized lesion, and these results suggest that the specificity of cellular polymerases provides an effective mechanism to avoid incorporating dSpTP lesions into DNA from the nucleotide pool.
Co-reporter:Kelly M. Schermerhorn and Sarah Delaney
Accounts of Chemical Research 2014 Volume 47(Issue 4) pp:1238
Publication Date(Web):March 19, 2014
DOI:10.1021/ar400275a
Our cellular genome is continuously exposed to a wide spectrum of exogenous and endogenous DNA damaging agents. These agents can lead to formation of an extensive array of DNA lesions including single- and double-stranded breaks, inter- and intrastrand cross-links, abasic sites, and modification of DNA nucleobases. Persistence of these DNA lesions can be both mutagenic and cytotoxic, and can cause altered gene expression and cellular apoptosis leading to aging, cancer, and various neurological disorders. To combat the deleterious effects of DNA lesions, cells have a variety of DNA repair pathways responsible for restoring damaged DNA to its canonical form. Here we examine one of those repair pathways, the base excision repair (BER) pathway, a highly regulated network of enzymes responsible for repair of modified nucleobase and abasic site lesions.The enzymes required to reconstitute BER in vitro have been identified, and the repair event can be considered to occur in two parts: (1) excision of the modified nucleobase by a DNA glycosylase, and (2) filling the resulting “hole” with an undamaged nucleobase by a series of downstream enzymes. DNA glycosylases, which initiate a BER event, recognize and remove specific modified nucleobases and yield an abasic site as the product. The abasic site, a highly reactive BER intermediate, is further processed by AP endonuclease 1 (APE1), which cleaves the DNA backbone 5′ to the abasic site, generating a nick in the DNA backbone. After action of APE1, BER can follow one of two subpathways, the short-patch (SP) or long-patch (LP) version, which differ based on the number of nucleotides a polymerase incorporates at the nick site. DNA ligase is responsible for sealing the nick in the backbone and regenerating undamaged duplex.Not surprisingly, and consistent with the idea that BER maintains genetic stability, deficiency and/or inactivity of BER enzymes can be detrimental and result in cancer. Intriguingly, this DNA repair pathway has also been implicated in causing genetic instability by contributing to the trinucleotide repeat expansion associated with several neurological disorders.Within this Account, we outline the chemistry of the human BER pathway with a mechanistic focus on the DNA glycosylases that initiate the repair event. Furthermore, we describe kinetic studies of many BER enzymes as a means to understand the complex coordination that occurs during this highly regulated event. Finally, we examine the pitfalls associated with deficiency in BER activity, as well as instances when BER goes awry.
Co-reporter:Kelly M. Schermerhorn and Sarah Delaney
Biochemistry 2013 Volume 52(Issue 43) pp:
Publication Date(Web):September 30, 2013
DOI:10.1021/bi401218r
Apurinic/apyrimidinic endonuclease 1 (APE1) is an Mg2+-dependent enzyme responsible for incising the DNA backbone 5′ to an apurinic/apyrimidinic (AP) site. Here, we use rapid quench flow (RQF) techniques to provide a comprehensive kinetic analysis of the strand-incision activity (kchemistry) of APE1 acting on an authentic AP site along with two widely used analogs, a reduced AP site and a tetrahydrofuran (THF) site. In the presence of biologically relevant Mg2+, APE1 incises all three substrates at a rate faster than the resolution of the RQF, ≥700 s–1. To obtain quantitative values of kchemistry and to facilitate a comparison of the authentic substrate versus the substrate analogs, we replaced Mg2+ with Mn2+ or Ni2+ or introduced a mismatch 5′ to the lesion site. Both strategies were sufficient to slow kchemistry and resulted in rates within the resolution of the RQF. In all cases where quantitative rates were obtained, kchemistry for the reduced AP site is indistinguishable from the authentic AP site. Notably, there is a small decrease, ∼1.5-fold, in kchemistry for the THF site relative to the authentic AP site. These results highlight a role in strand incision for the C1′ oxygen of the AP site and warrant consideration when designing experiments using substrate analogs.
Co-reporter:Craig J. Yennie and Sarah Delaney
Chemical Research in Toxicology 2012 Volume 25(Issue 8) pp:1732
Publication Date(Web):July 10, 2012
DOI:10.1021/tx300190a
Guanidinohydantoin (Gh) is a hyperoxidized DNA lesion produced by oxidation of 8-oxo-7,8-dihydroguanine (8-oxoG). Previous work has shown that Gh is potently mutagenic in both in vitro and in vivo coding for G → T and G → C transversion mutations. In this work, analysis by circular dichroism shows that the Gh lesion does not significantly alter the global structure of a 15-mer duplex and that the DNA remains in the B-form. However, we find that Gh causes a large decrease in the thermal stability, decreasing the duplex melting temperature by ∼17 °C relative to an unmodified duplex control. Using optical melting analysis and differential scanning calorimetry, the thermodynamic parameters describing duplex melting were also determined. We find that the Gh lesion causes a dramatic decrease in the enthalpic stability of the duplex. This enthalpic destabilization is somewhat tempered by entropic stabilization; yet, Gh results in an overall decrease in thermodynamic stability of the duplex relative to a control that lacks DNA damage, with a ΔΔG° of −7 kcal/mol. These results contribute to our understanding of the consequences of hyperoxidation of G and provide insight into how the thermal and thermodynamic destabilization caused by Gh may influence replication and/or repair of the lesion.
Co-reporter:Catherine B. Volle, Daniel A. Jarem, and Sarah Delaney
Biochemistry 2012 Volume 51(Issue 1) pp:
Publication Date(Web):December 7, 2011
DOI:10.1021/bi201552s
In the phenomenon of trinucleotide repeat (TNR) expansion, an important interplay exists between DNA damage repair of 8-oxo-7,8-dihydroguanine (8-oxoG) and noncanonical structure formation. We show that TNR DNA adapts its structure to accommodate 8-oxoG. Using chemical probe analysis, we find that CAG repeats composing the stem–loop arm of a three-way junction alter the population of structures in response to 8-oxoG by positioning the lesion at or near the loop. Furthermore, we find that oligonucleotides composed of odd-numbered repeat sequences, which form populations of two structures, will also alter their structure to place 8-oxoG in the loop. However, sequences with an even number of repeats do not display this behavior. Analysis by differential scanning calorimetry indicates that when the lesion is located within the loop, there are no significant changes to the thermodynamic parameters as compared to the DNA lacking 8-oxoG. This contrasts with the enthalpic destabilization observed when 8-oxoG is base-paired to C and indicates that positioning 8-oxoG in the loop avoids the thermodynamic penalty associated with 8-oxoG base-pairing. Since formation of stem–loop hairpins is proposed to facilitate TNR expansion, these results highlight the importance of defining the structural consequences of DNA damage.
Co-reporter:Catherine B. Volle and Sarah Delaney
Biochemistry 2012 Volume 51(Issue 49) pp:
Publication Date(Web):November 16, 2012
DOI:10.1021/bi301416v
Trinucleotide repeats (TNRs) occur throughout the genome, and their expansion has been linked to several neurodegenerative disorders, including Huntington’s disease. TNRs have been studied using both oligonucleotides and plasmids; however, less is know about how repetitive DNA responds to genomic packaging. Here, we investigate the behavior of CAG/CTG repeats incorporated into nucleosome core particles, the most basic unit of chromatin packaging. To assess the general interaction between CAG/CTG repeats and the histone core, we determined the efficiency with which various TNR-containing DNA substrates form nucleosomes, revealing that even short CAG/CTG tracts are robust incorporators. However, the presence of the Huntington gene flanking sequence (htt) decreases the rate of incorporation. Enzymatic and chemical probing revealed repositioning of the DNA in the nucleosome as the number of CAG/CTG repeats increased, regardless of the flanking sequence. Notably, the periodicity of the repeat tract remained unchanged as a function of length and is consistently 10.7 bp per helical turn. In contrast, the periodicity of the nonrepetitive flanking sequence varies and is smaller than the repeat tract at ∼10.0–10.5 bp per turn. Furthermore, while the CAG/CTG repeats remain as a canonical duplex in the nucleosome, nucleosome formation causes kinking in a secondary repeat tract in the htt gene, comprised of CCG/CGG repeats. This work highlights the innate ability of CAG/CTG repeats to incorporate and to position in nucleosomes and how that behavior is modulated by the htt flanking sequence. In addition, it illuminates the differences in packaging of healthy and diseased length repeat tracts within the genome.
Co-reporter:Amalia Ávila Figueroa, Douglas Cattie, and Sarah Delaney
Biochemistry 2011 Volume 50(Issue 21) pp:
Publication Date(Web):April 28, 2011
DOI:10.1021/bi200397b
Expansion of trinucleotide repeats (TNR) has been implicated in the emergence of neurodegenerative diseases. Formation of non-B conformations such as hairpins by these repeat sequences during DNA replication and/or repair has been proposed as a contributing factor to expansion. In this work we employed a combination of fluorescence, chemical probing, optical melting, and gel shift assays to characterize the structure of a series of (CTG)n sequences and the kinetic parameters describing their interaction with a complementary sequence. Our structure-based experiments using chemical probing reveal that sequences containing an even or odd number of CTG repeats adopt stem-loop hairpins that differ from one another by the absence or presence of a stem overhang. Furthermore, we find that this structural difference dictates the rate at which the TNR hairpins convert to duplex with a complementary CAG sequence. Indeed, the rate constant describing conversion to (CAG)10/(CTG)n duplex is slower for sequences containing an even number of CTG repeats than for sequences containing an odd number of repeats. Thus, when both the CAG and CTG hairpins have an even number of the repeats, they display a longer lifetime relative to when the CTG hairpin has an odd number of repeats. The difference in lifetimes observed for these TNR hairpins has implications toward their persistence during DNA replication or repair events and could influence their predisposition toward expansion. Taken together, these results contribute to our understanding of trinucleotide repeats and the factors that regulate persistence of hairpins in these repetitive sequences and conversion to canonical duplex.
Co-reporter:Daniel A. Jarem, Lauren V. Huckaby and Sarah Delaney
Biochemistry 2010 Volume 49(Issue 32) pp:
Publication Date(Web):July 15, 2010
DOI:10.1021/bi1007782
The trinucleotide repeat sequence CGG/CCG is known to expand in the human genome. This expansion is the primary pathogenic signature of fragile X syndrome, which is the most common form of inherited mental retardation. It has been proposed that formation of non-B conformations by the repetitive sequence contributes to the expansion mechanism. It is also known that the CGG/CCG repeat sequence of healthy individuals, which is not prone to expansion, contains AGG/CCT interruptions every 8−11 CGG/CCG repeats. Using DNA containing 19 or 39 CGG repeats, we have found that both the position and number of interruptions modulate the non-B conformation adopted by the repeat sequence. Analysis by chemical probes revealed larger loops and the presence of bulges for sequences containing interruptions. Additionally, using optical analysis and calorimetry, the effect of these structural changes on the thermodynamic stability of the conformation has been quantified. Notably, changing even one nucleotide, as occurs when CGG is replaced with an AGG interruption, causes a measurable decrease in the stability of the conformation adopted by the repeat sequence. These results provide insight into the role interruptions may play in preventing expansion in vivo and also contribute to our understanding of the relationship between non-B conformations and trinucleotide repeat expansion.
Co-reporter:Daniel A. Jarem, Nicole R. Wilson and Sarah Delaney
Biochemistry 2009 Volume 48(Issue 28) pp:
Publication Date(Web):June 15, 2009
DOI:10.1021/bi9007403
Triplet repeat sequences, such as CAG/CTG, expand in the human genome to cause several neurological disorders. As part of the expansion process the formation of non-B DNA conformations by the repeat sequence has previously been proposed. Furthermore, the base excision repair enzyme 7,8-dihydro-8-oxoguanine glycosylase (OGG1) has recently been implicated in the repeat expansion [Kovtun, I. V., Liu, Y., Bjoras, M., Klugland, A., Wilson, S. H., and McMurray, C. T. (2007) Nature 447, 447−452]. In this work we have found that the non-B conformation adopted by (CAG)10, a hairpin, is hypersusceptible to DNA damage relative to the (CAG)10/(CTG)10 duplex and, in particular, that a hot spot for DNA damage exists. Specifically, we find that a single guanine in the loop of the hairpin is susceptible to modification by peroxynitrite. Interestingly, we find that human OGG1 (hOGG1) is able to excise 7,8-dihydro-8-oxoguanine (8-oxoG) from the loop of a hairpin substrate, albeit with a marked decrease in efficiency relative to duplex substrates; the hOGG1 enzyme removes 8-oxoG from the loop of a hairpin with a rate that is ∼700-fold slower than that observed for DNA duplex. Thus, while damage is preferentially generated in the loop of the hairpin, DNA repair is less efficient. These observed structure-dependent patterns of DNA damage and repair may contribute to the OGG1-dependent mechanism of trinucleotide repeat expansion.



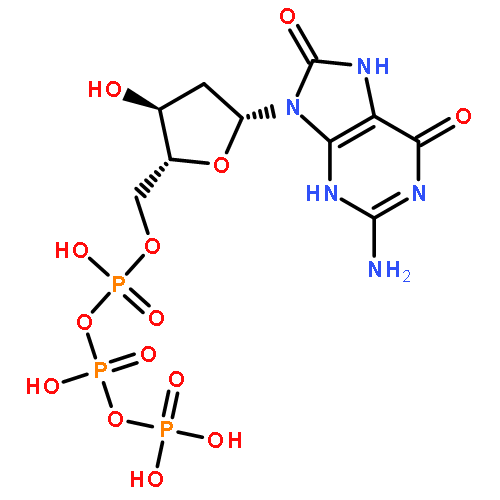
![3H-Imidazo[2,1-i]purine](http://img.cochemist.com/ccimg/13900/13875-63-3.png)
![3H-Imidazo[2,1-i]purine](http://img.cochemist.com/ccimg/13900/13875-63-3_b.png)


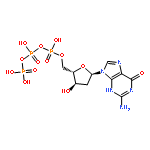



























![Guanidine, [3-(2-deoxy-β-D-erythro-pentofuranosyl)-2,5-dioxo-4-imidazolidinyl]-](/data/chemimg/119300/265986-31-0.png)
![Guanidine, [3-(2-deoxy-β-D-erythro-pentofuranosyl)-2,5-dioxo-4-imidazolidinyl]-](/data/chemimg/119300/265986-31-0_b.png)