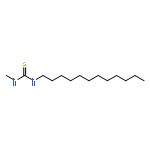
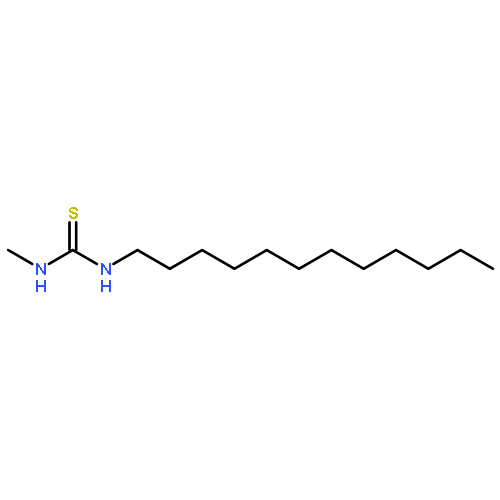
Co-reporter:Yiwu Zheng, Jing Ren, Yaqi Wu, Xiaoting Meng, Yibing Zhao, and Chuanliu Wu
Bioconjugate Chemistry October 18, 2017 Volume 28(Issue 10) pp:2620-2620
Publication Date(Web):September 18, 2017
DOI:10.1021/acs.bioconjchem.7b00471
Targeted prodrugs exploiting cleavable linkers capable of responding to endogenous stimuli have increasingly been explored for cancer therapy. Successful application of these prodrug designs relies on the manipulation of both stability and responsiveness of the cleavable linkers, which, however, are difficult to be finely regulated, particularly for acid-responsive acylhydrazone bonds. Here we developed a new class of peptide-bridged twin-acylhydrazone linkers (PTA linkers) displaying both an ultrahigh stability and a rapid responsiveness—highly stable in neutral and acidic conditions due to the effect of cooperativity between the two acylhydrazone bonds, easily cleavable in acidic conditions after enzymatically triggered unlocking of the two bonds. Moreover, our study shows the design of PTA-linked prodrugs and the proof-of-concept application of the PTA linkers for site-specific release of anticancer drugs into cancer cells.
Co-reporter:Wei Gao;Jinghui Wang;Tao Li;Yibing Zhao
Analytical Chemistry January 3, 2017 Volume 89(Issue 1) pp:937-944
Publication Date(Web):December 1, 2016
DOI:10.1021/acs.analchem.6b04096
Study on the processes of the thiol-mediated disulfide exchange reactions on the cell surface is not only important to our understanding of extracellular natural bioreduction processes but to the development of novel strategies for the intracellular delivery of synthetic bioactive molecules. However, disulfide-bonded probes have their intrinsic inferiority in exploring the detailed exchange pathway because of the bidirectional reactivity of disulfide bonds toward reactive thiols. In this work, we developed thioether-bonded fluorescent probes that enable us to explore thiol-mediated thioether (and disulfide) exchange reactions on the cell surface through fluorescence recovery and/or cell imaging. We demonstrated that our thioether-bonded probes can be efficiently cleaved through thiol-thioether exchanges with exofacial protein thiols and/or glutathione (GSH) efflux. The exchanges mainly take place on the cell surface, and GSH efflux-mediated exchange reactions can take place without the requirement of pre-exchanges of the probes with cell surface-associated protein thiols. On the basis of our founder methodology, for the first time we demonstrated the interplay of exofacial protein thiols and GSH efflux on the cleavage of external thioether-bonded compounds. Moreover, given that an understanding of the process of GSH efflux and the mechanism on which it relies is crucial to our understanding of the cellular redox homeostasis and the mechanism of multidrug resistance, we expect that our thioether-bonded probes and strategies would greatly benefit the fundamental study of GSH efflux in living cells.
Co-reporter:Tao Li, Wei Gao, Jingjing Liang, Mirao Zha, Yaqi Chen, Yibing Zhao, and Chuanliu Wu
Analytical Chemistry August 15, 2017 Volume 89(Issue 16) pp:8501-8501
Publication Date(Web):July 17, 2017
DOI:10.1021/acs.analchem.7b02084
In recent years, delivery systems based on the incorporation of thiols/disulfides have been extensively explored to promote the intracellular delivery of biological cargoes. However, it remains unclear about the detailed processes of thiol–disulfide exchanges taking place on the cell surface and how the exchange reactions promote the cellular uptake of cargoes bearing thiols or disulfide bonds. In this work, we report the rational design of biscysteine motif-containing peptide probes with substantially different ring-closing property and how these peptide probes were employed to explore the thiol–disulfide exchanges on the cell surface. Our results show that extensive thiol–disulfide exchanges between peptides and exofacial protein thiols/disulfides are involved in the cellular uptake of these peptide probes, and importantly glutathione (GSH) exported from the cytosols participates extensively in the exchange reactions. Cysteine−glycine−cysteine (CGC)-containing peptide probes can be more efficiently taken up by cells compared to other probes, and we suggested that the driving force for the superior cellular uptake arises from very likely the unique propensity of the CGC motif in forming doubly bridged disulfide bonds with exofacial proteins. Our probe-based strategy provides firsthand information on the detailed processes of the exchange reactions, which would be of great benefit to the development of delivery systems based on the extracellular thiol–disulfide exchanges for intracellular delivery of biologics.
Co-reporter:Yaqi Chen;Tao Li;Jianguo Li;Shiyan Cheng;Jinghui Wang;Chandra Verma;Yibing Zhao
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 8) pp:1921-1929
Publication Date(Web):2017/02/22
DOI:10.1039/C6OB02786E
Peptides have been promising molecular scaffolds for the development of potential therapeutics with high affinity and specificity to biomacromolecules. However, their inherent proteolytic instability significantly hampers their biological applications. Strategies that can stabilize peptides against proteolytic digestion on the basis of noncovalent interactions—without extensive manipulation of the sequence or use of unnatural residues—are greatly desired. In this work, we developed a general, convenient, and efficient strategy for the stabilization of peptides against proteolysis, which involves noncovalent π–π interactions between aromatic amino acid residues in peptides and synthetic electron-deficient aromatics (NDI), together with the implication of steric hindrance (from the bulky NDI moiety), and the enhancement of peptide α-helicity. This strategy is complementary in concept to the conventional well-established covalent approaches for peptide stabilization, and is thus promising for being utilized, in combination with the latter ones, to circumvent the problem of proteolytic instability of peptides. We envisioned that this study should provide invaluable guidelines to the design and synthesis of organic molecule–peptide hybrids with significantly improved proteolytic resistance, and benefit the development of peptide therapeutics and probes.
Co-reporter:Yiwu Zheng;Zhuoru Li;Jing Ren;Weidong Liu;Yaqi Wu;Yibing Zhao
Chemical Science (2010-Present) 2017 vol. 8(Issue 4) pp:2547-2552
Publication Date(Web):2017/03/28
DOI:10.1039/C6SC05710A
Disulfide-rich peptides are emerging as potential templates for drug design applications. However, the synthesis and reengineering of disulfide-rich peptides are challenging, owing to the complexity of the oxidative folding process involving a number of diverse isomeric structures. Novel disulfide-rich peptide scaffolds that are not besieged by their disulfide isomers are still greatly desired. In this work, we report the design and synthesis of a novel class of artificial disulfide-rich peptide scaffolds with precisely defined disulfide patterns and a minimized number of isomers. In theory, natural peptides with three disulfide bonds have 15 possible isomers. By rationally engineering the thiol-framework of a peptide containing six cysteines with penicillamines and a dithiol amino acid, we demonstrated, for the first time, that the total number of isomers formed after oxidative folding can be decreased to a minimum of two (i.e., from 15 to 2). As fewer isomeric folds are involved in the oxidative folding, the pathway of the folding becomes more concise and the yield of the artificial scaffolds is substantially increased compared to that of its six-cysteine-containing analogue, which makes the artificial disulfide-rich scaffolds (with only 2 predefined isomeric folds) extremely promising for being exploited as structurally complex templates for the design of peptide therapeutics and ligands.
Co-reporter:Jun Liu;Shunqing Zhou;Jing Ren;Yibing Zhao
Analyst (1876-Present) 2017 vol. 142(Issue 23) pp:4522-4528
Publication Date(Web):2017/11/20
DOI:10.1039/C7AN01280B
There is increasing evidence indicating that lysosomal H2O2 is closely related to autophagy and apoptotic pathways under both physiological and pathological conditions. Therefore, fluorescent probes that can be exploited to visualize H2O2 in lysosomes are potential tools for exploring diverse roles of H2O2 in cells. However, functional exploration of lysosomal H2O2 is limited by the lack of fluorescent probes capable of compatibly sensing H2O2 under weak acidic conditions (pH = 4.5) of lysosomes. Lower spatial resolution of the fluorescent visualization of lysosomal H2O2 might be caused by the interference of signals from cytosolic and mitochondrial H2O2, as well as the non-specific distribution of the probes in cells. In this work, we developed a lysosome-locating and acidic-pH-activatable fluorescent probe for the detection and visualization of H2O2 in lysosomes, which consists of a H2O2-responsive boronate unit, a lysosome-locating morpholine group, and a pH-activatable benzorhodol fluorophore. The response of the fluorescent probe to H2O2 is significantly more pronounced under acidic pH conditions than that under neutral pH conditions. Notably, the present probe enables the fluorescence sensing of endogenous lysosomal H2O2 in living cells without external stimulations, with signal interference from the cytoplasm and other intracellular organelles being negligible.
Co-reporter:Jinhong Gao;Zhuoru Li;Ouyang Zhang;Yibing Zhao
Analyst (1876-Present) 2017 vol. 142(Issue 7) pp:1084-1090
Publication Date(Web):2017/03/27
DOI:10.1039/C7AN00019G
Inspired by the primitive role of lipopolysaccharide (LPS) and taking advantage of the membrane-philic properties of amphiphilic gold nanoparticles (AuNPs), we established a facile and efficient fluorescence turn-on detection strategy for LPS. Upon binding onto the surface of liposomes, LPS can tailor the accessibility of liposomes towards AuNPs, reminiscent of its primitive function on the surface of bacteria. Thus, while the fluorescence of the dyes labeled on liposomes can be markedly quenched by the membrane-philic AuNPs, the quenching effect can be efficiently prevented by the surface-bound LPS. The de-quenching effect is highly selective to LPS, relative to other negatively charged bio-analytes, which is due to not only the extremely high affinity of LPS to lipid bilayers, but also the unique molecular structure of LPS. Furthermore, this easy-to-construct method offers a limit of detection of ∼0.65 nM, which is comparable to that obtained from the superb synthetic sensors for LPS reported in the literature. This study would open up a new route for the design of sensing systems for LPS exploiting its unique structural pattern and primitive function.
Co-reporter:Yunyang Ling, Jing Ren, Tao Li, Yibing Zhao and Chuanliu Wu
Chemical Communications 2016 vol. 52(Issue 24) pp:4533-4536
Publication Date(Web):25 Feb 2016
DOI:10.1039/C6CC00369A
POEGMA-based disulfide-containing fluorescent probes were designed and synthesized to enable the real-time and quantitative analysis of the (bio)reduction of disulfides within the polymeric backbones in complex redox media and in the presence of cells. This study lights up the way of exploiting bioreduction in the extracellular spaces for the delivery of hydrophobic drugs.
Co-reporter:Jun Liu, Jing Ren, Xiaojia Bao, Wei Gao, Chuanliu Wu, and Yibing Zhao
Analytical Chemistry 2016 Volume 88(Issue 11) pp:5865
Publication Date(Web):May 6, 2016
DOI:10.1021/acs.analchem.6b00654
Intracellular H2O2 plays an important role in regulating a variety of cellular functions. Fluorescent probes that can make response to intracellular levels of H2O2 would provide valuable tools for revealing the functions of H2O2 in living organisms. However, traditional pH-insensitive probes and lysosome-targetable probes can only provide spatially nonspecific visualization of intracellular H2O2 and specific sensing of lysosomal H2O2, respectively. In this work, we developed a H2O2-responsive and pH-switchable fluorescent probe (HP-L1) which can make response sequentially to intracellular H2O2 and lysosomal pH. The fluorescent probe is comprised of a H2O2-responsive boronate moiety and a pH-switchable spirobenzopyran fluorophore. When the probe was applied for intracellular H2O2 sensing, only fluorescent emission from lysosomes is visible, and the fluorescence from other regions is not able to be obviously detected, which is due to the pH-switchable property of the spirobenzopyran fluorophore. Thus, the developed fluorescence probe enables the spatially confined (i.e., lysosome-specific) visualization of the intracellular H2O2. We envisioned that this kind of fluorescent probe (or the proposed sensing strategy) would allow the visualization of the overall levels of intracellular H2O2 without interferences of possible fluorescent signals from other sources (e.g., dyes for cellular staining and multiplex analysis).
Co-reporter:Shuo Liu;Jianbin Chen;Xiaojia Bao;Tao Li;Yunyang Ling;Chunxiang Li;Dr. Chuanliu Wu;Dr. Yibing Zhao
Chemistry – An Asian Journal 2016 Volume 11( Issue 12) pp:1811-1820
Publication Date(Web):
DOI:10.1002/asia.201600250
Abstract
Herein, we report a strategy for exploiting nanoscale metal–organic frameworks (nano-MOFs) as templates for the layer-by-layer (LbL) assembly of polyelectrolytes. Because small-molecule drugs or imaging agents cannot be efficiently encapsulated by polyelectrolyte nanocapsules, we investigated two promising and biocompatible polymers (comb-shaped polyethylene glycol (PEG) and hyperbranched polyglycerol-based PEG) for the conjugation of model drugs and imaging agents, which were then encapsulated inside the nano-MOF-templated nanocapsules. Furthermore, we also systemically explored the release kinetics of the encapsulated conjugates, and examined how the encapsulation and/or release processes could be controlled by varying the composition and architecture of the polymers. We envision that our nano-MOFs-templated nanocapsules, through combining with small-molecule–polymer conjugates, will represent a new type of delivery system that could open up new opportunities for biomedical applications.
Co-reporter:Jinhong Gao, Ouyang Zhang, Jing Ren, Chuanliu Wu, and Yibing Zhao
Langmuir 2016 Volume 32(Issue 6) pp:1601-1610
Publication Date(Web):January 21, 2016
DOI:10.1021/acs.langmuir.6b00035
The presence of large hydrophobic aromatic residues in cell-penetrating peptides or proteins has been demonstrated to be advantageous for their cell penetration. This phenomenon has also been observed when AuNPs were modified with peptides containing aromatic amino acids. However, it is still not clear how the presence of hydrophobic and aromatic groups on the surface of anionic AuNPs affects their interaction with lipid bilayers. Here, we studied the interaction of a range of anionic amphiphilic AuNPs coated by different combinations of hydrophobic and anionic ligands with four different types of synthetic lipid vesicles. Our results demonstrated the important role of the surface aromatic or bulky groups, relative to the hydrocarbon chains, in the interaction of anionic AuNPs with lipid bilayers. Hydrophobic interaction itself arising from the insertion of aromatic/bulky ligands on the surface of AuNPs into lipid bilayers is sufficiently strong to cause overt disruption of lipid vesicles and cell membranes. Moreover, by comparing the results obtained from AuNPs coated with aromatic ligands and cyclohexyl ligands lacking aromaticity respectively, we demonstrated that the bulkiness of the terminal groups in hydrophobic ligands instead of the aromatic character might be more important to the interaction of AuNPs with lipid bilayers. Finally, we further correlated the observation on model liposomes with that on cell membranes, demonstrating that AuNPs that are more disruptive to the more negatively charged liposomes are also substantially more disruptive to cell membranes. In addition, our results revealed that certain cellular membrane domains that are more susceptible to disruption caused by hydrophobic interactions with nanoparticle surfaces might determine the threshold of AuNP-mediated cytotoxicity.
Co-reporter:Shuo Liu;Jianbin Chen;Xiaojia Bao;Tao Li;Yunyang Ling;Chunxiang Li;Dr. Chuanliu Wu;Dr. Yibing Zhao
Chemistry – An Asian Journal 2016 Volume 11( Issue 12) pp:
Publication Date(Web):
DOI:10.1002/asia.201600788
Co-reporter:Yiwu Zheng; Linxiang Zhai; Yibing Zhao
Journal of the American Chemical Society 2015 Volume 137(Issue 48) pp:15094-15097
Publication Date(Web):November 20, 2015
DOI:10.1021/jacs.5b10779
Precise disulfide pairing in synthetic peptides usually is achieved using orthogonal protecting group strategies or relies on primary sequence manipulation. Orthogonal disulfide pairing technology should be promising for directing the rational folding of multicyclic peptides from the fully reduced peptides. Here, we report a discovery on the orthogonality between heterodisulfide pairing of cysteine (Cys) and penicillamine (Pen) and formation of Cys-Cys/Pen-Pen homodisulfides. The orthogonal Cys-Pen disulfide pairing can be exploited for highly selective production of certain (multi)cyclic structures (or even a sole structure without isomers) through direct oxidation in air or thiol–disulfide exchanges in redox media. This strategy makes rational folding of multicyclic peptides without protecting groups, sequence manipulation, and complex synthetic reactions a reality, thus providing invaluable assets to peptide communities, and should greatly benefit the development of multicyclic peptide therapeutics and ligands.
Co-reporter:Yujie Li, Tao Li, Jinghui Wang, Xiaojia Bao, Yibing Zhao and Chuanliu Wu
Polymer Chemistry 2015 vol. 6(Issue 45) pp:7862-7870
Publication Date(Web):17 Sep 2015
DOI:10.1039/C5PY01080B
Direct conjugation of peptides to large polymers has been a promising strategy to overcome the intrinsic limitation of peptides in terms of circulation lifetime, proteolytic stability, and cell/tissue uptake efficiency. As a comb-shaped derivative of polyethylene glycol (PEG), poly(oligoethylene glycol monomethyl ether methacrylate) (pOEGMA) has recently attracted a great deal of interest, which leads to the development of new polymer–protein conjugates. However, pOEGMA has rarely been exploited for the construction of polymer–peptide conjugates. This study reports a new class of copolymer–peptide conjugates which exploits pOEGMA as the polymeric backbone, into which multiple copies of peptides are incorporated. Peptides that can efficiently modulate the protein interaction of p53 and Mdm2/Mdm4 were selected as a model to explore the effect of the composition of OEGMA-based copolymers, the crosslinker length, and especially the number of conjugated peptides on the binding affinity to the target protein. We demonstrated that the binding affinity of the conjugated peptides to the protein is slightly increased (or at least not compromised) even when a large number of peptides (up to 45) are displayed on copolymers because of the positive contribution of multivalent effects. Compared to linear or branched PEGs, OEGMA-based polymers offer superior flexibility in the structure for finely modulating the bioactivity of pOEGMA–peptide conjugates, which is of particular interest for further biomedicine applications. This study not only generates unique multivalent peptide-displayed copolymer conjugates with therapeutic potential, but also provides insight into the design of other therapeutic polymer–peptide conjugates.
Co-reporter:Yaqi Chen, Chaoqiong Yang, Tao Li, Miao Zhang, Yang Liu, Marc A. Gauthier, Yibing Zhao, and Chuanliu Wu
Biomacromolecules 2015 Volume 16(Issue 8) pp:
Publication Date(Web):July 9, 2015
DOI:10.1021/acs.biomac.5b00567
The contribution of noncovalent interactions to the stability of naturally occurring peptides and proteins has been generally acknowledged, though how these can be rationally manipulated to improve the proteolytic stability of synthetic peptides remains to be explored. In this study, a platform to enhance the proteolytic stability of peptides was developed by controllably dimerizing them into α-helical dimers, connected by two disulfide bonds. This platform not only directs peptides toward an α-helical conformation but permits control of the interfacial hydrophobic interactions between the peptides of the dimer. Using two model dimeric systems constructed from the N-terminal α-helix of RNase A and known inhibitors for the E3 ubiquitin ligase MDM2 (and its homologue MDMX), a deeper understanding into the interplay of disulfide bonds, α-helicity, and hydrophobic interactions on enhanced proteolytic stability was sought out. Results reveal that all three parameters play an important role on attaining ultrahigh proteolytic resistance, a concept that can be exploited for the development of future peptide therapeutics. The understanding gained through this study will enable this strategy to be tailored to new peptides because the proposed strategy displays substantial tolerance to sequence permutation. It thus appears promising for conveniently creating prodrugs composed entirely of the therapeutic peptide itself (i.e., in the form of a dimer).
Co-reporter:Shuo Liu, Linxiang Zhai, Chunxiang Li, Yujie Li, Xiangqun Guo, Yibing Zhao, and Chuanliu Wu
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 8) pp:5404
Publication Date(Web):April 3, 2014
DOI:10.1021/am500192b
Surface properties determine, to a great extent, the biologically relevant functions of various kinds of nanosized materials. Although the modification of the surface of traditional inorganic or polymeric nanoparticles can be routinely achieved through covalent or noncovalent manner or both, the surface modification of nanoscale metal–organic frameworks (nano-MOFs) is extremely challenging because of their rapid degradation in aqueous environments. In this work, we systematically studied the synergistic and dynamic noncovalent interactions between fluorescent probes and iron(III) carboxylate nano-MOFs (i.e., MIL-101-NH2 (Fe), one of the most prevalent MOFs used in drug delivery and imaging). We further examined the interplay between the surface binding of fluorescent probes and the degradation of MIL-101-NH2 (Fe) in aqueous medium. It was demonstrated that the surface binding of probes is not only of high affinity but also dynamic and nonsheddable, even during the degradation, a feature that is essentially different from the covalent conjugation. Subsequently, we developed a unique and straightforward strategy for the surface modification of MIL-101-NH2 (Fe) with polymer by exploiting the synergy of noncovalent interactions between functionalized copolymers and MIL-101-NH2 (Fe). We demonstrated that the binding of polymers onto MIL-101-NH2 (Fe) surface was very effective in aqueous solution and surprisingly nonsheddable during the process of degradation. Surface polymers can creep on the surface of MIL-101-NH2 (Fe), in a dynamic and real-time manner, to the new sites formed immediately after the degradation. In addition, the stability of MIL-101-NH2 (Fe) particles in aqueous environments can be improved to some extent by the surface polymer coating. The results presented herein constitute an important innovation for surface engineering of nano-MOFs, which would benefit the application of nano-MOFs as delivery systems in aqueous systems.Keywords: dynamic noncovalent chemistry; fluorescent probe; metal−organic framework; polymer; surface modification;
Co-reporter:Linxiang Zhai;Jingjing Liang;Dr. Xiangqun Guo;Dr. Yibing Zhao ;Dr. Chuanliu Wu
Chemistry - A European Journal 2014 Volume 20( Issue 52) pp:17507-17514
Publication Date(Web):
DOI:10.1002/chem.201404909
Abstract
Disulfide bonds have frequently been incorporated into synthetic materials to promote sensitivity of the systems towards different redox environments. Although many strategies have been developed to rationally tune the stability of disulfide linkers, methods to tune their responsiveness towards different redox environments remain elusive. In this work we have developed and explored a disulfide linker bearing two independent disulfide bonds, referred to as a twin-disulfide linker. We have demonstrated that the twin-disulfide linker displays an ultrahigh stability at lower concentrations of reducing agent or in weakly reducing environments without a significant compromise in the sensitivity of its response to highly reducing environments such as cytoplasm, a feature that is in remarkable contrast to the traditional single disulfide bonds. Such an extraordinary responsiveness arises from the cooperativity of the twin-disulfide bonds, which should be of particular interest for applications such as controlled drug delivery and sensing, because relatively large differences in disulfide stability in different redox environments is desired in these applications.
Co-reporter:Jinhong Gao, Yangwei Lai, Chuanliu Wu and Yibing Zhao
Nanoscale 2013 vol. 5(Issue 17) pp:8242-8248
Publication Date(Web):27 Jun 2013
DOI:10.1039/C3NR02490C
The sensing of lipopolysaccharide (LPS) relies on the synergy of multiple electrostatic and hydrophobic interactions between LPS and the sensor. However, how non-covalent interactions are coordinated to impel the recognition process still remains elusive, and the exploration of which would promote the development of LPS sensors with higher specificity and sensitivity. In this work, we hypothesize that Au NPs would provide a straightforward and flexible platform for studying the synergy of non-covalent interactions. The detailed mechanism of interactions between the designed fluorescent probes and Au NPs with two distinct surface properties was systematically explored. We demonstrated that only when the electrostatic attraction and hydrophobic stacking are both present, the binding of fluorescent probes onto Au NPs can be not only highly efficient, but also positively cooperative. After that, hybrid systems that consist of Au NPs and surface-assembled fluorescent probes were exploited for fluorescent turn-on sensing of LPS. The results show that the sensitivity and selectivity to LPS relies strongly on the binding affinity between fluorescent probes and Au NPs. Fluorescent probes assembled Au NPs thus provide an attractive platform for further optimization of the sensitivity/selectivity of LPS sensing.
Co-reporter:Yaqi Chen, Tao Li, Jianguo Li, Shiyan Cheng, Jinghui Wang, Chandra Verma, Yibing Zhao and Chuanliu Wu
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 8) pp:NaN1929-1929
Publication Date(Web):2017/02/01
DOI:10.1039/C6OB02786E
Peptides have been promising molecular scaffolds for the development of potential therapeutics with high affinity and specificity to biomacromolecules. However, their inherent proteolytic instability significantly hampers their biological applications. Strategies that can stabilize peptides against proteolytic digestion on the basis of noncovalent interactions—without extensive manipulation of the sequence or use of unnatural residues—are greatly desired. In this work, we developed a general, convenient, and efficient strategy for the stabilization of peptides against proteolysis, which involves noncovalent π–π interactions between aromatic amino acid residues in peptides and synthetic electron-deficient aromatics (NDI), together with the implication of steric hindrance (from the bulky NDI moiety), and the enhancement of peptide α-helicity. This strategy is complementary in concept to the conventional well-established covalent approaches for peptide stabilization, and is thus promising for being utilized, in combination with the latter ones, to circumvent the problem of proteolytic instability of peptides. We envisioned that this study should provide invaluable guidelines to the design and synthesis of organic molecule–peptide hybrids with significantly improved proteolytic resistance, and benefit the development of peptide therapeutics and probes.
Co-reporter:Yunyang Ling, Jing Ren, Tao Li, Yibing Zhao and Chuanliu Wu
Chemical Communications 2016 - vol. 52(Issue 24) pp:NaN4536-4536
Publication Date(Web):2016/02/25
DOI:10.1039/C6CC00369A
POEGMA-based disulfide-containing fluorescent probes were designed and synthesized to enable the real-time and quantitative analysis of the (bio)reduction of disulfides within the polymeric backbones in complex redox media and in the presence of cells. This study lights up the way of exploiting bioreduction in the extracellular spaces for the delivery of hydrophobic drugs.
Co-reporter:Yiwu Zheng;Zhuoru Li;Jing Ren;Weidong Liu;Yaqi Wu;Yibing Zhao
Chemical Science (2010-Present) 2017 - vol. 8(Issue 4) pp:
Publication Date(Web):2017/03/28
DOI:10.1039/C6SC05710A
Disulfide-rich peptides are emerging as potential templates for drug design applications. However, the synthesis and reengineering of disulfide-rich peptides are challenging, owing to the complexity of the oxidative folding process involving a number of diverse isomeric structures. Novel disulfide-rich peptide scaffolds that are not besieged by their disulfide isomers are still greatly desired. In this work, we report the design and synthesis of a novel class of artificial disulfide-rich peptide scaffolds with precisely defined disulfide patterns and a minimized number of isomers. In theory, natural peptides with three disulfide bonds have 15 possible isomers. By rationally engineering the thiol-framework of a peptide containing six cysteines with penicillamines and a dithiol amino acid, we demonstrated, for the first time, that the total number of isomers formed after oxidative folding can be decreased to a minimum of two (i.e., from 15 to 2). As fewer isomeric folds are involved in the oxidative folding, the pathway of the folding becomes more concise and the yield of the artificial scaffolds is substantially increased compared to that of its six-cysteine-containing analogue, which makes the artificial disulfide-rich scaffolds (with only 2 predefined isomeric folds) extremely promising for being exploited as structurally complex templates for the design of peptide therapeutics and ligands.