Co-reporter:Chunqing Guo, Jacob W. Fulp, Yuqi Jiang, Xia Li, Jeremy E. Chojnacki, Jingde Wu, Xiang-Yang Wang, and Shijun Zhang
ACS Chemical Neuroscience October 18, 2017 Volume 8(Issue 10) pp:2194-2194
Publication Date(Web):June 27, 2017
DOI:10.1021/acschemneuro.7b00124
In our efforts to develop novel small-molecule inhibitors for the NOD-like receptor family pyrin-domain-containing 3 (NLRP3) inflammasome as potential disease-modifying agents to treat neurological disorders including multiple sclerosis (MS), a hydroxyl sulfonamide analogue JC-171 has been rationally designed and biologically characterized both in vitro and in vivo. Our studies established that JC-171 dose dependently inhibited LPS/ATP-induced interleukin-1β (IL-1β) release from J774A.1 macrophages with an IC50 of 8.45 ± 1.56 μM. Selective inhibition of the NLRP3 inflammasome induced IL-1β release by this compound was also confirmed using mouse bone-marrow-derived macrophages and LPS-challenged mice in vivo. Furthermore, immunoprecipitation study revealed that JC-171 interfered with NLRP3/ASC interaction induced by LPS/ATP stimulation. More importantly, JC-171 treatment delayed the progression and reduced the severity of experimental autoimmune encephalomyelitis (EAE), a mouse model of MS, in both prophylactic and therapeutic settings. This coincided with blocking of IL-1β production and a pathogenic Th17 response. Collectively, these results suggest that JC-171 is a selective NLRP3 inflammasome inhibitor with biological activity in vivo, thus strongly encouraging further development of this lead compound as a potential therapeutic agent for human MS.Keywords: experimental autoimmune encephalomyelitis; multiple sclerosis; NLRP3 inflammasome; rational design; small molecule inhibitor;
Co-reporter:Gorka Gerenu, Kai Liu, Jeremy E. Chojnacki, John M. Saathoff, Pablo Martínez-Martín, George Perry, Xiongwei Zhu, Hyoung-gon Lee, and Shijun Zhang
ACS Chemical Neuroscience 2015 Volume 6(Issue 8) pp:1393
Publication Date(Web):April 20, 2015
DOI:10.1021/acschemneuro.5b00082
In our efforts to develop hybrid compounds of curcumin and melatonin as potential disease-modifying agents for Alzheimer’s disease (AD), a potent lead hybrid compound, Z-CM-I-1, has been recently identified and biologically characterized in vitro. In this work, we report the in vivo effects of Z-CM-I-1 on AD pathologies in an APP/PS1 transgenic AD model. Our studies demonstrated that Z-CM-I-1 significantly decreased the accumulation of Aβ in the hippocampus and cortex regions of the brain and reduced inflammatory responses and oxidative stress after treatment for 12 weeks at 50 mg/kg per dose via oral administration. Furthermore, Z-CM-I-1 significantly improved synaptic dysfunction evidenced by the increased expression of synaptic marker proteins, PSD95 and synaptophysin, indicating its protective effects on synaptic degeneration. Lastly, we demonstrated that Z-CM-I-1 significantly increased the expression level of complexes I, II, and IV of the mitochondria electron transport chain in the brain tissue of APP/PS1 mice. Collectively, these results clearly suggest that Z-CM-I-1 is orally available and exhibits multifunctional properties in vivo on AD pathologies, thus strongly encouraging further development of this lead compound as a potential disease-modifying agent for AD patients.Keywords: Alzheimer’s disease; curcumin; Hybrid compounds; melatonin; neuroprotectants
Co-reporter:Kai Liu and Shijun Zhang
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 8) pp:894
Publication Date(Web):July 10, 2015
DOI:10.1021/acsmedchemlett.5b00158
Carbazoles represent a family of tricyclic compounds that widely appeared in nature. Numerous studies have revealed a diverse array of bioactivity associated with this scaffold. In the present study, a novel and highly efficient methodology for preparing 1,3-dihydroxy-2-carboxycarbazole from indole-3-acetic acid and Meldrum’s acid was developed. Furthermore, biological characterization demonstrated that this multisubstituted carbazole analogue exhibited inhibitory activity on Aβ aggregation, antioxidative properties, and promising neuroprotective activities in a cellular model of Alzheimer’s disease, thus further supporting the valuable application of this synthetic methodology in search for effective neuroprotectants.Keywords: antioxidant; Aβ oligomers; Carbazole derivative; Meldrum’s acid; neuroprotection; synthesis
Co-reporter:Kai Liu, Shijun Zhang
Tetrahedron Letters 2014 Volume 55(Issue 40) pp:5566-5569
Publication Date(Web):1 October 2014
DOI:10.1016/j.tetlet.2014.08.064
Hydrogen sulfide (H2S) has been recently recognized as an important signaling molecule in biological systems. Herein, we report the development of a fluorescence turn-on probe based on the structure of pomalidomide, a FDA approved drug for the treatment of multiple myeloma. Various characterizations demonstrated high selectivity and sensitivity of this probe toward H2S. Furthermore, the application of this probe to detect H2S in living cells was confirmed by flow cytometry and fluorescence imaging studies.
Co-reporter:Kai Liu, Datong Zhang, Jeremy Chojnacki, Yuhong Du, Haian Fu, Steven Grant and Shijun Zhang
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 29) pp:4757-4763
Publication Date(Web):11 Jun 2013
DOI:10.1039/C3OB40595H
In our efforts to develop effective treatment agents for human multiple myeloma (MM), a series of hybrid molecules based on the structures of thalidomide (1) and curcumin (2) were designed, synthesized, and biologically characterized in human multiple myeloma MM1S, RPMI8226, U266 cells, and human lung cancer A549 cells. The biological results showed that two hybrid compounds, 5 and 7, exhibited significantly improved lethal effects towards all three human MM cell models compared to 1 or 2 alone, as well as the combination of 1 and 2. Furthermore, mechanistic studies in U266 cells demonstrated that 5 and 7 can induce the production of reactive oxygen species (ROS) and cause G1/S arrest, thus leading to apoptosis and cell death. Additionally, they exhibited inhibitory effects on NFκB activation in A549 cells. Collectively, the results obtained from these hybrid compounds strongly encourage their further optimization as new leads to develop effective treatment agents for human MM.
Co-reporter:Kai Liu, Jiangmin Chen, Jeremy Chojnacki, Shijun Zhang
Tetrahedron Letters 2013 Volume 54(Issue 16) pp:2070-2073
Publication Date(Web):17 April 2013
DOI:10.1016/j.tetlet.2013.02.015
A concise and one-pot cascade method has been developed to achieve the synthesis of difluoroboron-derivatized curcumins (BF2C). Treatment of 2,4-pentanedione with BF3·OEt2, followed by condensation with aldehydes in the presence of tributyl borate and butylamine at 65 °C in toluene furnished the corresponding symmetric (s-BF2C) and unsymmetric difluoroboron-derivatized curcumins (us-BF2C) in good (60–99%) and moderate yields (23–42%) within 6–12 h, respectively.
Co-reporter:Kai Liu, Tai L. Guo, Jeremy Chojnacki, Hyoung-Gon Lee, Xinglong Wang, Sandra L. Siedlak, Wei Rao, Xiongwei Zhu, and Shijun Zhang
ACS Chemical Neuroscience 2012 Volume 3(Issue 2) pp:141
Publication Date(Web):December 20, 2011
DOI:10.1021/cn200122j
A recently developed bivalent ligand BMAOI 14 (7) has been evaluated for its ability to label and detect aggregated β-amyloid (Aβ) peptide as a fluorescent probe. This probe contains curcumin as the Aβ recognition moiety and cholesterol as an anchor to the neuronal cell membrane–lipid rafts. The results demonstrate that 7 binds to the monomers, oligomers, and fibrils of Aβ42 with low micromolar to submicromolar binding affinities. This chemical probe also has many of the required optical properties for use in imaging and can rapidly cross the blood–brain barrier in vivo. Furthermore, 7 specifically binds to Aβ plaques in both Alzheimer's disease human patients and Aβ precursor protein transgenic mouse brain tissues. Collectively, these results suggest that 7 is a strong candidate as an Aβ imaging agent and encourage further optimization of 7 as a new lead for the development of the next generation of Aβ imaging probes.Keywords: Alzheimer’s disease; Aβ plaques; Bivalent ligands; fluorescent probes
Co-reporter:Kai Liu, Wei Rao, Hardik Parikh, Qianbin Li, Tai L. Guo, Steven Grant, Glen E. Kellogg, Shijun Zhang
European Journal of Medicinal Chemistry 2012 Volume 47() pp:125-137
Publication Date(Web):January 2012
DOI:10.1016/j.ejmech.2011.10.031
A series of 2,5-disubstituted-thiazolidine-2,4-dione analogs based on the newly identified lead 1, a potential anticancer agent via the inhibition of the Raf/MEK/extracellular signal regulated kinase (ERK) and phosphatidylinositol 3-kinase (PI3K)/Akt signaling cascades, were synthesized and biologically characterized. A new lead structure, 15, was identified to have improved anti-proliferative activities in U937 cells, to induce apoptosis in U937, M12 and DU145 cancer cells, and to arrest U937 cells at the S-phase. Furthermore, Western blot analysis demonstrated a correlation of the anti-proliferative activity and blockade of the Raf/MEK/ERK and PI3K/Akt signaling pathways. Collectively, these results strongly encourage further optimization of 15 as a new lead with multi-target properties to develop more potent compounds as anticancer agents.Highlights► Thiazolidine-2,4-dione analogs have anticancer activity. ► We designed and synthesized a series of 3,5-disubstituted-thiazolidine-2,4-dione analogs as anticancer candidates. ► Synthesized compounds were tested in various human cancer cell lines. ► Compound 15 was identified as a new lead compound to develop more potent analogs.
Co-reporter:Kai Liu, Ronak Gandhi, Jiangmin Chen, and Shijun Zhang
ACS Medicinal Chemistry Letters 2012 Volume 3(Issue 11) pp:942
Publication Date(Web):September 11, 2012
DOI:10.1021/ml300229y
In a continuing effort to develop multifunctional compounds as potential treatment agents for Alzheimer's disease (AD), a series of bivalent ligands containing curcumin and cholesterylamine were designed, synthesized, and biologically characterized. Biological characterization supported earlier results that the spacer length and its attachment position on curcumin are essential structural determinants for biological activity in this class. Compounds with a spacer length of 17–21 atoms exhibited optimal neuroprotection in human neuroblastoma MC65 cells with submicromolar potency. These compounds inhibited the formation of amyloid-β oligomers (AβOs) and exhibited antioxidative activities in MC65 cells. Bivalent ligand 8, with its spacer (length of 17 atoms) connected at the methylene carbon between the two carbonyls of the curcumin moiety, is the most potent with an EC50 of 0.083 ± 0.017 μM. In addition, 8 formed a complex with biometals, such as Cu, Fe, and Zn. Collectively, the results strongly support our assertion that these compounds are designed bivalent ligands with potential as multifunctional and neuroprotective agents.Keywords: Alzheimer's disease; antioxidant; Aβ oligomers; bivalent ligands; metal chelating; multifunctional
Co-reporter:James A. Lenhart ; Xiao Ling ; Ronak Gandhi ; Tai L. Guo ; Phillip M. Gerk ; Darlene H. Brunzell
Journal of Medicinal Chemistry 2010 Volume 53(Issue 16) pp:6198-6209
Publication Date(Web):July 28, 2010
DOI:10.1021/jm100601q
In our effort to develop multifunctional compounds that cotarget beta-amyloid oligomers (AβOs), cell membrane/lipid rafts (CM/LR), and oxidative stress, a series of bivalent multifunctional Aβ oligomerization inhibitors (BMAOIs) containing cholesterol and curcumin were designed, synthesized, and biologically characterized as potential treatments for Alzheimer’s disease (AD). The in vitro assay results established that the length of spacer that links cholesterol and curcumin and the attaching position of the spacer on curcumin are important structural determinants for their biological activities. Among the BMAOIs tested, 14 with a 21-atom-spacer was identified to localize to the CM/LR of human neuroblastoma MC65 cells, to inhibit the formation of AβOs in MC65 cells, to protect cells from AβOs-induced cytotoxicity, and to retain antioxidant properties of curcumin. Furthermore, 14 was confirmed to have the potential to cross the blood−brain barrier (BBB) as demonstrated in a Caco-2 cell model. Collectively, these results strongly encourage further optimization of 14 as a new hit to develop more potent BMAOIs.
Co-reporter:Qianbin Li, Jingde Wu, Hui Zheng, Kai Liu, Tai L. Guo, Yuying Liu, Scott T. Eblen, Steven Grant, Shijun Zhang
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 15) pp:4526-4530
Publication Date(Web):1 August 2010
DOI:10.1016/j.bmcl.2010.06.030
A thiazolidine-2,4-dione derivative, 3-(2-aminoethyl)-5-(3-phenyl-propylidene)-thiazolidine-2,4-dione (2), was identified as a dual inhibitor of the Raf/MEK/ extracellular signal-regulated kinase (ERK) and the phosphatidylinositol 3-kinase (PI3K)/Akt signaling cascades. The discovered compound inhibited cell proliferation, induced early apoptosis, and arrested cells in G0/G1 phase in human leukemia U937 cells. These results indicate its potential as a new lead compound to develop novel dual signaling pathway inhibitors and anticancer agents.The identification of 3-(2-aminoethyl)-5-(3-phenyl-propylidene)-thiazolidine-2,4-dione as a dual inhibitor of the Raf/MEK/ERK and PI3K/Akt signaling pathways is reported.
Co-reporter:Qianbin Li, Adnan Al-Ayoubi, Tailiang Guo, Hui Zheng, Aurijit Sarkar, Tri Nguyen, Scott T. Eblen, Steven Grant, Glen E. Kellogg, Shijun Zhang
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 21) pp:6042-6046
Publication Date(Web):1 November 2009
DOI:10.1016/j.bmcl.2009.09.057
A series of analogs of 3-(2-amino-ethyl)-5-(4-ethoxy-benzylidene)-thiazolidine-2,4-dione, a putative substrate-specific ERK1/2 inhibitor, were synthesized and biologically characterized in human leukemia U937 cells to define its pharmacophore. It was discovered that shift of ethoxy substitution from the 4- to the 2-position on the phenyl ring significantly improved functional activities of inhibiting cell proliferation and inducing apoptosis. This may provide access to a new lead for developing ERK1/2 substrate-specific inhibitors.Preliminary structure–activity relationship studies of 3-(2-amino-ethyl)-5-(4-ethoxy-benzylidene)-thiazolidine-2,4-dione, a putative substrate-specific ERK1/2 inhibitor, is reported.
Co-reporter:Kai Liu, Datong Zhang, Jeremy Chojnacki, Yuhong Du, Haian Fu, Steven Grant and Shijun Zhang
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 29) pp:NaN4763-4763
Publication Date(Web):2013/06/11
DOI:10.1039/C3OB40595H
In our efforts to develop effective treatment agents for human multiple myeloma (MM), a series of hybrid molecules based on the structures of thalidomide (1) and curcumin (2) were designed, synthesized, and biologically characterized in human multiple myeloma MM1S, RPMI8226, U266 cells, and human lung cancer A549 cells. The biological results showed that two hybrid compounds, 5 and 7, exhibited significantly improved lethal effects towards all three human MM cell models compared to 1 or 2 alone, as well as the combination of 1 and 2. Furthermore, mechanistic studies in U266 cells demonstrated that 5 and 7 can induce the production of reactive oxygen species (ROS) and cause G1/S arrest, thus leading to apoptosis and cell death. Additionally, they exhibited inhibitory effects on NFκB activation in A549 cells. Collectively, the results obtained from these hybrid compounds strongly encourage their further optimization as new leads to develop effective treatment agents for human MM.

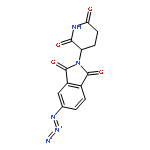
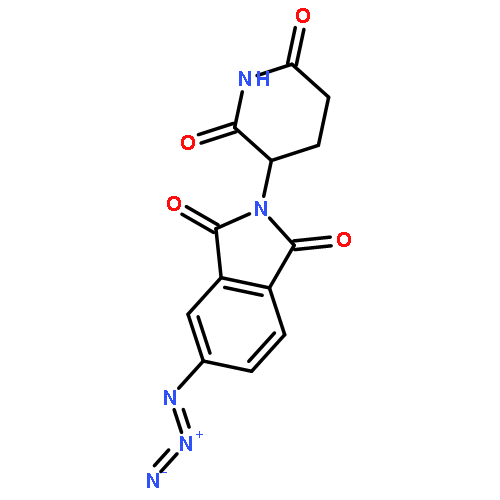


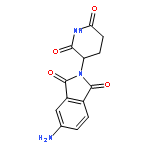
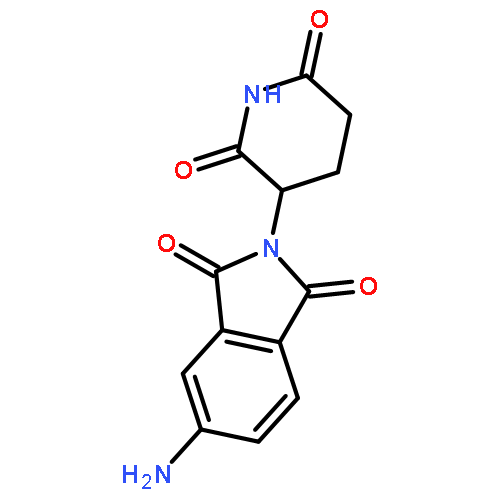









![Ethanamine, 2-[2-(2-azidoethoxy)ethoxy]-](/data/chemimg/124000/166388-57-4.png)
![Ethanamine, 2-[2-(2-azidoethoxy)ethoxy]-](/data/chemimg/124000/166388-57-4_b.png)




![Ethanol, 2-[2-(2-azidoethoxy)ethoxy]-](/data/chemimg/964400/86520-52-7.png)
![Ethanol, 2-[2-(2-azidoethoxy)ethoxy]-](/data/chemimg/964400/86520-52-7_b.png)

![N'-[(1Z)-1-(3-PYRIDINYL)ETHYLIDENE]NICOTINOHYDRAZIDE](http://img.cochemist.com/ccimg/71100/71031-15-7.png)
![N'-[(1Z)-1-(3-PYRIDINYL)ETHYLIDENE]NICOTINOHYDRAZIDE](http://img.cochemist.com/ccimg/71100/71031-15-7_b.png)