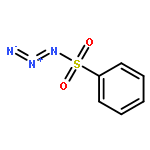
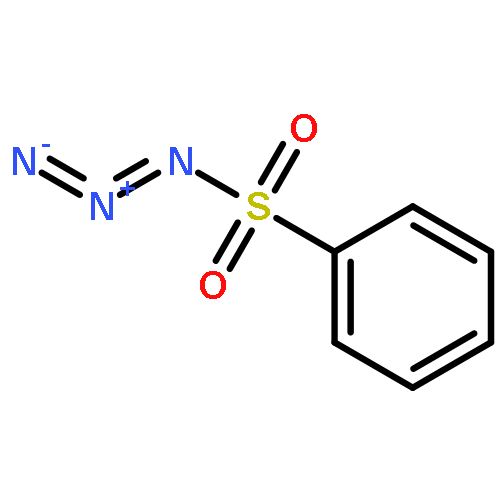
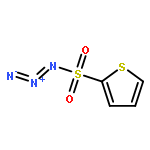
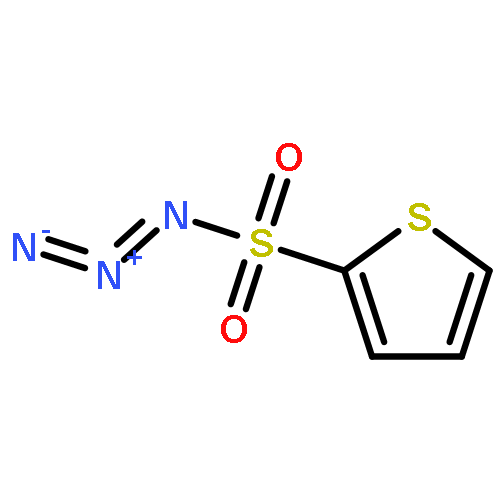
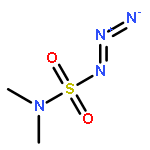
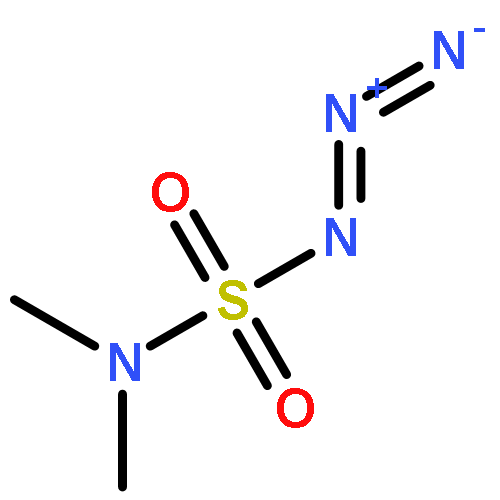
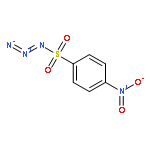
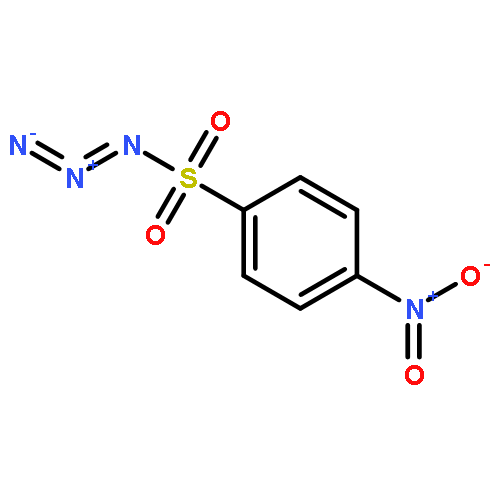
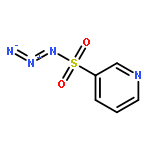
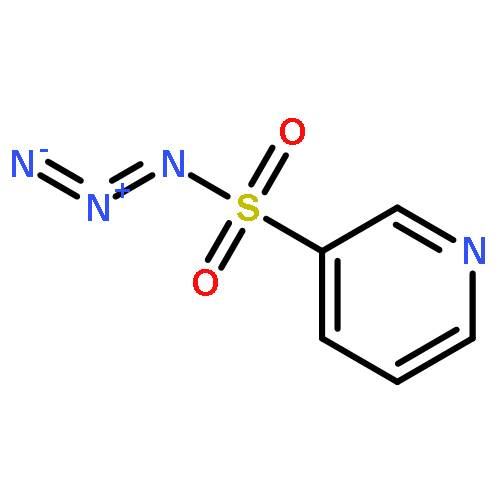
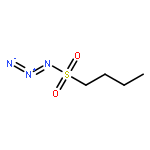
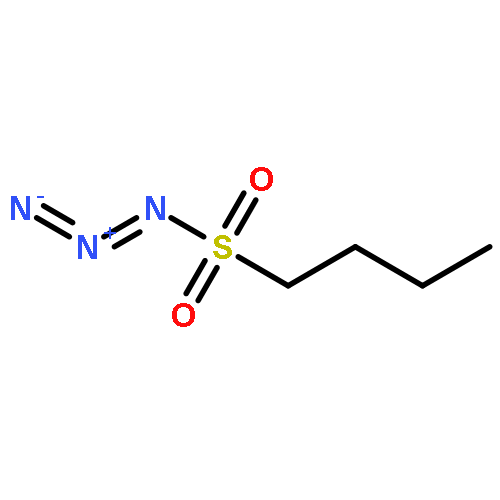
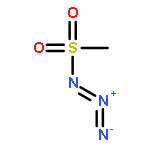
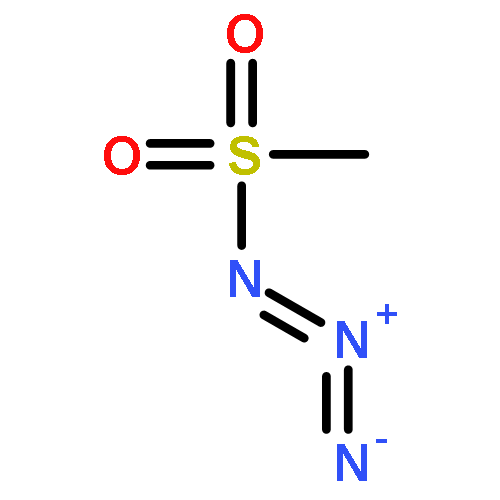
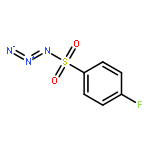
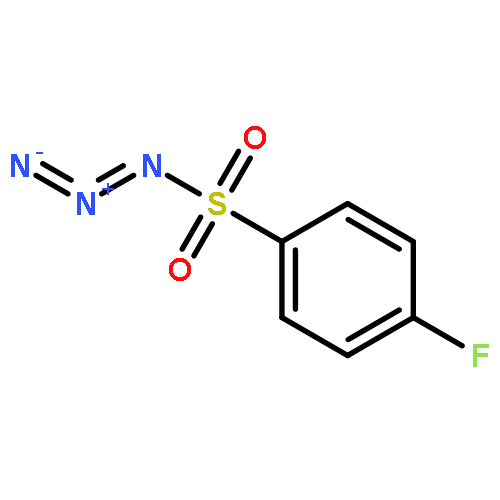
Co-reporter:Cheng-Jie Yang, Zi-Long Song, Masuo Goto, Ying-Qian Liu, Kan-Yen Hsieh, Susan L. Morris-Natschke, Yong-Long Zhao, Jun-Xiang Zhang, Kuo-Hsiung Lee
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 17(Issue 17) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.bmcl.2017.07.078
In our continuing search for camptothecin (CPT)-derived antitumor drugs, novel 7-substituted CPT derivatives incorporating piperazinyl-sulfonylamidine moieties were designed, synthesized and evaluated for cytotoxicity against five tumor cell lines (A-549, MDA-MB-231, MCF-7, KB, and KB-VIN). All of the derivatives showed promising in vitro cytotoxic activity against the tested tumor cell lines, and were more potent than irinotecan. Remarkably, most of the compounds exhibited comparable cytotoxicity against the multidrug-resistant (MDR) KB-VIN and parental KB tumor cell lines, while irinotecan lost activity completely against KB-VIN. Especially, compounds 13r and 13p (IC50 0.38 and 0.85 μM, respectively) displayed the greatest cytotoxicity against the MDR KB-VIN cell line and merit further development into preclinical and clinical drug candidates for treating cancer, including MDR phenotype.Download high-res image (73KB)Download full-size image
Co-reporter:Cheng-Jie Yang, Zi-Long Song, Masuo Goto, Pei-Ling Hsu, Xiao-Shuai Zhang, Qian-Ru Yang, Ying-Qian Liu, Mei-Juan Wang, Susan L. Morris-Natschke, Xiao-Fei Shang, Kuo-Hsiung Lee
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 20(Issue 20) pp:
Publication Date(Web):15 October 2017
DOI:10.1016/j.bmcl.2017.09.012
Fluorination is a well-known strategy for improving the bioavailability of bioactive molecules in the lead optimization phase of drug discovery projects. In an attempt to improve the antitumor activity of camptothecins (CPTs), novel 10-fluoro-CPT derivatives were designed, synthesized and evaluated for cytotoxicity against five human cancer cell lines (A-549, MDA-MB-231, KB, KB-VIN and MCF-7). All of the derivatives showed more potent in vitro cytotoxic activity than the clinical CPT-derived drug irinotecan against the tumor cell lines tested, and most of them showed comparable or superior potency to topotecan. Remarkably, compounds 16b (IC50, 67.0 nM) and 19b (IC50, 99.2 nM) displayed the highest cytotoxicity against the multidrug-resistant (MDR) KB-VIN cell line and merit further development as preclinical drug candidates for treating cancer, including MDR phenotype. Our study suggested that incorporation of a fluorine atom into position 10 of CPT is an effective method for discovering new potent CPT derivatives.A novel type of 10-fluorocamptothecin derivatives with excellent cytotoxicity activity were designed and synthesized.Download high-res image (168KB)Download full-size image
Co-reporter:Gao-Xiang Zhu, Pi-Le Cheng, Masuo Goto, Na Zhang, Susan L. Morris-Natschke, Kan-Yen Hsieh, Guan-Zhou Yang, Qian-Ru Yang, Ying-Qian Liu, Hai-Le Chen, Xiao-Shuai Zhang, Kuo-Hsiung Lee
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 8(Issue 8) pp:
Publication Date(Web):15 April 2017
DOI:10.1016/j.bmcl.2017.02.066
In an effort to discover potent camptothecin-derived antitumor agents, novel camptothecin analogues with sulfonylpiperazinyl motifs at position-7 were designed and synthesized. They were evaluated for in vitro cytotoxicity with the sulforhodamine-B (SRB) method in five types of human tumor cell lines, A-549, MDA-MB-231, KB, KB-VIN and MCF-7. With IC50 values in the low μM to nM level, most of the new analogues showed greater cytotoxicity activity than the reference compounds irinotecan and topotecan. Furthermore, compounds 12l (IC50, 1.2 nM) and 12k (IC50, 20.2 nM) displayed the highest cytotoxicity against the multidrug-resistant (MDR) KB-VIN cell line and merit further development as preclinical drug candidates for treating cancer, including MDR phenotype. Our study suggested that integration of sulfonylpiperazinyl motifs into position-7 of camptothecin is an effective strategy for discovering new potent cytotoxic camptothecin derivatives.Download high-res image (141KB)Download full-size image
Co-reporter:Zi-Long Song, Mei-Juan Wang, Lanlan Li, Dan Wu, Yu-Han Wang, Li-Ting Yan, Susan L. Morris-Natschke, Ying-Qian Liu, Yong-Long Zhao, Chih-Ya Wang, Huanxiang Liu, Masuo Goto, Heng Liu, Gao-Xiang Zhu, Kuo-Hsiung Lee
European Journal of Medicinal Chemistry 2016 Volume 115() pp:109-120
Publication Date(Web):10 June 2016
DOI:10.1016/j.ejmech.2016.02.070
•20(S)-Sulfonylamidine CPT-derivatives were prepared and tested for cytotoxicity.•Several analogs showed superior cytotoxic activity compared to irinotecan.•Key structural features related to cytotoxicity were identified by SAR analysis.•Compounds 9 and 15c interacted with Topo I-DNA by a different binding mode from CPT.•These compounds are new generation CPT-derived antitumor agents.In an ongoing investigation of 20-sulfonylamidine derivatives (9, YQL-9a) of camptothecin (1) as potential anticancer agents directly and selectively inhibiting topoisomerase (Topo) I, the sulfonylamidine pharmacophore was held constant, and a camptothecin derivatives with various substitution patterns were synthesized. The new compounds were evaluated for antiproliferative activity against three human tumor cell lines, A-549, KB, and multidrug resistant (MDR) KB subline (KBvin). Several analogs showed comparable or superior antiproliferative activity compared to the clinically prescribed 1 and irinotecan (3). Significantly, the 20-sulfonylamidine derivatives exhibited comparable cytotoxicity against KBvin, while 1 and 3 were less active against this cell line. Among them, compound 15c displayed much better cytotoxic activity than the controls 1, 3, and 9. Novel key structural features related to the antiproliferative activities were identified by structure–activity relationship (SAR) analysis. In a molecular docking model, compounds 9 and 15c interacted with Topo I-DNA through a different binding mode from 1 and 3. The sulfonylamidine side chains of 9 and 15c could likely form direct hydrogen bonds with Topo I, while hydrophobic interaction with Topo I and π–π stacking with double strand DNA were also confirmed as binding driving forces. The results from docking models were consistent with the SAR conclusions. The introduction of bulky substituents at the 20-position contributed to the altered binding mode of the compound by allowing them to form new interactions with Topo I residues. The information obtained in this study will be helpful for the design of new derivatives of 1 with most promising anticancer activity.CPT (green), 9 (magenta), and 15c (blue) in the binding site of DNA-Topo-I.
Co-reporter:Jian Zhang, Xiang Nan, Hai-Tao Yu, Pi-Le Cheng, Yan Zhang, Ying-Qian Liu, Shao-Yong Zhang, Guan-Fang Hu, Huanxiang Liu, An-Liang Chen
European Journal of Medicinal Chemistry 2016 Volume 121() pp:422-432
Publication Date(Web):4 October 2016
DOI:10.1016/j.ejmech.2016.05.056
•Three new series of avermectin derivatives were synthesized and bioassayed.•All the compounds showed potent lethal activities against three insect species.•Some of compounds exhibited higher lethal activity than avermectin.•The preliminary SAR correlations were formulated.•The built QSAR model provide insight to develop potential avermectin derivatives.In an effort to discover new molecules with good insecticidal activities, more than 40 new avermectin derivatives were synthesized and evaluated for their biological activities against three species of arachnids, insects and nematodes, namely, Tetranychus Cinnabarinus, Aphis craccivora and Bursaphelenchus xylophilus. All the tested compounds showed potent inhibitory activities against three insect species. Notably, the majority of compounds exhibited high selectivity against T. cinnabarinus, some of which were much better in comparison with avermectin. Especially compounds 9j (LC50: 0.005 μM) and 16d (LC50: 0.002 μM) were 2.5- and 4.7-fold more active than avermectin (LC50: 0.013 μM), respectively, against T. cinnabarinus. Moreover, compounds 9b, 9d–f, 9h, 9j, 9l, 9n, 9p, 9r, 9v and 17d showed superior activities with LC50 values of 2.959–5.013 μM compared to that of 1 (LC50: 6.746 μM) against B. xylophilus. Meanwhile, the insecticidal activities of compounds 9f, 9g, 9h, and 9m against A. craccivora were 7–8 times better than that of avermectin, with LC50 values of 7.744, 5.634, 6.809, 7.939 and 52.234 μM, respectively. Furthermore, QSAR analysis showed that the molecular shape, size, connectivity degree and electronic distribution of avermectin analogues had substantial effects on insecticidal potency. These preliminary results provided useful insight in guiding further modifications of avermectin in the development of potential new insecticides.Three new series of avermectin derivatives were synthesized. All the compounds showed potent lethal activities against three insect species, and some of them exhibited higher lethal activity than avermectin.
Co-reporter:Zi-Long Song, Hai-Le Chen, Yu-Han Wang, Masuo Goto, Wen-Jing Gao, Pi-Le Cheng, Susan L. Morris-Natschke, Ying-Qian Liu, Gao-Xiang Zhu, Mei-Juan Wang, Kuo-Hsiung Lee
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 13) pp:2690-2693
Publication Date(Web):1 July 2015
DOI:10.1016/j.bmcl.2015.04.060
In our continuing search for camptothecin (CPT)-derived antitumor drugs, novel structurally diverse PEG-based 20(S)-CPT sulfonylamidine derivatives were designed, synthesized via a Cu-multicomponent reaction (MCR), and evaluated for cytotoxicity against four human tumor cell lines (A-549, MDA-MB-231, KB, and KBvin). All of the derivatives showed promising in vitro cytotoxic activity against the tested tumor cell lines, and were more potent than irinotecan. Significantly, these derivatives exhibited comparable cytotoxicity against KBvin, while irinotecan was less active against this cell line. With a concise efficient synthesis and potent cytotoxic profiles, especially significant activity towards KBvin, these compounds merit further development as a new generation of CPT-derived PEG-conjugated drug candidates.
Co-reporter:Mei-Juan Wang ; Ying-Qian Liu ; Ling-Chu Chang ; Chih-Ya Wang ; Yong-Long Zhao ; Xiao-Bo Zhao ; Keduo Qian ; Xiang Nan ; Liu Yang ; Xiao-Ming Yang ; Hsin-Yi Hung ; Jai-Sing Yang ⊗; Daih-Huang Kuo ; Masuo Goto ; Susan L. Morris-Natschke ; Shiow-Lin Pan ▽; Che-Ming Teng ; Sheng-Chu Kuo ; Tian-Shung Wu ◆; Yang-Chang Wu ;Kuo-Hsiung Lee
Journal of Medicinal Chemistry 2014 Volume 57(Issue 14) pp:6008-6018
Publication Date(Web):July 8, 2014
DOI:10.1021/jm5003588
Twelve novel 20-sulfonylamidine derivatives (9a–9l) of camptothecin (1) were synthesized via a Cu-catalyzed three-component reaction. They showed similar or superior cytotoxicity compared with that of irinotecan (3) against A-549, DU-145, KB, and multidrug-resistant (MDR) KBvin tumor cell lines. Compound 9a demonstrated better cytotoxicity against MDR cells compared with that of 1 and 3. Mechanistically, 9a induced significant DNA damage by selectively inhibiting Topoisomerase (Topo) I and activating the ATM/Chk related DNA damage-response pathway. In xenograft models, 9a demonstrated significant activity without overt adverse effects at 5 and 10 mg/kg, comparable to 3 at 100 mg/kg. Notably, 9a at 300 mg/kg (i.p.) showed no overt toxicity in contrast to 1 (LD50 56.2 mg/kg, i.p.) and 3 (LD50 177.5 mg/kg, i.p.). Intact 9a inhibited Topo I activity in a cell-free assay in a manner similar to that of 1, confirming that 9a is a new class of Topo I inhibitor. 20-Sulfonylamidine 1-derivative 9a merits development as an anticancer clinical trial candidate.
Co-reporter:Liang Kou, Mei-Juan Wang, Li-Ting Wang, Xiao-Bo Zhao, Xiang Nan, Liu Yang, Ying-Qian Liu, Susan L. Morris-Natschke, Kuo-Hsiung Lee
European Journal of Medicinal Chemistry 2014 Volume 75() pp:282-288
Publication Date(Web):21 March 2014
DOI:10.1016/j.ejmech.2014.01.038
•New spin-labeled podophyllotoxin analogs were prepared and tested for cytotoxicity.•The synthesis used an isocyanide multicomponent coupling reaction.•Potent cytotoxicity was found against A-549, DU-145, KB and KBvin cancer cell lines.•Two compounds (12e, 12h) showed superior potency to etoposide.Spin-labeled podophyllotoxins have elicited widespread interest due to their far superior antitumor activity compared to podophyllotoxin. To extend our prior studies in this research area, we synthesized a new generation of spin-labeled podophyllotoxin analogs via isocyanide multicomponent reactions and evaluated their cytotoxicity against four human cancer cell lines (A-549, DU-145, KB and KBvin). Most of the compounds exhibited potent cytotoxic activity against all four cell lines, notably against the drug resistant KBvin cancer cell line. Among the new analogs, compounds 12e (IC50: 0.60–0.75 μM) and 12h (IC50: 1.12–2.03 μM) showed superior potency to etoposide (IC50: 2.03 to >20 μM), a clinically available anticancer drug. With a concise efficient synthesis and potent cytotoxic profiles, compounds 12e and 12h merit further development as a new generation of epipodophyllotoxin-derived antitumor clinical trial candidates.
Co-reporter:Zhi-Jun Zhang, Jing Tian, Li-Ting Wang, Mei-Juan Wang, Xiang Nan, Liu Yang, Ying-Qian Liu, Susan L. Morris-Natschke, Kuo-Hsiung Lee
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 1) pp:204-210
Publication Date(Web):1 January 2014
DOI:10.1016/j.bmc.2013.11.035
Three series of novel sulfonylurea podophyllotoxin derivatives were designed, synthesized, and evaluated for in vitro cytotoxicity against four tumor cell lines (A-549, DU-145, KB and KBvin). Compounds 14c (IC50: 1.41–1.76 μM) and 14e (IC50: 1.72–2.01 μM) showed superior cytotoxic activity compared with etoposide (IC50: 2.03 to >20 μM), a clinically available anticancer drug. Significantly, most of the compounds exhibited comparable cytotoxicity against the drug-resistant tumor cell line KBvin, while etoposide lost activity completely. Preliminary structure–activity relationship (SAR) correlations indicated that the 4′-O-methyl functionality in podophyllotoxin analogues may be essential to maintain cytotoxic activity, while an arylsulfonylurea side chain at podophyllotoxin’s 4β position can significantly improve cytotoxic activity.
Co-reporter:Xiao-Bo Zhao, Dan Wu, Mei-Juan Wang, Masuo Goto, Susan L. Morris-Natschke, Ying-Qian Liu, Xiao-Bing Wu, Zi-Long Song, Gao-Xiang Zhu, Kuo-Hsiung Lee
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 22) pp:6453-6458
Publication Date(Web):15 November 2014
DOI:10.1016/j.bmc.2014.09.035
In our continuing search for natural product-based spin-labeled antitumor drugs, 20 novel spin-labeled camptothecin derivatives were synthesized via a Cu-catalyzed one pot reaction and evaluated for cytotoxicity against four human tumor cell lines (A-549, MDA-MB-231, KB, and KBvin). Eighteen of the target compounds (9a, 9b, 9d–9k, 9m–9t) exhibited significant in vitro antiproliferative activity against these four tested tumor cell lines. Compounds 9e and 9j (IC50 0.057 and 0.072 μM, respectively) displayed the greatest cytotoxicity against the multidrug-resistant (MDR) KBvin cell line and merit further development into preclinical and clinical drug candidates for treating cancer including MDR phenotype.
Co-reporter:Jing Zhang, Li-Ting Yan, En-Lin Yuan, Hai-Xin Ding, Huo-Chun Ye, Zheng-Ke Zhang, Chao Yan, Ying-Qian Liu, and Gang Feng
Journal of Agricultural and Food Chemistry 2014 Volume 62(Issue 21) pp:4905-4910
Publication Date(Web):May 13, 2014
DOI:10.1021/jf500968b
Cortex Pseudolaricis is the root bark of Pseudolarix amabilis Rehder, found only in China, and has been widely used in folk antifungal remedies in traditional Chinese medicine. In order to find the natural antifungal agents against mango anthracnose, eight compounds, namely pseudolaric acid A (1), ethyl pseudolaric acid B (2), pseudolaric acid B (3), pseudolaric acid B—O-β-d-glucoside (4), piperonylic acid (5), propionic acid (6), 3-hydroxy-4-methoxybenzoic acid (7), and 4-(3-formyl-5-methoxyphenyl) butanoic acid (8) were isolated from the ethanol extracts of Cortex Pseudolaricis by bioassay-guided fractionation and evaluated for in vitro antifungal activity against Colletotrichum gloeosporioides Penz. Results demonstrated that all of the eight compounds inhibited the mycelial growth of C. gloeosporioides at 5 μg/mL. Among them, pseudolaric acid B and pseudolaric acid A showed the strongest inhibition with the EC50 values of 1.07 and 1.62 μg/mL, respectively. Accordingly, both Pseudolaric acid B and Pseudolaric acid A highly inhibited spore germination and germ tube elongation of C. gloeosporioides. Dipping 100 μg/mL pseudolaric acid B treatment exhibited more effective suppression on postharvest anthracnose in mango fruit when compared to the same concentration of carbendazim. Scanning electron microscopy observations revealed that pseudolaric acid B caused alterations in the hyphal morphology of C. gloeosporioides, including distortion, swelling, and collapse. Pseudolaric acid B caused the mycelial apexes to show an abnormal growth in dimensions with multiple ramifications in subapical expanded areas with irregular shape. These findings warrant further investigation into optimization of pseudolaric acid B to explore a potential antifungal agent for crop protection.
Co-reporter:Mei-Juan Wang, Xiao-Bo Zhao, Dan Wu, Ying-Qian Liu, Yan Zhang, Xiang Nan, Huanxiang Liu, Hai-Tao Yu, Guan-Fang Hu, and Li-Ting Yan
Journal of Agricultural and Food Chemistry 2014 Volume 62(Issue 24) pp:5429-5442
Publication Date(Web):May 17, 2014
DOI:10.1021/jf501108j
Three novel series of N3-substituted imidacloprid derivatives were designed and synthesized, and their structures were identified on the basis of satisfactory analytical and spectral (1H NMR, 13C NMR, MS, elemental analysis, and X-ray) data. Preliminary bioassays indicated that all of the derivatives exhibited significant insecticidal activities against Aphis craccivora, with LC50 values ranging from 0.00895 to 0.49947 mmol/L, and the insecticidal activities of some of them were comparable to those of the control imidacloprid. Some key structural features related to their insecticidal activities were identified, and the binding modes between target compounds and nAChR model were also further explored by molecular docking. By comparing the interaction features of imidacloprid and compound 26 with highest insecticidal activity, the origin of the high insecticidal activity of compound 26 was identified. On the basis of the conformations generated by molecular docking, a satisfactory 2D-QSAR model with six selected descriptors was built using genetic algorithm–multiple linear regression (GA-MLR) method. The analysis of the built model showed the molecular size, shape, and the ability to form hydrogen bond were important for insecticidal potency. The information obtained in the study will be very helpful for the design of new derivatives with high insecticidal activities.
Co-reporter:Xiao-Bo Zhao, Masuo Goto, Zi-Long Song, Susan L. Morris-Natschke, Yu Zhao, Dan Wu, Liu Yang, Shu-Gang Li, Ying-Qian Liu, Gao-Xiang Zhu, Xiao-Bing Wu, Kuo-Hsiung Lee
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 16) pp:3850-3853
Publication Date(Web):15 August 2014
DOI:10.1016/j.bmcl.2014.06.060
A series of novel 7-(N-substituted-methyl)-camptothecin derivatives was designed, synthesized, and evaluated for in vitro cytotoxicity against four human tumor cell lines, A-549, MDA-MB-231, KB, and KBvin. All of the derivatives showed promising in vitro cytotoxic activity against the tested tumor cell lines, with IC50 values ranging from 0.0023 to 1.11 μM, and were as or more potent than topotecan. Compounds 9d, 9e, and 9r exhibited the highest antiproliferative activity among all prepared derivatives. Furthermore, all of the compounds were more potent than paclitaxel against the multidrug-resistant (MDR) KBvin subline. With a concise efficient synthesis and potent cytotoxic profiles, especially significant activity towards KBvin, compounds 9d, 9e, and 9r merit further development as a new generation of camptothecin-derived anticancer clinical trial candidates.
Co-reporter:Liu Yang;Mei-Juan Wang;Zhi-Jun Zhang
Medicinal Chemistry Research 2014 Volume 23( Issue 7) pp:3269-3273
Publication Date(Web):2014 July
DOI:10.1007/s00044-013-0906-8
Chemotherapy is a general treatment option for various cancers, including lung cancer. In order to find compounds with superior bioactivity and less toxicity against lung cancer, novel spin-labeled 5-fluorouracil (5-FU) derivatives (3a–f) were synthesized and evaluated against four human tumor cell lines (A-549, DU-145, KB, and KBvin). Two promising compounds 3d and 3f exhibited IC50 values of 2.76 and 2.38 μM, respectively, against non-small cell lung carcinoma cell line A-549. These compounds were twofold more cytotoxic than 5-FU and less toxic against other tested cell lines. Compound 3f exhibited seven times more selective cytotoxicity against A-549 than 5-FU. Our results suggest that compounds 3d and 3f merit further investigation for development into clinical trial candidates for non-small cell lung cancer.
Co-reporter:Liu Yang;Yong-Long Zhao;Chun-Yan Zhao;Huan-Huan Li
Medicinal Chemistry Research 2014 Volume 23( Issue 12) pp:5043-5057
Publication Date(Web):2014 December
DOI:10.1007/s00044-014-1046-5
Two novel series of sulfonylamidine-derived neonicotinoid analogues were designed and synthesized via a Cu-catalyzed one pot reaction. The structures of the target compounds were characterized by 1H-NMR, 13C-NMR, HR-MS and the single-crystal structure of 14k was further determined by X-ray diffraction crystallography. In preliminary bioassays, many of the target compounds exhibited significant activity against Tetranychuscinnabarinus (carmine spider mite) and Brevicorynebrassicae (cabbage aphid). Some structural features important for potency were observed, and a model of the ligand–receptor complex was also generated with a molecular docking study using representative active analogues. The molecular binding mode observed at the active site of the nicotinic acetylcholine receptor model should provide useful information for future structure-based design of novel sulfonylamidine-derived neonicotinoid analogues.
Co-reporter:Xiu-Qing Lv;Gang Feng;Xiang Nan;Liu Yang
Medicinal Chemistry Research 2014 Volume 23( Issue 7) pp:3347-3352
Publication Date(Web):2014/07/01
DOI:10.1007/s00044-014-0917-0
The insecticidal activity of a natural compound combretastatin A-4 (CA-4) was assessed in in vivo assays against Spodoptera litura. CA-4, mixed into an artificial diet, caused various effects, depending on the concentration used. Compared with controls, food intake was lower, larval growth was reduced and larval development prolonged. Pupae weight was reduced, and adult morphology was also affected. Relative to the untreated group, treated groups showed higher mortality at larval and pupal stages, which was generally caused by moult disruption. Results of this study with CA-4 resemble those found in treatment with insect growth regulators. This study indicated that CA-4 merits further study as a lead compound in a novel class of potential insect-control agents or for managing field populations of Lepidoptera pests on cruciferous crops.
Co-reporter:Ying-Qian Liu, Xiao-Jing Li, Chun-Yan Zhao, Xiang Nan, Jing Tian, Susan L. Morris-Natschke, Zhi-Jun Zhang, Xiao-Ming Yang, Liu Yang, Lin-Hai Li, Xing-Wen Zhou, Kuo-Hsiung Lee
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 5) pp:1248-1256
Publication Date(Web):1 March 2013
DOI:10.1016/j.bmc.2012.12.046
Two series (14a–d and 21a–h) of novel spin-labeled combretastatin derivatives were synthesized and evaluated for cytotoxicity against four tumor cell lines (K562, SGC-7901, Hela and HepG-2). Simultaneously, a representative compound 21a was selected to investigate the antitumor mechanisms of these synthetic compounds. The results indicated that some of the compounds showed significant cytotoxicity against four tumor cell lines in vitro and were more active than etoposide, a clinically available anticancer drug. Among the newly synthesized compounds, 21a, 21b and 21c displayed the greatest cytotoxicity against three tested tumor cell lines (HEPG-2, BGC-832 and Hela), with IC50 values ranging from 0.15 to 1.05 μM, compared with values of 0.014–0.403 μM for 3-amino-deoxycombretastatin A-4 (3). In addition, the mechanistic analysis revealed that compound 21a effectively interfered with tubulin dynamics to prevent mitosis in cancer cells, leading to cell cycle arrest and, eventually, dose dependent apoptosis.
Co-reporter:Wen-Qun Li, Xu-Li Wang, Keduo Qian, Ying-Qian Liu, Chih-Ya Wang, Liu Yang, Jin Tian, Susan L. Morris-Natschke, Xing-Wen Zhou, Kuo-Hsiung Lee
Bioorganic & Medicinal Chemistry 2013 21(8) pp: 2363-2369
Publication Date(Web):
DOI:10.1016/j.bmc.2013.01.069
Co-reporter:Ying-Qian Liu;Xiao-Jing Li;Chun-Yan Zhao;Yan Lu
Medicinal Chemistry Research 2013 Volume 22( Issue 5) pp:2196-2206
Publication Date(Web):2013 May
DOI:10.1007/s00044-012-0212-x
Continuing our search for natural product-based compounds for the control of B. longissima Larvae, 25 stilbene analogs were synthesized and evaluated for insect antifeedant activity against third-instar larvae of B. longissima for the first time. Among all the tested compounds, especially compounds 3a, 3c, and 6 showed pronounced antifeedant activities with AFC50 values of 0.218, 0.327, and 0.226 mg/mL, respectively. The different antifeedant activity ranges of these compounds indicated that variation of chemical structures in the stilbene skeleton markedly affected the activity profiles of this compound class, and some important SAR information has been revealed from it. In addition, to understand the structural requirements for antifeedant activities of the 25 synthesized stilbene analogs, a comparative molecular field analysis (CoMFA) model, which yielded the leave-one-out (LOO) cross-validated correlation coefficient (q2) of 0.533 and a non-cross-validated correlation coefficient (r2) of 0.929, was constructed. Together, these preliminary results may be useful in guiding further modification of stilbenes in the development of potential new antifeedants.
Co-reporter:Ying-Qian Liu, Wei Dai, Chih-Ya Wang, Susan L. Morris-Natschke, Xing-Wen Zhou, Liu Yang, Xiao-Ming Yang, Wen-Qun Li, Kuo-Hsiung Lee
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 24) pp:7659-7661
Publication Date(Web):15 December 2012
DOI:10.1016/j.bmcl.2012.10.002
New 7-acyl camptothecin derivatives were designed and synthesized from camptothecin in a one-pot reaction through a Minisci type-reaction and were evaluated for cytotoxicity against four tumor cell lines, A-549, DU-145, KB, and KB-vin. All of the new compounds showed significant inhibition of human tumor cell growth, with IC50 values ranging from 0.01538 to 13.342 μM. Most of the derivatives were more cytotoxic than irinotecan, and the (7a) and 7-propionyl (7b) analogs exhibited the highest cytotoxic activity against the tumor cell lines tested. This compound class merits further development as anticancer clinical trial candidates.
Co-reporter:Ying-Qian Liu;Wei Dai;Jing Tian;Liu Yang;Gang Feng;Xing-Wen Zhou;Liang Kou;Yong-Long Zhao;Wen-Qun Li;Lin-Hai Li;Hong-Yu Li
Heteroatom Chemistry 2011 Volume 22( Issue 6) pp:687-691
Publication Date(Web):
DOI:10.1002/hc.20734
Abstract
Continuing our search for natural product based compounds for the control of Brontispa longissima larvae, eight spin-labeled camptothecin derivatives 7a–h and the intermediate 2 were first tested for their insecticidal activities against fifth-instar larvae of Brontispa longissima. Among all the tested compounds, especially compounds 7a and 2 showed promising insecticidal activities with the corrected mortality rates of 69.55% and 74.07% against fifth-instar larvae of B. longissima, respectively. The different insecticidal activity ranges of these compounds indicated that the variation of the structures of l-amino acids in these compounds markedly affected the activity profiles of this compound class, and some important SAR information has been revealed from it. © 2011 Wiley Periodicals, Inc. Heteroatom Chem 22:687–691, 2011; View this article online at wileyonlinelibrary.com. DOI 10.1002/hc.20734
Co-reporter:Ying-Qian Liu, Xuan Tian, Liu Yang, Zong-Cheng Zhan
European Journal of Medicinal Chemistry 2008 Volume 43(Issue 11) pp:2610-2614
Publication Date(Web):November 2008
DOI:10.1016/j.ejmech.2008.01.008
In an effort to improve the stability of labile lactone ring and water solubility of camptothecin, five novel spin-labeled camptothecin derivatives were synthesized in quantitative yield by a simple modification of the carbodiimide method using the combination of scandium triflate (Sc(OTf)3) and 4-dimethylaminopyridine (DMAP), and the in vitro pharmacokinetic determination of the lactones of representative compound 13a showed that the biological life span of their lactone forms in human and mouse plasma significantly increased when compared with their mother compound camptothecin. Also, the in vitro cytotoxicity of compounds 13a–13e against human bladder cancer T-24 showed either similar or better activity than that of the parent drug, camptothecin, and clinically available drug, irinotecan. Five novel spin-labeled camptothecin derivatives were synthesized and the in vitro pharmacokinetic determination of the lactones of representative compound 13a showed that the biological life span of their lactone forms in human and mouse plasma significantly increased when compared with their mother compound camptothecin. Also, the in vitro cytotoxicity of compounds 13a–13e against human bladder cancer T-24 showed either similar or better activity than that of the parent drug, camptothecin, and clinically available drug, irinotecan.
Co-reporter:Ying-Qian Liu, Yanqing Liu, Hang Xiao, Rong Gao, Xuan Tian
Pesticide Biochemistry and Physiology (June 2008) Volume 91(Issue 2) pp:116-121
Publication Date(Web):1 June 2008
DOI:10.1016/j.pestbp.2008.03.002
In order to find the biorational pesticides, eight novel 4β-substituted phenoxyaniline derivatives of podophyllotoxin have been synthesized with significant stereoselectivity and improved yields by employing the BF3·Et2O/NaI reagent system and evaluated for their antifeedant effect against 5th instar larvae of Pieris rapae and effect on the development of 5th instar larvae of P. rapae as well as for insecticidal activity against P. rapae. The antifeedant activities showed that these compounds exhibited less potent than podophyllotoxin. Though these derivatives showed less potent antifeedant activities than podophyllotoxin, they acted as growth development inhibitor to 5th instar larvae of P. rapae, which were found that the animals treated with podophyllotoxin and its derivatives showed moulting disturbances and/or deformities. Also, the insecticidal activity results show that all of these derivatives of PPT showed delayed insecticidal activity, which differs from traditional neurotoxic insecticides. Among them, compounds possessing a 4β-phenoxyaniline moiety substituted (CO2C2H5, Cl and OH) at para position exhibited greater insecticidal activity against P. rapae than podophyllotoxin.
Co-reporter:Ying-Qian Liu, Yong-Long Zhao, Liu Yang, Xing-Wen Zhou, Gang Feng
Pesticide Biochemistry and Physiology (January 2012) Volume 102(Issue 1) pp:11-18
Publication Date(Web):1 January 2012
DOI:10.1016/j.pestbp.2011.09.012
In an attempt to find the effective phytopesticides, a series of novel podophyllotoxin derivatives were firstly synthesized and preliminarily tested for their antifeedant and insecticidal effects against the fifth-instar larvae of Brontispa longissima. The different antifeedant and insecticidal activity ranges of compounds 3a-l showed that variations of NR1R2 groups in the 4-position of podophyllotoxin skeleton markedly affected the activity profiles of this compound class, and some important SAR information has been revealed from it. To clarify their mode of action of insecticidal activity, the docking models as well as tubulin inhibitory effect of representative compound 3i were also investigated, and the result indicated that the insecticidal activity of these compounds was due to the tubulin inhibitory effect of these derivatives, thereby possibly providing some useful information for rational designs of novel podophyllotoxin-based insecticides.Graphical abstractA series of novel podophyllotoxin derivatives were firstly synthesized via a Cu-catalyzed one pot reaction for their insecticidal activity studies.Download full-size imageHighlights► Insecticidal activity of 4β-O-sulfonyl amidine podophyllotoxin derivative against Brontispa longissima was firstly reported. ► Their insecticidal activity results are encouraging and important SAR clues has revealed from it. ► Molecular docking and study of mechanisms contributed to a better development of podophyllotoxin-derived insecticides.
Co-reporter:Ying-Qian Liu, Chun-Yan Zhao, Liu Yang, Yong-Long Zhao, Ting-Ting Liu
Pesticide Biochemistry and Physiology (January 2011) Volume 99(Issue 1) pp:39-44
Publication Date(Web):1 January 2011
DOI:10.1016/j.pestbp.2010.10.002
Twenty podophyllotoxin analogues were first tested for their insecticidal activity against the fifth-instar larvae of Brontispa longissima in vivo. Among them, compounds 6–9 and 19 showed more promising and pronounced insecticidal activity than toosendanin, a commercial insecticide derived from Melia azedarach. The different insecticidal activity ranges of compounds 1–20 indicated that variation of chemical structures in the podophyllotoxin skeleton markedly affected the activity profiles of this compound class, and some important SAR information has been revealed from it. The results obtained from SAR analysis show good correlation with the docking models as well as with QSAR studies, which allows for the rational design of more potent podophyllotoxin derivatives in the development of potential new insecticides.Graphical abstractTwenty podophyllotoxin analogues were first tested for their insecticidal activity against the fifth-instar larvae of Brontispa longissima in vivo. The results obtained from SAR analysis show good correlation with the docking models as well as with QSAR studies, which allows for the rational design of more potent podophyllotoxin derivatives in the development of potential new insecticides.Download full-size imageResearch highlights► Insecticidal activity of compounds 1–20 against B. longissima was firstly reported. ► Their biological results are encouraging and important SAR clues has revealed from it. ► QSAR and molecular docking contributed to further optimization of podophyllotoxin derivatives.

![4(S)-Ethyl-4-(propionyloxy)-3,4,12,14-tetrahydro-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,14-dione](http://img.cochemist.com/ccimg/194500/194414-69-2.png)
![4(S)-Ethyl-4-(propionyloxy)-3,4,12,14-tetrahydro-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,14-dione](http://img.cochemist.com/ccimg/194500/194414-69-2_b.png)
![[5R-(5ALPHA,5ABETA,8AALPHA,9BETA)]-9-[2-[N-[2-(DIMETHYLAMINO)ETHYL]-N-METHYLAMINO]ETHYL]-5-(4-HYDROXY-3,5-DIMETHOXYPHENYL)-5,5A,6,8,8A,9-HEXAHYDROFURO[3',4':6,7]NAPHTHO[2,3-D]-1,3-DIOXOL-6-ONE](http://img.cochemist.com/ccimg/148300/148262-19-5.png)
![[5R-(5ALPHA,5ABETA,8AALPHA,9BETA)]-9-[2-[N-[2-(DIMETHYLAMINO)ETHYL]-N-METHYLAMINO]ETHYL]-5-(4-HYDROXY-3,5-DIMETHOXYPHENYL)-5,5A,6,8,8A,9-HEXAHYDROFURO[3',4':6,7]NAPHTHO[2,3-D]-1,3-DIOXOL-6-ONE](http://img.cochemist.com/ccimg/148300/148262-19-5_b.png)









![1-Piperidinyloxy, 2,2,6,6-tetramethyl-4-[(2-propynyloxy)methyl]-](http://img.cochemist.com/ccimg/147100/147045-22-5.png)
![1-Piperidinyloxy, 2,2,6,6-tetramethyl-4-[(2-propynyloxy)methyl]-](http://img.cochemist.com/ccimg/147100/147045-22-5_b.png)


![Furo[3',4':6,7]naphtho[2,3-d]-1,3-dioxol-6(5aH)-one,5,8,8a,9-tetrahydro-5-(4-hydroxy-3,5-dimethoxyphenyl)-9-[(4-nitrophenyl)amino]-,(5R,5aR,8aS,9S)-](http://img.cochemist.com/ccimg/127900/127882-73-9.png)
![Furo[3',4':6,7]naphtho[2,3-d]-1,3-dioxol-6(5aH)-one,5,8,8a,9-tetrahydro-5-(4-hydroxy-3,5-dimethoxyphenyl)-9-[(4-nitrophenyl)amino]-,(5R,5aR,8aS,9S)-](http://img.cochemist.com/ccimg/127900/127882-73-9_b.png)
![Furo[3',4':6,7]naphtho[2,3-d]-1,3-dioxol-6(5aH)-one,9-[(4-fluorophenyl)amino]-5,8,8a,9-tetrahydro-5-(4-hydroxy-3,5-dimethoxyphenyl)-,(5R,5aR,8aS,9S)-](http://img.cochemist.com/ccimg/125900/125830-36-6.png)
![Furo[3',4':6,7]naphtho[2,3-d]-1,3-dioxol-6(5aH)-one,9-[(4-fluorophenyl)amino]-5,8,8a,9-tetrahydro-5-(4-hydroxy-3,5-dimethoxyphenyl)-,(5R,5aR,8aS,9S)-](http://img.cochemist.com/ccimg/125900/125830-36-6_b.png)








![N-[1-[(6-CHLOROPYRIDIN-3-YL)METHYL]-4,5-DIHYDROIMIDAZOL-2-YL]NITRAMIDE](http://img.cochemist.com/ccimg/105900/105827-78-9.png)
![N-[1-[(6-CHLOROPYRIDIN-3-YL)METHYL]-4,5-DIHYDROIMIDAZOL-2-YL]NITRAMIDE](http://img.cochemist.com/ccimg/105900/105827-78-9_b.png)







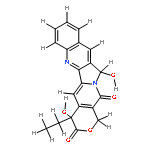
![(4S)-4-ethyl-4-hydroxy-3,14-dioxo-3,4,12,14-tetrahydro-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-11-carbaldehyde](http://img.cochemist.com/ccimg/80800/80758-83-4.png)
![(4S)-4-ethyl-4-hydroxy-3,14-dioxo-3,4,12,14-tetrahydro-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-11-carbaldehyde](http://img.cochemist.com/ccimg/80800/80758-83-4_b.png)
![Methanesulfonic acid, 1,1,1-trifluoro-, 4-[(4-chlorophenyl)hydrazinylidenemethyl]phenyl ester](/data/chemimg/1326400/78930-87-7.png)
![Methanesulfonic acid, 1,1,1-trifluoro-, 4-[(4-chlorophenyl)hydrazinylidenemethyl]phenyl ester](/data/chemimg/1326400/78930-87-7_b.png)
![[4-(4-CHLOROBENZOYL)PHENYL] TRIFLUOROMETHANESULFONATE](http://img.cochemist.com/ccimg/79000/78930-82-2.png)
![[4-(4-CHLOROBENZOYL)PHENYL] TRIFLUOROMETHANESULFONATE](http://img.cochemist.com/ccimg/79000/78930-82-2_b.png)

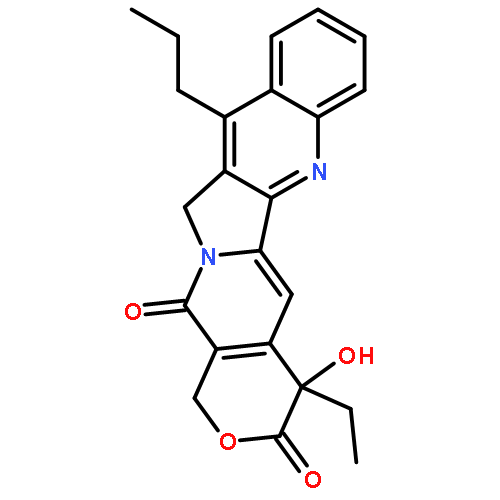
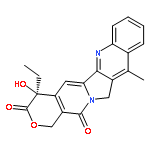
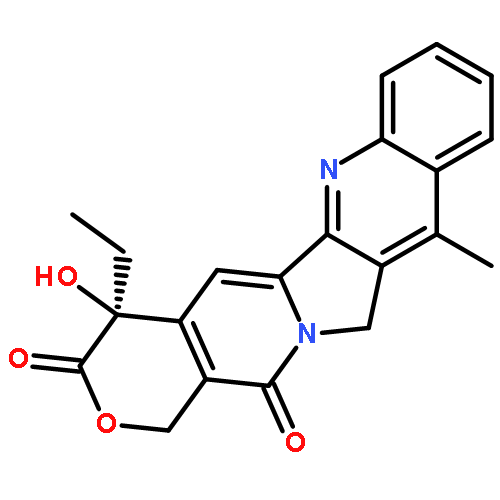
![(4S)-4-ethyl-4-hydroxy-3,14-dioxo-3,4,12,14-tetrahydro-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-11-carboxylic acid](http://img.cochemist.com/ccimg/78300/78287-24-8.png)
![(4S)-4-ethyl-4-hydroxy-3,14-dioxo-3,4,12,14-tetrahydro-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-11-carboxylic acid](http://img.cochemist.com/ccimg/78300/78287-24-8_b.png)

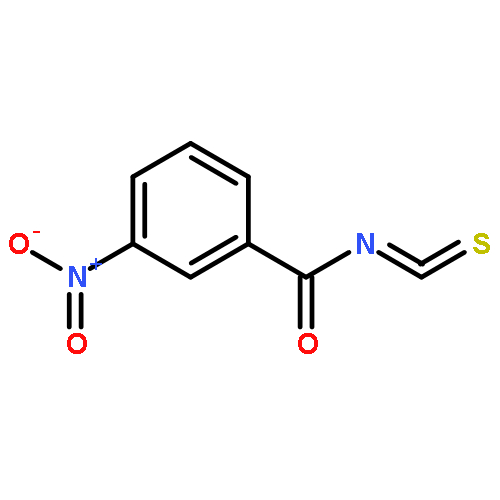
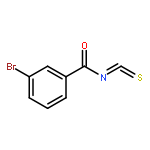








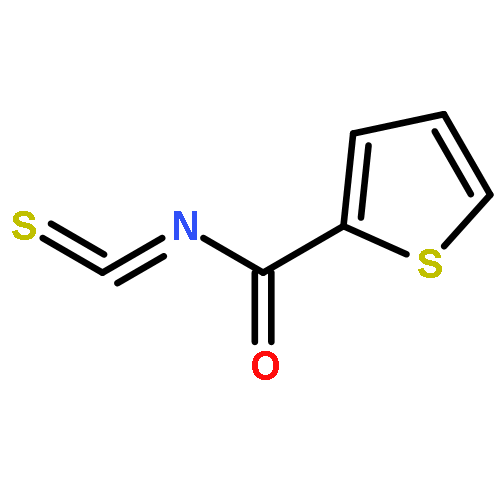
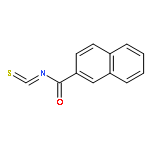
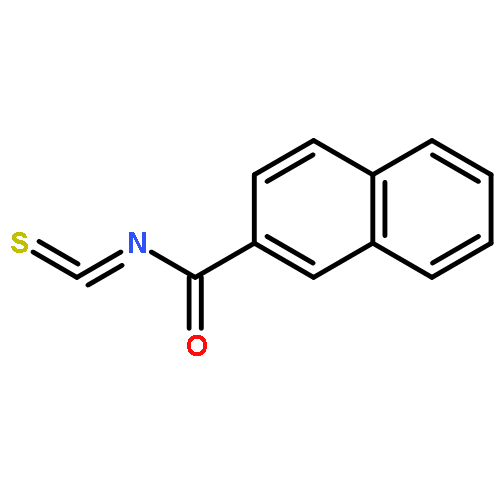



![Indolizino[1,2-b]quinolin-9(11H)-one,8-methyl-7-(1-oxopropyl)-](http://img.cochemist.com/ccimg/55900/55854-89-2.png)
![Indolizino[1,2-b]quinolin-9(11H)-one,8-methyl-7-(1-oxopropyl)-](http://img.cochemist.com/ccimg/55900/55854-89-2_b.png)










![Carbamic acid, [(4-methoxyphenyl)sulfonyl]-, ethyl ester](http://img.cochemist.com/ccimg/14000/13945-54-5.png)
![Carbamic acid, [(4-methoxyphenyl)sulfonyl]-, ethyl ester](http://img.cochemist.com/ccimg/14000/13945-54-5_b.png)



![1H-PYRANO[3',4'_6,7]INDOLIZINO[1,2-B]QUINOLINE-3,14(4H,12H)- DIONE, 4-(ACETYLOXY)-4-ETHYL-, (S)-](http://img.cochemist.com/ccimg/7700/7688-64-4.png)
![1H-PYRANO[3',4'_6,7]INDOLIZINO[1,2-B]QUINOLINE-3,14(4H,12H)- DIONE, 4-(ACETYLOXY)-4-ETHYL-, (S)-](http://img.cochemist.com/ccimg/7700/7688-64-4_b.png)
![9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydro-[2]benzofuro[5,6-f][1,3]benzodioxol-5-ol](http://img.cochemist.com/ccimg/6400/6320-12-3.png)
![9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydro-[2]benzofuro[5,6-f][1,3]benzodioxol-5-ol](http://img.cochemist.com/ccimg/6400/6320-12-3_b.png)








![ethyl [(4-chlorophenyl)sulfonyl]carbamate](http://img.cochemist.com/ccimg/14000/13945-53-4.png)
![ethyl [(4-chlorophenyl)sulfonyl]carbamate](http://img.cochemist.com/ccimg/14000/13945-53-4_b.png)