Co-reporter:Hikaru Takano;Tetsuo Narumi;Wataru Nomura
The Journal of Organic Chemistry March 3, 2017 Volume 82(Issue 5) pp:2739-2744
Publication Date(Web):February 2, 2017
DOI:10.1021/acs.joc.6b02656
By screening of reaction conditions and evaluation of its fluorescent properties, an environmentally sensitive fluorescent group, 8-aza-7-hydroxy-4-methylcoumarin, was synthesized in 70% yield using MgBr2 as a Lewis acid under microwave irradiation. It has a high fluorescent quantum yield, is adequately soluble in water, and produces fluorescent emission in protic solvents and at neutral pH. Therefore, it could be useful in biosensors that are required to emit in hydrophilic environments such as cells.
Co-reporter:Takuya Kobayakawa, Hirokazu Tamamura
Tetrahedron 2017 Volume 73, Issue 30(Issue 30) pp:
Publication Date(Web):27 July 2017
DOI:10.1016/j.tet.2017.06.003
An efficient method for an atom-economical synthesis of Xaa-Yaa type (Z)-chloroalkene dipeptide isosteres (CADIs) in high yield and diastereoselectivity has been developed. Reaction of an allylic gem-dichloride compound with organocopper reagents prepared from catalyst quantity of 30 mol% CuCl and organozinc reagents causes formation of (Z)-chloroalkenes corresponding to (L,D)-type Xaa-Yaa CADIs by diastereoselective allyic alkylation. Using (E)-enoates of substrates in place of (Z)-enoates in similar reactions with organocopper reagents, which prepared from 140 mol% CuCl and organozinc reagents, provides various (L,L)-type CADIs.Download high-res image (223KB)Download full-size image
Co-reporter:Kei Toyama, Takaaki Mizuguchi, Wataru Nomura, Hirokazu Tamamura
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 16) pp:3406-3412
Publication Date(Web):15 August 2016
DOI:10.1016/j.bmc.2016.05.026
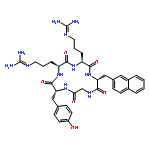
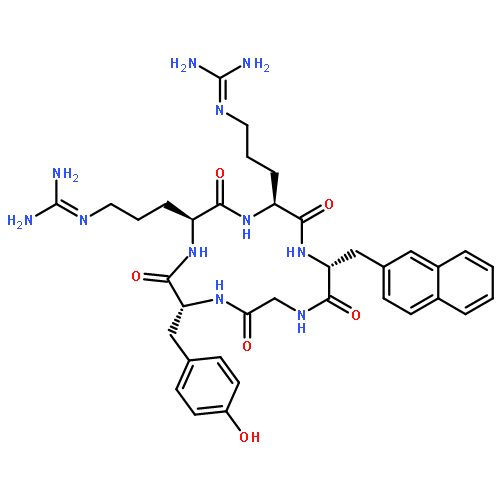
A cyclic decapeptide (1, ), which acts on the extracellular region of the EGF receptor, preventing it from dimerizing, has been developed. Peptide 2, which was labeled with fluorescein at the N-terminus of peptide 1, was synthesized based on structure–activity relationship studies. Peptide 2 essentially retained the inhibitory activity of peptide 1 against the receptor autophosphorylation. Confocal microscopy studies revealed that in carcinoma cells, the fluorescence of peptide 2 was localized inside some vesicles. Treatment of intact cells by peptide 1 in combination with peptide 2 decreased the fluorescence intensity significantly compared to treatment with only peptide 2. These results indicate that peptide 2 competes with peptide 1 for binding to the cellular surface. Six derivatives of peptide 2, in which constituent amino acids, with the exception of two cysteines and proline were randomized, were synthesized and used to treat the cells. Peptides 6 and 9 showed the highest fluorescence intensity in cells. From the results of the EGF receptor autophosphorylation assay, these two derivatives were proven to have higher inhibitory activity than peptide 2, which would therefore be a useful delivery peptide and fluorescent probe to find new inhibitors against the EGF receptor. Peptides 6 and 9 are promising leads for EGF receptor inhibitors.
Co-reporter:Takaaki Mizuguchi, Shigeyoshi Harada, Tomoyuki Miura, Nami Ohashi, Tetsuo Narumi, Hiromi Mori, Yu Irahara, Yuko Yamada, Wataru Nomura, Shuzo Matsushita, Kazuhisa Yoshimura, Hirokazu Tamamura
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 2) pp:397-400
Publication Date(Web):15 January 2016
DOI:10.1016/j.bmcl.2015.11.103
Several CD4 mimics have been reported as HIV-1 entry inhibitors which can block the interaction between the viral envelope glycoprotein gp120 and the cell surface protein CD4. We previously found a lead compound 2 (YYA-021) with high anti-HIV activity and low cytotoxicity. Pharmacokinetic analysis however showed compound 2 to have wide tissue distribution and relatively high distribution volumes in rats and rhesus macaques. In the present study we searched for more hydrophilic CD4 mimics with a view to reducing tissue distribution. A new compound (5) with a 1,3-benzodioxolyl moiety was found to have relatively high anti-HIV activity and no significant cytotoxicity. Compound 5 is more hydrophilic than compound 2 and the pharmacokinetics of the intravenous administration of compound 5 in a rhesus macaque showed that compound 5 has lower tissue distribution than compound 2, suggesting that compound 5 possesses a better profile.A low-cytotoxic CD4 mimic molecule with an appropriate pharmacokinetic profile as an HIV entry inhibitor is reported.
Co-reporter:Takuya Kobayakawa, Hirokazu Tamamura
Tetrahedron 2016 Volume 72(Issue 32) pp:4968-4971
Publication Date(Web):11 August 2016
DOI:10.1016/j.tet.2016.06.080
A highly stereoselective synthesis of (Z)-chloroalkene dipeptide isosteres (CADIs) of the Xaa-Gly type has been achieved. Treatment of an allylic gem-dichloride with an organocuprate provided (Z)-chloroalkene formation corresponding to an Xaa-Gly type CADI through reductive dechlorination in high yield with (Z)-selectivity.
Co-reporter:Takuya Kobayakawa, Tetsuo Narumi, and Hirokazu Tamamura
Organic Letters 2015 Volume 17(Issue 10) pp:2302-2305
Publication Date(Web):May 7, 2015
DOI:10.1021/acs.orglett.5b00611
Highly diastereoselective synthesis of (Z)-chloroalkene dipeptide isosteres has been achieved by 1,4-asymmetric induction in the organocuprate-mediated allylic alkylation adjacent to the chiral center of allylic gem-dichlorides. The reaction proceeds with a variety of heterocuprates prepared from CuCN and various organometallic reagents. It allows rapid construction of valuable architectures of l,d-type and l,l-type (Z)-chloroalkene dipeptide isosteres from the corresponding (E)- and (Z)-allylic gem-dichlorides in high yields, with excellent (Z)-selectivity and diastereoselectivity.
Co-reporter:Hikaru Takano, Tetsuo Narumi, Wataru Nomura, Toshiaki Furuta, and Hirokazu Tamamura
Organic Letters 2015 Volume 17(Issue 21) pp:5372-5375
Publication Date(Web):October 15, 2015
DOI:10.1021/acs.orglett.5b02720
A remarkable improvement of the photochemical properties of coumarin-type photolabile protecting groups was achieved by iodine substitution. The newly identified 7-hydroxy-6-iodo-8-azacoumarin (8-aza-Ihc)-caged acetate showed excellent photolytic efficiency, significantly higher than that of the corresponding bromine-containing coumarin- and azacoumarin-type caging groups. The results provide a solid approach to improving the photosensitivity of photolabile protecting groups.
Co-reporter:Wataru Nomura, Nami Ohashi, Atsumi Mori, and Hirokazu Tamamura
Bioconjugate Chemistry 2015 Volume 26(Issue 6) pp:1080
Publication Date(Web):May 4, 2015
DOI:10.1021/acs.bioconjchem.5b00131
Fluorogenic probes are useful as molecular tools in chemical biology because they can overcome noise associated with background emission. Previously, using a leucine zipper assembly, we developed a fluorogenically active ZIP tag–probe pair. A probe peptide was designed as an α-helical peptide containing 4-nitrobenzo-2-oxa-1,3-diazole, a solvatochromic fluorescent dye. Tag peptides were designed as antiparallel 2 α-helical peptides, and the tag and probe together form the 3 α-helical bundle structure of the leucine zipper. The use of the system was limited to membrane proteins or targets on the cellular surface because the probe peptide was not compatible with cell penetration. In this study, a challenge for the fluorescent imaging of proteins inside the cells was conducted by development of the ZIP tag–probe system as the second generation. To enable the cell penetration of the probe peptide, the addition of a cell penetrating peptide sequence was tested and a probe peptide with a C-terminal octa-arginine was shown to have high affinity for the tag peptide. In addition to attachment of a CPP structure, pretreatment of cells by 1-pyrenebutyrate enhanced distribution of the probe peptide into the cytosol. Observed colocalization of fluorescence of monomer Kusabira Orange and 4-nitrobenzo-2-oxa-1,3-diazole indicates our fluorogenic tag–probe system can be utilized with tagged proteins. Following stimulation by phorbol ester, the translocation of protein kinase C was tracked by the fluorescence of 4-nitrobenzo-2-oxa-1,3-diazole, suggesting the formation of the noncovalently assembled tag–probe pairing is maintained during the translocation, even when the concentration of the probe peptide is reduced to 0.1 μM. The results indicated that the dynamic change of the protein localization by chemical stimulations can be revealed by the ZIP tag–probe system. Above all, the system is simple to handle and highly compatible with virtually any protein inside the cells.
Co-reporter:Wataru Nomura, Taisuke Koseki, Nami Ohashi, Takaaki Mizuguchi and Hirokazu Tamamura
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 32) pp:8734-8739
Publication Date(Web):09 Jul 2015
DOI:10.1039/C5OB00891C
The assembly status of G protein-coupled receptors (GPCR) on the cell surface is of interest because the multimerization of GPCR could play pivotal roles in cellular functions. A bivalent ligand with polyproline linkers for CXCR4 has been shown to serve as a “molecular ruler” as a result of the rigid structure of polyproline helices. To expand the utility of the ligands with rigid linkers and explore the possible multimeric forms of GPCR, trivalent ligands with polyproline helices were newly designed and synthesized. The binding affinities of the trivalent ligands for CXCR4 suggested that the ligands recognize the dimeric form of CXCR4 on the cell surface. The fluorescence imaging and analysis by flow cytometry revealed that the ligand with 9 proline linkers binds to CXCR4 with remarkable specificity. The results of the present study suggest that the ligand design with rigid linkers is useful in the multimeric form, but the design of trivalent ligands requires different strategic approaches.
Co-reporter:Takaaki Mizuguchi, Nami Ohashi, Wataru Nomura, Mao Komoriya, Chie Hashimoto, Naoki Yamamoto, Tsutomu Murakami, Hirokazu Tamamura
Bioorganic & Medicinal Chemistry 2015 23(15) pp: 4423-4427
Publication Date(Web):
DOI:10.1016/j.bmc.2015.06.020
Co-reporter:Tetsuo Narumi, Hikaru Takano, Nami Ohashi, Akinobu Suzuki, Toshiaki Furuta, and Hirokazu Tamamura
Organic Letters 2014 Volume 16(Issue 4) pp:1184-1187
Publication Date(Web):February 4, 2014
DOI:10.1021/ol5000583
Described is the development of 8-azacoumarin-4-ylmethyl groups as aqueous photolabile protecting groups. A key feature of the strategy is the isosteric replacement of the C7–C8 enol double bond of the Bhc derivative with an amide bond, resulting in conversion of the chromophore from coumarin to 8-azacoumarin. This strategy makes dramatically enhanced water solubility and facile photocleavage possible.
Co-reporter:Dr. Tetsuo Narumi;Dr. Seiji Tsuzuki;Dr. Hirokazu Tamamura
Asian Journal of Organic Chemistry 2014 Volume 3( Issue 4) pp:497-503
Publication Date(Web):
DOI:10.1002/ajoc.201400026
Abstract
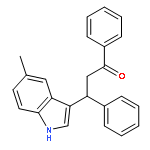
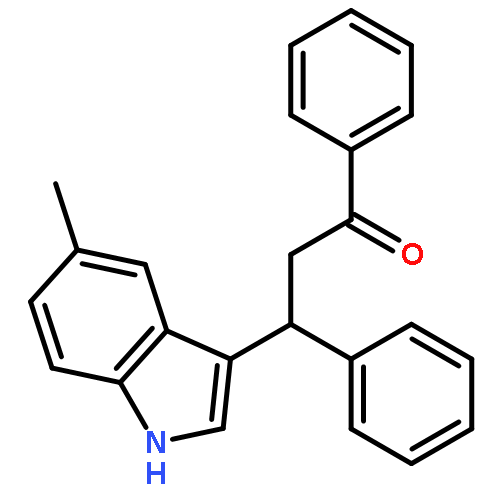
We describe the facile synthesis of 3-substituted indoles by Friedel–Crafts-type conjugate addition catalyzed by imidazolium salts. The reactions proceed under mild conditions with no base, solvent, or N-heterocyclic carbene. Mechanistic studies suggest the potential mechanism operates through the dual activation of indoles involving cation–π interactions of imidazolium cations with indoles and Lewis base activation by chloride anions derived from the imidazolium salts. High-level ab initio molecular orbital calculations reveal that there is a strong attraction between the imidazolium cation and the indole, and that the electrostatic and induction interactions strongly contributes to the attraction in the indole/imidazolium complex, which is a characteristic feature of the cation–π interaction.
Co-reporter:Hikaru Takano, Tetsuo Narumi, Nami Ohashi, Akinobu Suzuki, Toshiaki Furuta, Wataru Nomura, Hirokazu Tamamura
Tetrahedron 2014 70(29) pp: 4400-4404
Publication Date(Web):
DOI:10.1016/j.tet.2014.04.063
Co-reporter:Wataru Nomura, Haruo Aikawa, Nami Ohashi, Emiko Urano, Mathieu Métifiot, Masayuki Fujino, Kasthuraiah Maddali, Taro Ozaki, Ami Nozue, Tetsuo Narumi, Chie Hashimoto, Tomohiro Tanaka, Yves Pommier, Naoki Yamamoto, Jun A. Komano, Tsutomu Murakami, and Hirokazu Tamamura
ACS Chemical Biology 2013 Volume 8(Issue 10) pp:2235
Publication Date(Web):July 30, 2013
DOI:10.1021/cb400495h
HIV-1 integrase (IN) is an enzyme which is indispensable for the stable infection of host cells because it catalyzes the insertion of viral DNA into the genome and thus is an attractive target for the development of anti-HIV agents. Earlier, we found Vpr-derived peptides with inhibitory activity against HIV-1 IN. These Vpr-derived peptides are originally located in an α-helical region of the parent Vpr protein. Addition of an octa-arginyl group to the inhibitory peptides caused significant inhibition against HIV replication associated with an increase in cell permeability but also relatively high cytotoxicity. In the current study, stapled peptides, a new class of stabilized α-helical peptidomimetics were adopted to enhance the cell permeability of the above lead peptides. A series of stapled peptides, which have a hydrocarbon link formed by a ruthenium-catalyzed ring-closing metathesis reaction between successive turns of α-helix, were designed, synthesized, and evaluated for biological activity. In cell-based assays some of the stapled peptides showed potent anti-HIV activity comparable with that of the original octa-arginine-containing peptide (2) but with lower cytotoxicity. Fluorescent imaging experiments revealed that these stapled peptides are significantly cell permeable, and CD analysis showed they form α-helical structures, whereas the unstapled congeners form β-sheet structures. The application of this stapling strategy to Vpr-derived IN inhibitory peptides led to a remarkable increase in their potency in cells and a significant reduction of their cytotoxicity.
Co-reporter:Wataru Nomura, Chie Hashimoto, Takaharu Suzuki, Nami Ohashi, Masayuki Fujino, Tsutomu Murakami, Naoki Yamamoto, Hirokazu Tamamura
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 15) pp:4452-4458
Publication Date(Web):1 August 2013
DOI:10.1016/j.bmc.2013.05.060
To date, several HIV-1 fusion inhibitors based on the carboxy-terminal leucine/isoleucine heptad repeat (CHR) region of an HIV-1 envelope protein gp41 have been discovered. We have shown that a synthetic peptide mimetic of a trimer form of the CHR-derived peptide C34 has potent inhibitory activity against the HIV-1 fusion mechanism, compared to a monomer C34 peptide. The present study revealed that a dimeric form of C34 is evidently structurally critical for fusion inhibitors, and that the activity of multimerized CHR-derived peptides in fusion inhibition is affected by the properties of the unit peptides C34, SC34EK, and T20. The fluorescence-based study suggested that the N36-interactive sites of the C34 trimer, including hydrophobic residues, are exposed outside the trimer and that trimerization of C34 caused a remarkable increase in fusion inhibitory activity. The present results could be useful in the design of fusion inhibitors against viral infections which proceed via membrane fusion with host cells.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Chie Hashimoto, Tetsuo Narumi, Hiroyuki Otsuki, Yuki Hirota, Hiroshi Arai, Kazuhisa Yoshimura, Shigeyoshi Harada, Nami Ohashi, Wataru Nomura, Tomoyuki Miura, Tatsuhiko Igarashi, Shuzo Matsushita, Hirokazu Tamamura
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 24) pp:7884-7889
Publication Date(Web):15 December 2013
DOI:10.1016/j.bmc.2013.10.005
To date, several small molecules of CD4 mimics, which can suppress competitively the interaction between an HIV-1 envelope glycoprotein gp120 and a cellular surface protein CD4, have been reported as viral entry inhibitors. A lead compound 2 (YYA-021) with relatively high potency and low cytotoxicity has been identified previously by SAR studies. In the present study, the pharmacokinetics of the intravenous administration of compound 2 in rats and rhesus macaques is reported. The half-lives of compound 2 in blood in rats and rhesus macaques suggest that compound 2 shows wide tissue distribution and relatively high distribution volumes. A few hours after the injection, both plasma concentrations of compound 2 maintained micromolar levels, indicating it might have promise for intravenous administration when used combinatorially with anti-gp120 monoclonal antibodies.
Co-reporter:Chie Hashimoto, Wataru Nomura, Tetsuo Narumi, Masayuki Fujino, Toru Nakahara, Naoki Yamamoto, Tsutomu Murakami, Hirokazu Tamamura
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 22) pp:6878-6885
Publication Date(Web):15 November 2013
DOI:10.1016/j.bmc.2013.09.037
Despite almost 30 years since the identification of the human immunodeficiency virus type I (HIV-1), development of effective AIDS vaccines has been hindered by the high mutability of HIV-1. The HIV-1 co-receptors CCR5 and CXCR4 are genetically stable, but viral proteins may mutate rapidly during the course of infection. CXCR4 is a seven transmembrane G protein-coupled receptor, possessing an N-terminal region (NT) and three extracellular loops (ECL1-3). Previous studies have shown that the CXCR4-ED-derived peptides inhibit the entry of HIV-1 by interacting with gp120, an HIV-1 envelope glycoprotein. In the present study, antigenicity of CXCR4-derived peptides has been investigated and the anti-HIV-1 effects of induced antisera have been assessed. It was found that CXCR4-ED-derived antigen molecules immunize mice, showing that the linear peptides have higher antigenicity than the cyclic peptides. The L1- and L2-induced antisera inhibited the HIV-1 entry significantly, while anti-N1 antibodies have no inhibitory activity. This study produced promising examples for the design of AIDS vaccines which target the human protein and can overcome mutability of HIV-1.
Co-reporter:Tetsuo Narumi, Hiroshi Arai, Kazuhisa Yoshimura, Shigeyoshi Harada, Yuki Hirota, Nami Ohashi, Chie Hashimoto, Wataru Nomura, Shuzo Matsushita, Hirokazu Tamamura
Bioorganic & Medicinal Chemistry 2013 21(9) pp: 2518-2526
Publication Date(Web):
DOI:10.1016/j.bmc.2013.02.041
Co-reporter:Dr. Tetsuo Narumi;Dr. Haruo Aikawa;Dr. Tomohiro Tanaka;Chie Hashimoto;Dr. Nami Ohashi;Dr. Wataru Nomura;Takuya Kobayakawa;Hikaru Takano;Yuki Hirota;Dr. Tsutomu Murakami; Naoki Yamamoto; Hirokazu Tamamura
ChemMedChem 2013 Volume 8( Issue 1) pp:118-124
Publication Date(Web):
DOI:10.1002/cmdc.201200390
Abstract
Low-molecular-weight CXCR4 ligands based on known lead compounds including the 14-mer peptide T140, the cyclic pentapeptide FC131, peptide mimetics, and dipicolylamine-containing compounds were designed and synthesized. Three types of aromatic spacers, 1,4-phenylenedimethanamine, naphthalene-2,6-diyldimethanamine, and [1,1′-biphenyl]-4,4′-diyldimethanamine, were used to build four pharmacophore groups. As pharmacophore groups, 2-pyridylmethyl and 1-naphthylmethyl are present in all of the compounds, and several aromatic groups and a cationic group from 1-propylguanidine and 1,1,3,3-tetramethyl-2-propylguanidine were also used. Several compounds showed significant CXCR4 binding affinity, and zinc(II) complexation of bis(pyridin-2-ylmethyl)amine moieties resulted in a remarkable increase in CXCR4 binding affinity.
Co-reporter:Dr. Chie Hashimoto;Dr. Wataru Nomura;Dr. Tetsuo Narumi;Dr. Masayuki Fujino;Dr. Hiroshi Tsutsumi;Masaki Haseyama; Naoki Yamamoto;Dr. Tsutomu Murakami; Hirokazu Tamamura
ChemMedChem 2013 Volume 8( Issue 10) pp:1668-1672
Publication Date(Web):
DOI:10.1002/cmdc.201300289
Abstract
The human immunodeficiency virus type 1 (HIV-1) uses CD4 and the co-receptor CCR5 or CXCR4 in the process of cell entry. The negatively charged extracellular domains of CXCR4 (CXCR4-ED) interact with positive charges on the V3 loop of gp120, facilitating binding via electrostatic interactions. The presence of highly conserved positively charged residues in the V3 loop suggests that CXCR4-ED-derived inhibitors might be broadly effective inhibitors. Synthetic peptide derivatives were evaluated for anti-HIV-1 activity. The 39-mer extracellular N-terminal region (NT) was divided into three fragments with 10-mer overlapping sites (N1–N3), and these linear peptides were synthesized. Peptide N1 contains Met 1–Asp 20 and shows significant anti-HIV-1 activity. Extracellular loops 1 and 2 (ECL1 and 2) were mimicked by cyclic peptides C1 and C2, which were synthesized by chemoselective cyclization. Cyclic peptides C1 and C2 show higher anti-HIV-1 activity than their linear peptide counterparts, L1 and L2. The cytotoxicities of C1 and C2 are lower than those of L1 and L2. These results indicate that Met 1–Asp 20 segments of the NT and cyclic peptides of ECL1 and ECL2 are potent anti-HIV-1 drug candidates.
Co-reporter:Tetsuo Narumi, Takuya Kobayakawa, Haruo Aikawa, Shunsuke Seike, and Hirokazu Tamamura
Organic Letters 2012 Volume 14(Issue 17) pp:4490-4493
Publication Date(Web):August 17, 2012
DOI:10.1021/ol301988d
A robust and efficient method for the synthesis of trisubstituted (Z)-chloroalkenes is described. A one-pot reaction of γ,γ-dichloro-α,β-enoyl sultams involving organocuprate-mediated reduction/asymmetric alkylation affords α-chiral (Z)-chloroalkene derivatives in moderate to high yields with excellent diastereoselectivity, and allylic alkylation of internal allylic gem-dichlorides is also demonstrated. This study provides the first examples of the use of allylic gem-dichlorides adjacent to the chiral center for novel 1,4-asymmetric induction.
Co-reporter:Chie Hashimoto, Wataru Nomura, Aki Ohya, Emiko Urano, Kosuke Miyauchi, Tetsuo Narumi, Haruo Aikawa, Jun A. Komano, Naoki Yamamoto, Hirokazu Tamamura
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 10) pp:3287-3291
Publication Date(Web):15 May 2012
DOI:10.1016/j.bmc.2012.03.050
An artificial antigen forming the C34 trimeric structure targeting membrane-fusion mechanism of HIV-1 has been evaluated as an HIV vaccine. The C34 trimeric molecule was previously designed and synthesized using a novel template with C3-symmetric linkers by us. The antiserum produced by immunization of the C34 trimeric form antigen showed 23-fold higher binding affinity for the C34 trimer than for the C34 monomer and showed significant neutralizing activity. The present results suggest effective strategies of the design of HIV vaccines and anti-HIV agents based on the native structure mimic of proteins targeting dynamic supramolecular mechanisms in HIV fusion.
Co-reporter:Tetsuo Narumi, Mao Komoriya, Chie Hashimoto, Honggui Wu, Wataru Nomura, Shintaro Suzuki, Tomohiro Tanaka, Joe Chiba, Naoki Yamamoto, Tsutomu Murakami, Hirokazu Tamamura
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 4) pp:1468-1474
Publication Date(Web):15 February 2012
DOI:10.1016/j.bmc.2011.12.055
Compounds which inhibit the HIV-1 replication cycle have been found amongst fragment peptides derived from an HIV-1 matrix (MA) protein. Overlapping peptide libraries covering the whole sequence of MA were designed and constructed with the addition of an octa-arginyl group to increase their cell membrane permeability. Imaging experiments with fluorescent-labeled peptides demonstrated these peptides with an octa-arginyl group can penetrate cell membranes. The fusion of an octa-arginyl group was proven to be an efficient way to find active peptides in cells such as HIV-inhibitory peptides.
Co-reporter:Wataru Nomura, Akemi Masuda, Kenji Ohba, Arisa Urabe, Nobutoshi Ito, Akihide Ryo, Naoki Yamamoto, and Hirokazu Tamamura
Biochemistry 2012 Volume 51(Issue 7) pp:
Publication Date(Web):January 23, 2012
DOI:10.1021/bi201878x
Artificial zinc finger proteins (ZFPs) consist of Cys2-His2-type modules composed of ∼30 amino acids with a ββα structure that coordinates a zinc ion. ZFPs that recognize specific DNA target sequences can substitute for the binding domains of enzymes that act on DNA to create designer enzymes with programmable sequence specificity. The most studied of these engineered enzymes are zinc finger nucleases (ZFNs). ZFNs have been widely used to model organisms and are currently in human clinical trials with an aim of therapeutic gene editing. Difficulties with ZFNs arise from unpredictable mutations caused by nonhomologous end joining and off-target DNA cleavage and mutagenesis. A more recent strategy that aims to address the shortcomings of ZFNs involves zinc finger recombinases (ZFRs). A thorough understanding of ZFRs and methods for their modification promises powerful new tools for gene manipulation in model organisms as well as in gene therapy. In an effort to design efficient and specific ZFRs, the effects of the DNA binding affinity of the zinc finger domains and the linker sequence between ZFPs and recombinase catalytic domains have been assessed. A plasmid system containing ZFR target sites was constructed for evaluation of catalytic activities of ZFRs with variable linker lengths and numbers of zinc finger modules. Recombination efficiencies were evaluated by restriction enzyme analysis of isolated plasmids after reaction in Escherichia coli and changes in EGFP fluorescence in mammalian cells. The results provide information relevant to the design of ZFRs that will be useful for sequence-specific genome modification.
Co-reporter:Tetsuo Narumi, Tomohiro Tanaka, Chie Hashimoto, Wataru Nomura, Haruo Aikawa, Akira Sohma, Kyoko Itotani, Miyako Kawamata, Tsutomu Murakami, Naoki Yamamoto, Hirokazu Tamamura
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 12) pp:4169-4172
Publication Date(Web):15 June 2012
DOI:10.1016/j.bmcl.2012.04.032
Low molecular weight CXCR4 ligands were developed based on the peptide T140, which has previously been identified as a potent CXCR4 antagonist. Some compounds with naphthyl, fluorobenzyl and pyridyl moieties as pharmacophore groups in the molecule showed significant CXCR4-binding activity and anti-HIV activity. Structure–activity relationships were studied and characteristics of each of these three moieties necessary for CXCR4 binding were defined. In this way, CXCR4 ligands with two types of recognition modes for CXCR4 have been found.SAR studies of low molecular weight CXCR4 ligands are reported.
Co-reporter:Dr. Wataru Nomura;Chie Hashimoto;Aki Ohya;Dr. Kosuke Miyauchi;Dr. Emiko Urano;Dr. Tomohiro Tanaka;Dr. Tetsuo Narumi;Toru Nakahara;Dr. Jun A. Komano;Dr. Naoki Yamamoto;Dr. Hirokazu Tamamura
ChemMedChem 2012 Volume 7( Issue 2) pp:205-208
Publication Date(Web):
DOI:10.1002/cmdc.201100542
Co-reporter:Nami Ohashi, Wataru Nomura, Tetsuo Narumi, Nancy E. Lewin, Kyoko Itotani, Peter M. Blumberg, and Hirokazu Tamamura
Bioconjugate Chemistry 2011 Volume 22(Issue 1) pp:82
Publication Date(Web):December 22, 2010
DOI:10.1021/bc100414a
Protein kinase C (PKC) is a critical cell signaling pathway involved in many disorders such as cancer and Alzheimer-type dementia. To date, evaluation of PKC ligand binding affinity has been performed by competitive studies against radiolabeled probes that are problematic for high-throughput screening. In the present study, we have developed a fluorescent-based binding assay system for identifying ligands that target the PKC ligand binding domain (C1 domain). An environmentally sensitive fluorescent dye (solvatochromic fluorophore), which has been used in multiple applications to assess protein-binding interactions, was inserted in proximity to the binding pocket of a novel PKCδ C1b domain. These resultant fluorescent-labeled δC1b domain analogues underwent a significant change in fluorescent intensity upon ligand binding, and we further demonstrate that the fluorescent δC1b domain analogues can be used to evaluate ligand binding affinity.
Co-reporter:Wataru Nomura, Nami Ohashi, Yoshiaki Okuda, Tetsuo Narumi, Teikichi Ikura, Nobutoshi Ito, and Hirokazu Tamamura
Bioconjugate Chemistry 2011 Volume 22(Issue 5) pp:923
Publication Date(Web):March 25, 2011
DOI:10.1021/bc100567k
A novel fluorescence-quenching screening method for protein kinase C (PKC) ligands was developed utilizing solvatochromic fluorophores. Solvatochromic dyes, highly sensitive to the presence or the absence of competitive ligands in their binding to the C1b domain of PKCδ (δC1b), were combined with a known pharmacophoric moiety of 1,2-diacylglycerol (DAG) lactones, PKC ligands. Addition of δC1b to the fluorescent compounds caused a gradual increase in the fluorescent intensity in proportion to the increase of δC1b. As a competitive ligand was added to the complex of δC1b domain and fluorescent compounds, a gradual decrease in the fluorescent intensity was observed. The relative binding affinities of known ligands were successfully determined by this fluorescent method and corresponded well to the Ki values measured by a radioisotope method. These results indicate that washing, which is a laborious step in binding evaluations, is not required for this environmentally sensitive fluorophore based system. Screening with the system was performed for 2560 preselected library compounds with possible pharmacophores, and some lead compounds were found. This fluorescence-based method could be applied widely to known ligand−receptor combinations.
Co-reporter:Tetsuo Narumi, Hiroshi Arai, Kazuhisa Yoshimura, Shigeyoshi Harada, Wataru Nomura, Shuzo Matsushita, Hirokazu Tamamura
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 22) pp:6735-6742
Publication Date(Web):15 November 2011
DOI:10.1016/j.bmc.2011.09.045
Derivatives of CD4 mimics were designed and synthesized to interact with the conserved residues of the Phe43 cavity in gp120 to investigate their anti-HIV activity, cytotoxicity, and CD4 mimicry effects on conformational changes of gp120. Significant potency gains were made by installation of bulky hydrophobic groups into the piperidine moiety, resulting in discovery of a potent compound with a higher selective index and CD4 mimicry. The current study identified a novel lead compound 11 with significant anti-HIV activity and lower cytotoxicity than those of known CD4 mimics.
Co-reporter:Tomohiro Tanaka;Dr. Tetsuo Narumi;Taro Ozaki;Akira Sohma;Nami Ohashi;Chie Hashimoto;Kyoko Itotani;Dr. Wataru Nomura;Dr. Tsutomu Murakami; Naoki Yamamoto; Hirokazu Tamamura
ChemMedChem 2011 Volume 6( Issue 5) pp:834-839
Publication Date(Web):
DOI:10.1002/cmdc.201000548
Abstract
The chemokine receptor CXCR4 is a member of the seven transmembrane GPCR family, which is implicated in multiple diseases, including HIV infection, cancers, and rheumatoid arthritis. Low-molecular-weight nonpeptidic compounds, including AMD3100 and various pyridyl macrocyclic zinc(II) complexes, have been identified as selective antagonists of CXCR4. In the present study, structure–activity relationship studies were performed by combining the common structural features of alkylamino and pyridiyl macrocyclic antagonists. Several new zinc(II) or copper(II) complexes demonstrated potent anti-HIV activity, strong CXCR4-binding activity, and significant inhibitory activity against Ca2+ mobilization induced by CXCL12 stimulation. These results may prove useful in the design of novel CXCR4 antagonists, and the compounds described could potentially be developed as therapeutics against CXCR4-relevant diseases or chemical probes to study the biological activity of CXCR4.
Co-reporter:Dr. Hiroshi Tsutsumi ;Seiichiro Abe ;Tomoaki Mino ;Dr. Wataru Nomura; Hirokazu Tamamura
ChemBioChem 2011 Volume 12( Issue 5) pp:691-694
Publication Date(Web):
DOI:10.1002/cbic.201000692
Co-reporter:Dr. Wataru Nomura;Dr. Tetsuo Narumi;Nami Ohashi;Yuki Serizawa;Nancy E. Lewin;Dr. Peter M. Blumberg; Toshiaki Furuta ; Hirokazu Tamamura
ChemBioChem 2011 Volume 12( Issue 4) pp:535-539
Publication Date(Web):
DOI:10.1002/cbic.201000670
Co-reporter:Tomohiro Tanaka ; Wataru Nomura ; Tetsuo Narumi ; Akemi Masuda
Journal of the American Chemical Society 2010 Volume 132(Issue 45) pp:15899-15901
Publication Date(Web):October 25, 2010
DOI:10.1021/ja107447w
To date, challenges in the design of bivalent ligands for G protein-coupled receptors (GPCRs) have revealed difficulties stemming from lack of knowledge of the state of oligomerization of the GPCR. The synthetic bivalent ligands with rigid linkers that are presented here can predict the dimer form of CXCR4 and be applied to molecular probes in cancerous cells. This “molecular ruler” approach would be useful in elucidating the details of CXCR4 oligomer formation.
Co-reporter:Shintaro Suzuki ; Emiko Urano ; Chie Hashimoto ; Hiroshi Tsutsumi ; Toru Nakahara ; Tomohiro Tanaka ; Yuta Nakanishi ; Kasthuraiah Maddali ; Yan Han ; Makiko Hamatake ; Kosuke Miyauchi ; Yves Pommier ; John A. Beutler ; Wataru Sugiura ; Hideyoshi Fuji ; Tyuji Hoshino ; Kyoko Itotani ; Wataru Nomura ; Tetsuo Narumi ; Naoki Yamamoto ; Jun A. Komano
Journal of Medicinal Chemistry 2010 Volume 53(Issue 14) pp:5356-5360
Publication Date(Web):June 29, 2010
DOI:10.1021/jm1003528
Anti-HIV peptides with inhibitory activity against HIV-1 integrase (IN) have been found in overlapping peptide libraries derived from HIV-1 gene products. In a strand transfer assay using IN, inhibitory active peptides with certain sequential motifs related to Vpr- and Env-derived peptides were found. The addition of an octa-arginyl group to the inhibitory peptides caused a remarkable inhibition of the strand transfer and 3′-end-processing reactions catalyzed by IN and significant inhibition against HIV replication.
Co-reporter:Toru Nakahara, Wataru Nomura, Kenji Ohba, Aki Ohya, Tomohiro Tanaka, Chie Hashimoto, Tetsuo Narumi, Tsutomu Murakami, Naoki Yamamoto and Hirokazu Tamamura
Bioconjugate Chemistry 2010 Volume 21(Issue 4) pp:709
Publication Date(Web):April 1, 2010
DOI:10.1021/bc900502z
A synthetic antigen targeting membrane-fusion mechanism of HIV-1 has a newly designed template with C3-symmetric linkers mimicking N36 trimeric form. The antiserum produced by immunization of the N36 trimeric form antigen showed structural preference in binding to N36 trimer and stronger inhibitory activity against HIV-1 infection than the N36 monomer. Our results suggest an effective strategy of HIV vaccine design based on a relationship to the native structure of proteins involved in HIV fusion mechanisms.
Co-reporter:Shintaro Suzuki, Kasthuraiah Maddali, Chie Hashimoto, Emiko Urano, Nami Ohashi, Tomohiro Tanaka, Taro Ozaki, Hiroshi Arai, Hiroshi Tsutsumi, Tetsuo Narumi, Wataru Nomura, Naoki Yamamoto, Yves Pommier, Jun A. Komano, Hirokazu Tamamura
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 18) pp:6771-6775
Publication Date(Web):15 September 2010
DOI:10.1016/j.bmc.2010.07.050
Structure–activity relationship studies were conducted on HIV integrase (IN) inhibitory peptides which were found by the screening of an overlapping peptide library derived from HIV-1 gene products. Since these peptides located in the second helix of Vpr are considered to have an α-helical conformation, Glu-Lys pairs were introduced into the i and i + 4 positions to increase the helicity of the lead compound possessing an octa-arginyl group. Ala-scan was also performed on the lead compound for the identification of the amino acid residues responsible for the inhibitory activity. The results indicated the importance of an α-helical structure for the expression of inhibitory activity, and presented a binding model of integrase and the lead compound.SAR studies of peptidic integrase inhibitors derived from HIV gene products are reported.
Co-reporter:Tetsuo Narumi, Chihiro Ochiai, Kazuhisa Yoshimura, Shigeyoshi Harada, Tomohiro Tanaka, Wataru Nomura, Hiroshi Arai, Taro Ozaki, Nami Ohashi, Shuzo Matsushita, Hirokazu Tamamura
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 19) pp:5853-5858
Publication Date(Web):1 October 2010
DOI:10.1016/j.bmcl.2010.07.106
Small molecules behaving as CD4 mimics were previously reported as HIV-1 entry inhibitors that block the gp120–CD4 interaction and induce a conformational change in gp120, exposing its co-receptor-binding site. A structure–activity relationship (SAR) study of a series of CD4 mimic analogs was conducted to investigate the contribution from the piperidine moiety of CD4 mimic 1 to anti-HIV activity, cytotoxicity, and CD4 mimicry effects on conformational changes of gp120. In addition, several hybrid molecules based on conjugation of a CD4 mimic analog with a selective CXCR4 antagonist were also synthesized and their utility evaluated.SAR studies of CD4 mimics and their conjugation with a CXCR4 antagonist are reported.
Co-reporter:Yuko Yamada, Chihiro Ochiai, Kazuhisa Yoshimura, Tomohiro Tanaka, Nami Ohashi, Tetsuo Narumi, Wataru Nomura, Shigeyoshi Harada, Shuzo Matsushita, Hirokazu Tamamura
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 1) pp:354-358
Publication Date(Web):1 January 2010
DOI:10.1016/j.bmcl.2009.10.098
A structure–activity relationship study was conducted of several CD4 mimicking small molecules which block the interaction between HIV-1 gp120 and CD4. These CD4 mimics induce a conformational change in gp120, exposing its co-receptor-binding site. This induces a highly synergistic interaction in the use in combination with a co-receptor CXCR4 antagonist and reveals a pronounced effect on the dynamic supramolecular mechanism of HIV-1 entry.CD4 mimic small molecules interacting with a large cavity of HIV-1 gp120, which are targeted for the dynamic supramolecular mechanism of HIV entry, are reported.
Co-reporter:Tomohiro Tanaka, Wataru Nomura, Tetsuo Narumi, Ai Esaka, Shinya Oishi, Nami Ohashi, Kyoko Itotani, Barry J. Evans, Zi-xuan Wang, Stephen C. Peiper, Nobutaka Fujii and Hirokazu Tamamura
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 18) pp:3805-3809
Publication Date(Web):20 Jul 2009
DOI:10.1039/B908286G
Previously, downsizing of a 14-residue peptidic CXCR4 antagonist 1 has led to the development of a highly potent CXCR4 antagonist 2 [cyclo(-D-Tyr1-Arg2-Arg3-Nal4-Gly5-)]. In the present study, cyclic pentapeptide libraries that were designed by substitutions of several amino acids for D-Tyr1 and Arg2 in peptide 2 were prepared and screened to evaluate binding activity for CXCR4. The above structure-activity relationship study led to the finding of several potent CXCR4 ligands.
Co-reporter:Hiroshi Tsutsumi Dr.;Wataru Nomura Dr.;Seiichiro Abe;Tomoaki Mino;Akemi Masuda;Nami Ohashi;Tomohiro Tanaka;Kenji Ohba Dr.;Naoki Yamamoto ;Kazunari Akiyoshi
Angewandte Chemie 2009 Volume 121( Issue 48) pp:9328-9330
Publication Date(Web):
DOI:10.1002/ange.200903183
Co-reporter:Hiroshi Tsutsumi Dr.;Wataru Nomura Dr.;Seiichiro Abe;Tomoaki Mino;Akemi Masuda;Nami Ohashi;Tomohiro Tanaka;Kenji Ohba Dr.;Naoki Yamamoto ;Kazunari Akiyoshi
Angewandte Chemie International Edition 2009 Volume 48( Issue 48) pp:9164-9166
Publication Date(Web):
DOI:10.1002/anie.200903183
Co-reporter:Wataru Nomura, Yasuaki Tanabe, Hiroshi Tsutsumi, Tomohiro Tanaka, Kenji Ohba, Naoki Yamamoto and Hirokazu Tamamura
Bioconjugate Chemistry 2008 Volume 19(Issue 9) pp:1917
Publication Date(Web):August 16, 2008
DOI:10.1021/bc800216p
Development of CXCR4-specific ligands is an important issue in chemotherapy of HIV infection, cancer metastasis, and rheumatoid arthritis, and numerous potential ligands have been developed to date. However, it is difficult to assess their binding mode and specificity because of uncertainties in the structure of the CXCR4−ligand complexes. To address this problem, we have synthesized fluorophore labeled Ac-TZ14011, which is derived from T140, a powerful CXCR4 antagonist. Binding of Ac-TZ14011 to CXCR4 on the cell membrane was observed by fluorescence microscope, and analysis of the binding data produced IC50 values of several ligands comparable to those obtained in RI-based assays. This fluorescence-based assay is applicable to explore new pharmacophores of CXCR4-specific ligands with high-throughput screening and also to screening of the other GPCR binding ligands.
Co-reporter:Tomohiro Tanaka, Hiroshi Tsutsumi, Wataru Nomura, Yasuaki Tanabe, Nami Ohashi, Ai Esaka, Chihiro Ochiai, Jun Sato, Kyoko Itotani, Tsutomu Murakami, Kenji Ohba, Naoki Yamamoto, Nobutaka Fujii and Hirokazu Tamamura
Organic & Biomolecular Chemistry 2008 vol. 6(Issue 23) pp:4374-4377
Publication Date(Web):17 Oct 2008
DOI:10.1039/B812029C
A highly potent CXCR4 antagonist 2 [cyclo (-D-Tyr1-Arg2-Arg3-Nal4-Gly5-)] has previously been identified by screening cyclic pentapeptide libraries that were designed based on pharmacophore residues of a 14-residue peptidic CXCR4 antagonist 1. In the present study, D-Tyr and Arg in peptide 2 were replaced by a bicyclic aromatic amino acid and a cationic amino acid, respectively, and their binding activity for CXCR4 was evaluated for identification of the novel pharmacophore.
Co-reporter:Hirokazu Tamamura, Hiroshi Tsutsumi
Chemistry & Biology 2006 Volume 13(Issue 1) pp:8-10
Publication Date(Web):January 2006
DOI:10.1016/j.chembiol.2005.12.005
Chemokine receptors have attracted a good deal of public attention as important therapeutic targets for many diseases and disorders. In this issue of Chemistry & Biology, Kumar and colleagues propose a new concept of synthetic modular modifications to generate unnatural chemokines, which exhibit high receptor selectivity [1].
Co-reporter:Hirokazu Tamamura, Hiroshi Tsutsumi, Hiroyuki Masuno, Satoko Mizokami, Kenichi Hiramatsu, Zixuan Wang, John O. Trent, Hideki Nakashima, Naoki Yamamoto, Stephen C. Peiper and Nobutaka Fujii
Organic & Biomolecular Chemistry 2006 vol. 4(Issue 12) pp:2354-2357
Publication Date(Web):2006/05/12
DOI:10.1039/B603818B
A linear type of several low molecular weight CXCR4 antagonists were developed based on T140 analogs, which were previously found to be strong CXCR4 antagonists that block X4-HIV-1 entry and have inhibitory activities against cancer metastasis/progression and rheumatoid arthritis.
Co-reporter:Tomohiro Tanaka, Hiroshi Tsutsumi, Wataru Nomura, Yasuaki Tanabe, Nami Ohashi, Ai Esaka, Chihiro Ochiai, Jun Sato, Kyoko Itotani, Tsutomu Murakami, Kenji Ohba, Naoki Yamamoto, Nobutaka Fujii and Hirokazu Tamamura
Organic & Biomolecular Chemistry 2008 - vol. 6(Issue 23) pp:NaN4377-4377
Publication Date(Web):2008/10/17
DOI:10.1039/B812029C
A highly potent CXCR4 antagonist 2 [cyclo (-D-Tyr1-Arg2-Arg3-Nal4-Gly5-)] has previously been identified by screening cyclic pentapeptide libraries that were designed based on pharmacophore residues of a 14-residue peptidic CXCR4 antagonist 1. In the present study, D-Tyr and Arg in peptide 2 were replaced by a bicyclic aromatic amino acid and a cationic amino acid, respectively, and their binding activity for CXCR4 was evaluated for identification of the novel pharmacophore.
Co-reporter:Wataru Nomura, Taisuke Koseki, Nami Ohashi, Takaaki Mizuguchi and Hirokazu Tamamura
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 32) pp:NaN8739-8739
Publication Date(Web):2015/07/09
DOI:10.1039/C5OB00891C
The assembly status of G protein-coupled receptors (GPCR) on the cell surface is of interest because the multimerization of GPCR could play pivotal roles in cellular functions. A bivalent ligand with polyproline linkers for CXCR4 has been shown to serve as a “molecular ruler” as a result of the rigid structure of polyproline helices. To expand the utility of the ligands with rigid linkers and explore the possible multimeric forms of GPCR, trivalent ligands with polyproline helices were newly designed and synthesized. The binding affinities of the trivalent ligands for CXCR4 suggested that the ligands recognize the dimeric form of CXCR4 on the cell surface. The fluorescence imaging and analysis by flow cytometry revealed that the ligand with 9 proline linkers binds to CXCR4 with remarkable specificity. The results of the present study suggest that the ligand design with rigid linkers is useful in the multimeric form, but the design of trivalent ligands requires different strategic approaches.
Co-reporter:Tomohiro Tanaka, Wataru Nomura, Tetsuo Narumi, Ai Esaka, Shinya Oishi, Nami Ohashi, Kyoko Itotani, Barry J. Evans, Zi-xuan Wang, Stephen C. Peiper, Nobutaka Fujii and Hirokazu Tamamura
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 18) pp:NaN3809-3809
Publication Date(Web):2009/07/20
DOI:10.1039/B908286G
Previously, downsizing of a 14-residue peptidic CXCR4 antagonist 1 has led to the development of a highly potent CXCR4 antagonist 2 [cyclo(-D-Tyr1-Arg2-Arg3-Nal4-Gly5-)]. In the present study, cyclic pentapeptide libraries that were designed by substitutions of several amino acids for D-Tyr1 and Arg2 in peptide 2 were prepared and screened to evaluate binding activity for CXCR4. The above structure-activity relationship study led to the finding of several potent CXCR4 ligands.

![2-(2,4,6-trimethylphenyl)-2,5,6,7-tetrahydropyrrolo[2,1-c][1,2,4]triazol-4-ium chloride](http://img.cochemist.com/ccimg/862900/862893-81-0.png)
![2-(2,4,6-trimethylphenyl)-2,5,6,7-tetrahydropyrrolo[2,1-c][1,2,4]triazol-4-ium chloride](http://img.cochemist.com/ccimg/862900/862893-81-0_b.png)
















![Ethanone, 1-[1-(phenylmethyl)-1H-pyrrol-3-yl]-](http://img.cochemist.com/ccimg/129000/128942-91-6.png)
![Ethanone, 1-[1-(phenylmethyl)-1H-pyrrol-3-yl]-](http://img.cochemist.com/ccimg/129000/128942-91-6_b.png)











![1-Propanone, 3-[(4-chlorophenyl)amino]-1,3-diphenyl-](http://img.cochemist.com/ccimg/94900/94864-08-1.png)
![1-Propanone, 3-[(4-chlorophenyl)amino]-1,3-diphenyl-](http://img.cochemist.com/ccimg/94900/94864-08-1_b.png)


![4-methyl-2H-Pyrano[2,3-b]pyridine-2,7(8H)-dione](http://img.cochemist.com/ccimg/58000/57980-05-9.png)
![4-methyl-2H-Pyrano[2,3-b]pyridine-2,7(8H)-dione](http://img.cochemist.com/ccimg/58000/57980-05-9_b.png)
![1-Propanone, 3-[(4-methylphenyl)amino]-1,3-diphenyl-](http://img.cochemist.com/ccimg/38000/37904-94-2.png)
![1-Propanone, 3-[(4-methylphenyl)amino]-1,3-diphenyl-](http://img.cochemist.com/ccimg/38000/37904-94-2_b.png)















![4-Nitrobenzo[c][1,2,5]oxadiazole](http://img.cochemist.com/ccimg/16400/16322-19-3.png)
![4-Nitrobenzo[c][1,2,5]oxadiazole](http://img.cochemist.com/ccimg/16400/16322-19-3_b.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)

