Co-reporter:Tina L. Mistry, Lena Truong, Arun K. Ghosh, Michael E. Johnson, and Shahila Mehboob
ACS Infectious Diseases 2017 Volume 3(Issue 1) pp:
Publication Date(Web):October 18, 2016
DOI:10.1021/acsinfecdis.6b00123
The enoyl-ACP reductase (FabI) enzyme is a well validated target for anti-staphylococcal drug discovery and development. With the goal of finding alternate therapeutics for drug-resistant strains of Staphylococcus aureus, such as methicillin-resistant S. aureus (MRSA), our previously published series of benzimidazole-based inhibitors of the FabI enzyme from Francisella tularensis (FtFabI) have been evaluated against FabI from S. aureus (SaFabI). We report here the preliminary structure–activity relationship of this series and the prioritization of compounds toward lead optimization. Mutational studies have identified key residues that contribute toward stabilizing the inhibitors in the active site of FabI. Mutations that do not significantly impact enzyme function but destabilize inhibitor binding are more likely to occur in nature as organisms evolve to evade the action of antibiotics leading to resistance. Identifying these residues provides guidance for minimizing susceptibility to resistance. Additionally, we have identified compounds that elicit antibacterial activity through off-target effects and observe that close analogs can display differing modes of action (on-target vs off-target) and need to be individually evaluated early on to prioritize compounds for lead optimization. Overall, our data suggest that the benzimidazole scaffold is a promising scaffold for anti-staphylococcal drug development.Keywords: benzimidazole inhibitor; enoyl-ACP reductase; FabI; mode of action; Staphylococcus aureus;
Co-reporter:Amy J. Rice, Hao Lei, Bernard D. Santarsiero, Hyun Lee, Michael E. Johnson
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 19) pp:4536-4543
Publication Date(Web):1 October 2016
DOI:10.1016/j.bmc.2016.07.055
Dihydroorotase (DHOase) is the third enzyme in the de novo pyrimidine synthesis pathway and is responsible for the reversible cyclization of carbamyl-aspartate (Ca-asp) to dihydroorotate (DHO). DHOase is further divided into two classes based on several structural characteristics, one of which is the length of the flexible catalytic loop that interacts with the substrate, Ca-asp, regulating the enzyme activity. Here, we present the crystal structure of Class I Bacillus anthracis DHOase with Ca-asp in the active site, which shows the peptide backbone of glycine in the shorter loop forming the necessary hydrogen bonds with the substrate, in place of the two threonines found in Class II DHOases. Despite the differences in the catalytic loop, the structure confirms that the key interactions between the substrate and active site residues are similar between Class I and Class II DHOase enzymes, which we further validated by mutagenesis studies. B. anthracis DHOase is also a potential antibacterial drug target. In order to identify prospective inhibitors, we performed high-throughput screening against several libraries using a colorimetric enzymatic assay and an orthogonal fluorescence thermal binding assay. Surface plasmon resonance was used for determining binding affinity (KD) and competition analysis with Ca-asp. Our results highlight that the primary difference between Class I and Class II DHOase is the catalytic loop. We also identify several compounds that can potentially be further optimized as potential B. anthracis inhibitors.
Co-reporter:Hao Lei, Christopher Jones, Tian Zhu, Kavankumar Patel, Nina M. Wolf, Leslie W.-M. Fung, Hyun Lee, Michael E. Johnson
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 4) pp:596-605
Publication Date(Web):15 February 2016
DOI:10.1016/j.bmc.2015.12.029
The de novo purine biosynthesis pathway is an attractive target for antibacterial drug design, and PurE from this pathway has been identified to be crucial for Bacillus anthracis survival in serum. In this study we adopted a fragment-based hit discovery approach, using three screening methods—saturation transfer difference nucleus magnetic resonance (STD-NMR), water-ligand observed via gradient spectroscopy (WaterLOGSY) NMR, and surface plasmon resonance (SPR), against B. anthracis PurE (BaPurE) to identify active site binding fragments by initially testing 352 compounds in a Zenobia fragment library. Competition STD NMR with the BaPurE product effectively eliminated non-active site binding hits from the primary hits, selecting active site binders only. Binding affinities (dissociation constant, KD) of these compounds varied between 234 and 301 μM. Based on test results from the Zenobia compounds, we subsequently developed and applied a streamlined fragment screening strategy to screen a much larger library consisting of 3000 computationally pre-selected fragments. Thirteen final fragment hits were confirmed to exhibit binding affinities varying from 14 μM to 700 μM, which were categorized into five different basic scaffolds. All thirteen fragment hits have ligand efficiencies higher than 0.30. We demonstrated that at least two fragments from two different scaffolds exhibit inhibitory activity against the BaPurE enzyme.
Co-reporter:Hyun Lee, Hao Lei, Bernard D. Santarsiero, Joseph L. Gatuz, Shuyi Cao, Amy J. Rice, Kavankumar Patel, Michael Z. Szypulinski, Isabel Ojeda, Arun K. Ghosh, and Michael E. Johnson
ACS Chemical Biology 2015 Volume 10(Issue 6) pp:1456
Publication Date(Web):March 6, 2015
DOI:10.1021/cb500917m
The Middle East Respiratory Syndrome coronavirus (MERS-CoV) papain-like protease (PLpro) blocking loop 2 (BL2) structure differs significantly from that of SARS-CoV PLpro, where it has been proven to play a crucial role in SARS-CoV PLpro inhibitor binding. Four SARS-CoV PLpro lead inhibitors were tested against MERS-CoV PLpro, none of which were effective against MERS-CoV PLpro. Structure and sequence alignments revealed that two residues, Y269 and Q270, responsible for inhibitor binding to SARS-CoV PLpro, were replaced by T274 and A275 in MERS-CoV PLpro, making critical binding interactions difficult to form for similar types of inhibitors. High-throughput screening (HTS) of 25 000 compounds against both PLpro enzymes identified a small fragment-like noncovalent dual inhibitor. Mode of inhibition studies by enzyme kinetics and competition surface plasmon resonance (SPR) analyses suggested that this compound acts as a competitive inhibitor with an IC50 of 6 μM against MERS-CoV PLpro, indicating that it binds to the active site, whereas it acts as an allosteric inhibitor against SARS-CoV PLpro with an IC50 of 11 μM. These results raised the possibility that inhibitor recognition specificity of MERS-CoV PLpro may differ from that of SARS-CoV PLpro. In addition, inhibitory activity of this compound was selective for SARS-CoV and MERS-CoV PLpro enzymes over two human homologues, the ubiquitin C-terminal hydrolases 1 and 3 (hUCH-L1 and hUCH-L3).
Co-reporter:Shahila Mehboob, Jinhua Song, Kirk E. Hevener, Pin-Chih Su, Teuta Boci, Libby Brubaker, Lena Truong, Tina Mistry, Jiangping Deng, James L. Cook, Bernard D. Santarsiero, Arun K. Ghosh, Michael E. Johnson
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 6) pp:1292-1296
Publication Date(Web):15 March 2015
DOI:10.1016/j.bmcl.2015.01.048
Francisella tularensis, the causative agent of tularemia, presents a significant biological threat and is a Category A priority pathogen due to its potential for weaponization. The bacterial FASII pathway is a viable target for the development of novel antibacterial agents treating Gram-negative infections. Here we report the advancement of a promising series of benzimidazole FabI (enoyl-ACP reductase) inhibitors to a second-generation using a systematic, structure-guided lead optimization strategy, and the determination of several co-crystal structures that confirm the binding mode of designed inhibitors. These compounds display an improved low nanomolar enzymatic activity as well as promising low microgram/mL antibacterial activity against both F. tularensis and Staphylococcus aureus and its methicillin-resistant strain (MRSA). The improvements in activity accompanying structural modifications lead to a better understanding of the relationship between the chemical structure and biological activity that encompasses both enzymatic and whole-cell activity.
Co-reporter:Anuradha Mittal, Michael E. Johnson
Journal of Molecular Graphics and Modelling 2015 Volume 55() pp:115-122
Publication Date(Web):February 2015
DOI:10.1016/j.jmgm.2014.11.004
•FabH homologs show distinct conformational preferences of binding pocket residues.•Computational solvent mapping identifies hot spots.•Distinct conformational properties result in different binding hot spots.The molecular basis of variable substrate and inhibitor specificity of the highly conserved bacterial fatty acid synthase enzyme, FabH, across different bacterial species remains poorly understood. In the current work, we explored the conformational diversity of FabH enzymes to understand the determinants of diverse interaction specificity across Gram-positive and Gram-negative bacteria. Atomistic molecular dynamics simulations reveal that FabH from E. coli and E. faecalis exhibit distinct native state conformational ensembles and dynamic behaviors. Despite strikingly similar substrate binding pockets, hot spot assessment using computational solvent mapping identified quite different favorable binding interactions between the two homologs. Our data suggest that FabH utilizes protein dynamics and seemingly minor sequence and structural differences to modulate its molecular recognition and substrate specificity across bacterial species. These insights will potentially facilitate the rational design and development of antibacterial inhibitors against FabH enzymes.
Co-reporter:Hyun Lee, Anuradha Mittal, Kavankumar Patel, Joseph L. Gatuz, Lena Truong, Jaime Torres, Debbie C. Mulhearn, Michael E. Johnson
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 1) pp:167-177
Publication Date(Web):1 January 2014
DOI:10.1016/j.bmc.2013.11.041
We have used a combination of virtual screening (VS) and high-throughput screening (HTS) techniques to identify novel, non-peptidic small molecule inhibitors against human SARS-CoV 3CLpro. A structure-based VS approach integrating docking and pharmacophore based methods was employed to computationally screen 621,000 compounds from the ZINC library. The screening protocol was validated using known 3CLpro inhibitors and was optimized for speed, improved selectivity, and for accommodating receptor flexibility. Subsequently, a fluorescence-based enzymatic HTS assay was developed and optimized to experimentally screen approximately 41,000 compounds from four structurally diverse libraries chosen mainly based on the VS results. False positives from initial HTS hits were eliminated by a secondary orthogonal binding analysis using surface plasmon resonance (SPR). The campaign identified a reversible small molecule inhibitor exhibiting mixed-type inhibition with a Ki value of 11.1 μM. Together, these results validate our protocols as suitable approaches to screen virtual and chemical libraries, and the newly identified compound reported in our study represents a promising structural scaffold to pursue for further SARS-CoV 3CLpro inhibitor development.
Co-reporter:Tian Zhu ; Shuyi Cao ; Pin-Chih Su ; Ram Patel ; Darshan Shah ; Heta B. Chokshi ; Richard Szukala ; Michael E. Johnson ;Kirk E. Hevener
Journal of Medicinal Chemistry 2013 Volume 56(Issue 17) pp:6560-6572
Publication Date(Web):May 20, 2013
DOI:10.1021/jm301916b
A critical analysis of virtual screening results published between 2007 and 2011 was performed. The activity of reported hit compounds from over 400 studies was compared to their hit identification criteria. Hit rates and ligand efficiencies were calculated to assist in these analyses, and the results were compared with factors such as the size of the virtual library and the number of compounds tested. A series of promiscuity, druglike, and ADMET filters were applied to the reported hits to assess the quality of compounds reported, and a careful analysis of a subset of the studies that presented hit optimization was performed. These data allowed us to make several practical recommendations with respect to selection of compounds for experimental testing, definition of hit identification criteria, and general virtual screening hit criteria to allow for realistic hit optimization. A key recommendation is the use of size-targeted ligand efficiency values as hit identification criteria.
Co-reporter:Tian Zhu, Hyun Lee, Hao Lei, Christopher Jones, Kavankumar Patel, Michael E. Johnson, and Kirk E. Hevener
Journal of Chemical Information and Modeling 2013 Volume 53(Issue 3) pp:560-572
Publication Date(Web):February 22, 2013
DOI:10.1021/ci300502h
We have developed a rigorous computational screening protocol to identify novel fragment-like inhibitors of N5-CAIR mutase (PurE), a key enzyme involved in de novo purine synthesis that represents a novel target for the design of antibacterial agents. This computational screening protocol utilizes molecular docking, graphics processing unit (GPU)-accelerated molecular dynamics, and Molecular Mechanics/Poisson–Boltzmann Surface Area (MM/PBSA) free energy estimations to investigate the binding modes and energies of fragments in the active sites of PurE. PurE is a functional octamer comprised of identical subunits. The octameric structure, with its eight active sites, provided a distinct advantage in these studies because, for a given simulation length, we were able to place eight separate fragment compounds in the active sites to increase the throughput of the MM/PBSA analysis. To validate this protocol, we have screened an in-house fragment library consisting of 352 compounds. The theoretical results were then compared with the results of two experimental fragment screens, Nuclear Magnetic Resonance (NMR) and Surface Plasmon Resonance (SPR) binding analyses. In these validation studies, the protocol was able to effectively identify the competitive binders that had been independently identified by experimental testing, suggesting the potential utility of this method for the identification of novel fragments for future development as PurE inhibitors.
Co-reporter:Amy J. Rice, Lena Truong, Michael E. Johnson, Hyun Lee
Analytical Biochemistry 2013 Volume 441(Issue 1) pp:87-94
Publication Date(Web):1 October 2013
DOI:10.1016/j.ab.2013.05.035
Abstract
Dihydroorotase (DHOase) is the third enzyme in the de novo pyrimidine biosynthesis pathway and is a potential new antibacterial drug target. No target-based high-throughput screening (HTS) assay for this enzyme has been reported to date. Here, we optimized two colorimetric-based enzymatic assays that detect the ureido moiety of the DHOase substrate, carbamyl-aspartate (Ca-asp). Each assay was developed in a 40-μl assay volume using 384-well plates with a different color mix, diacetylmonoxime (DAMO)–thiosemicarbazide (TSC) or DAMO–antipyrine. The sensitivity and color interference of both color mixes were compared in the presence of common HTS buffer additives, including dimethyl sulfoxide, reducing agents, detergents, and bovine serum albumin. DAMO–TSC (Z′-factors 0.7–0.8) was determined to be superior to DAMO–antipyrine (Z′-factors 0.5–0.6) with significantly less variability within replicates. An HTS pilot screening with 29,552 compounds from four structurally diverse libraries confirmed the quality of our newly optimized colorimetric assay with DAMO–TSC. This robust method has no heating requirement, which was the main obstacle to applying previous assays to HTS. More important, this well-optimized HTS assay for DHOase, the first of its kind, should make it possible to screen large-scale compound libraries to develop new inhibitors against any enzymes that produce ureido functional groups.
Co-reporter:Dr. Hyun Lee;Shuyi Cao; Kirk E. Hevener;Lena Truong;Joseph L. Gatuz;Kavankumar Patel; Arun K. Ghosh; Michael E. Johnson
ChemMedChem 2013 Volume 8( Issue 8) pp:1361-1372
Publication Date(Web):
DOI:10.1002/cmdc.201300134
Abstract
We previously developed two potent chemical classes that inhibit the essential papain-like protease (PLpro) of severe acute respiratory syndrome coronavirus. In this study, we applied a novel approach to identify small fragments that act synergistically with these inhibitors. A fragment library was screened in combination with four previously developed lead inhibitors by fluorescence-based enzymatic assays. Several fragment compounds synergistically enhanced the inhibitory activity of the lead inhibitors by approximately an order of magnitude. Surface plasmon resonance measurements showed that three fragments bind specifically to the PLpro enzyme. Mode of inhibition, computational solvent mapping, and molecular docking studies suggest that these fragments bind adjacent to the binding site of the lead inhibitors and further stabilize the inhibitor-bound state. We propose potential next-generation compounds based on a computational fragment-merging approach. This approach provides an alternative strategy for lead optimization for cases in which direct co-crystallization is difficult.
Co-reporter:Shahila Mehboob ; Kirk E. Hevener ; Kent Truong ; Teuta Boci ; Bernard D. Santarsiero
Journal of Medicinal Chemistry 2012 Volume 55(Issue 12) pp:5933-5941
Publication Date(Web):May 29, 2012
DOI:10.1021/jm300489v
Because of structural and mechanistic differences between eukaryotic and prokaryotic fatty acid synthesis enzymes, the bacterial pathway, FAS-II, is an attractive target for the design of antimicrobial agents. We have previously reported the identification of a novel series of benzimidazole compounds with particularly good antibacterial effect against Francisella tularensis, a Category A biowarfare pathogen. Herein we report the crystal structure of the F. tularensis FabI enzyme in complex with our most active benzimidazole compound bound with NADH. The structure reveals that the benzimidazole compounds bind to the substrate site in a unique conformation that is distinct from the binding motif of other known FabI inhibitors. Detailed inhibition kinetics have confirmed that the compounds possess a novel inhibitory mechanism that is unique among known FabI inhibitors. These studies could have a strong impact on future antimicrobial design efforts and may reveal new avenues for the design of FAS-II active antibacterial compounds.
Co-reporter:Kirk E. Hevener ; Shahila Mehboob ; Pin-Chih Su ; Kent Truong ; Teuta Boci ; Jiangping Deng ; Mahmood Ghassemi ; James L. Cook
Journal of Medicinal Chemistry 2012 Volume 55(Issue 1) pp:268-279
Publication Date(Web):November 18, 2011
DOI:10.1021/jm201168g
Enoyl-acyl carrier protein (ACP) reductase, FabI, is a key enzyme in the bacterial fatty acid biosynthesis pathway (FAS II). FabI is an NADH-dependent oxidoreductase that acts to reduce enoyl-ACP substrates in a final step of the pathway. The absence of this enzyme in humans makes it an attractive target for the development of new antibacterial agents. FabI is known to be unresponsive to structure-based design efforts due to a high degree of induced fit and a mobile flexible loop encompassing the active site. Here we discuss the development, validation, and careful application of a ligand-based virtual screen used for the identification of novel inhibitors of the Francisella tularensis FabI target. In this study, four known classes of FabI inhibitors were used as templates for virtual screens that involved molecular shape and electrostatic matching. The program ROCS was used to search a high-throughput screening library for compounds that matched any of the four molecular shape queries. Matching compounds were further refined using the program EON, which compares and scores compounds by matching electrostatic properties. Using these techniques, 50 compounds were selected, ordered, and tested. The tested compounds possessed novel chemical scaffolds when compared to the input query compounds. Several hits with low micromolar activity were identified and follow-up scaffold-based searches resulted in the identification of a lead series with submicromolar enzyme inhibition, high ligand efficiency, and a novel scaffold. Additionally, one of the most active compounds showed promising whole-cell antibacterial activity against several Gram-positive and Gram-negative species, including the target pathogen. The results of a preliminary structure–activity relationship analysis are presented.
Co-reporter:Rima Chaudhuri, Hyun Lee, Lena Truong, Jaime Torres, Kavankumar Patel, and Michael E. Johnson
Journal of Chemical Information and Modeling 2012 Volume 52(Issue 8) pp:2245-2256
Publication Date(Web):June 14, 2012
DOI:10.1021/ci300177p
Drug discovery and design for inhibition of the Hepatitis C Virus (HCV) NS3/4A serine protease is a major challenge. The broad, shallow, and generally featureless nature of the active site makes it a difficult target for “hit” selection especially using standard docking programs. There are several macrocyclic NS3/4A protease inhibitors that have been approved or are in clinical trials to treat chronic HCV (alone or as combination therapy), but most of the current therapies for HCV infection have untoward side effects, indicating a continuing medical need for the discovery of novel therapeutics with improved efficacy. In this study, we designed and implemented a two-tiered and progressive docking regime that successfully identified five non-macrocyclic small molecules that show inhibitory activity in the low micromolar range. Of these, four compounds show varying inhibition against HCV subgenotypes 1b, 1a, 2a, and 4d. The top inhibitor (3) has an IC50 value of 15 μM against both subgenotypes 1b and 2a of the NS3/4A protease enzyme. Another inhibitor, 1, inhibits all four subgenotypes with moderate activity, showing highest activity for genotype 2a (24 μM). The five inhibitors presented in this study could be valuable candidates for future hit to lead optimization. Additionally, enzyme–inhibitor interaction models presented herein provide key information regarding structural differences between the active sites of the NS3/4A protease of the HCV subgenotype 1a and 1b that might explain the variable inhibitory activity between subgenotypes of the small molecule inhibitors identified here.
Co-reporter:Hyun Lee, Jaime Torres, Lena Truong, Rima Chaudhuri, Anuradha Mittal, Michael E. Johnson
Analytical Biochemistry 2012 Volume 423(Issue 1) pp:46-53
Publication Date(Web):1 April 2012
DOI:10.1016/j.ab.2012.01.006
High-throughput screening (HTS) of large compound libraries has become a commonly used method for the identification of drug leads, and nonphysiological reducing agents have been widely used for HTS. However, a comparison of the difference in the HTS results based on the choice of reducing agent used and potency comparisons of selected inhibitors has not been done with the physiological reducing agent reduced glutathione (GSH). Here, we compared the effects of three reducing agents—dithiothreitol (DTT), β-mercaptoethanol (β-MCE), and tris(2-carboxyethyl)phosphine (TCEP)—as well as GSH against three drug target proteins. Approximately 100,000 compounds were computationally screened for each target protein, and experimental testing of high-scoring compounds (∼560 compounds) with the four reducing agents surprisingly produced many nonoverlapping hits. More importantly, we found that various reducing agents altered inhibitor potency (IC50) from approximately 10 μM with one reducing agent to complete loss (IC50 > 200 μM) of inhibitory activity with another reducing agent. Therefore, the choice of reducing agent in an HTS is critical because this may lead to the pursuit of falsely identified active compounds or failure to identify the true active compounds. We demonstrate the feasibility of using GSH for in vitro HTS assays with these three target enzymes.
Co-reporter:Shahila Mehboob, Liang Guo, Wentao Fu, Anuradha Mittal, Tiffany Yau, Kent Truong, Mary Johlfs, Fei Long, Leslie W.-M. Fung and Michael E. Johnson
Biochemistry 2009 Volume 48(Issue 29) pp:
Publication Date(Web):June 24, 2009
DOI:10.1021/bi9005072
Glutamate racemase (RacE) is a bacterial enzyme that converts l-glutamate to d-glutamate, an essential precursor for peptidoglycan synthesis. In prior work, we have shown that both isoforms cocrystallize with d-glutamate as dimers, and the enzyme is in a closed conformation with limited access to the active site [May, M., et al. (2007) J. Mol. Biol. 371, 1219−1237]. The active site of RacE2 is especially restricted. We utilize several computational and experimental approaches to understand the overall conformational dynamics involved during catalysis when the ligand enters and the product exits the active site. Our steered molecular dynamics simulations and normal-mode analysis results indicate that the monomeric form of the enzyme is more flexible than the native dimeric form. These results suggest that the monomeric enzyme might be more active than the dimeric form. We thus generated site-specific mutations that disrupt dimerization and find that the mutants exhibit significantly higher catalytic rates in the d-Glu to l-Glu reaction direction than the native enzyme. Low-resolution models restored from solution X-ray scattering studies correlate well with the first six normal modes of the dimeric form of the enzyme, obtained from NMA. Thus, along with the local active site residues, global domain motions appear to be implicated in the catalytically relevant structural dynamics of this enzyme and suggest that increased flexibility may accelerate catalysis. This is a novel observation that residues distant from the catalytic site restrain catalytic activity through formation of the dimer structure.
Co-reporter:Suresh K. Tipparaju, Sipak Joyasawal, Sara Forrester, Debbie C. Mulhearn, Scott Pegan, Michael E. Johnson, Andrew D. Mesecar, Alan P. Kozikowski
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 12) pp:3565-3569
Publication Date(Web):15 June 2008
DOI:10.1016/j.bmcl.2008.05.004
Enoyl-ACP reductase (ENR), the product of the FabI gene, from Bacillus anthracis (BaENR) is responsible for catalyzing the final step of bacterial fatty acid biosynthesis. A number of novel 2-pyridone derivatives were synthesized and shown to be potent inhibitors of BaENR.Novel 2-pyridone derivatives were synthesized and shown to be potent inhibitors of enoyl-ACP reductase from Bacillus anthracis.
Co-reporter:Sochanchingwung Rumthao, Oukseub Lee, Qi Sheng, WenTao Fu, Debbie C. Mulhearn, David Crich, Andrew D. Mesecar, Michael E. Johnson
Bioorganic & Medicinal Chemistry Letters 2004 Volume 14(Issue 20) pp:5165-5170
Publication Date(Web):18 October 2004
DOI:10.1016/j.bmcl.2004.07.054
We have designed, synthesized, and evaluated the factor Xa inhibitory activities of p-amidinophenyl-sulfones, amines, and alcohols intended to take advantage of the polarity and hydrogen-bonding potential of the oxyanion hole region of the S1 specificity pocket. We demonstrate that placement of an anionic group within the oxyanion hole region of the catalytic site substantially enhances activity, with small flexible groups favored over bulkier ones. Ab initio pKa calculations suggest that the hydroxyl substituent frequently used for benzamidine moieties may be ionized to form an anionic group, consistent with the general trend. One nonamidine based substituent also shows promising activity.Placement of an anionic group within the oxyanion hole region of the Factor Xa catalytic site substantially enhances activity, with small flexible groups favored over bulkier ones.
Co-reporter:Jian Cui, David Crich, Donald Wink, Matthew Lam, Arnold L Rheingold, David A Case, WenTao Fu, Yasheen Zhou, Mohan Rao, Arthur J Olson, Michael E Johnson
Bioorganic & Medicinal Chemistry 2003 Volume 11(Issue 16) pp:3379-3392
Publication Date(Web):5 August 2003
DOI:10.1016/S0968-0896(03)00332-8
Two conformationally constrained templates have been designed to provide selective inhibitors of the coagulation cascade serine protease, Factor Xa (FXa). The most active inhibitor, 2,7-bis[(Z)-p-amidinobenzylidene)]-3,3,6,6-tetramethylcycloheptanone, exhibits a Ki of 42 nM against FXa, with strong selectivity against thrombin (1000-fold), trypsin (300-fold) and plasmin (900-fold). With only two freely rotatable bonds, molecular modeling suggests that one amidine group is positioned into the S1 pocket, forming hydrogen bonds with the side chain of Asp189, similar to other amidine-based inhibitors, with the second benzamidine positioned into the S4 pocket in a position to form strong cation–pi bonding with the S4 aryl cage. We suggest that this interaction plays an important role in the specificity of these inhibitors against other serine proteases.Compound 34 exhibited ∼40 nN activity against Factor Xa, and good selectivity against thrombin, trypsin and plasmin.
Co-reporter:Jian Cui, Fatima Marankan, Wentao Fu, David Crich, Andrew Mesecar, Michael E Johnson
Bioorganic & Medicinal Chemistry 2002 Volume 10(Issue 1) pp:41-46
Publication Date(Web):January 2002
DOI:10.1016/S0968-0896(01)00259-0
p-amidinophenylmethylphosphinic acid (AMPA) was designed, synthesized and crystallized in complex with trypsin to study interactions with the oxyanion hole at the S1 site. In comparison to benzamidine, AMPA shows improved activity, which the crystal structure demonstrates to result from hydrogen bonds between the negatively charged phosphinic acid group and the catalytic residues at the oxyanion hole.The synthesis and crystal structure of p-amidinophenylmethylphosphinic acid (AMPA) complexed with trypsin is reported.
Co-reporter:Lena Truong, Kirk E. Hevener, Amy J. Rice, Kavankumar Patel, Michael E. Johnson, Hyun Lee
Protein Expression and Purification (March 2013) Volume 88(Issue 1) pp:98-106
Publication Date(Web):1 March 2013
DOI:10.1016/j.pep.2012.11.018
Staphylococcus aureus is a pathogenic bacterium that causes a variety of mild to lethal human diseases. The rapid spread of multidrug-resistant strains makes the discovery of new antimicrobial agents critical. Dihydroorotase (PyrC), the third enzyme in the bacterial pyrimidine biosynthesis pathway, is structurally and mechanistically distinct from its mammalian counterpart. It has been confirmed to be essential in S. aureus making it an attractive antibacterial drug target. No protocol to express and purify S. aureus PyrC (SaPyrC) has been reported. To obtain the SaPyrC enzyme and overcome anticipated solubility problems, the SaPyrC gene was cloned into the pET-SUMO vector. The N-terminal His-SUMO fused SaPyrC was expressed in Escherichia coli BL21 (DE3) with an HRV 3C protease recognition site inserted between the SUMO tag and SaPyrC to allow for improved cleavage by HRV protease. Purification of cleaved protein using HisTrap affinity and gel filtration columns resulted in native SaPyrC with estimated 95% purity and 40% yield. Both His-SUMO tagged and native SaPyrC form dimers, and enzyme characterization studies have shown that the His-SUMO tag affects enzyme activity slightly. Forward and reverse kinetic rate constants for both tagged and native SaPyrC were determined, and pH profiling studies revealed the optimal pH values for forward and reverse reactions.Highlights► The purification of the Staphylococcus aureus PyrC enzyme using a His-SUMO tag is described. ► Protein solubility and yield were high with the use of the His-SUMO tag. ► Improved tag cleavage was achieved with HRV 3C protease over SUMO protease. ► Forward and reverse kinetic rate constants for SaPyrC were calculated experimentally. ► Optimal pH values for forward and reverse reactions were determined.
Co-reporter:Rima Chaudhuri, Sishi Tang, Guijun Zhao, Hui Lu, ... Michael E. Johnson
Journal of Molecular Biology (25 November 2011) Volume 414(Issue 2) pp:272-288
Publication Date(Web):25 November 2011
DOI:10.1016/j.jmb.2011.09.030
The human severe acute respiratory syndrome coronavirus (SARS-CoV) and the NL63 coronaviruses are human respiratory pathogens for which no effective antiviral treatment exists. The papain-like cysteine proteases encoded by the coronavirus (SARS-CoV: PLpro; NL63: PLP1 and PLP2) represent potential targets for antiviral drug development. Three recent inhibitor-bound PLpro structures highlight the role of an extremely flexible six-residue loop in inhibitor binding. The high binding site plasticity is a major challenge in computational drug discovery/design efforts. From conventional molecular dynamics and accelerated molecular dynamics (aMD) simulations, we find that with conventional molecular dynamics simulation, PLpro translationally samples the open and closed conformation of BL2 loop on a picosecond–nanosecond timescale but does not reproduce the peptide bond inversion between loop residues Tyr269 and Gln270 that is observed on inhibitor GRL0617 binding. Only aMD simulation, starting from the closed loop conformation, reproduced the 180° ϕ–ψ dihedral rotation back to the open loop state. The Tyr–Gln peptide bond inversion appears to involve a progressive conformational change of the full loop, starting at one side, and progressing to the other. We used the SARS-CoV apo X-ray structure to develop a model of the NL63-PLP2 catalytic site. Superimposition of the PLP2 model on the PLpro X-ray structure identifies binding site residues in PLP2 that contribute to the distinct substrate cleavage site specificities between the two proteases. The topological and electrostatic differences between the two protease binding sites also help explain the selectivity of non-covalent PLpro inhibitors.Download high-res image (132KB)Download full-size imageHighlights► aMD reproduces inhibitor induced-fit conformational change in PLpro. ► SARS-CoV PLpro and NL63-PLP2 differing substrate and inhibitor specificities are correlated with electrostatic and topological catalytic site differences. ► Broad-spectrum coronavirus papain-like protease inhibitor discovery must compensate for enzymatic structural and electrostatic variation.

![1-(8-methoxy-1,3,4,5-tetrahydropyrido[4,3-b]indol-2-yl)ethanone](http://img.cochemist.com/ccimg/1040700/1040695-96-2.png)
![1-(8-methoxy-1,3,4,5-tetrahydropyrido[4,3-b]indol-2-yl)ethanone](http://img.cochemist.com/ccimg/1040700/1040695-96-2_b.png)




![1-Ethyl-1,2,3,4-tetrahydropyrrolo[1,2-a]pyrazine](http://img.cochemist.com/ccimg/119000/118959-62-9.png)
![1-Ethyl-1,2,3,4-tetrahydropyrrolo[1,2-a]pyrazine](http://img.cochemist.com/ccimg/119000/118959-62-9_b.png)
![1-[5-(2-CHLOROPHENYL)-1,2-OXAZOL-3-YL]METHANAMINE HYDROCHLORIDE (1:1)](http://img.cochemist.com/ccimg/1160300/1160245-59-9.png)
![1-[5-(2-CHLOROPHENYL)-1,2-OXAZOL-3-YL]METHANAMINE HYDROCHLORIDE (1:1)](http://img.cochemist.com/ccimg/1160300/1160245-59-9_b.png)
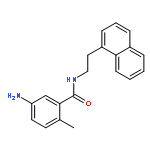
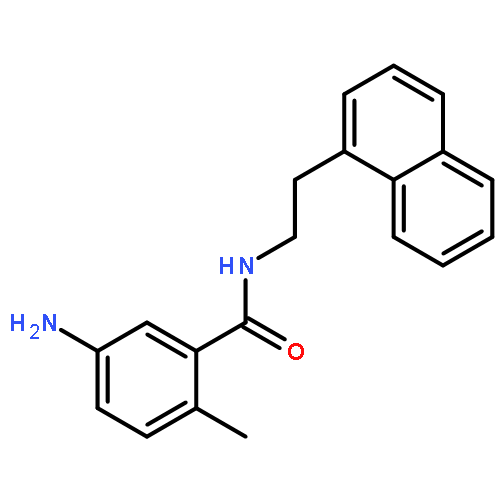
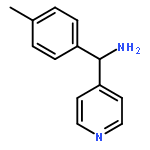
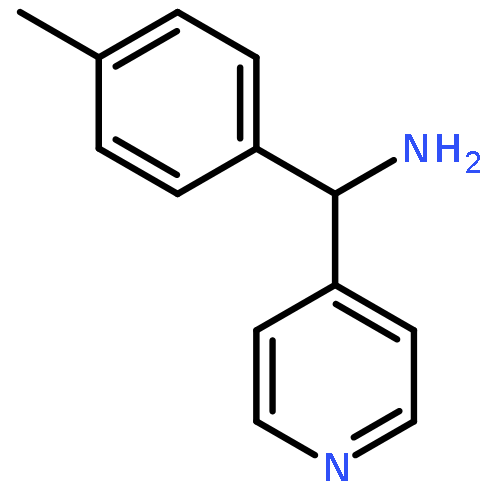
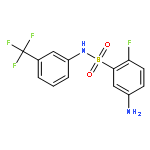
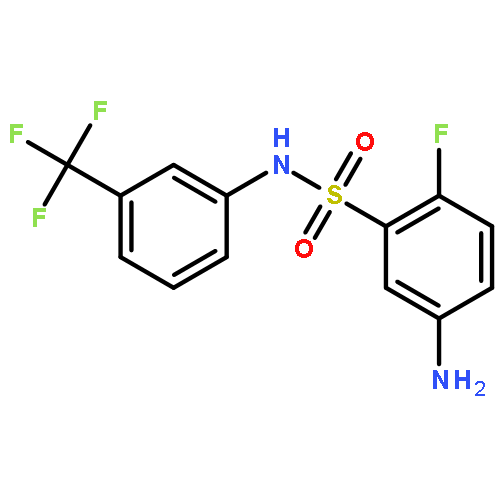
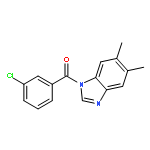
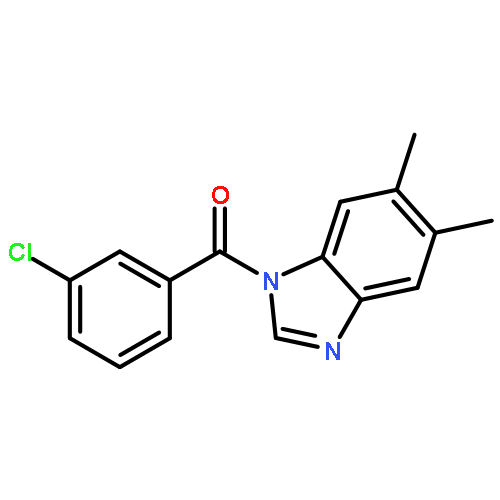


![1-(4-Chlorobenzyl)-1H-benzo[d]imidazol-2-amine](http://img.cochemist.com/ccimg/109700/109635-38-3.png)
![1-(4-Chlorobenzyl)-1H-benzo[d]imidazol-2-amine](http://img.cochemist.com/ccimg/109700/109635-38-3_b.png)




![1H-Benzimidazole, 1-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/46900/46885-71-6.png)
![1H-Benzimidazole, 1-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/46900/46885-71-6_b.png)


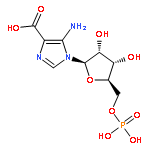
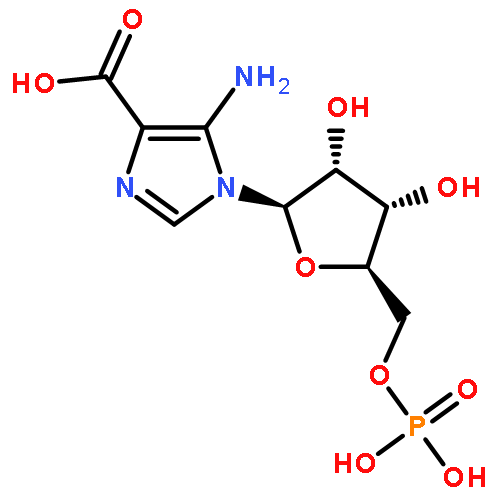
![N-{2-[(4-FLUOROBENZYL)AMINO]-2-OXOETHYL}-N-(2-METHOXYBENZYL)-1,2,3-THIADIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/5000/4981-92-4.png)
![N-{2-[(4-FLUOROBENZYL)AMINO]-2-OXOETHYL}-N-(2-METHOXYBENZYL)-1,2,3-THIADIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/5000/4981-92-4_b.png)
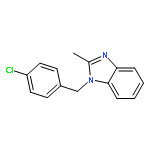
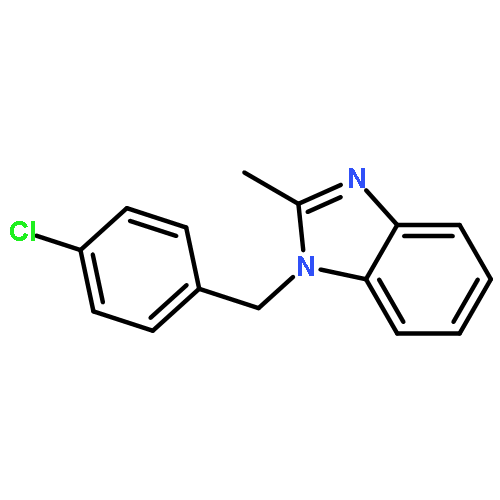


![3-Phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-6-amine](http://img.cochemist.com/ccimg/3200/3176-53-2.png)
![3-Phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-6-amine](http://img.cochemist.com/ccimg/3200/3176-53-2_b.png)
![L-Aspartic acid,N-[[5-amino-1-(5-O-phosphono-b-D-ribofuranosyl)-1H-imidazol-4-yl]carbonyl]-](http://img.cochemist.com/ccimg/3100/3031-95-6.png)
![L-Aspartic acid,N-[[5-amino-1-(5-O-phosphono-b-D-ribofuranosyl)-1H-imidazol-4-yl]carbonyl]-](http://img.cochemist.com/ccimg/3100/3031-95-6_b.png)





![1-(4-Chlorobenzyl)-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/107500/107456-42-8.png)
![1-(4-Chlorobenzyl)-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/107500/107456-42-8_b.png)


![1H-Benzimidazole, 1-[(4-bromophenyl)methyl]-](http://img.cochemist.com/ccimg/73800/73798-61-5.png)
![1H-Benzimidazole, 1-[(4-bromophenyl)methyl]-](http://img.cochemist.com/ccimg/73800/73798-61-5_b.png)