Co-reporter:Hyowon Seo, Aofei Liu, and Timothy F. Jamison
Journal of the American Chemical Society October 11, 2017 Volume 139(Issue 40) pp:13969-13969
Publication Date(Web):September 27, 2017
DOI:10.1021/jacs.7b05942
The direct β-selective hydrocarboxylation of styrenes under atmospheric pressure of CO2 has been developed using photoredox catalysis in continuous flow. The scope of this methodology was demonstrated with a range of functionalized terminal styrenes, as well as α-substituted and β-substituted styrenes.
Co-reporter:Anne-Catherine Bédard, Ashley R. Longstreet, Joshua Britton, Yuran Wang, Hideki Moriguchi, Robert W. Hicklin, William H. Green, Timothy F. Jamison
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 23(Issue 23) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.bmc.2017.02.002
Minimizing the waste stream associated with the synthesis of active pharmaceutical ingredients (APIs) and commodity chemicals is of high interest within the chemical industry from an economic and environmental perspective. In exploring solutions to this area, we herein report a highly optimized and environmentally conscious continuous-flow synthesis of two APIs identified as essential medicines by the World Health Organization, namely diazepam and atropine. Notably, these approaches significantly reduced the E-factor of previously published routes through the combination of continuous-flow chemistry techniques, computational calculations and solvent minimization. The E-factor associated with the synthesis of atropine was reduced by 94-fold (about two orders of magnitude), from 2245 to 24, while the E-factor for the synthesis of diazepam was reduced by 4-fold, from 36 to 9.Download high-res image (176KB)Download full-size image
Co-reporter:Amanda C. Wicker;Frank A. Leibfarth
Polymer Chemistry (2010-Present) 2017 vol. 8(Issue 37) pp:5786-5794
Publication Date(Web):2017/09/26
DOI:10.1039/C7PY01204G
Monodisperse oligomers are important intermediates for studying structure–property relationships in soft materials but are traditionally laborious to synthesize. A semi-automated synthetic system that combines the benefits of telescoped reactions in continuous flow with iterative exponential growth (IEG) greatly expedites this process and makes the rapid synthesis of structurally diverse oligomer libraries practical. Herein, the coupling chemistry in the Flow-IEG system has been upgraded and expanded to include both 1,4- and 1,5-triazole linkages between monomers through an improved copper-catalyzed azide–alkyne cycloaddition (CuAAC) and a newly-optimized ruthenium-catalyzed azide–alkyne cycloaddition (RuAAC), respectively. Improvements to the Flow-IEG framework enabled the library synthesis of monodisperse oligomers with variations in triazole connectivity. These discrete oligomers allowed the systematic evaluation of the consequences of triazole sequence on material properties. The crystallization properties of these macromolecules were highly dependent on both their monomer sequence and triazole substitution pattern.
Co-reporter:Joshua Britton
European Journal of Organic Chemistry 2017 Volume 2017(Issue 44) pp:6566-6574
Publication Date(Web):2017/12/01
DOI:10.1002/ejoc.201700992
Multi-step continuous flow synthesis enables a parallel approach to obtain agrochemicals and pharmaceuticals containing 3-fluoroalkyl pyrazole cores. In this system, fluorinated amines are transformed into pyrazole cores through a telescoped in situ generation and consumption of diazoalkanes. Once synthesized, additional continuous flow and batch reactions add complexity to the pyrazole core via C–N arylation and methylation, TMS cleavage, and amidation. Using this modular assembly line approach, Bixafen and Fluxapyroxad were synthesized in 38 % yield over four continuous flow steps in an overall reaction time of 56 min. Finally, coupling selected continuous flow processes with an offline (batch) Ullmann coupling afforded Celecoxib, Mavacoxib, and SC-560 in 33–54 % yield over two to three steps.
Co-reporter:Dr. Hongkun Lin;Dr. Chunhui Dai; Timothy F. Jamison; Klavs F. Jensen
Angewandte Chemie International Edition 2017 Volume 56(Issue 30) pp:8870-8873
Publication Date(Web):2017/07/17
DOI:10.1002/anie.201703812
AbstractWithin a total residence time of 9 min, the sodium salt of ciprofloxacin was prepared from simple building blocks via a linear sequence of six chemical reactions in five flow reactors. Sequential offline acidifications and filtrations afforded ciprofloxacin and ciprofloxacin hydrochloride. The overall yield of the eight-step sequence was 60 %. No separation of intermediates was required throughout the synthesis when a single acylation reaction was applied to remove the main byproduct, dimethylamine.
Co-reporter:Dr. Joshua Britton; Dr. Timothy F. Jamison
Angewandte Chemie International Edition 2017 Volume 56(Issue 30) pp:8823-8827
Publication Date(Web):2017/07/17
DOI:10.1002/anie.201704529
AbstractA rapid and modular continuous flow synthesis of highly functionalized fluorinated pyrazoles and pyrazolines has been developed. Flowing fluorinated amines through sequential reactor coils mediates diazoalkane formation and [3+2] cycloaddition to generate more than 30 azoles in a telescoped fashion. Pyrazole cores are then sequentially modified through additional reactor modules performing N-alkylation and arylation, deprotection, and amidation to install broad molecular diversity in short order. Continuous flow synthesis enables the safe handling of diazoalkanes at elevated temperatures, and the use of aryl alkyne dipolarphiles under catalyst-free conditions. This assembly-line synthesis provides a flexible approach for the synthesis of agrochemicals and pharmaceuticals, as demonstrated by a four-step, telescoped synthesis of measles therapeutic, AS-136A, in a total residence time of 31.7 min (1.76 g h−1).
Co-reporter:Dr. Hongkun Lin;Dr. Chunhui Dai; Timothy F. Jamison; Klavs F. Jensen
Angewandte Chemie 2017 Volume 129(Issue 30) pp:8996-8999
Publication Date(Web):2017/07/17
DOI:10.1002/ange.201703812
AbstractWithin a total residence time of 9 min, the sodium salt of ciprofloxacin was prepared from simple building blocks via a linear sequence of six chemical reactions in five flow reactors. Sequential offline acidifications and filtrations afforded ciprofloxacin and ciprofloxacin hydrochloride. The overall yield of the eight-step sequence was 60 %. No separation of intermediates was required throughout the synthesis when a single acylation reaction was applied to remove the main byproduct, dimethylamine.
Co-reporter:Dr. Joshua Britton; Dr. Timothy F. Jamison
Angewandte Chemie 2017 Volume 129(Issue 30) pp:8949-8953
Publication Date(Web):2017/07/17
DOI:10.1002/ange.201704529
AbstractA rapid and modular continuous flow synthesis of highly functionalized fluorinated pyrazoles and pyrazolines has been developed. Flowing fluorinated amines through sequential reactor coils mediates diazoalkane formation and [3+2] cycloaddition to generate more than 30 azoles in a telescoped fashion. Pyrazole cores are then sequentially modified through additional reactor modules performing N-alkylation and arylation, deprotection, and amidation to install broad molecular diversity in short order. Continuous flow synthesis enables the safe handling of diazoalkanes at elevated temperatures, and the use of aryl alkyne dipolarphiles under catalyst-free conditions. This assembly-line synthesis provides a flexible approach for the synthesis of agrochemicals and pharmaceuticals, as demonstrated by a four-step, telescoped synthesis of measles therapeutic, AS-136A, in a total residence time of 31.7 min (1.76 g h−1).
Co-reporter:Dr. Peter D. Morse; Dr. Timothy F. Jamison
Angewandte Chemie International Edition 2017 Volume 56(Issue 45) pp:13999-14002
Publication Date(Web):2017/11/06
DOI:10.1002/anie.201706157
AbstractWe report a method for overcoming the low stability of nitroalkynes through the development of nitrated vinyl silyltriflate equivalents. Because of their instability, nitroalkynes have only rarely been utilized in synthesis. The reactivity of these silyltriflates, which are prepared in situ, is exemplified by dipolar cycloaddition reactions with nitrones to give highly substituted 4-nitro-4-isoxazolines in high yields. This approach has proven general for several different alkyl and aryl substituted alkynes. In order to minimize the accumulation of potentially hazardous reaction intermediates, we have also developed a continuous flow variant of this method that is capable of carrying out the entire reaction sequence in a good yield and a short residence time.
Co-reporter:Andrea Adamo;Rachel L. Beingessner;Eve M. Revalor;Klavs F. Jensen;Torsten Stelzer;Jie Chen;Allan S. Myerson;Mohsen Behnam;Nopphon Weeranoppanant;Jean-Christophe M. Monbaliu;David R. Snead;Shin Yee Wong;Ping Zhang
Science 2016 Volume 352(Issue 6281) pp:61-67
Publication Date(Web):01 Apr 2016
DOI:10.1126/science.aaf1337
Drug manufacturing in a fridge-sized box
Commodity chemicals tend to be manufactured in a continuous fashion. However, the preparation of pharmaceuticals still proceeds batch by batch, partly on account of the complexity of their molecular structures. Adamo et al. now present an apparatus roughly the size of a household refrigerator that can synthesize and purify pharmaceuticals under continuous-flow conditions (see the Perspective by Martin). The integrated set of modules can produce hundreds to thousands of accumulated doses in a day, delivered in aqueous solution.
Science, this issue p. 61; see also p. 44
Co-reporter:Eric A. Standley, Sarah Z. Tasker, Kim L. Jensen, and Timothy F. Jamison
Accounts of Chemical Research 2015 Volume 48(Issue 5) pp:1503
Publication Date(Web):April 23, 2015
DOI:10.1021/acs.accounts.5b00064
Nickel(0) catalysts have proven to be powerful tools for multicomponent coupling reactions in our laboratories over the past 15 years. This interest was originally sparked by the ubiquity of allylic alcohol motifs in natural products, such as (–)-terpestacin, which we envisioned assembling by the coupling of two π components (alkyne and aldehyde) with concomitant reduction. Mechanistic investigations allowed us to elucidate several modes of controlling the regioselectivity and stereoselectivity in the oxidative cyclization, and these insights enabled us to leverage combinations of alkenes and phosphine ligands to direct regioselective outcomes. The initial success in developing the first intermolecular reductive alkyne–aldehyde coupling reaction launched a series of methodological investigations that rapidly expanded to include coupling reactions of alkynes with other electrophilic π components, such as imines and ketones, as well as electrophilic σ components, such as epoxides. Aziridines proved to be more challenging substrates for reductive coupling, but we were recently able to demonstrate that cross-coupling of aziridines and alkylzinc reagents is smoothly catalyzed by a zero-valent nickel/phenanthroline system. Moreover, the enantioselective alkyne–aldehyde coupling and the development of novel P-chiral ferrocenyl ligands enabled the total synthesis of (–)-terpestacin, amphidinolides T1 and T4, (–)-gloeosporone, and pumiliotoxins 209F and 251D.We subsequently determined that alkenes could be used in place of alkynes in several nickel-catalyzed reactions when a silyl triflate activating agent was added. We reason that such an additive functions largely to enhance the electrophilicity of the metal center by coordination to the electrophilic π component, such that less nucleophilic alkene π donors can undergo productive combination with nickel complexes. This activation manifold was further demonstrated to be effective for alkene–aldehyde couplings. In a related manner, electrophilic promoters were also successfully employed for allylic substitution reactions of allylic carbonates with simple alkenes and in the Mizoroki–Heck reaction of both benzyl and aryl electrophiles. In these instances, it is proposed that counterion exchange from a more strongly coordinating anion to the weakly or noncoordinating triflate counterion enables reaction at an electrophilic Ni(II) center rather than by coordination to one of the coupling components. Mechanistic insights also played an important role in the development of mixed N-heterocyclic carbene/phosphite ligand systems to overcome challenges in regioselective alkene–aldehyde coupling reactions.We hope that, taken together, the body of work summarized in this Account demonstrates the constructive interplay among total synthesis, methodological development, and mechanistic investigation that has driven our research program.
Co-reporter:Kurt W. Armbrust; Matthew G. Beaver
Journal of the American Chemical Society 2015 Volume 137(Issue 21) pp:6941-6946
Publication Date(Web):May 18, 2015
DOI:10.1021/jacs.5b03570
[Rh(CO)2Cl]2 is as an effective catalyst for endo-selective cyclizations and cascades of epoxy-(E)-enoate alcohols, thus enabling the synthesis of oxepanes and oxepane-containing polyethers from di- and trisubstituted epoxides. Syntheses of the ABC and EF ring systems of (−)-brevisin via all endo-diepoxide-opening cascades using this method constitute a formal total synthesis and demonstrate the utility of this methodology in the context of the synthesis of marine ladder polyether natural products.
Co-reporter:Sarah Z. Tasker
Journal of the American Chemical Society 2015 Volume 137(Issue 30) pp:9531-9534
Publication Date(Web):July 21, 2015
DOI:10.1021/jacs.5b05597
Nickel/photoredox catalysis is used to synthesize indolines in one step from iodoacetanilides and alkenes. Very high regioselectivity for 3-substituted indoline products is obtained for both aliphatic and styrenyl olefins. Mechanistic investigations indicate that oxidation to Ni(III) is necessary to perform the difficult C–N bond-forming reductive elimination, producing a Ni(I) complex, which in turn is reduced to Ni(0). This process serves to further demonstrate the utility of photoredox catalysts as controlled single electron transfer agents in multioxidation state nickel catalysis.
Co-reporter:Charles E. Ocampo, Doris Lee, and Timothy F. Jamison
Organic Letters 2015 Volume 17(Issue 4) pp:820-823
Publication Date(Web):February 9, 2015
DOI:10.1021/ol503612h
Described herein is a novel Lewis acid catalyzed rearrangement–coupling of oxygen heterocycles and bis(diethylamino)chlorophosphine that provides direct formation of the phosphonomethyl ether functionality found in several important antiretroviral agents. A wide range of dioxolanes and 1,3-dioxanes may be employed, furnishing the desired products in good yield. The utility of this method is demonstrated in a novel synthesis of tenofovir, an antiretroviral drug used in the treatment of HIV/AIDS and hepatitis B.
Co-reporter:Lara C. Czabaniuk and Timothy F. Jamison
Organic Letters 2015 Volume 17(Issue 4) pp:774-777
Publication Date(Web):February 3, 2015
DOI:10.1021/ol503400j
A new and highly selective method for the synthesis of hydroxyl-substituted tetrahydropyrans is described. This method utilizes titanium(IV) isopropoxide and diethyl tartrate to perform a diastereoselective epoxidation followed by in situ epoxide activation and highly selective endo-cyclization to form the desired tetrahydropyran ring. The HIJ ring fragment of the marine ladder polyether yessotoxin was synthesized using this two-stage tactic that proceeds with high efficiency and excellent regioselectivity.
Co-reporter:Dr. David R. Snead ;Dr. Timothy F. Jamison
Angewandte Chemie International Edition 2015 Volume 54( Issue 3) pp:983-987
Publication Date(Web):
DOI:10.1002/anie.201409093
Abstract
In a total residence time of three minutes, ibuprofen was assembled from its elementary building blocks with an average yield of above 90 % for each step. A scale-up of this five-stage process (3 bond-forming steps, one work-up, and one in-line liquid–liquid separation) provided ibuprofen at a rate of 8.09 g h−1 (equivalent to 70.8 kg y−1) using a system with an overall footprint of half the size of a standard laboratory fume hood. Aside from the high throughput, several other aspects of this synthesis expand the capabilities of continuous-flow processing, including a Friedel–Crafts acylation run under neat conditions and promoted by AlCl3, an exothermic in-line quench of high concentrations of precipitation-prone AlCl3, liquid–liquid separations run at or above 200 psi to provide solvent-free product, and the use of highly aggressive oxidants, such as iodine monochloride. The use of simple, inexpensive, and readily available reagents thus affords a practical synthesis of this important generic pharmaceutical.
Co-reporter:Dr. Kim L. Jensen;Dennis U. Nielsen ;Dr. Timothy F. Jamison
Chemistry - A European Journal 2015 Volume 21( Issue 20) pp:7379-7383
Publication Date(Web):
DOI:10.1002/chem.201500886
Abstract
In this communication, we report a straightforward synthesis of enantiomerically pure 2-alkyl azetidines. The protocol is based on a highly regioselective nickel-catalyzed cross-coupling of aliphatic organozinc reagents with an aziridine that features a tethered thiophenyl group. Activation by methylation transforms the sulfide into an excellent leaving group and triggers the formation of the 2-substituted azetidine core structure by cyclization. In addition, we have expanded this concept to the synthesis of enantiomerically pure, terminal alkyl aziridines. Coupling of a TMS-protected aziridine alcohol, followed by acidic work-up to remove the silyl group, provides 1,2-amino alcohol products that are readily cyclized to aziridines. Both of these sequences display excellent functional group tolerance and deliver the desired azetidine and aziridine products in good to excellent yields.
Co-reporter:Frank A. Leibfarth;Jeremiah A. Johnson
PNAS 2015 Volume 112 (Issue 34 ) pp:10617-10622
Publication Date(Web):2015-08-25
DOI:10.1073/pnas.1508599112
We report a semiautomated synthesis of sequence and architecturally defined, unimolecular macromolecules through a marriage
of multistep flow synthesis and iterative exponential growth (Flow-IEG). The Flow-IEG system performs three reactions and
an in-line purification in a total residence time of under 10 min, effectively doubling the molecular weight of an oligomeric
species in an uninterrupted reaction sequence. Further iterations using the Flow-IEG system enable an exponential increase
in molecular weight. Incorporating a variety of monomer structures and branching units provides control over polymer sequence
and architecture. The synthesis of a uniform macromolecule with a molecular weight of 4,023 g/mol is demonstrated. The user-friendly
nature, scalability, and modularity of Flow-IEG provide a general strategy for the automated synthesis of sequence-defined,
unimolecular macromolecules. Flow-IEG is thus an enabling tool for theory validation, structure–property studies, and advanced
applications in biotechnology and materials science.
Co-reporter:Dr. David R. Snead ;Dr. Timothy F. Jamison
Angewandte Chemie 2015 Volume 127( Issue 3) pp:997-1001
Publication Date(Web):
DOI:10.1002/ange.201409093
Abstract
In a total residence time of three minutes, ibuprofen was assembled from its elementary building blocks with an average yield of above 90 % for each step. A scale-up of this five-stage process (3 bond-forming steps, one work-up, and one in-line liquid–liquid separation) provided ibuprofen at a rate of 8.09 g h−1 (equivalent to 70.8 kg y−1) using a system with an overall footprint of half the size of a standard laboratory fume hood. Aside from the high throughput, several other aspects of this synthesis expand the capabilities of continuous-flow processing, including a Friedel–Crafts acylation run under neat conditions and promoted by AlCl3, an exothermic in-line quench of high concentrations of precipitation-prone AlCl3, liquid–liquid separations run at or above 200 psi to provide solvent-free product, and the use of highly aggressive oxidants, such as iodine monochloride. The use of simple, inexpensive, and readily available reagents thus affords a practical synthesis of this important generic pharmaceutical.
Co-reporter:Kim L. Jensen ; Eric A. Standley
Journal of the American Chemical Society 2014 Volume 136(Issue 31) pp:11145-11152
Publication Date(Web):July 23, 2014
DOI:10.1021/ja505823s
Herein, we report the first ligand-controlled, nickel-catalyzed cross-coupling of aliphatic N-tosylaziridines with aliphatic organozinc reagents. The reaction protocol displays complete regioselectivity for reaction at the less hindered C–N bond, and the products are furnished in good to excellent yield for a broad selection of substrates. Moreover, we have developed an air-stable nickel(II) chloride/ligand precatalyst that can be handled and stored outside a glovebox. In addition to increasing the activity of this catalyst system, this also greatly improves the practicality of this reaction, as the use of the very air-sensitive Ni(cod)2 is avoided. Finally, mechanistic investigations, including deuterium-labeling studies, show that the reaction proceeds with overall inversion of configuration at the terminal position of the aziridine by way of aziridine ring opening by Ni (inversion), transmetalation (retention), and reductive elimination (retention).
Co-reporter:Jie Wu, Jennifer A. Kozak, Fritz Simeon, T. Alan Hatton and Timothy F. Jamison
Chemical Science 2014 vol. 5(Issue 3) pp:1227-1231
Publication Date(Web):27 Jan 2014
DOI:10.1039/C3SC53422G
A mechanism-guided design of a multi-step flow system enabled an efficient general process for the synthesis of cyclic carbonates from alkenes and CO2. The flow system proved to be an ideal platform for multicomponent reactions because it was straightforward to introduce reagents at specific stages without their interacting with each other or with reaction intermediates prone to destruction by them. This system exhibited superior reactivity, increased yield, and broader substrate scope relative to conventional batch conditions and suppressed the formation of undesired byproducts, such as, epoxides and 1,2-dibromoalkanes.
Co-reporter:Xiaoqing Yang, Jie Wu, Xianwen Mao, Timothy F. Jamison and T. Alan Hatton
Chemical Communications 2014 vol. 50(Issue 24) pp:3245-3248
Publication Date(Web):06 Feb 2014
DOI:10.1039/C4CC00252K
An effective transformation of alkenes into cyclic carbonates has been achieved using NaHCO3 as the C1 source in acetone–water under microwave heating, with selectivities and yields significantly surpassing those obtained using conventional heating.
Co-reporter:Leandro H. Andrade, Wolfgang Kroutil, and Timothy F. Jamison
Organic Letters 2014 Volume 16(Issue 23) pp:6092-6095
Publication Date(Web):November 13, 2014
DOI:10.1021/ol502712v
E. coli cells containing overexpressed (R)-selective ω-transaminase and the cofactor PLP were immobilized on methacrylate beads suitable for continuous flow applications. The use of an organic solvent suppresses leaching of PLP from the cells; no additional cofactor was required after setting up the packed-bed reactor containing the biocatalyst (ω-TA-PLP). Non-natural ketone substrates were transformed in flow with excellent enantioselectivity (>99% ee). Features of this novel system include high-throughput (30–60 min residence time), clean production (no quench, workup, or purification required), high enzyme stability (the packed-bed reactor can be continuously operated for 1–10 days), and excellent mass recovery.
Co-reporter:Ping Zhang, M. Grace Russell, and Timothy F. Jamison
Organic Process Research & Development 2014 Volume 18(Issue 11) pp:1567-1570
Publication Date(Web):August 27, 2014
DOI:10.1021/op500166n
Small molecules bearing 1,2,3-triazole functionalities are important intermediates and pharmaceuticals. Common methods to access the triazole moiety generally require the generation and isolation of organic azide intermediates. Continuous flow synthesis provides the opportunity to synthesize and consume the energetic organoazides, without accumulation thereof. In this report, we described a continuous synthesis of the antiseizure medication rufinamide. This route is convergent and features copper tubing reactor-catalyzed cycloaddition reaction. Each of the three chemical steps enjoys significant benefits and has several advantages by being conducted in flow. The total average residence time of the synthesis is approximately 11 min, and rufinamide is obtained in 92% overall yield.
Co-reporter:Sarah Z. Tasker;Dr. Alicia C. Gutierrez ; Timothy F. Jamison
Angewandte Chemie International Edition 2014 Volume 53( Issue 7) pp:1858-1861
Publication Date(Web):
DOI:10.1002/anie.201308391
Abstract
Achieving high selectivity in the Heck reaction of electronically unbiased alkenes has been a longstanding challenge. Using a nickel-catalyzed cationic Heck reaction, we were able to achieve excellent selectivity for branched products (≥19:1 in all cases) over a wide range of aryl electrophiles and aliphatic olefins. A bidentate ligand with a suitable bite angle and steric profile was key to obtaining high branched/linear selectivity, whereas the appropriate base suppressed alkene isomerization of the product. Although aryl triflates are traditionally used to access the cationic Heck pathway, we have shown that, by using triethylsilyl trifluoromethanesulfonate, we can effect a counterion exchange of the catalytic nickel complex, such that cheaper and more stable aryl chlorides, mesylates, tosylates, and sulfamates can be used to yield the same branched products with high selectivity.
Co-reporter:Dr. Zhi He ;Dr. Timothy F. Jamison
Angewandte Chemie International Edition 2014 Volume 53( Issue 13) pp:3353-3357
Publication Date(Web):
DOI:10.1002/anie.201310572
Abstract
Phenols are important compounds in chemical industry. An economical and green approach to phenol preparation by the direct oxidation of aryl Grignard reagents using compressed air in continuous gas-liquid segmented flow systems is described. The process tolerates a broad range of functional groups, including oxidation-sensitive functionalities such as alkenes, amines, and thioethers. By integrating a benzyne-mediated in-line generation of arylmagnesium intermediates with the aerobic oxidation, a facile three-step, one-flow process, capable of preparing 2-functionalized phenols in a modular fashion, is established.
Co-reporter:Dr. Jie Wu;Dr. Xiaoqing Yang;Dr. Zhi He;Dr. Xianwen Mao;Dr. T. Alan Hatton;Dr. Timothy F. Jamison
Angewandte Chemie International Edition 2014 Volume 53( Issue 32) pp:8416-8420
Publication Date(Web):
DOI:10.1002/anie.201405014
Abstract
We describe an efficient continuous flow synthesis of ketones from CO2 and organolithium or Grignard reagents that exhibits significant advantages over conventional batch conditions in suppressing undesired symmetric ketone and tertiary alcohol byproducts. We observed an unprecedented solvent-dependence of the organolithium reactivity, the key factor in governing selectivity during the flow process. A facile, telescoped three-step–one-flow process for the preparation of ketones in a modular fashion through the in-line generation of organometallic reagents is also established.
Co-reporter:Dr. Zhi He;Minwoo Bae;Dr. Jie Wu ;Dr. Timothy F. Jamison
Angewandte Chemie International Edition 2014 Volume 53( Issue 52) pp:14451-14455
Publication Date(Web):
DOI:10.1002/anie.201408522
Abstract
A mild and facile method for preparing highly functionalized pyrrolo[1,2-a]quinoxalines and other nitrogen-rich heterocycles, each containing a quinoxaline core or an analogue thereof, has been developed. The novel method features a visible-light-induced decarboxylative radical coupling of ortho-substituted arylisocyanides and radicals generated from phenyliodine(III) dicarboxylate reagents and exhibits excellent functional group compatibility. A wide range of quinoxaline heterocycles have been prepared. Finally, a telescoped preparation of these polycyclic compounds by integration of the in-line isocyanide formation and photochemical cyclization has been established in a three-step continuous-flow system.
Co-reporter:Eric A. Standley, Stacey J. Smith, Peter Müller, and Timothy F. Jamison
Organometallics 2014 Volume 33(Issue 8) pp:2012-2018
Publication Date(Web):April 16, 2014
DOI:10.1021/om500156q
A series of air-stable nickel complexes of the form L2Ni(aryl) X (L = monodentate phosphine, X = Cl, Br) and LNi(aryl)X (L = bis-phosphine) have been synthesized and are presented as a library of precatalysts suitable for a wide variety of nickel-catalyzed transformations. These complexes are easily synthesized from low-cost NiCl2·6H2O or NiBr2·3H2O and the desired ligand followed by addition of 1 equiv of Grignard reagent. A selection of these complexes were characterized by single-crystal X-ray diffraction, and an analysis of their structural features is provided. A case study of their use as precatalysts for the nickel-catalyzed carbonyl-ene reaction is presented, showing superior reactivity in comparison to reactions using Ni(cod)2. Furthermore, as the precatalysts are all stable to air, no glovebox or inert-atmosphere techniques are required to make use of these complexes for nickel-catalyzed reactions.
Co-reporter:Dr. Zhi He ;Dr. Timothy F. Jamison
Angewandte Chemie 2014 Volume 126( Issue 13) pp:3421-3425
Publication Date(Web):
DOI:10.1002/ange.201310572
Abstract
Phenols are important compounds in chemical industry. An economical and green approach to phenol preparation by the direct oxidation of aryl Grignard reagents using compressed air in continuous gas-liquid segmented flow systems is described. The process tolerates a broad range of functional groups, including oxidation-sensitive functionalities such as alkenes, amines, and thioethers. By integrating a benzyne-mediated in-line generation of arylmagnesium intermediates with the aerobic oxidation, a facile three-step, one-flow process, capable of preparing 2-functionalized phenols in a modular fashion, is established.
Co-reporter:Dr. Zhi He;Minwoo Bae;Dr. Jie Wu ;Dr. Timothy F. Jamison
Angewandte Chemie 2014 Volume 126( Issue 52) pp:14679-14683
Publication Date(Web):
DOI:10.1002/ange.201408522
Abstract
A mild and facile method for preparing highly functionalized pyrrolo[1,2-a]quinoxalines and other nitrogen-rich heterocycles, each containing a quinoxaline core or an analogue thereof, has been developed. The novel method features a visible-light-induced decarboxylative radical coupling of ortho-substituted arylisocyanides and radicals generated from phenyliodine(III) dicarboxylate reagents and exhibits excellent functional group compatibility. A wide range of quinoxaline heterocycles have been prepared. Finally, a telescoped preparation of these polycyclic compounds by integration of the in-line isocyanide formation and photochemical cyclization has been established in a three-step continuous-flow system.
Co-reporter:Sarah Z. Tasker;Dr. Alicia C. Gutierrez ; Timothy F. Jamison
Angewandte Chemie 2014 Volume 126( Issue 7) pp:1889-1892
Publication Date(Web):
DOI:10.1002/ange.201308391
Abstract
Achieving high selectivity in the Heck reaction of electronically unbiased alkenes has been a longstanding challenge. Using a nickel-catalyzed cationic Heck reaction, we were able to achieve excellent selectivity for branched products (≥19:1 in all cases) over a wide range of aryl electrophiles and aliphatic olefins. A bidentate ligand with a suitable bite angle and steric profile was key to obtaining high branched/linear selectivity, whereas the appropriate base suppressed alkene isomerization of the product. Although aryl triflates are traditionally used to access the cationic Heck pathway, we have shown that, by using triethylsilyl trifluoromethanesulfonate, we can effect a counterion exchange of the catalytic nickel complex, such that cheaper and more stable aryl chlorides, mesylates, tosylates, and sulfamates can be used to yield the same branched products with high selectivity.
Co-reporter:Jennifer A. Kozak ; Jie Wu ; Xiao Su ; Fritz Simeon ; T. Alan Hatton
Journal of the American Chemical Society 2013 Volume 135(Issue 49) pp:18497-18501
Publication Date(Web):November 20, 2013
DOI:10.1021/ja4079094
A continuous method for the formation of cyclic carbonates from epoxides and carbon dioxide (CO2) is described. The catalysts used are inexpensive and effective in converting the reagents to the products in a residence time (tR) of 30 min. The cyclic carbonate products are obtained in good to excellent yield (51–92%). On the basis of a series of kinetics experiments, we propose a reaction mechanism involving epoxide activation by electrophilic bromine and CO2 activation by an amide.
Co-reporter:Eric A. Standley
Journal of the American Chemical Society 2013 Volume 135(Issue 4) pp:1585-1592
Publication Date(Web):January 14, 2013
DOI:10.1021/ja3116718
The synthesis and characterization of the air-stable nickel(II) complex trans-(PCy2Ph)2Ni(o-tolyl)Cl is described in conjunction with an investigation of its use for the Mizoroki–Heck-type, room temperature, internally selective coupling of substituted benzyl chlorides with terminal alkenes. This reaction, which employs a terminal alkene as an alkenylmetal equivalent, provides rapid, convergent access to substituted allylbenzene derivatives in high yield and with regioselectivity greater than 95:5 in nearly all cases. The reaction is operationally simple, can be carried out on the benchtop with no purification or degassing of solvents or reagents, and requires no exclusion of air or water during setup. Synthesis of the precatalyst is accomplished through a straightforward procedure that employs inexpensive, commercially available reagents, requires no purification steps, and proceeds in high yield.
Co-reporter:David R. Snead and Timothy F. Jamison
Chemical Science 2013 vol. 4(Issue 7) pp:2822-2827
Publication Date(Web):21 May 2013
DOI:10.1039/C3SC50859E
A continuous end-to-end synthesis and purification of diphenhydramine hydrochloride featuring atom economy and waste minimization is described. Combining a 1:1 molar ratio of the two starting material streams (chlorodiphenylmethane and N,N-dimethylaminoethanol) in the absence of additional solvent at high temperature gives the target compound directly as a molten salt (ionic liquid above 168 °C) in high yield. This represents the first example of continuous active pharmaceutical ingredient (API) production in this manner. Six of the twelve principles of green chemistry as defined by the American Chemical Society are achieved, most prominently waste minimization and atom economy.
Co-reporter:Andrew S. Kleinke and Timothy F. Jamison
Organic Letters 2013 Volume 15(Issue 3) pp:710-713
Publication Date(Web):January 22, 2013
DOI:10.1021/ol400051n
The first continuous hydrogenation that requires neither H2 nor metal catalysis generates diimide by a novel reagent combination. The simple flow reactor employed minimizes residence time by enabling safe operation at elevated temperature.
Co-reporter:Dr. James J. Mousseau;Dr. Christopher J. Morten ;Dr. Timothy F. Jamison
Chemistry - A European Journal 2013 Volume 19( Issue 30) pp:10004-10016
Publication Date(Web):
DOI:10.1002/chem.201300845
Abstract
Ladder polyether natural products are a class of natural products denoted by their high functional-group density and large number of well-defined stereocenters. They comprise the toxic component of harmful algal blooms (HABs), having significant negative economic and environmental ramifications. However, their mode of action, namely blocking various cellular ion channels, also denotes their promise as potential anticancer agents. Understanding their potential mode of biosynthesis will not only help with developing ways to limit the damage of HABs, but would also facilitate the synthesis of a range of analogs with interesting biological activity. 1,3-Dioxan-5-ol substrates display remarkable ‘enhanced template effects’ in water-promoted epoxide cyclization processes en route to the synthesis of these ladder polyether natural products. In many cases, they provide near complete endo-to-exo selectivity in the cyclization of epoxy alcohols, thereby strongly favoring the formation of tetrahydropyran (THP) over tetrahydrofuran (THF) rings. The effects of various Brønsted and Lewis acidic and basic conditions are explored to demonstrate the superior selectivity of the template over the previously reported THP-based epoxy alcohols. In addition, the consideration of other synthetic routes are also considered with the goal of gaining rapid access to a plethora of potential starting materials applicable towards the synthesis of ladder polyethers. Finally, cascade sequences with polyepoxides are investigated, further demonstrating the versatility of this new reaction template.
Co-reporter:Brian S. Underwood, Jessica Tanuwidjaja, Sze-Sze Ng, Timothy F. Jamison
Tetrahedron 2013 69(25) pp: 5205-5220
Publication Date(Web):
DOI:10.1016/j.tet.2013.04.041
Co-reporter:Jeffery A. Byers
PNAS 2013 Volume 110 (Issue 42 ) pp:16724-16729
Publication Date(Web):2013-10-15
DOI:10.1073/pnas.1311133110
Despite the myriad of selective enzymatic reactions that occur in water, chemists have rarely capitalized on the unique properties
of this medium to govern selectivity in reactions. Here we report detailed mechanistic investigations of a water-promoted
reaction that displays high selectivity for what is generally a disfavored product. A combination of structural and kinetic
data indicates not only that synergy between substrate and water suppresses undesired pathways but also that water promotes
the desired pathway by stabilizing charge in the transition state, facilitating proton transfer, doubly activating the substrate
for reaction, and perhaps most remarkably, reorganizing the substrate into a reactive conformation that leads to the observed
product. This approach serves as an outline for a general strategy of exploiting solvent-solute interactions to achieve unusual
reactivity in chemical reactions. These findings may also have implications in the biosynthesis of the ladder polyether natural
products, such as the brevetoxins and ciguatoxins.
Co-reporter:Dr. Yuan Zhang;Dr. Melissa L. Blackman;Dr. Andrew B. Leduc ;Dr. Timothy F. Jamison
Angewandte Chemie 2013 Volume 125( Issue 15) pp:4345-4349
Publication Date(Web):
DOI:10.1002/ange.201300504
Co-reporter:Dr. Yuan Zhang;Dr. Melissa L. Blackman;Dr. Andrew B. Leduc ;Dr. Timothy F. Jamison
Angewandte Chemie International Edition 2013 Volume 52( Issue 15) pp:4251-4255
Publication Date(Web):
DOI:10.1002/anie.201300504
Co-reporter:Bo Shen and Timothy F. Jamison
Organic Letters 2012 Volume 14(Issue 13) pp:3348-3351
Publication Date(Web):June 13, 2012
DOI:10.1021/ol301324g
A general, green, and efficient Brønsted acid-catalyzed glycosylation serves as a key step in the one-flow, multistep syntheses of several important 5′-deoxyribonucleoside pharmaceuticals.
Co-reporter:Damien Webb and Timothy F. Jamison
Organic Letters 2012 Volume 14(Issue 2) pp:568-571
Publication Date(Web):December 29, 2011
DOI:10.1021/ol2031872
A continuous flow system for the multiparameter (flow rate, temperature, residence time, stoichiometry) optimization of the DIBALH reduction of esters to aldehydes is described. Incorporating an in-line quench (MeOH), these transformations are generally complete in fewer than 60 s. Mixing of the DIBALH and ester solutions was observed to be an exceptionally critical parameter for optimum results. This system thus provides general guidelines based on the structure of the ester for selective reduction of an ester without overreduction.
Co-reporter:Bo Shen, Matthew W. Bedore, Adam Sniady and Timothy F. Jamison
Chemical Communications 2012 vol. 48(Issue 60) pp:7444-7446
Publication Date(Web):22 Jun 2012
DOI:10.1039/C2CC33356B
A unique photochemical flow reactor featuring quartz tubing, an aluminum mirror and temperature control has been developed for the photo-induced electron-transfer deoxygenation reaction to produce 2′-deoxy and 2′,3′-dideoxynucleosides. The continuous flow format significantly increased the efficiency and selectivity of the reaction.
Co-reporter:Damien Webb and Timothy F. Jamison
Organic Letters 2012 Volume 14(Issue 10) pp:2465-2467
Publication Date(Web):April 30, 2012
DOI:10.1021/ol300722e
A continuous protocol for the two-carbon homologation of esters to α,β-unsaturated esters is described. This multireactor homologation telescopes an ester reduction, phosphonate deprotonation, and Horner–Wadsworth–Emmons olefination, thus converting a three-operation procedure into a single, uninterrupted system that eliminates the need for isolation or purification of the aldehyde intermediates. The homologated products are obtained in high yield and selectivity.
Co-reporter:Andrew B. Leduc and Timothy F. Jamison
Organic Process Research & Development 2012 Volume 16(Issue 5) pp:1082-1089
Publication Date(Web):June 15, 2011
DOI:10.1021/op200118h
We report a method for the oxidation of a range of alcohols and aldehydes utilizing a simple flow system of alcohols in EtOAc with a stream of 12.5% NaOCl and catalytic Bu4NBr. Secondary alcohols are oxidized to ketones, aldehydes are oxidized directly to methyl esters in the presence of methanol, and benzylic alcohols are oxidized to either benzaldehydes or methyl esters, depending on the conditions used. The reaction conditions are mild and generally provide complete conversion in 5–30 min.
Co-reporter:Andrew S. Kleinke, Damien Webb, Timothy F. Jamison
Tetrahedron 2012 68(35) pp: 6999-7018
Publication Date(Web):
DOI:10.1016/j.tet.2012.05.081
Co-reporter:Ryosuke Matsubara ; Alicia C. Gutierrez
Journal of the American Chemical Society 2011 Volume 133(Issue 47) pp:19020-19023
Publication Date(Web):November 8, 2011
DOI:10.1021/ja209235d
Nickel-catalyzed intermolecular benzylation and heterobenzylation of unactivated alkenes to provide functionalized allylbenzene derivatives are described. A wide range of both the benzyl chloride and alkene coupling partners are tolerated. In contrast to analogous palladium-catalyzed variants of this process, all reactions described herein employ electronically unbiased aliphatic olefins (including ethylene), proceed at room temperature, and provide 1,1-disubstituted olefins over the more commonly observed 1,2-disubstituted olefins with very high selectivity.
Co-reporter:Christopher J. Morten ; Jeffery A. Byers
Journal of the American Chemical Society 2011 Volume 133(Issue 6) pp:1902-1908
Publication Date(Web):January 14, 2011
DOI:10.1021/ja1088748
A detailed kinetic study of the endo-selective epoxide-opening cascade reaction of a diepoxy alcohol in neutral water was undertaken using 1H NMR spectroscopy. The observation of monoepoxide intermediates resulting from initial endo and exo cyclization indicated that the cascade proceeds via a stepwise mechanism rather than through a concerted one. Independent synthesis and cyclization of these monoepoxide intermediates demonstrated that they are chemically and kinetically competent intermediates in the cascade. Analysis of each step of the reaction revealed that both the rate and regioselectivity of cyclization improve as the cascade reaction proceeds. In the second step, cyclization of an epoxy alcohol substrate templated by a fused diad of two tetrahydropyran rings proceeds with exceptionally high regioselectivity (endo:exo = 19:1), the highest we have measured in the opening of a simple trans-disubstituted epoxide. The origins of these observations are discussed.
Co-reporter:Nikolay Zaborenko, Matthew W. Bedore, Timothy F. Jamison, and Klavs F. Jensen
Organic Process Research & Development 2011 Volume 15(Issue 1) pp:131-139
Publication Date(Web):December 14, 2010
DOI:10.1021/op100252m
A continuous-flow microreactor is applied for a kinetic study of a model β-amino alcohol formation by epoxide aminolysis. A large number of experiments are performed in a short time with minimal reagent consumption. The kinetics of formation of secondary aminolysis between starting epoxide and product are decoupled from the primary synthesis, constructing a complete model for desired product formation. The activation energy for the formation of desired product is observed to be higher than those for regioisomer formation and for secondary aminolysis, indicating that increasing temperature improves selectivity in addition to accelerating the reaction. A set of optimized conditions is then selected for best reaction performance, and the process is scaled up to a 100-fold larger reactor volume with model predictions in good agreement with measured process performance.
Co-reporter:Dr. Ryosuke Matsubara ;Dr. Timothy F. Jamison
Chemistry – An Asian Journal 2011 Volume 6( Issue 7) pp:1860-1875
Publication Date(Web):
DOI:10.1002/asia.201000875
Abstract
This report describes a nickel-catalyzed allylic substitution process of simple alkenes whereby an important structural motif, a 1,4-diene, was prepared. The key to success is the use of an appropriate nickel–phosphine complex and a stoichiometric amount of silyl triflate. Reactions of 1-alkyl-substituted alkenes consistently provided 1,1-disubstituted alkenes with high selectivity. Insight into the reaction mechanism as well as miscellaneous application of the developed catalytic process is also documented.
Co-reporter:Dr. Prakash B. Palde ;Dr. Timothy F. Jamison
Angewandte Chemie International Edition 2011 Volume 50( Issue 15) pp:3525-3528
Publication Date(Web):
DOI:10.1002/anie.201006272
Co-reporter:Dr. Adam Sniady;Dr. Matthew W. Bedore ;Dr. Timothy F. Jamison
Angewandte Chemie International Edition 2011 Volume 50( Issue 9) pp:2155-2158
Publication Date(Web):
DOI:10.1002/anie.201006440
Co-reporter:Dr. Adam Sniady;Dr. Matthew W. Bedore ;Dr. Timothy F. Jamison
Angewandte Chemie 2011 Volume 123( Issue 9) pp:2203-2206
Publication Date(Web):
DOI:10.1002/ange.201006440
Co-reporter:Dr. Prakash B. Palde ;Dr. Timothy F. Jamison
Angewandte Chemie 2011 Volume 123( Issue 15) pp:3587-3590
Publication Date(Web):
DOI:10.1002/ange.201006272
Co-reporter:Peng Liu ; Patrick McCarren ; Paul Ha-Yeon Cheong ; Timothy F. Jamison ;K. N. Houk
Journal of the American Chemical Society 2010 Volume 132(Issue 6) pp:2050-2057
Publication Date(Web):January 22, 2010
DOI:10.1021/ja909562y
The origins of reactivity and regioselectivity in nickel-catalyzed reductive coupling reactions of alkynes and aldehydes were investigated with density functional calculations. The regioselectivities of reactions of simple alkynes are controlled by steric effects, while conjugated enynes and diynes are predicted to have increased reactivity and very high regioselectivities, placing alkenyl or alkynyl groups distal to the forming C−C bond. The reactions of enynes and diynes involve 1,4-attack of the Ni−carbonyl complex on the conjugated enyne or diyne. The consequences of these conclusions on reaction design are discussed.
Co-reporter:Ryosuke Matsubara
Journal of the American Chemical Society 2010 Volume 132(Issue 20) pp:6880-6881
Publication Date(Web):April 30, 2010
DOI:10.1021/ja101186p
Nickel-catalyzed intermolecular allylic substitution of simple alkenes (ethylene and alpha olefins) is described. This method is the first catalytic intermolecular process for direct allylation of nonconjugated, nonstrained simple alkenes. Catalyst loadings as low as 2.5 mol % Ni afford the desired product in high yield in both gram-scale and smaller scale coupling reactions.
Co-reporter:Damien Webb and Timothy F. Jamison
Chemical Science 2010 vol. 1(Issue 6) pp:675-680
Publication Date(Web):23 Sep 2010
DOI:10.1039/C0SC00381F
Using continuous flow techniques for multi-step synthesis enables multiple reaction steps to be combined into a single continuous operation. In this mini-review we discuss the current state of the art in this field and highlight recent progress and current challenges facing this emerging area.
Co-reporter:Azusa Kondoh and Timothy F. Jamison
Chemical Communications 2010 vol. 46(Issue 6) pp:907-909
Publication Date(Web):05 Jan 2010
DOI:10.1039/B921387B
A rhodium-catalyzed dehydrogenative borylation of cyclic alkenes is described. This reaction provides direct access to cyclic 1-alkenylboronic acid pinacol esters, useful intermediates in organic synthesis. Suzuki–Miyaura cross-coupling applications are also presented.
Co-reporter:Matthew W. Bedore, Nikolay Zaborenko, Klavs F. Jensen and Timothy F. Jamison
Organic Process Research & Development 2010 Volume 14(Issue 2) pp:432-440
Publication Date(Web):January 26, 2010
DOI:10.1021/op9003136
The use of a continuous flow microreactor for β-amino alcohol formation by epoxide aminolysis is evaluated. Comparison to microwave batch reactions reveals that conditions obtainable in the microreactor can match or improve yields in many cases. By increasing the pressure of the system, maximum temperatures can also exceed those accessible using a microwave unit. The use of a microreactor for epoxide aminolysis reactions in the synthesis of two pharmaceutical relevant compounds is described.
Co-reporter:Megan A. Foley and Timothy F. Jamison
Organic Process Research & Development 2010 Volume 14(Issue 5) pp:1177-1181
Publication Date(Web):August 20, 2010
DOI:10.1021/op1001269
A rapid carboxylic acid-promoted lactone aminolysis is reported. A number of carboxylic acids were found to promote this amide bond-forming transformation, with aliphatic acids being the most efficient. This reaction is an equilibrium process (Keq ≈ 1.8), and mechanistic investigations are consistent with mediation of a kinetically important proton-transfer step by the carboxylate, i.e., the conjugate base of the acid employed.
Co-reporter:Christopher J. Morten, Jeffery A. Byers, Aaron R. Van Dyke, Ivan Vilotijevic and Timothy F. Jamison
Chemical Society Reviews 2009 vol. 38(Issue 11) pp:3175-3192
Publication Date(Web):16 Sep 2009
DOI:10.1039/B816697H
This tutorial review traces the development of endo-regioselective epoxide-opening reactions in water. Templated, water-promoted epoxide-opening cyclization reactions can offer rapid access to subunits of the ladder polyethers, a fascinating and complex family of natural products. This review may be of interest to those curious about the ladder polyethers and their hypothesized biogenesis, about organic reactions in water, and about the development and application of cascade reactions in organic synthesis.
Co-reporter:Jessica Tanuwidjaja ; Sze-Sze Ng
Journal of the American Chemical Society 2009 Volume 131(Issue 34) pp:12084-12085
Publication Date(Web):August 10, 2009
DOI:10.1021/ja9052366
In the first total synthesis of ent-dioxepandehydrothyrsiferol, the signature trans-anti-trans 7,7,6-fused tricyclic polyether framework was constructed in a single bromonium-initiated epoxide-opening cascade that incorporates both endo- and exo-selective epoxide openings, each directed by the substitution pattern of the epoxide (methyl groups). This study thus demonstrates the feasibility of a possible biogenesis.
Co-reporter:Jeffery A. Byers
Journal of the American Chemical Society 2009 Volume 131(Issue 18) pp:6383-6385
Publication Date(Web):April 22, 2009
DOI:10.1021/ja9004909
A regioselective epoxy alcohol cyclization promoted by the combination of neutral water and a tetrahydropyran template was investigated through a series of mechanistic experiments carried out on an epoxy alcohol containing a tetrahydropyran ring (1a) and its carbocyclic congener (1b). In contrast to 1a, cyclizations of 1b were unselective and displayed significantly faster reaction rates suggesting that the tetrahydropyran oxygen in 1a is requisite for regioselective cyclization. Reactions for both substrates were shown to occur in solution and under kinetic control without significant influence from hydrophobic effects. Kinetic measurements carried out in water/dimethyl sulfoxide mixtures suggest that 1b reacts exclusively through an unselective pathway requiring one water molecule more than what is required to solvate the epoxy alcohol. Similar experiments for 1a suggest a competition between an unselective and a selective pathway requiring one and two water molecules in excess of those required to solvate 1a, respectively. The selective pathway observed for 1a but not in 1b is rationalized by electronic and conformational differences between the two compounds.
Co-reporter:Christopher J. Morten
Journal of the American Chemical Society 2009 Volume 131(Issue 19) pp:6678-6679
Publication Date(Web):April 29, 2009
DOI:10.1021/ja9025243
Water is an effective promoter of the endo-selective opening of trisubstituted epoxides, enabling related cascades leading to a variety of substituted ladder polyether structures. When used in conjunction with a tetrahydropyran-templated nucleophile, water can overcome the powerful electronic directing effect of a methyl substituent at either site of the epoxide, making water a uniquely versatile medium and promoter of epoxide opening.
Co-reporter:P. R. McCarren ; Peng Liu ; Paul Ha-Yeon Cheong ; Timothy F. Jamison ;K. N. Houk
Journal of the American Chemical Society 2009 Volume 131(Issue 19) pp:6654-6655
Publication Date(Web):April 28, 2009
DOI:10.1021/ja900701g
The mechanism of nickel-catalyzed reductive alkyne−aldehyde coupling reactions has been investigated using density functional theory. The preferred mechanism involves oxidative cyclization to form the nickeladihydrofuran intermediate followed by transmetalation and reductive elimination. The rate- and selectivity-determining oxidative cyclization transition state is analyzed in detail. The d → π*⊥ back-donation stabilizes the transition state and leads to higher reactivity for alkynes than alkenes. Strong Lewis acids accelerate the couplings with both alkynes and alkenes by coordinating with the aldehyde oxygen in the transition state.
Co-reporter:Brian A. Sparling, Graham L. Simpson, Timothy F. Jamison
Tetrahedron 2009 65(16) pp: 3270-3280
Publication Date(Web):
DOI:10.1016/j.tet.2008.11.086
Co-reporter:Christopher J. Morten, Timothy F. Jamison
Tetrahedron 2009 65(33) pp: 6648-6655
Publication Date(Web):
DOI:10.1016/j.tet.2009.05.074
Co-reporter:AaronR. VanDyke ;TimothyF. Jamison
Angewandte Chemie 2009 Volume 121( Issue 24) pp:4494-4496
Publication Date(Web):
DOI:10.1002/ange.200900924
Co-reporter:JamesD. Trenkle Dr.;TimothyF. Jamison Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 29) pp:5366-5368
Publication Date(Web):
DOI:10.1002/anie.200902079
Co-reporter:AaronR. VanDyke ;TimothyF. Jamison
Angewandte Chemie International Edition 2009 Volume 48( Issue 24) pp:4430-4432
Publication Date(Web):
DOI:10.1002/anie.200900924
Co-reporter:Ivan Vilotijevic ;TimothyF. Jamison Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 29) pp:5250-5281
Publication Date(Web):
DOI:10.1002/anie.200900600
Co-reporter:JamesD. Trenkle Dr.;TimothyF. Jamison Dr.
Angewandte Chemie 2009 Volume 121( Issue 29) pp:5470-5472
Publication Date(Web):
DOI:10.1002/ange.200902079
Co-reporter:Chun-Yu Ho Dr.;Hirohisa Ohmiya Dr.;TimothyF. Jamison Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 10) pp:1893-1895
Publication Date(Web):
DOI:10.1002/anie.200705163
Co-reporter:Chun-Yu Ho Dr.;Hirohisa Ohmiya Dr.;TimothyF. Jamison Dr.
Angewandte Chemie 2008 Volume 120( Issue 10) pp:1919-1921
Publication Date(Web):
DOI:10.1002/ange.200705163
Co-reporter:Ryan M. Moslin, Karen Miller-Moslin and Timothy F. Jamison
Chemical Communications 2007 (Issue 43) pp:4441-4449
Publication Date(Web):03 Jul 2007
DOI:10.1039/B707737H
Nickel-catalysed reductive coupling reactions of alkynes have emerged as powerful synthetic tools for the selective preparation of functionalized alkenes. One of the greatest challenges associated with these transformations is control of regioselectivity. Recent work from our laboratory has provided an improved understanding of several of the factors governing regioselectivity in these reactions, and related studies have revealed that the reaction mechanism can differ substantially depending on the ligand employed. A discussion of stereoselective transformations and novel applications of nickel catalysis in coupling reactions of alkynes is also included.
Co-reporter:Timothy F. Jamison;Ivan Vilotijevic
Science 2007 Volume 317(Issue 5842) pp:1189-1192
Publication Date(Web):31 Aug 2007
DOI:10.1126/science.1146421
Abstract
Selectivity rules in organic chemistry have been inferred largely from nonaqueous environments. In contrast, enzymes operate in water, and the chemical effect of the medium change remains only partially understood. Structural characterization of the “ladder” polyether marine natural products raised a puzzle that persisted for 20 years: Although the stereochemistry of adjacent tetrahydropyran (THP) cycles would seem to arise from a biosynthetic cascade of epoxide-opening reactions, experience in organic solvents argued consistently that such a pathway would be kinetically disfavored. We report that neutral water acts as an optimal promoter for the requisite ring-opening selectivity, once a single templating THP is appended to a chain of epoxides. This strategy offers a high-yielding route to the naturally occurring ladder core and highlights the likely importance of aqueous-medium effects in underpinning certain noteworthy enzymatic selectivities.
Co-reporter:Chun-Yu Ho Dr.;Timothy F. Jamison
Angewandte Chemie International Edition 2007 Volume 46(Issue 5) pp:
Publication Date(Web):8 DEC 2006
DOI:10.1002/anie.200603907
Give and take: Both a strong electron donor (1) and a strong electron acceptor (P(OPh)3) are necessary for a highly selective, nickel-catalyzed coupling reaction between alkenes, aldehydes, and silyl triflates (see scheme; cod=cycloocta-1,5-diene, Tf=trifluoromethanesulfonyl). This synergistic effect may also be useful in other transformations catalyzed by NHC–metal complexes (NHC=N-heterocyclic carbene).
Co-reporter:Chun-Yu Ho Dr.;Timothy F. Jamison
Angewandte Chemie International Edition 2007 Volume 46(Issue 8) pp:
Publication Date(Web):5 FEB 2007
DOI:10.1002/anie.200790025
Co-reporter:Chun-Yu Ho Dr.;Timothy F. Jamison
Angewandte Chemie 2007 Volume 119(Issue 8) pp:
Publication Date(Web):5 FEB 2007
DOI:10.1002/ange.200790025
Co-reporter:Chun-Yu Ho Dr.;Timothy F. Jamison
Angewandte Chemie 2007 Volume 119(Issue 5) pp:
Publication Date(Web):8 DEC 2006
DOI:10.1002/ange.200603907
Ein Geben und Nehmen: Der starke Elektronendonor 1 und der starke Elektronenacceptor P(OPh)3 sind für die hochselektive nickelkatalysierte Kupplung von Alkenen, Aldehyden und Silyltriflaten erforderlich (siehe Schema; cod=Cycloocta-1,5-dien, Tf=Trifluormethansulfonyl). Dieser Synergieeffekt könnte auch für andere durch NHC-Metall-Komplexe katalysierte Umsetzungen nützlich sein (NHC=N-heterocyclisches Carben).
Co-reporter:Karen M. Miller;Elizabeth A. Colby;Katrina S. Woodin;Timothy F. Jamison
Advanced Synthesis & Catalysis 2005 Volume 347(Issue 11-13) pp:
Publication Date(Web):19 OCT 2005
DOI:10.1002/adsc.200505175
Nickel-catalyzed reductive coupling reactions of 1,3-enynes and aromatic aldehydes efficiently afford conjugated dienols in excellent regioselectivity and modest enantioselectivity when conducted in the presence of catalytic amounts of a monodentate, P-chiral ferrocenyl phosphine ligand. 1-(Trimethylsilyl)-substituted enynes are shown to be effective coupling partners in these reactions, and the dienol products thus formed readily undergo protiodesilylation under mild conditions.
Co-reporter:Elizabeth A. Colby and Timothy F. Jamison
Organic & Biomolecular Chemistry 2005 vol. 3(Issue 15) pp:2675-2684
Publication Date(Web):01 Jul 2005
DOI:10.1039/B507315B
In this article we compare and contrast the strategies and tactics used in the syntheses of the amphidinolide T family of natural products that have been reported by Fürstner, Ghosh and ourselves. Similar approaches to the trisubstituted THF ring present in the targets are utilized in all of the syntheses, but each strategy showcases a different means of macrocyclization.
Co-reporter:Carmela Molinaro Dr. Dr.
Angewandte Chemie International Edition 2005 Volume 44(Issue 1) pp:
Publication Date(Web):15 DEC 2004
DOI:10.1002/anie.200461705
Several lines of evidence suggest that reduction of the carbonyl group occurs after epoxide-ring opening in the first examples of catalytic, reductive coupling of epoxides and aldehydes (see scheme). The hydroxyether products are obtained with >95:5 regioselectivity in all cases, independent of the steric and electronic nature of R1 and R2.
Co-reporter:Carmela Molinaro Dr. Dr.
Angewandte Chemie 2005 Volume 117(Issue 1) pp:
Publication Date(Web):15 DEC 2004
DOI:10.1002/ange.200461705
Mehrere Belege sprechen dafür, dass in den ersten katalytischen reduktiven Kupplungen von Epoxiden mit Aldehyden (siehe Schema) die Carbonylgruppe nach der Epoxidringöffnung reduziert wird. Die Hydroxyetherprodukte bildeten sich in allen Fällen mit >95:5 Regioselektivität, unabhängig vom sterischen und elektronischen Charakter von R1 und R2.
Co-reporter:Sejal J. Patel Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 30) pp:
Publication Date(Web):20 JUL 2004
DOI:10.1002/anie.200460044
The aryl substituent on the catalyst is central to the success of the title reaction (see scheme) which affords allylic amines in up to 89 % ee and 91 % yield with a catalyst derived from [Ni(cod)2] and a P-chiral ferrocenyl phosphane (e.g. 1). The coupling products are easily deprotected to enantiomerically enriched, tetrasubstituted primary allylic amines, which can be recrystallized to optical purity.
Co-reporter:Sejal J. Patel Dr.
Angewandte Chemie 2004 Volume 116(Issue 30) pp:
Publication Date(Web):20 JUL 2004
DOI:10.1002/ange.200460044
Der Arylsubstituent des Katalysators ist für den Erfolg der Titelreaktion entscheidend (siehe Schema), die mit einem von [Ni(cod)2] und einem P-chiralen Ferrocenylphosphan (z. B. 1) abgeleiteten Katalysator allylische Amine in bis zu 89 % ee und 91 % Ausbeute liefert. Die Kupplungsprodukte lassen sich zu enantiomerenangereicherten, tetrasubstituierten primären allylischen Aminen entschützen, die durch Umkristallisieren optisch rein erhalten werden können.
Co-reporter:Sejal J. Patel Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 12) pp:
Publication Date(Web):26 MAR 2003
DOI:10.1002/anie.200390349
An unusual degree of functional group compatibility for imine addition reactions was observed in the assembly of allylic amines from alkynes, imines, and organoboron reagents (boronic acids or boranes) by using a catalyst derived from [Ni(cod)2] and (c-C5H9)3P—this catalytic three-component process (see scheme) is tolerant of ketones, esters, and hydroxylic solvents.
Co-reporter:Sejal J. Patel Dr.
Angewandte Chemie 2003 Volume 115(Issue 12) pp:
Publication Date(Web):26 MAR 2003
DOI:10.1002/ange.200390321
Die Toleranz funktioneller Gruppen ist ungewöhnlich hoch für eine Addition an Imine bei der hier beschriebenen Synthese von Allylaminen aus Alkinen, Iminen und Organoborreagentien (Boronsäuren oder Boranen) mithilfe eines Katalysatorsystems aus [Ni(cod)2] und (c-C5H9)3P: Bei dieser Drei-Komponenten-Reaktion (siehe Schema) können Substrate eingesetzt werden, die Keton- oder Estercarbonylgruppen oder auch freie Hydroxygruppen enthalten.
Co-reporter:Matthew G. Beaver
Organic Letters () pp:
Publication Date(Web):June 30, 2011
DOI:10.1021/ol201702a
A Ni-catalyzed reductive coupling of alkynes and epoxides using Ni(II) salts and simple alcohol reducing agents is described. Whereas previously reported conditions relied on Ni(cod)2 and Et3B, this system has several advantages including the use of air-stable and inexpensive Ni(II) precatalysts (e.g., NiBr2·3H2O) as the source of Ni(0) and simple alcohols (e.g., 2-propanol) as the reducing agent. Deuterium-labeling experiments are consistent with oxidative addition of an epoxide C–O bond that occurs with inversion of configuration.
Co-reporter:David R. Snead and Timothy F. Jamison
Chemical Science (2010-Present) 2013 - vol. 4(Issue 7) pp:NaN2827-2827
Publication Date(Web):2013/05/21
DOI:10.1039/C3SC50859E
A continuous end-to-end synthesis and purification of diphenhydramine hydrochloride featuring atom economy and waste minimization is described. Combining a 1:1 molar ratio of the two starting material streams (chlorodiphenylmethane and N,N-dimethylaminoethanol) in the absence of additional solvent at high temperature gives the target compound directly as a molten salt (ionic liquid above 168 °C) in high yield. This represents the first example of continuous active pharmaceutical ingredient (API) production in this manner. Six of the twelve principles of green chemistry as defined by the American Chemical Society are achieved, most prominently waste minimization and atom economy.
Co-reporter:Damien Webb and Timothy F. Jamison
Chemical Science (2010-Present) 2010 - vol. 1(Issue 6) pp:NaN680-680
Publication Date(Web):2010/09/23
DOI:10.1039/C0SC00381F
Using continuous flow techniques for multi-step synthesis enables multiple reaction steps to be combined into a single continuous operation. In this mini-review we discuss the current state of the art in this field and highlight recent progress and current challenges facing this emerging area.
Co-reporter:Jie Wu, Jennifer A. Kozak, Fritz Simeon, T. Alan Hatton and Timothy F. Jamison
Chemical Science (2010-Present) 2014 - vol. 5(Issue 3) pp:NaN1231-1231
Publication Date(Web):2014/01/27
DOI:10.1039/C3SC53422G
A mechanism-guided design of a multi-step flow system enabled an efficient general process for the synthesis of cyclic carbonates from alkenes and CO2. The flow system proved to be an ideal platform for multicomponent reactions because it was straightforward to introduce reagents at specific stages without their interacting with each other or with reaction intermediates prone to destruction by them. This system exhibited superior reactivity, increased yield, and broader substrate scope relative to conventional batch conditions and suppressed the formation of undesired byproducts, such as, epoxides and 1,2-dibromoalkanes.
Co-reporter:Azusa Kondoh and Timothy F. Jamison
Chemical Communications 2010 - vol. 46(Issue 6) pp:NaN909-909
Publication Date(Web):2010/01/05
DOI:10.1039/B921387B
A rhodium-catalyzed dehydrogenative borylation of cyclic alkenes is described. This reaction provides direct access to cyclic 1-alkenylboronic acid pinacol esters, useful intermediates in organic synthesis. Suzuki–Miyaura cross-coupling applications are also presented.
Co-reporter:Xiaoqing Yang, Jie Wu, Xianwen Mao, Timothy F. Jamison and T. Alan Hatton
Chemical Communications 2014 - vol. 50(Issue 24) pp:NaN3248-3248
Publication Date(Web):2014/02/06
DOI:10.1039/C4CC00252K
An effective transformation of alkenes into cyclic carbonates has been achieved using NaHCO3 as the C1 source in acetone–water under microwave heating, with selectivities and yields significantly surpassing those obtained using conventional heating.
Co-reporter:Bo Shen, Matthew W. Bedore, Adam Sniady and Timothy F. Jamison
Chemical Communications 2012 - vol. 48(Issue 60) pp:NaN7446-7446
Publication Date(Web):2012/06/22
DOI:10.1039/C2CC33356B
A unique photochemical flow reactor featuring quartz tubing, an aluminum mirror and temperature control has been developed for the photo-induced electron-transfer deoxygenation reaction to produce 2′-deoxy and 2′,3′-dideoxynucleosides. The continuous flow format significantly increased the efficiency and selectivity of the reaction.
Co-reporter:Ryan M. Moslin, Karen Miller-Moslin and Timothy F. Jamison
Chemical Communications 2007(Issue 43) pp:NaN4449-4449
Publication Date(Web):2007/07/03
DOI:10.1039/B707737H
Nickel-catalysed reductive coupling reactions of alkynes have emerged as powerful synthetic tools for the selective preparation of functionalized alkenes. One of the greatest challenges associated with these transformations is control of regioselectivity. Recent work from our laboratory has provided an improved understanding of several of the factors governing regioselectivity in these reactions, and related studies have revealed that the reaction mechanism can differ substantially depending on the ligand employed. A discussion of stereoselective transformations and novel applications of nickel catalysis in coupling reactions of alkynes is also included.
Co-reporter:Christopher J. Morten, Jeffery A. Byers, Aaron R. Van Dyke, Ivan Vilotijevic and Timothy F. Jamison
Chemical Society Reviews 2009 - vol. 38(Issue 11) pp:NaN3192-3192
Publication Date(Web):2009/09/16
DOI:10.1039/B816697H
This tutorial review traces the development of endo-regioselective epoxide-opening reactions in water. Templated, water-promoted epoxide-opening cyclization reactions can offer rapid access to subunits of the ladder polyethers, a fascinating and complex family of natural products. This review may be of interest to those curious about the ladder polyethers and their hypothesized biogenesis, about organic reactions in water, and about the development and application of cascade reactions in organic synthesis.
.jpg)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]-2-(phenylmethoxy)propoxy]methyl]-α-[[[1-[(2R)-2-(phenylmethoxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721800/1809614-24-1.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]-2-(phenylmethoxy)propoxy]methyl]-α-[[[1-[(2R)-2-(phenylmethoxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721800/1809614-24-1_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-3-[4-[[(2R)-2-(acetyloxy)-3-azidopropoxy]methyl]-1H-1,2,3-triazol-1-yl]-2-(phenylmethoxy)propoxy]methyl]-α-[[[1-[(2R)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]-2-(phenylmethoxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721800/1809614-23-0.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-3-[4-[[(2R)-2-(acetyloxy)-3-azidopropoxy]methyl]-1H-1,2,3-triazol-1-yl]-2-(phenylmethoxy)propoxy]methyl]-α-[[[1-[(2R)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]-2-(phenylmethoxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721800/1809614-23-0_b.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[1-[(2R)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]-2-(phenylmethoxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-4-[[(2R)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]-2-(phenylmethoxy)propoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721800/1809614-22-9.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[1-[(2R)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]-2-(phenylmethoxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-4-[[(2R)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]-2-(phenylmethoxy)propoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721800/1809614-22-9_b.png)
![1H-1,2,3-Triazole, 4-[[(2R)-2-oxiranylmethoxy]methyl]-1-[(2R)-2-(phenylmethoxy)-3-(2-propyn-1-yloxy)propyl]-](/data/chemimg/3721700/1809614-21-8.png)
![1H-1,2,3-Triazole, 4-[[(2R)-2-oxiranylmethoxy]methyl]-1-[(2R)-2-(phenylmethoxy)-3-(2-propyn-1-yloxy)propyl]-](/data/chemimg/3721700/1809614-21-8_b.png)
![2-Propanol, 1-azido-3-[[1-[(2R)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]-2-(phenylmethoxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]-, 2-acetate, (2R)-](/data/chemimg/3721700/1809614-20-7.png)
![2-Propanol, 1-azido-3-[[1-[(2R)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]-2-(phenylmethoxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]-, 2-acetate, (2R)-](/data/chemimg/3721700/1809614-20-7_b.png)
![1H-1,2,3-Triazole, 1-[(2R)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]-2-(phenylmethoxy)propyl]-4-[[(2R)-2-oxiranylmethoxy]methyl]-](/data/chemimg/3721700/1809614-19-4.png)
![1H-1,2,3-Triazole, 1-[(2R)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]-2-(phenylmethoxy)propyl]-4-[[(2R)-2-oxiranylmethoxy]methyl]-](/data/chemimg/3721700/1809614-19-4_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-[[1-[(2R)-2-(acetyloxy)-3-[[1-[(2R)-2-(acetyloxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-15-0.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-[[1-[(2R)-2-(acetyloxy)-3-[[1-[(2R)-2-(acetyloxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-15-0_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-12-7.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-12-7_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-(acetyloxy)-3-azidopropoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-11-6.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-(acetyloxy)-3-azidopropoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-11-6_b.png)


![1H-1,2,3-Triazole-1-ethanol, α-[[[1-[(2R)-2-(acetyloxy)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-10-5.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[1-[(2R)-2-(acetyloxy)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-4-[[(2R)-2-(acetyloxy)-3-[4-[[(2R)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-10-5_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-oxiranylmethoxy]methyl]-α-[(2-propyn-1-yloxy)methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-09-2.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-oxiranylmethoxy]methyl]-α-[(2-propyn-1-yloxy)methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-09-2_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-azidopropoxy]methyl]-α-[[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-08-1.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-azidopropoxy]methyl]-α-[[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-08-1_b.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]methyl]-4-[[(2R)-2-oxiranylmethoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-07-0.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]methyl]-4-[[(2R)-2-oxiranylmethoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809614-07-0_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-(acetyloxy)-3-[4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-[[1-[(2S)-2-(acetyloxy)-3-[[1-[(2R)-2-(acetyloxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αS)-](/data/chemimg/3721700/1809614-01-4.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-(acetyloxy)-3-[4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-[[1-[(2S)-2-(acetyloxy)-3-[[1-[(2R)-2-(acetyloxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αS)-](/data/chemimg/3721700/1809614-01-4_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αS)-](/data/chemimg/3721700/1809613-98-6.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-(2-propyn-1-yloxy)propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αS)-](/data/chemimg/3721700/1809613-98-6_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-(acetyloxy)-3-azidopropoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αS)-](/data/chemimg/3721700/1809613-97-5.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-(acetyloxy)-3-azidopropoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-α-[[[1-[(2R)-2-(acetyloxy)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-, 1-acetate, (αS)-](/data/chemimg/3721700/1809613-97-5_b.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[1-[(2R)-2-(acetyloxy)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-, 1-acetate, (αS)-](/data/chemimg/3721700/1809613-96-4.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[1-[(2R)-2-(acetyloxy)-3-[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]propyl]-1H-1,2,3-triazol-4-yl]methoxy]methyl]-4-[[(2R)-2-(acetyloxy)-3-[4-[[(2S)-2-oxiranylmethoxy]methyl]-1H-1,2,3-triazol-1-yl]propoxy]methyl]-, 1-acetate, (αS)-](/data/chemimg/3721700/1809613-96-4_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2S)-2-oxiranylmethoxy]methyl]-α-[(2-propyn-1-yloxy)methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809613-95-3.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2S)-2-oxiranylmethoxy]methyl]-α-[(2-propyn-1-yloxy)methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809613-95-3_b.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2S)-2-(acetyloxy)-3-azidopropoxy]methyl]-α-[[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809613-94-2.png)
![1H-1,2,3-Triazole-1-ethanol, 4-[[(2S)-2-(acetyloxy)-3-azidopropoxy]methyl]-α-[[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809613-94-2_b.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]methyl]-4-[[(2S)-2-oxiranylmethoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809613-93-1.png)
![1H-1,2,3-Triazole-1-ethanol, α-[[[3-[(1,1-dimethylethyl)dimethylsilyl]-2-propyn-1-yl]oxy]methyl]-4-[[(2S)-2-oxiranylmethoxy]methyl]-, 1-acetate, (αR)-](/data/chemimg/3721700/1809613-93-1_b.png)
![PIPERIDINE, 1-[[4-(1,1-DIMETHYLETHYL)PHENYL]METHYL]-](http://img.cochemist.com/ccimg/870800/870721-99-6.png)
![PIPERIDINE, 1-[[4-(1,1-DIMETHYLETHYL)PHENYL]METHYL]-](http://img.cochemist.com/ccimg/870800/870721-99-6_b.png)
![Azetidine, 2-hexyl-1-[(4-methylphenyl)sulfonyl]-, (2R)-](http://img.cochemist.com/ccimg/825700/825601-61-4.png)
![Azetidine, 2-hexyl-1-[(4-methylphenyl)sulfonyl]-, (2R)-](http://img.cochemist.com/ccimg/825700/825601-61-4_b.png)




![2-Aziridinemethanol, 1-[(4-methylphenyl)sulfonyl]-, (2R)-](http://img.cochemist.com/ccimg/188900/188816-36-6.png)
![2-Aziridinemethanol, 1-[(4-methylphenyl)sulfonyl]-, (2R)-](http://img.cochemist.com/ccimg/188900/188816-36-6_b.png)
![2-Hexen-1-ol, 6-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-3-methyl-, (2E)-](http://img.cochemist.com/ccimg/187400/187336-22-7.png)
![2-Hexen-1-ol, 6-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-3-methyl-, (2E)-](http://img.cochemist.com/ccimg/187400/187336-22-7_b.png)
![2-Hepten-1-ol, 7-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-, (E)-](http://img.cochemist.com/ccimg/177000/176907-55-4.png)
![2-Hepten-1-ol, 7-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-, (E)-](http://img.cochemist.com/ccimg/177000/176907-55-4_b.png)






![1(3aH)-Cyclopentacyclopentadecenone,4,7,8,9,12,13,16,16a-octahydro-2,7-dihydroxy-3-[(1S)-2-hydroxy-1-methylethyl]-6,10,14,16a-tetramethyl-,(3aR,5E,7S,10E,14E,16aS)-](http://img.cochemist.com/ccimg/146500/146436-22-8.png)
![1(3aH)-Cyclopentacyclopentadecenone,4,7,8,9,12,13,16,16a-octahydro-2,7-dihydroxy-3-[(1S)-2-hydroxy-1-methylethyl]-6,10,14,16a-tetramethyl-,(3aR,5E,7S,10E,14E,16aS)-](http://img.cochemist.com/ccimg/146500/146436-22-8_b.png)


![Aziridine, 2-hexyl-1-[(4-methylphenyl)sulfonyl]-, (R)-](http://img.cochemist.com/ccimg/140500/140421-16-5.png)
![Aziridine, 2-hexyl-1-[(4-methylphenyl)sulfonyl]-, (R)-](http://img.cochemist.com/ccimg/140500/140421-16-5_b.png)
![1-Butanol, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/130400/130372-07-5.png)
![1-Butanol, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/130400/130372-07-5_b.png)




![1-Pentanol, 5-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/121700/121671-76-9.png)
![1-Pentanol, 5-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/121700/121671-76-9_b.png)





![2-Hexen-1-ol, 6-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-, (2E)-](http://img.cochemist.com/ccimg/111700/111649-71-9.png)
![2-Hexen-1-ol, 6-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-, (2E)-](http://img.cochemist.com/ccimg/111700/111649-71-9_b.png)




![1,3-Dioxolane, 4-[(phenylmethoxy)methyl]-](http://img.cochemist.com/ccimg/98300/98238-47-2.png)
![1,3-Dioxolane, 4-[(phenylmethoxy)methyl]-](http://img.cochemist.com/ccimg/98300/98238-47-2_b.png)








![Piperidine, 1-[(2-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/59600/59507-48-1.png)
![Piperidine, 1-[(2-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/59600/59507-48-1_b.png)
![Piperidine, 1-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/59600/59507-42-5.png)
![Piperidine, 1-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/59600/59507-42-5_b.png)

![5-(2-Chlorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine](http://img.cochemist.com/ccimg/55200/55142-85-3.png)
![5-(2-Chlorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine](http://img.cochemist.com/ccimg/55200/55142-85-3_b.png)
![Piperidine, 1-[(2-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/51200/51180-63-3.png)
![Piperidine, 1-[(2-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/51200/51180-63-3_b.png)

![PIPERIDINE, 1-[(4-METHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/46500/46441-11-6.png)
![PIPERIDINE, 1-[(4-METHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/46500/46441-11-6_b.png)
![7-Benzyl-1,4-dioxa-7-aza-spiro[4.5]decane](http://img.cochemist.com/ccimg/38000/37943-54-7.png)
![7-Benzyl-1,4-dioxa-7-aza-spiro[4.5]decane](http://img.cochemist.com/ccimg/38000/37943-54-7_b.png)





































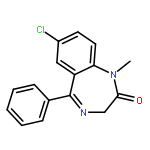
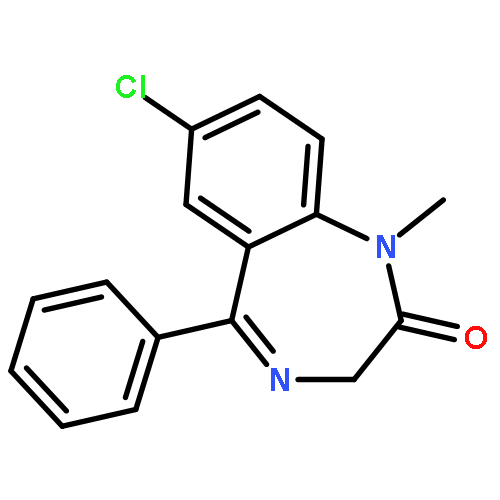






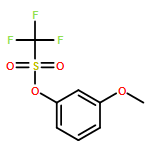
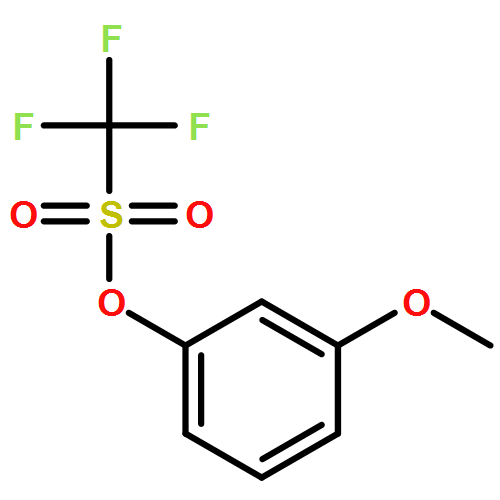
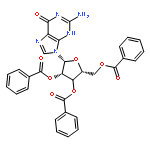
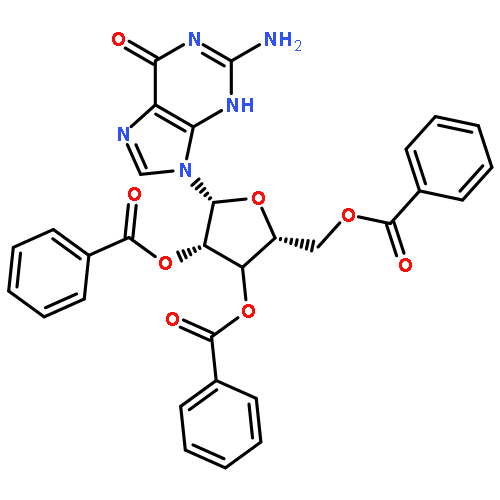
![Benzo[b]thiophene, 3-(2-propenyl)-](http://img.cochemist.com/ccimg/65900/65880-13-9.png)
![Benzo[b]thiophene, 3-(2-propenyl)-](http://img.cochemist.com/ccimg/65900/65880-13-9_b.png)





![2-Propanol, 1-[(1,1-dimethylethyl)amino]-3-phenoxy-](http://img.cochemist.com/ccimg/65000/64980-40-1.png)
![2-Propanol, 1-[(1,1-dimethylethyl)amino]-3-phenoxy-](http://img.cochemist.com/ccimg/65000/64980-40-1_b.png)
![1-(Benzo[d][1,3]dioxol-5-yl)pentan-1-one](http://img.cochemist.com/ccimg/63800/63740-98-7.png)
![1-(Benzo[d][1,3]dioxol-5-yl)pentan-1-one](http://img.cochemist.com/ccimg/63800/63740-98-7_b.png)
![SILANE, [(1E)-1,3-BUTADIENYLOXY]TRIMETHYL-](http://img.cochemist.com/ccimg/63400/63383-46-0.png)
![SILANE, [(1E)-1,3-BUTADIENYLOXY]TRIMETHYL-](http://img.cochemist.com/ccimg/63400/63383-46-0_b.png)




![Cyclopropyl-[4-(trifluoromethyl)phenyl]methanone](http://img.cochemist.com/ccimg/62600/62587-07-9.png)
![Cyclopropyl-[4-(trifluoromethyl)phenyl]methanone](http://img.cochemist.com/ccimg/62600/62587-07-9_b.png)


![1-Propanone,1-[4-(2-methylpropyl)phenyl]-](http://img.cochemist.com/ccimg/59800/59771-24-3.png)
![1-Propanone,1-[4-(2-methylpropyl)phenyl]-](http://img.cochemist.com/ccimg/59800/59771-24-3_b.png)


![Benzene, [(4-pentynyloxy)methyl]-](http://img.cochemist.com/ccimg/57700/57618-47-0.png)
![Benzene, [(4-pentynyloxy)methyl]-](http://img.cochemist.com/ccimg/57700/57618-47-0_b.png)
![BENZENEMETHANAMINE, N-[(4-METHYLPHENYL)METHYLENE]-, N-OXIDE](http://img.cochemist.com/ccimg/55700/55606-41-2.png)
![BENZENEMETHANAMINE, N-[(4-METHYLPHENYL)METHYLENE]-, N-OXIDE](http://img.cochemist.com/ccimg/55700/55606-41-2_b.png)







![BENZO[B]THIOPHENE, 2-(2-PROPENYL)-](http://img.cochemist.com/ccimg/54500/54470-30-3.png)
![BENZO[B]THIOPHENE, 2-(2-PROPENYL)-](http://img.cochemist.com/ccimg/54500/54470-30-3_b.png)












![Oxirane,2-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/51500/51410-45-8.png)
![Oxirane,2-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/51500/51410-45-8_b.png)
![Methanamine, N-[(3-methoxyphenyl)methylene]-, N-oxide](http://img.cochemist.com/ccimg/51400/51357-27-8.png)
![Methanamine, N-[(3-methoxyphenyl)methylene]-, N-oxide](http://img.cochemist.com/ccimg/51400/51357-27-8_b.png)
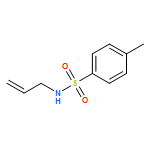
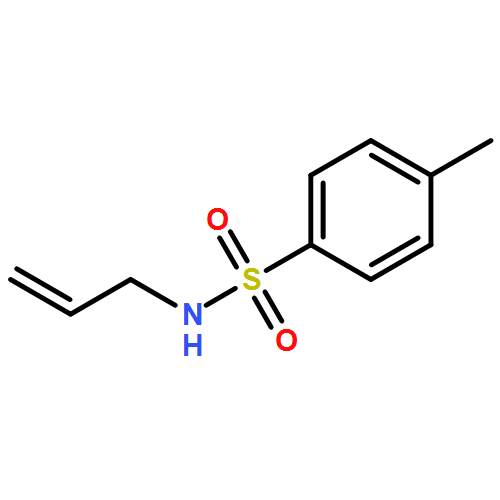









![N-[2-(1H-imidazol-1-yl)phenyl]-Formamide](http://img.cochemist.com/ccimg/38400/38346-48-4.png)
![N-[2-(1H-imidazol-1-yl)phenyl]-Formamide](http://img.cochemist.com/ccimg/38400/38346-48-4_b.png)













![Pyrimidine,5-ethyl-2,4-bis[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/31200/31167-05-2.png)
![Pyrimidine,5-ethyl-2,4-bis[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/31200/31167-05-2_b.png)

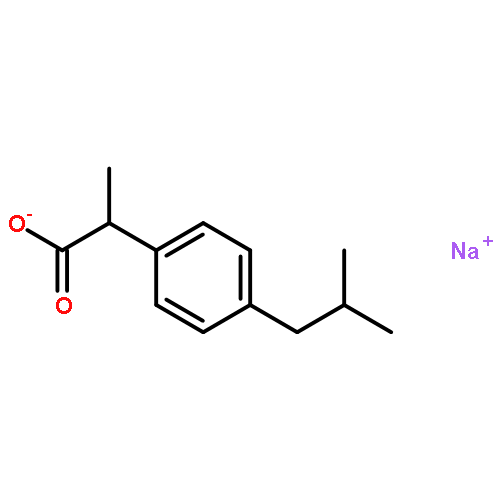
![Benzenemethanol, α-[(phenylamino)methyl]-](/data/chemimg/1185000/31121-09-2.png)
![Benzenemethanol, α-[(phenylamino)methyl]-](/data/chemimg/1185000/31121-09-2_b.png)




![1-Pentanone, 1-[4-(dimethylamino)phenyl]-](http://img.cochemist.com/ccimg/26700/26672-80-0.png)
![1-Pentanone, 1-[4-(dimethylamino)phenyl]-](http://img.cochemist.com/ccimg/26700/26672-80-0_b.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/25900/25856-77-3.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/25900/25856-77-3_b.png)



![L-Alanine, N-[(phenylmethoxy)carbonyl]-L-phenylalanyl-, methyl ester](http://img.cochemist.com/ccimg/25500/25422-44-0.png)
![L-Alanine, N-[(phenylmethoxy)carbonyl]-L-phenylalanyl-, methyl ester](http://img.cochemist.com/ccimg/25500/25422-44-0_b.png)






![Methanone, bis[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/21300/21221-91-0.png)
![Methanone, bis[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/21300/21221-91-0_b.png)














![Benzenemethanol, a-[[(1,1-dimethylethyl)amino]methyl]-](http://img.cochemist.com/ccimg/18400/18366-40-0.png)
![Benzenemethanol, a-[[(1,1-dimethylethyl)amino]methyl]-](http://img.cochemist.com/ccimg/18400/18366-40-0_b.png)


![Furan, tetrahydro-2-[(1E)-2-phenylethenyl]-](/data/chemimg/3767100/18138-78-8.png)
![Furan, tetrahydro-2-[(1E)-2-phenylethenyl]-](/data/chemimg/3767100/18138-78-8_b.png)


![L-Serine, N-[(4-methylphenyl)sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/17200/17136-46-8.png)
![L-Serine, N-[(4-methylphenyl)sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/17200/17136-46-8_b.png)





![Methanamine, N-[(4-bromophenyl)methylene]-, N-oxide](http://img.cochemist.com/ccimg/16100/16089-65-9.png)
![Methanamine, N-[(4-bromophenyl)methylene]-, N-oxide](http://img.cochemist.com/ccimg/16100/16089-65-9_b.png)
![Methanamine, N-[(4-methylphenyl)methylene]-, N-oxide](http://img.cochemist.com/ccimg/16100/16089-63-7.png)
![Methanamine, N-[(4-methylphenyl)methylene]-, N-oxide](http://img.cochemist.com/ccimg/16100/16089-63-7_b.png)






![Acetamide, N,N'-[1,1'-biphenyl]-2,2'-diylbis[2,2,2-trifluoro-](http://img.cochemist.com/ccimg/15000/14925-33-8.png)
![Acetamide, N,N'-[1,1'-biphenyl]-2,2'-diylbis[2,2,2-trifluoro-](http://img.cochemist.com/ccimg/15000/14925-33-8_b.png)

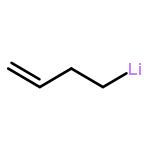

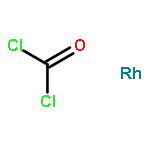
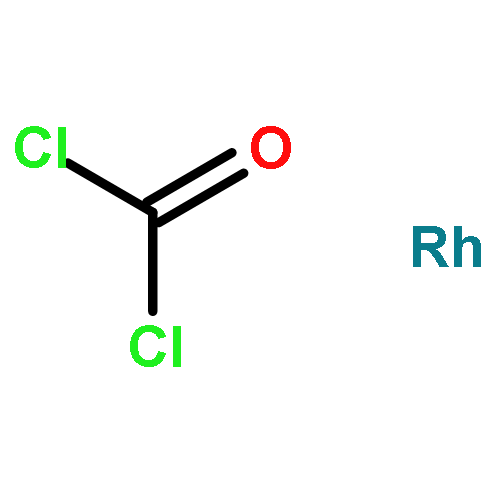







![Lithium,[4-(dimethylamino)phenyl]-](http://img.cochemist.com/ccimg/13200/13190-50-6.png)
![Lithium,[4-(dimethylamino)phenyl]-](http://img.cochemist.com/ccimg/13200/13190-50-6_b.png)






















![Benzene,[(pentyloxy)methyl]-](http://img.cochemist.com/ccimg/6400/6382-14-5.png)
![Benzene,[(pentyloxy)methyl]-](http://img.cochemist.com/ccimg/6400/6382-14-5_b.png)












![6-phenyl-7-oxabicyclo[4.1.0]heptane](http://img.cochemist.com/ccimg/4900/4829-01-0.png)
![6-phenyl-7-oxabicyclo[4.1.0]heptane](http://img.cochemist.com/ccimg/4900/4829-01-0_b.png)























![Lithium, [4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/2800/2786-01-8.png)
![Lithium, [4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/2800/2786-01-8_b.png)


![Naphth[2,3-b]oxirene,1a,2,7,7a-tetrahydro-](http://img.cochemist.com/ccimg/2500/2461-35-0.png)
![Naphth[2,3-b]oxirene,1a,2,7,7a-tetrahydro-](http://img.cochemist.com/ccimg/2500/2461-35-0_b.png)


![Benzo[b]thiophene, 2-(chloromethyl)-](http://img.cochemist.com/ccimg/2100/2076-88-2.png)
![Benzo[b]thiophene, 2-(chloromethyl)-](http://img.cochemist.com/ccimg/2100/2076-88-2_b.png)
![4-methyl-Pyrrolo[1,2-a]quinoxaline](http://img.cochemist.com/ccimg/2000/1931-32-4.png)
![4-methyl-Pyrrolo[1,2-a]quinoxaline](http://img.cochemist.com/ccimg/2000/1931-32-4_b.png)




![(3-[(3-TRIFLUOROMETHYL)PHENYL]-1-PROPENE )](http://img.cochemist.com/ccimg/1900/1813-96-3.png)
![(3-[(3-TRIFLUOROMETHYL)PHENYL]-1-PROPENE )](http://img.cochemist.com/ccimg/1900/1813-96-3_b.png)







![1,3-Dioxolan-2-one,4-[(phenylmethoxy)methyl]-](http://img.cochemist.com/ccimg/1000/949-97-3.png)
![1,3-Dioxolan-2-one,4-[(phenylmethoxy)methyl]-](http://img.cochemist.com/ccimg/1000/949-97-3_b.png)















![Benzenemethanol, α,α'-[[(2,3-dihydro-1H-inden-2-yl)imino]bis(methylene)]bis-](/data/chemimg/160200/1207760-26-6.png)
![Benzenemethanol, α,α'-[[(2,3-dihydro-1H-inden-2-yl)imino]bis(methylene)]bis-](/data/chemimg/160200/1207760-26-6_b.png)
![2-[(trimethylsilyl)oxy]-1H-Benzimidazole](http://img.cochemist.com/ccimg/873400/873332-72-0.png)
![2-[(trimethylsilyl)oxy]-1H-Benzimidazole](http://img.cochemist.com/ccimg/873400/873332-72-0_b.png)






![2-Penten-1-ol, 5-[(4-methoxyphenyl)methoxy]-, (2E)-](http://img.cochemist.com/ccimg/158900/158817-21-1.png)
![2-Penten-1-ol, 5-[(4-methoxyphenyl)methoxy]-, (2E)-](http://img.cochemist.com/ccimg/158900/158817-21-1_b.png)
![2-Penten-1-ol, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2E)-](http://img.cochemist.com/ccimg/158300/158213-53-7.png)
![2-Penten-1-ol, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2E)-](http://img.cochemist.com/ccimg/158300/158213-53-7_b.png)


![2-Penten-1-ol, 5-[(4-methoxyphenyl)methoxy]-, (Z)-](http://img.cochemist.com/ccimg/143600/143521-62-4.png)
![2-Penten-1-ol, 5-[(4-methoxyphenyl)methoxy]-, (Z)-](http://img.cochemist.com/ccimg/143600/143521-62-4_b.png)
![Benzene, [(1E)-3-methyl-1,4-pentadienyl]-](http://img.cochemist.com/ccimg/109900/109898-19-3.png)
![Benzene, [(1E)-3-methyl-1,4-pentadienyl]-](http://img.cochemist.com/ccimg/109900/109898-19-3_b.png)
![Benzene, [(1E)-3-[(trimethylsilyl)oxy]-1-propen-1-yl]-](/data/chemimg/1663000/109283-53-6.png)
![Benzene, [(1E)-3-[(trimethylsilyl)oxy]-1-propen-1-yl]-](/data/chemimg/1663000/109283-53-6_b.png)




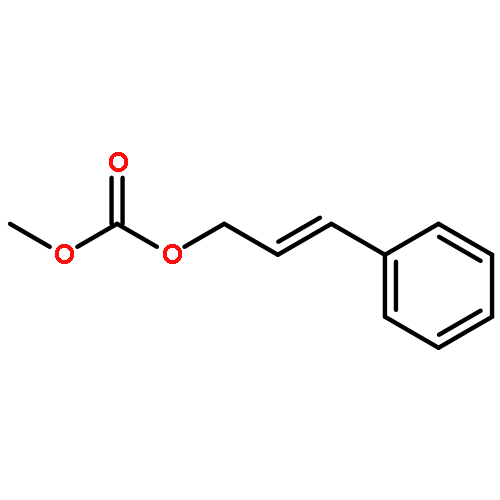
![Benzene, [(1E)-5-bromo-1-pentenyl]-](http://img.cochemist.com/ccimg/57300/57238-67-2.png)
![Benzene, [(1E)-5-bromo-1-pentenyl]-](http://img.cochemist.com/ccimg/57300/57238-67-2_b.png)


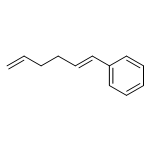

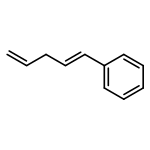

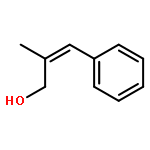
![Pyrimidine, 5-nitro-2,4-bis[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/52600/52522-97-1.png)
![Pyrimidine, 5-nitro-2,4-bis[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/52600/52522-97-1_b.png)


![[(2r,3s,5r)-3-benzoyloxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methyl Benzoate](http://img.cochemist.com/ccimg/35900/35898-30-7.png)
![[(2r,3s,5r)-3-benzoyloxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methyl Benzoate](http://img.cochemist.com/ccimg/35900/35898-30-7_b.png)
![1,3-DIOXASPIRO[4.5]DECAN-2-ONE](http://img.cochemist.com/ccimg/34300/34277-32-2.png)
![1,3-DIOXASPIRO[4.5]DECAN-2-ONE](http://img.cochemist.com/ccimg/34300/34277-32-2_b.png)
![Benzene, [(1E)-3-methoxy-1-propen-1-yl]-](/data/chemimg/1798100/22688-03-5.png)
![Benzene, [(1E)-3-methoxy-1-propen-1-yl]-](/data/chemimg/1798100/22688-03-5_b.png)





![Pyridine, 2-[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/18300/18292-04-1.png)
![Pyridine, 2-[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/18300/18292-04-1_b.png)




![TRIMETHYL-[5-(TRIFLUOROMETHYL)-2-TRIMETHYLSILYLOXYPYRIMIDIN-4-YL]OXYSILANE](http://img.cochemist.com/ccimg/7100/7057-43-4.png)
![TRIMETHYL-[5-(TRIFLUOROMETHYL)-2-TRIMETHYLSILYLOXYPYRIMIDIN-4-YL]OXYSILANE](http://img.cochemist.com/ccimg/7100/7057-43-4_b.png)
![BENZENE, [(1E)-3-ETHOXY-1-PROPENYL]-](http://img.cochemist.com/ccimg/5900/5882-84-8.png)
![BENZENE, [(1E)-3-ETHOXY-1-PROPENYL]-](http://img.cochemist.com/ccimg/5900/5882-84-8_b.png)
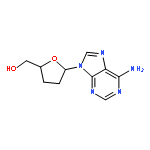





![Propanal, 3-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/128500/128461-65-4.png)
![Propanal, 3-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/128500/128461-65-4_b.png)













{kind=link}