Co-reporter:Chuan Deng, Xianggui Xue, Yu Chi, Hongzhen Li, Xinping Long, and Chaoyang Zhang
The Journal of Physical Chemistry C June 8, 2017 Volume 121(Issue 22) pp:12101-12101
Publication Date(Web):May 22, 2017
DOI:10.1021/acs.jpcc.7b04518
It is extensively deemed that the increased self-heating ability of defects relative to perfect crystals increases the sensitivity, or reduces the safety, of energetic materials. Nevertheless, the nature of such increased self-heating ability remains unclear. The present work provides insight into the origin of such ability by ReaxFF reactive molecular dynamics simulations on the thermal decay of perfect and twinned β-octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) under three typical heating conditions—constant-temperature, programmed, and adiabatic—that represent various rates of heat exchange between the HMX crystal and environment. As a result, it is found that the enhanced self-heating ability stemmed from the high internal energy of the molecules around the defects, and such ability is remarkably exhibited with low heat-exchange rates between the energetic materials EMs and environment. Adiabatic heating is an extreme to exhibit the most remarkable such ability, as the superiority of the high internal energy of the molecules around the defects cannot be lowered without heat exchange. Thereby, the twin-induced shock sensitivity enhancement of HMX and a small difference in differential scanning calorimetric measurement values between perfect and twinned HMX can well be understood by means of the insight.
Co-reporter:Liya Meng;Yu Ma;Xianggui Xue;Zhipeng Lu;Fude Nie
Crystal Growth & Design December 7, 2016 Volume 16(Issue 12) pp:7231-7239
Publication Date(Web):November 10, 2016
DOI:10.1021/acs.cgd.6b01409
The energy and performance of energetic materials can be improved by increasing their crystal packing density. Thus, we propose a strategy involving salification with hydroxylammonium cations (HA+) to increase the packing coefficients (PCs) and packing densities of energetic ionic salts (EISs). Structural analyses and theoretical calculations of the observed EISs indicate that the strong intermolecular hydrogen bonds (HBs) between HA+ and anions are primarily responsible for the increase in EIS density. Such strong HBs usually exist in HA+-based energetic salts and rarely in other EISs but are absent in energetic crystals with neutral molecules. Such HBs induce high PCs and relatively high crystal packing densities by compensating for the relatively lower molecular density of HA+ compared with other cations. Moreover, in combination with HBs in common explosives, we find a simple dependence showing that the shorter the strongest HB corresponds to the higher PC, suggesting that the strongest HB can be regarded as a simple indicator of PC. This study proposes that enhancing intermolecular HBs is the main strategy to increase compactness because H atoms usually exist in currently available energetic materials.
Co-reporter:Xianggui Xue, Liya Meng, Yu Ma, and Chaoyang Zhang
The Journal of Physical Chemistry C April 6, 2017 Volume 121(Issue 13) pp:7502-7502
Publication Date(Web):March 21, 2017
DOI:10.1021/acs.jpcc.7b00294
Hydrocarbon pyrolysis is the main way to achieve carbonaceous materials, while most related conversion mechanisms still remain unclear. This work images pyrolysis of methane at various temperatures and densities by molecular reactive force field (ReaxFF) simulations. First, it is interesting to find that the methane decay is dominated by intermolecular collision displacement instead of direct molecular decomposition. Second, a conversion of 1200 methane molecules into a regular carbon nanocavity (CNC) is realized at 3500 K temperature and 0.1 g/cm3 density after a simulation lasting for 10 ns, with 923 carbon atoms and a diameter of 3.4 nm. Such CNC is a perfect precursor of carbon nanotubes, which is confirmed by a sequent simulation on a larger system of 2400 methane molecules and in agreement with several experimental observations. It is found that the CNC growth obeys a polyyne model, without any single aromatic ring formed in the growth. Furthermore, the complex CNC growth appears in some successive stages: primary methane decay, chain elongation and branching, cyclization and condensation, and final sheeting and curling. The regular rearrangement of CNC is thought to be attributed to the limited active centers formed at the initial cyclization and condensation stage; that is, it is a key to control the primary active centers to form regular carbonaceous materials. Polyyne is found in the pyrolysis of both methane and acetylene at high temperatures, suggesting that carbyne, a novel valuable carbonaceous material, may be obtained by hydrocarbon pyrolysis.
Co-reporter:Zhipeng Lu, Xianggui Xue, Liya Meng, Qun Zeng, Yu Chi, Guijuan Fan, Hongzhen Li, Zengming Zhang, Fude Nie, and Chaoyang Zhang
The Journal of Physical Chemistry C April 20, 2017 Volume 121(Issue 15) pp:8262-8262
Publication Date(Web):April 1, 2017
DOI:10.1021/acs.jpcc.7b00086
Dihydroxylammonium 5,5′-bistetrazole-1,1′-diolate (TKX-50) is an attractive energetic ionic salt and outperforms many common explosives. We report a solid–solid phase transition of TKX-50 to a new metastable phase (Meta-TKX-50) at ∼180 °C by Raman spectroscopy and thermogravimetry–differential scanning calorimetry (TGA–DSC) measurements, in combination with ab initio calculations. Meta-TKX-50 is formed by the rotation of NH3OH+ with a small volume expanding of 3% and a crystal symmetry reduction from P21/C to P-1. In addition, the phase transition features a second order with a slight change of 0.3 J·K–1·mol–1 in specific heat capacity. This slight change of specific heat capacity is the main reason that the phase transition was overlooked in the past measurements. Besides, it is found that the phase transition facilitates the H transfer from NH3OH+ to C2O2N82– and the N–O bond dissociation of NH3OH+ to produce final small stable molecules including NH3 and H2O. Thus, the phase transition is expected to promote the decomposition of TKX-50 and deteriorate its thermal stability.
Co-reporter:Zhipeng Lu;Qun Zeng;Xianggui Xue;Zengming Zhang;Fude Nie
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 34) pp:23309-23317
Publication Date(Web):2017/08/30
DOI:10.1039/C7CP04015F
Performances and behaviors under high temperature–high pressure conditions are fundamentals for many materials. We study in the present work the pressure effect on the thermal decomposition of a new energetic ionic salt (EIS), TKX-50, by confining samples in a diamond anvil cell, using Raman spectroscopy measurements and ab initio simulations. As a result, we find a quadratic increase in decomposition temperature (Td) of TKX-50 with increasing pressure (P) (Td = 6.28P2 + 12.94P + 493.33, Td and P in K and GPa, respectively, and R2 = 0.995) and the decomposition under various pressures initiated by an intermolecular H-transfer reaction (a bimolecular reaction). Surprisingly, this finding is contrary to a general observation about the pressure effect on the decomposition of common energetic materials (EMs) composed of neutral molecules: increasing pressure will impede the decomposition if it starts from a bimolecular reaction. Our results also demonstrate that increasing pressure impedes the H-transfer via the enhanced long-range electrostatic repulsion of H+δ⋯H+δ of neighboring NH3OH+, with blue shifts of the intermolecular H-bonds. And the subsequent decomposition of the H-transferred intermediates is also suppressed, because the decomposition proceeds from a bimolecular reaction to a unimolecular one, which is generally prevented by compression. These two factors are the basic root for which the decomposition retarded with increasing pressure of TKX-50. Therefore, our finding breaks through the previously proposed concept that, for the condensed materials, increasing pressure will accelerate the thermal decomposition initiated by bimolecular reactions, and reveals a distinct mechanism of the pressure effect on thermal decomposition. That is to say, increasing pressure does not always promote the condensed material decay initiated through bimolecular reactions. Moreover, such a mechanism may be feasible to other EISs due to the similar intermolecular interactions.
Co-reporter:Zhipeng Lu;Xianggui Xue
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 46) pp:31054-31062
Publication Date(Web):2017/11/29
DOI:10.1039/C7CP06363F
Dihydroxylammonium 5,5′-bistetrazole-1,1′-diolate (TKX-50) is a new and attractive energetic material that outperforms numerous common explosives because of its excellent properties and performance, and is thus a promising candidate to replace some of them. Nevertheless, knowledge of its physico-chemical properties, in particular, the underlying mechanism for it undergoing external stimuli for complete decay still remains poor. In the present study, we ascertain a preferred slip system of (010)/[101] and a shear-induced phase transition of TKX-50 with the aid of theoretical calculations. In other words, a new phase of TKX-50, γ-TKX-50, is observed to be formed by shearing TKX-50 along a slip system of (010)/[101] or (010)/[10] with a space group of P21/a, elevated energy of 9.4 kcal mol−1 and a unit cell expanded 4%, relative to the original TKX-50. Moreover, γ-TKX-50 can most readily be formed by shearing TKX-50 along (010)/[101] with a lowest energy barrier of 18.6 kcal mol−1, which is much below that for TKX-50 decay. The predicted elastic constants of γ-TKX-50 verify its mechanical stability with decreased mechanical anisotropy relative to the original TKX-50. In addition, we find that, after phase transition, the hydrogen bonding is weakened, while the electrostatic repulsion of Hδ+⋯Hδ+ increases, which disfavors the proton transfer from NH3OH+ to C2O2N82− to facilitate the thermal decay of TKX-50. This suggests that the shear-induced transition from TKX-50 to γ-TKX-50 can enhance thermal stability by elevating the energy barrier for proton transfer, potentially contributing to the low mechanical sensitivity of TKX-50. Hopefully, this study would enrich the insight into the underlying mechanism of TKX-50 against external thermal–mechanical stimuli. Moreover, in combination with the newly found heat-induced phase, the shear-induced phase observed in the present study and the original one, there are at least three phases for TKX-50.
Co-reporter:Yu Ma;Xudong He;Liya Meng;Xianggui Xue
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 45) pp:30933-30944
Publication Date(Web):2017/11/22
DOI:10.1039/C7CP03801A
Energetic ionic salts (EISs) are attracting extensive attention because of their ready preparation and some excellent properties and performances that are comparable to those of common explosives with neutral molecules. Hydroxylamine (HA) is protonated or ionized as H-HA+ and preferred to be introduced into EISs to form HA-based EISs with almost all kinds of anions since these EISs possess higher packing densities and thus more excellent detonation performances than others with the same anions. Moreover, relative to that of pure HA, the thermal stability of HA-based EISs is significantly enhanced. This significantly enhanced thermal stability can extend the application of HA via deprotonation of H-HA+ back to HA; however, the mechanism for stabilization of HA by salification remains unclear. Herein, we employed thermodynamic and kinetic calculations and molecular dynamics simulations to reveal the thermal stability mechanisms of many currently synthesized HA-based EISs and some previously reported EISs with inorganic anions as well as those of pure HA and its aqueous solution. As a result, we have found that the enhanced stability of HA-based EISs is mainly due to the ionization and separation of HA molecules themselves. That is, H-HA+, as an ionized product, is more molecularly stable than HA, with significantly strengthened covalent bonds. The separation of H-HA+ ions or HA molecules makes decomposition more difficult as decomposition initiation varies from bimolecular to unimolecular reactions of HA, with a significant increase in the energy barrier. We have, therefore, proposed a strategy for the stabilization of unstable systems, such as neutral N-rich energetic compounds, by ionization and separation to strengthen these systems and change the decomposition mechanism by increasing the energy barriers of trigger steps such that these barriers become more difficult to overcome, respectively.
Co-reporter:Xianggui Xue, Yu Ma, Qun Zeng, and Chaoyang Zhang
The Journal of Physical Chemistry C 2017 Volume 121(Issue 9) pp:
Publication Date(Web):February 14, 2017
DOI:10.1021/acs.jpcc.7b00698
Energetic cocrystallization, by combining existing molecules together, is thought to be new strategy for creating energetic materials. Nevertheless, the underlying mechanism of its influences on properties and performances in comparison with their pure components remains unclear. The present work reveals the cocrystallization influence of a typical energetic cocrystal of CL-20/HMX on thermal stability, by ReaxFF molecular reactive dynamic simulations and kinetics calculations on the pure and cocrystals. As a result, we find that the cocrystal mediates the thermal stability of pure crystalsand this is in agreement with experimental observations. The initial decay steps in pure crystals remain still in the cocrystal, that is, the independent and intramolecular reactions of N–N bond cleavage governing the initial decay of the pure CL-20 and HMX crystals also dominate in the cocrystal of CL-20/HMX. Meanwhile, during the thermal decomposition of the cocrystal, CL-20 releases heat faster than HMX, thus the heat is transferred from CL-20 to HMX, and further the decay rate of HMX increases while that of CL-20 decreases, relative to the pure crystals. This leads to a moderate decay rate of the cocrystal and a small difference in decay barrier after cocrystallization. Besides, the moderated decay rate is also attributed to the small variation in intermolecular interactions after cocrystallization and the intrinsic weak stability of both component molecules of CL-20 and HMX. Thus, the intrinsic molecular stability of components and intermolecular interactions should be noted as two main factors in a strategy for increasing stability by energetic cocrystallization.
Co-reporter:Yu Ma;Liya Meng;Hongzhen Li
CrystEngComm (1999-Present) 2017 vol. 19(Issue 23) pp:3145-3155
Publication Date(Web):2017/06/13
DOI:10.1039/C7CE00529F
Creating new energetic materials (EMs) of high energy and high safety (low sensitivity) is one of the most important objectives of research related to EMs. In addition to the synthesis of new compounds, constructing new EMs by crystal engineering is currently recognized to be a breakthrough. In the present work, we reveal the underlying mechanism of the observed impact sensitivity (IS) variations of the cocrystal series of diacetone diperoxide—(DADP)/1,3,5-trichloro-2,4,6-trinitrobenzene (TCTNB), DADP/1,3,5-tribromo-2,4,6-trinitrobenzene (TBTNB), and DADP/1,3,5-triiodo-2,4,6-trinitrobenzene (TITNB)—in comparison to their pure components, from the viewpoint of their crystal packing structures. We find that: the remarkable IS improvement of DADP/TITNB results from increased intermolecular interactions and the enhanced anisotropy of these interactions (i.e., the enhanced differentiation between intra- and interlayered interactions); the difficulty in measurement of the IS of DADP/TBTNB stems from its thermodynamic instability (weakened intermolecular interactions); and DADP/TCTNB shows an IS intermediate between those of its two pure components, as it does little to improve the interactions and anisotropy. Therefore, we propose the enhancement of intermolecular interactions and their anisotropy as a strategy for building low-impact-sensitivity EMs in crystal engineering, such as by cocrystallization.
Co-reporter:Xudong He;Xianfeng Wei;Yu Ma;Zhipeng Lu
CrystEngComm (1999-Present) 2017 vol. 19(Issue 19) pp:2644-2652
Publication Date(Web):2017/05/15
DOI:10.1039/C7CE00489C
The dependence of discrete crystal density (dc) of a series of cubane and its nitro derivatives on the nitro group amount was understood via analogically analyzing their crystal packing. It was found that dc does not successively increase with the increase in the hydrogen atoms replacement by nitro groups because the intermolecular interactions that strongly influence the crystal packing vary from H⋯H to H⋯O and O⋯O interactions, with weak to strong and weak strength, respectively. The relatively low dc of octanitrocubane (less than that of 1,2,3,4,5,6,8-heptanitrocubane) is thought to be clearly due to the replacement of all the hydrogen atoms by nitro groups, rendering O⋯O interactions to be completely predominant and unfavorable for closer packing, i.e., they have a lower packing coefficient. This deduction from the crystal packing of a series of analogues of nitrocubane derivatives to understand the observed dc can be seen as a prototype to understand the crystal packing and provide a foundation for designing new CHNO explosives, in which H⋯O and O⋯O interactions are usually prevalent. Moreover, it was proposed that a specific quantity of hydrogen atoms is usually indispensable to achieve a high dc value for CHNO energetic crystals.
Co-reporter:Qun Zeng;Yu Ma;Jinshan Li
CrystEngComm (1999-Present) 2017 vol. 19(Issue 19) pp:2687-2694
Publication Date(Web):2017/05/15
DOI:10.1039/C6CE02373H
Energetic co-crystals (ECCs) are now thriving and becoming alternatives to energetic materials. Thereby, it is important to understand the intermolecular interactions present in ECCs to obtain knowledge about ECC engineering. However, the physical sources of the interactions remain unclear, even though the interactions have already been understood as the three conventional basic interaction kinds, or the three main traditional engineering motifs of organic crystals, namely hydrogen bonding, π-stacking and halogen bonding. Twelve typical molecular pairs extracted from five observed EECs covering all the three interaction kinds are selected to partition the intermolecular interaction energy and discuss the physical sources, by density functional theory calculations and block located wavefunction energy decomposition analyses (BLW-EDA). We find that, after carefully examining all observed ECCs, each conventional interaction motif in energetic–energetic molecular pairs is always weak, and dominated by a frozen effect, i.e., van der Waals and electrostatic interactions. The rather strong hydrogen bonding exists in the molecular pairs with one non-energetic molecule at least, and is predominated by the polarization and charge transfer effects. Meanwhile, we find small bond order variations caused by the crystal packing of energetic–energetic co-crystals (EECCs), thereby showing small molecular stability variation. It suggests that it is difficult to increase the molecular stability of the energetics by cocrystallization to improve safety; while the safety of EECCs will benefit from the enhanced intermolecular interactions and the improved crystal packing mode favoring ready shear slip.
Co-reporter:Liya Meng, Zhipeng Lu, Xianfeng Wei, Xianggui Xue, Yu Ma, Qun Zeng, Guijuan Fan, Fude Nie and Chaoyang Zhang
CrystEngComm 2016 vol. 18(Issue 13) pp:2258-2267
Publication Date(Web):09 Feb 2016
DOI:10.1039/C5CE02089A
Dihydroxylammonium 5,5′-bistetrazole-1,1′-diolate (TKX-50) is a recently synthesized energetic ionic salt (EISs) with some excellent properties including high energy content, high density, low impact sensitivity and low toxicity, and therefore is a promising alternative energetic material. In contrast to commonly applied energetic CHON materials, TKX-50 features strong intermolecular hydrogen bonds (HBs). However, the effects of these strong HBs on its important properties remain unclear. We report herein the two-sided effects of the strong HBs on the stability of TKX-50, through ab initio simulations on shear deformation and thermal decomposition. That is, on the one hand, the strong HBs in TKX-50 lead to a layer-like arrangement of the cations and dianions in the (010) planes with the adjacent layers connected only by two types of weakest HBs, which facilitates the conversion of external kinetic energy into interlayer sliding, and contributes to low impact sensitivity; on the other hand, the extensive HBs in TKX-50 act as a potential source to facilitate H-transfer (including proton transfer), which accelerates subsequent thermal decomposition, and thus deteriorates the thermal stability of TKX-50 relative to its notably low impact sensitivity. These two-sided effects of the strong HBs in a TKX-50 crystal should be useful to understand properties of other EISs, in which strong HBs are universal, and enrich the knowledge of the sensitivity mechanism of energetic materials.
Co-reporter:Chaoyang Zhang, Yushi Wen, Xianggui Xue, Jian Liu, Yu Ma, Xudong He, and Xinping Long
The Journal of Physical Chemistry C 2016 Volume 120(Issue 44) pp:25237-25245
Publication Date(Web):October 21, 2016
DOI:10.1021/acs.jpcc.6b08227
The fundamental core of chemistry is to create new substances, and numerous complex reactions may be involved in chemical conversions. Nevertheless, clarifying the mechanisms of these complex reactions remains challenging, thereby causing insufficiencies in the fundamentals to guide new substance creation. This work proposes and emphasizes a strategy of sequential molecular dynamics simulations (SMDSs) toward complex chemical reactions. The strategy is successfully demonstrated by clarifying a complex graphitization process of 1,3,5-triamino-2,4,6-trinitrobenzene (TATB), whose mechanism has not been imaged by a single simulation alone. We conducted SMDSs with a molecular reactive force field, ReaxFF, to resemble the cook-off of TATB, i.e., a sequence of heating, expansion, and cooling acting on TATB. Graphitization is found to sequentially undergo TATB molecular decay, clustering, cluster enlargement to C sheets (sheeting), and layered stacking of C sheets, along with phase separation. Moreover, the structures graphitized from TATB can be imaged only when simulations are conducted in the sequence of heating, expansion, and cooling, in accordance with the actual conditions of cooking TATB. This successful exemplification shows that a large number of complex reaction mechanisms can be revealed using the SMDS strategy and computation ability promotion, in combination with the clarified experimental conditions. This strategy exhibits considerable potential for future use.
Co-reporter:Xianggui Xue, Yushi Wen, and Chaoyang Zhang
The Journal of Physical Chemistry C 2016 Volume 120(Issue 38) pp:21169-21177
Publication Date(Web):September 2, 2016
DOI:10.1021/acs.jpcc.6b05228
ε-2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (CL-20) is currently the most powerful explosive commercially available. Nevertheless, the early decay events of shocked ε-CL-20 still remain unclear. We perform quantum based self-consistent charge density-functional tight-binding molecular dynamics simulations, in combination with the multiscale shock simulation technique, to reveal the events with four specified shock velocities (Us) of 8 to 11 km/s. We find that the temperature and pressure increases and that the volume reduction is enhanced with increasing shock strength. The ring opening is observed to trigger molecular decay at all four shock conditions; while the sufficient NO2 fission is observed at Us = 8 and 9 km/s, and strongly inhibited at Us = 10 and 11 km/s. Moreover, the evolution of main chemical species, such as active intermediates, stable products, and clusters, is strongly dependent on the shock strength. NO2 and H are dominant in the primary intermediates, responsible for weak and strong shock, respectively; CO2 and N2, as well as water, are the main stable products with a population gradation determined by the shock strength; and the bigger clusters with longer durations are found to be caused by the stronger shock, and their fast dissociation mainly undergoes through the ring opening. Besides, it is found that ε-CL-20 possesses weak anisotropy in the above-specified Us range. This work will enrich the knowledge of shocked energetic materials, in particular, the important energetic materials.
Co-reporter:Yushi Wen, Xianggui Xue, Xinping Long, and Chaoyang Zhang
The Journal of Physical Chemistry A 2016 Volume 120(Issue 22) pp:3929-3937
Publication Date(Web):May 16, 2016
DOI:10.1021/acs.jpca.6b03795
We carried out reactive molecular dynamics simulations by ReaxFF to study the initial events of an insensitive high explosive 1,3,5-triamino-2,4,6-trinitrobenzene (TATB) against various thermal stimuli including constant-temperature heating, programmed heating, and adiabatic heating to simulate TATB suffering from accidental heating in reality. Cluster evolution at the early stage of the thermal decomposition of condensed TATB was the main focus as cluster formation primarily occurs when TATB is heated. The results show that cluster formation is the balance of the competition of intermolecular collision and molecular decomposition of TATB, that is, an appropriate temperature and certain duration are required for cluster formation and preservation. The temperature in the range of 2000–3000 K was found to be optimum for fast formation and a period of preservation. Besides, the intra- and intermolecular H transfers are always favorable, whereas the C–NO2 partition was favorable at high temperature. The simulation results are helpful to deepen the insight into the thermal properties of condensed TATB.
Comparative Study of Experiments and Calculations on the Polymorphisms of 2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (CL-20) Precipitated by Solvent/Antisolvent Method
Co-reporter:Xianfeng Wei
The Journal of Physical Chemistry C 2016 Volume 120(Issue 9) pp:5042-5051
Publication Date(Web):February 10, 2016
DOI:10.1021/acs.jpcc.6b00304
2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (CL-20) is the most powerful explosive. However, the application of this compound is limited by its high sensitivity and serious polymorphic transformations. Thus, elucidating the mechanism of crystallization and polymorphic transformation of CL-20 is crucial. This work presents a comparative study of experiments and calculations to clarify the mechanism of CL-20 precipitation using an solvent/antisolvent method. Calculations show that the β-formed CL-20 conformations are always the most energetically favored. These conformations have generally the highest content in solutions, and the intermolecular conformational transformations in solutions have low energy barriers. In addition, it is predicted that the β-CL-20 crystal possesses the lowest lattice energy among all polymorphs. The calculated results are successfully applied to explain the experimental observations, as β-CL-20 crystal is initially precipitated from most of the highly supersaturated solutions and then converted into ε-CL-20 crystal. This precipitation is kinetically controlled by the dominance of β-CL-20 molecules in a metastable phase and rapid crystallization. The final conversion into ε-CL-20 crystal is attributed to its low energy barrier for polymorphic transformation and stability, that is, the conversion is dynamically dominated. Furthermore, calculated coherent energy densities (CEDs) of various CL-20 polymorphs, including hydrates with different hydration degrees, agree well with the thermal stabilities, as the higher CED corresponds to the higher thermal stability. Therefore, the complex crystallization of CL-20 is elucidated by combining experimental observations with theoretical calculations and simulations.
Co-reporter:Xiaoqing Zhou
The Journal of Physical Chemistry C 2016 Volume 120(Issue 25) pp:13434-13442
Publication Date(Web):June 7, 2016
DOI:10.1021/acs.jpcc.6b04612
The mechanical anisotropy of the wavelike π-stacked energetic crystal of 1,1-diamino-2,2-dinitroethylene (FOX-7) is investigated by nanoindentation experiments and density functional theory (DFT) calculations. The FOX-7 crystal exhibits distinct mechanical anisotropy when indented on different faces. The elastic modulus and hardness of the (020), (−101), and (002) faces change in a decreasing order. The indentation on the (020) face induces the largest depth and the highest pile-up around all three edges of the indenter without causing crack formation. By contrast, the indentations on the (−101) and (002) faces are similar and induce a small indentation depth, low pile-up with a small distribution, and crack formation. Mechanical anisotropy is essentially determined by the wavelike π stacking of FOX-7 along the (020) face with the support of intermolecular hydrogen bonds; i.e., the molecular orientations and intermolecular spaces along different faces vary distinctly. This is also supported by the DFT calculations on uniaxial compression and shear sliding. In this work, the nature of the wavelike π stacking responsible for the low impact sensitivity of FOX-7 is discussed and compared with that of other explosives with different packing structures.
Co-reporter:Jia Yuan, Xinping Long, and Chaoyang Zhang
The Journal of Physical Chemistry A 2016 Volume 120(Issue 47) pp:9446-9457
Publication Date(Web):November 14, 2016
DOI:10.1021/acs.jpca.6b08852
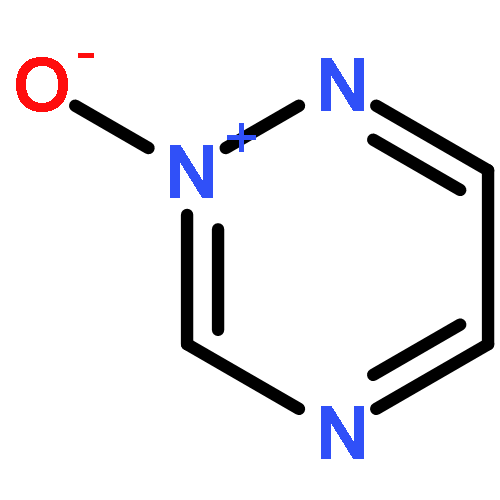
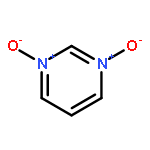
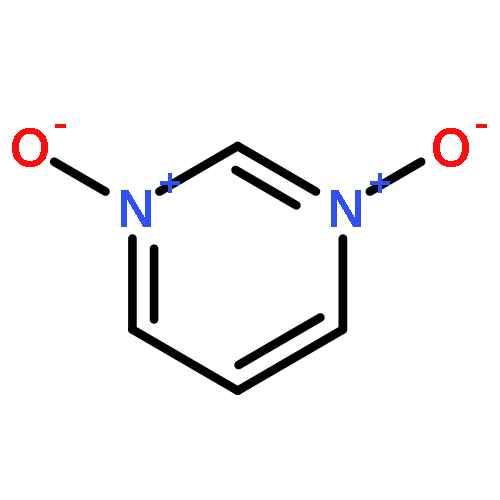
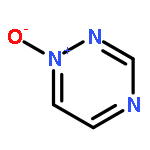
N-Oxidization is an important strategy for enhancing the density and energy of energetic materials. Nevertheless, the influence of N+–O– introduction on molecular stability remains relatively unknown. Thus, the present work comprehensively studied 102 basic N-rich ring structures, including azoles, furazans, and azines, as well as their N-oxides by quantum chemical calculations. The introduction of N+–O– weakens molecular stability in most cases because the process elongates chemical bonds, decreases ring aromaticity, narrows the gaps between the highest occupied and lowest unoccupied molecular orbitals, and increases the photochemical reactivity. Besides, the easy H transfer to the neighboring O atom, which forms a N–OH isomer in azoles, renders the stabilization by N-oxide introduction ineffective. However, N-oxide introduction can enhance the molecular stability of 1,2,3,4-tetrazine-1,3-dioxide and tetrazino-tetrazine 1,3,6,8-tetraoxide by promoting σ–π separation and relieving lone-pair repulsion. Moreover, the alternate arrangement of positive and negative charges is another factor stabilizing the 1,2,3,4-tetrazine ring by 1,3-dioxidation. Finally, we assess the accessibility of N-oxidized azoles and azines by regarding N2O and H2O2 as oxidizers. We find that all the oxidations were exothermic, thermodynamically spontaneous, and kinetically feasible. After an overall evaluation, we propose 19 N-oxides as basic structures for high-energy materials with considerable stability.
Co-reporter:Jian Liu, Qun Zeng, Yalin Zhang, and Chaoyang Zhang
The Journal of Physical Chemistry C 2016 Volume 120(Issue 28) pp:
Publication Date(Web):July 7, 2016
DOI:10.1021/acs.jpcc.6b04256
Modeling plastic deformation of crystalline materials by all-atomistic methods remains a challenge, and large-scale methods, such as coarse-grained (CG) methods, are highly desirable. To overcome the difficulty in constructing CG potentials for stiff molecular crystals by conventional approaches, we propose a limited-sample coarse-grained (LSCG) strategy. We construct a CG potential of α-1,3,5-trinitro-1,3,5-triazinane (α-RDX), a widely used energetic material, and perform coarse-grained molecular dynamics (CGMD) simulations to validate the LSCG potential. We compare the calculated mechanical properties with other reported results. The results show that the LSCG method is effective when compared with the all-atomic methods and provides adequate insight into systems with larger scales. Therefore, through the LSCG method, the deformation mechanisms of α-RDX crystals under nanoindentation conditions are revealed by a series of CGMD simulations that resemble nanoindentation on their (100) surface, with nanoholes sited differently. Valuable results are obtained and understood. That is, the stress around the nanohole can trigger void collapses when the nanohole is located at a shallow position directly beneath the indentation surface. At a location deeper than 4 times the maximum impression depth, the stress around the hole is extremely weak to cause void collapse. Most of the dislocation loops are found to be parallel to the (001) plane, which is attributed to the low slip threshold of the (010) [100] slip system. This result shows that the LSCG strategy can deal with much larger systems and reveal mechanisms on the mesoscale.
Co-reporter:Xianfeng Wei, Yu Ma, Xinping Long and Chaoyang Zhang
CrystEngComm 2015 vol. 17(Issue 37) pp:7150-7159
Publication Date(Web):13 Aug 2015
DOI:10.1039/C5CE01355K
Energetic–energetic cocrystals (EECCs) are promising alternatives to high-energy and low-sensitivity explosives, the development of which is still a challenge in the field of energetic materials due to their intrinsic energy-sensitivity contradiction (high energy usually accompanies high sensitivity). We propose a strategy to combine highly energetic but unstable hydrogen-free molecules with hydrogenous energetic molecules to form stable EECCs and maintain their high energy, developed by analyzing the crystal packing of all observed BTF-based EECCs. That is, in contrast to the pure BTF crystal, which is very sensitive to mechanics and shock, the increased intermolecular hydrogen bonding consolidates the EECCs, leading to largely enhanced cohesive energy densities. Furthermore, hydrogen bonds are formed regardless of coformer molecular geometry, suggesting a large number of potential coformer molecules and EECCs. The thermodynamics driving the EECC formation is discussed, and the increased lattice energy and increased entropy are thought to be the driving force for EECC formation. This strategy for consolidating crystals to stabilize unstable molecules by increasing intermolecular hydrogen bonding will renew the interest in some highly energetic compounds that have been overlooked for a long time due to their poor environmental compatibility.
Co-reporter:Chaoyang Zhang, Chi Zhang, Yu Ma and Xianggui Xue
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 17) pp:11469-11480
Publication Date(Web):24 Mar 2015
DOI:10.1039/C5CP00926J
C black is a class of substantial materials with a long history of applications. However, apart from some descriptions of primary reactions, subsequent processes leading up to the final formation mechanism remain unclear. This mechanism is also crucial for understanding the formation of other carbonaceous materials. In this work, we visualize C black formation by acetylene pyrolysis using molecular dynamics simulations with a molecular reactive force field named ReaxFF. We find that the formation undergoes four stages: (1) chain elongation by H abstraction and polymerization of small C species, (2) chain branching, (3) cyclization and ring densification, and (4) condensed ring folding. The simulated C black particle possesses a structure of folded graphite layers, which is in good accordance with experimental observations. Cyclization and condensation are derived from fusion between neighboring chains, significantly varying from common experimental observations at relatively low temperatures that abide by the mechanism of H abstraction and C2H2 addition. Moreover, polyyne and polyene are usually found during acetylene pyrolysis, suggesting that the pyrolysis of acetylene and other hydrocarbons may be a feasible method of obtaining carbyne, a novel carbonaceous material with a high value.
Co-reporter:Yushi Wen, Chaoyang Zhang, Xianggui Xue and Xinping Long
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 18) pp:12013-12022
Publication Date(Web):01 Apr 2015
DOI:10.1039/C5CP00006H
Clustering is experimentally and theoretically verified during the complicated processes involved in heating high explosives, and has been thought to influence their detonation properties. However, a detailed description of the clustering that occurs has not been fully elucidated. We used molecular dynamic simulations with an improved reactive force field, ReaxFF_lg, to carry out a comparative study of cluster evolution during the early stages of heating for three representative explosives: 1,3,5-triamino-2,4,6-trinitrobenzene (TATB), β-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) and pentaerythritol tetranitrate (PETN). These representatives vary greatly in their oxygen balance (OB), molecular structure, stability and experimental sensitivity. We found that when heated, TATB, HMX and PETN differ in the size, amount, proportion and lifetime of their clusters. We also found that the clustering tendency of explosives decreases as their OB becomes less negative. We propose that the relationship between OB and clustering can be attributed to the role of clustering in detonation. That is, clusters can form more readily in a high explosive with a more negative OB, which retard its energy release, secondary decomposition, further decomposition to final small molecule products and widen its detonation reaction zone. Moreover, we found that the carbon content of the clusters increases during clustering, in accordance with the observed soot, which is mainly composed of carbon as the final product of detonation or deflagration.
Co-reporter:Xianfeng Wei, Anbang Zhang, Yu Ma, Xianggui Xue, Junhong Zhou, Yuanqiang Zhu and Chaoyang Zhang
CrystEngComm 2015 vol. 17(Issue 47) pp:9037-9047
Publication Date(Web):26 Oct 2015
DOI:10.1039/C5CE02009C
Energetic cocrystallization is a promising crystal engineering method for energetic materials. However, the current yield of energetic–energetic cocrystals (EECCs) remains very limited largely as a result of inefficient EECC screening. Therefore, the crystallization thermodynamics of EECCs must be predicted; this process is the core of the screening procedure. The present work provides insight into the intermolecular interactions of and variations in lattice energy, enthalpy, and Gibbs free energy following the crystallization of observed and supposed EECCs. Moreover, this research clarifies the difference in the solubility parameters of each pair of coformers. As a result, formation is predicted to be thermodynamically favored for most observed and supposed EECCs. The dominance of entropy is more sufficient than that of enthalpy; this dominance is mainly caused by the structural similarity in energetic molecules that either produce little heat or absorb heat if an intermolecular rearrangement is observed to transform pure crystals into cocrystals. Implicitly, EECCs can be formed efficiently when confusion degrees or spatial molecular configurations increase, thus guaranteeing entropy dominance.
Co-reporter:Xianggui Xue
The Journal of Physical Chemistry C 2015 Volume 119(Issue 24) pp:13735-13742
Publication Date(Web):May 27, 2015
DOI:10.1021/acs.jpcc.5b03298
Molecular dynamics simulations of the chemical responses of shocked dislocation-contained and perfect (p) 1,3,5-trinitro-1,3,5-triazinane (RDX) crystals were performed using the ReaxFF force field combined with the multiscale shock technique. The shear dynamics of four types of dislocated RDX crystals are also modeled. The predicted mobilities of the crystals decrease in the order of (010) [001]/screw (s2) > (010) [001]/edge (e2) > (010) 1/2[100]/screw (s1) > (010)1/2[100]/edge (e1) according to their shear stress barriers, thus revealing the initial driving force required to activate a slip system. In view of the evolution of temperatures, pressures, and reactant decay rates of the shocked perfect and dislocated RDX, we confirm that shock sensitivity follows the order of e2 > e1 > s1 ≈ s2 > p. In particular, all dislocations enhance the shock sensitivity of RDX; in particular, edge dislocations enhance shock sensitivity significantly, whereas screw dislocations enhance it slightly. Shock sensitivity is not proportional to the shear stress barrier, which implies other factors influence shock initiation besides shear.
Co-reporter:Chaoyang Zhang, Yushi Wen, and Xianggui Xue
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 15) pp:12235
Publication Date(Web):July 23, 2014
DOI:10.1021/am501562m
Functionalized graphene sheet (FGS) is a promising additive that enhances fuel/propellant combustion, and the determination of its mechanism has attracted much interest. In the present study, a series of molecular dynamic simulations based on a reactive force field (ReaxFF) are performed to explore the catalytic activity (CA) of FGS in the thermal decay of nitromethane (NM, CH3NO2). FGSs and pristine graphene sheets (GSs) are oxidized in hot NM liquid to increase their functionalities and subsequently show self-enhanced CAs during the decay. The CAs result from the interatomic exchanges between the functional groups on the sheets and the NM liquid, i.e., mainly between H and O atoms. CA is dependent on the density of NM, functionalities of sheets, and temperature. The GSs and FGSs that originally exhibit different functionalities tend to possess similar functionalities and consequently similar CAs as temperature increases. Other carbon materials and their oxides can accelerate combustion of other fuels/propellants similar to NM, provided that they can be dispersed and their key reaction steps in combustion are similar to NM.Keywords: combustion; functionalized graphene; molecular dynamics simulations; nitromethane; ReaxFF; self-enhanced catalytic activity
Co-reporter:Chaoyang Zhang, Zongwei Yang, Xiaoqing Zhou, Chenghua Zhang, Yu Ma, Jinjiang Xu, Qi Zhang, Fude Nie, and Hongzhen Li
Crystal Growth & Design 2014 Volume 14(Issue 8) pp:3923-3928
Publication Date(Web):June 18, 2014
DOI:10.1021/cg500796r
We report two kinds of evident hydrogen bonded chains constructing two binary cocrystals of 2,4,6,8,10,12-hexanitrohexaazaisowurtzitane (CL-20) with para-benzoquinone (1) and 1,4-naphthoquinone (2): one kind is the CL-20 molecule chains linked by R22(6) hydrogen bonds, and the other is connected by CL-20 and coformer (1 or 2) molecules alternately through R21(5) hydrogen bonds. All chains extend to the entire cocrystals CL-20/1 and CL-20/2 with crossing points of CL-20 molecules. In contrast to the unremarkable intermolecular interactions in observed CL-20 polymorphs and cocrystals, these two kinds of chains in CL-20/1 and CL-20/2 are evident and can be readily understood using the definition of supramolecular synthons. Moreover, the thermal behaviors, impact sensitivity, and detonation properties of these two energetic cocrystals are reported.
Co-reporter:Yu Ma, Anbang Zhang, Chenghua Zhang, Daojian Jiang, Yuanqiang Zhu, and Chaoyang Zhang
Crystal Growth & Design 2014 Volume 14(Issue 9) pp:4703-4713
Publication Date(Web):July 25, 2014
DOI:10.1021/cg501048v
Low-sensitivity and high-energy explosives (LSHEs) are highly desired for their comprehensive superiority of safety and energy. Crystal packing is crucial to both the safety and energy, and therefore becomes of interest in energetic crystal engineering. This work carries out systemic analyses on the crystal packing of 11 existing LSHEs with both energy and safety close or superior to TNT. As a result, we find that the LSHE crystals wholly feature π–π stacking with the aid of intermolecular hydrogen bonding. Each LSHE molecule is π-bonded with a big conjugated structure composed of all non-hydrogen atoms in the entire molecule. Intramolecular hydrogen bonding exists in most LSHE molecules with strongly active hydrogen bond (HB) donors of amino and hydroxyl groups, and various strength. These big π-conjugated structures and intramolecular HBs lead to planar molecules with high stability, settling a base of π–π stacking in crystals. With the help of intermolecular HBs, the π–π stacking holding the LSHE crystals appears in four modes. Among them, the face-to-face stacking (always offset) gives rationally the smallest steric hindrance when interlayer slide occurs in crystal, which is the reason for very low impact sensitivity. This work suggests that the planar conjugated molecular structure and intermolecular hydrogen bonding supporting the π–π stacking are necessary to the crystal engineering of LSHEs.
Co-reporter:Yu Ma, Anbang Zhang, Xianggui Xue, Daojian Jiang, Yuanqiang Zhu, and Chaoyang Zhang
Crystal Growth & Design 2014 Volume 14(Issue 11) pp:6101-6114
Publication Date(Web):October 1, 2014
DOI:10.1021/cg501267f
Molecular and crystal designs are crucial to the engineering of high-energy explosives, which are a class of substantial materials usually with high costs and high risks. Understanding their structures, properties, and performances, and the relationships among them is the basis for the design. As a continuation of a systemic analysis of the crystal packing of low-sensitivity and high-energy explosives (LSHEs) ( Cryst. Growth Des. 2014, 14, 4703−4713), we present in this work another analysis of 10 existing impact-sensitive high-energy explosives (SHEs), which possess both velocities of detonation and impact sensitivity close to or higher than those of RDX. We find that SHE molecules are usually less stable than LSHE ones, due to the deficiencies of big π-conjugated molecular structures, and adequate and strong intramolecular hydrogen bonds (HBs) even though H atoms are contained. The intermolecular HBs cannot be formed sometimes in H-contained SHE crystals, and the noncovalent O···O interactions dominate the connection of SHE molecules to build a three-dimensional network and hold crystals, generally, with the strength above intermolecular HBs. The absence of single-atom-layered stacking in SHE crystals makes the intermolecular sliding difficult or even unallowed when against impact, which leads to inefficiency of energy buffering and ease of molecular decay, hot spot formation, and final combustion or detonation. In contrast to LSHEs, SHEs are disadvantageous on dual structural levels causing their high sensitivity: molecules with low stability and crystals without HB-aided single-atom-layered stacking. It re-verifies that the intermolecular HB-aided π–π stacking is necessary for crystal engineering of LSHEs, which are highly desired currently.
Co-reporter:Chaoyang Zhang, Xianggui Xue, Yaofeng Cao, Junhong Zhou, Anbang Zhang, Hongzhen Li, Yang Zhou, Ruijuan Xu and Tao Gao
CrystEngComm 2014 vol. 16(Issue 26) pp:5905-5916
Publication Date(Web):21 May 2014
DOI:10.1039/C4CE00584H
2,4,6,8-Hexanitro-2,4,6,8,10,12-hexaazatetracyclododecane (CL-20) is the most powerful explosive applied, and CL-20-based energetic–energetic co-crystals are promising new alternative explosives with tunable power and safety, resulting in much interest in them. This work discusses the structural, electronic and energetic features of three CL-20 polymorphs, β, γ and ε forms, and three CL-20-based energetic–energetic co-crystals, CL-20/TNT, CL-20/HMX and CL-20/BTF. As a result, we find that, relative to the pure CL-20 polymorphs, the co-crystallization of CL-20 with HMX, TNT and BTF cause little molecular deformation except from some torsion of its nitro groups, and the narrower band gaps. And dominantly, the O⋯O, O⋯H and O⋯N interactions hold all the crystal packing. There is possibly thermodynamic and kinetic dominance in the CL-20/TNT and CL-20/HMX, and CL-20/BTF co-crystallization, respectively, in terms of their formation energy. Further, a rough criterion for predicting energetic co-crystal formation is obtained, as the solubility parameter difference of two coformers of a binary energetic co-crystal is less than 8 MPa0.5.
Co-reporter:Yushi Wen, Chaoyang Zhang, Xianggui Xue and Xinping Long
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 18) pp:NaN12022-12022
Publication Date(Web):2015/04/01
DOI:10.1039/C5CP00006H
Clustering is experimentally and theoretically verified during the complicated processes involved in heating high explosives, and has been thought to influence their detonation properties. However, a detailed description of the clustering that occurs has not been fully elucidated. We used molecular dynamic simulations with an improved reactive force field, ReaxFF_lg, to carry out a comparative study of cluster evolution during the early stages of heating for three representative explosives: 1,3,5-triamino-2,4,6-trinitrobenzene (TATB), β-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) and pentaerythritol tetranitrate (PETN). These representatives vary greatly in their oxygen balance (OB), molecular structure, stability and experimental sensitivity. We found that when heated, TATB, HMX and PETN differ in the size, amount, proportion and lifetime of their clusters. We also found that the clustering tendency of explosives decreases as their OB becomes less negative. We propose that the relationship between OB and clustering can be attributed to the role of clustering in detonation. That is, clusters can form more readily in a high explosive with a more negative OB, which retard its energy release, secondary decomposition, further decomposition to final small molecule products and widen its detonation reaction zone. Moreover, we found that the carbon content of the clusters increases during clustering, in accordance with the observed soot, which is mainly composed of carbon as the final product of detonation or deflagration.
Co-reporter:Chaoyang Zhang, Chi Zhang, Yu Ma and Xianggui Xue
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 17) pp:NaN11480-11480
Publication Date(Web):2015/03/24
DOI:10.1039/C5CP00926J
C black is a class of substantial materials with a long history of applications. However, apart from some descriptions of primary reactions, subsequent processes leading up to the final formation mechanism remain unclear. This mechanism is also crucial for understanding the formation of other carbonaceous materials. In this work, we visualize C black formation by acetylene pyrolysis using molecular dynamics simulations with a molecular reactive force field named ReaxFF. We find that the formation undergoes four stages: (1) chain elongation by H abstraction and polymerization of small C species, (2) chain branching, (3) cyclization and ring densification, and (4) condensed ring folding. The simulated C black particle possesses a structure of folded graphite layers, which is in good accordance with experimental observations. Cyclization and condensation are derived from fusion between neighboring chains, significantly varying from common experimental observations at relatively low temperatures that abide by the mechanism of H abstraction and C2H2 addition. Moreover, polyyne and polyene are usually found during acetylene pyrolysis, suggesting that the pyrolysis of acetylene and other hydrocarbons may be a feasible method of obtaining carbyne, a novel carbonaceous material with a high value.

![1,3,4,6-TETRANITRO-2,3A,5,6A-TETRAHYDROIMIDAZO[4,5-D]IMIDAZOLE](http://img.cochemist.com/ccimg/473800/473796-67-7.png)
![1,3,4,6-TETRANITRO-2,3A,5,6A-TETRAHYDROIMIDAZO[4,5-D]IMIDAZOLE](http://img.cochemist.com/ccimg/473800/473796-67-7_b.png)

![3-nitro-4-[(4-nitro-1,2,5-oxadiazol-3-yl)-NNO-azoxy]-1,2,5-oxadiazole](http://img.cochemist.com/ccimg/152900/152845-82-4.png)
![3-nitro-4-[(4-nitro-1,2,5-oxadiazol-3-yl)-NNO-azoxy]-1,2,5-oxadiazole](http://img.cochemist.com/ccimg/152900/152845-82-4_b.png)
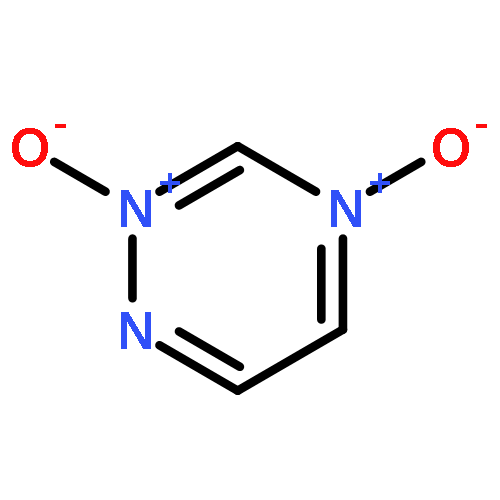
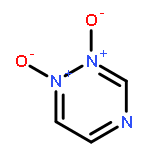


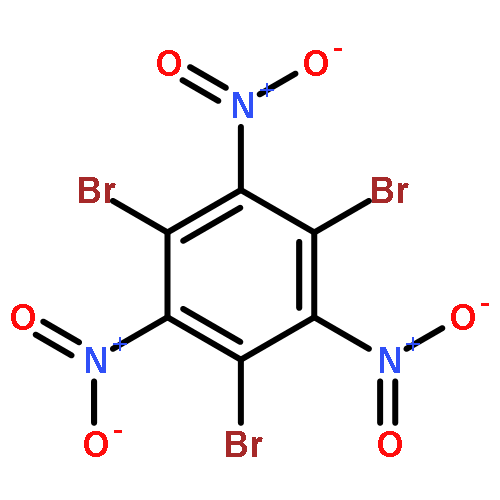
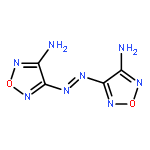
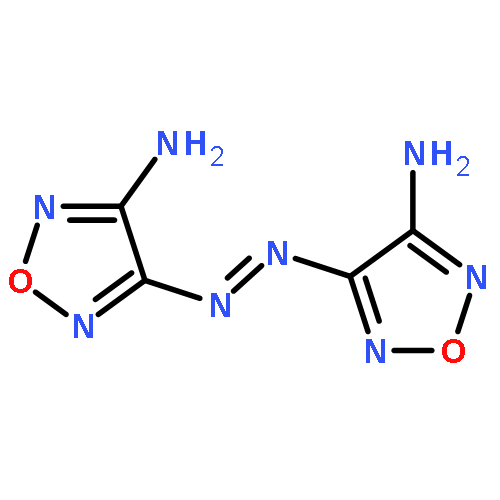
![(4-AMINO-1,2,5-OXADIAZOL-3-YL)-[(4-AMINO-1,2,5-OXADIAZOL-3-YL)IMINO]-OXIDOAZANIUM](http://img.cochemist.com/ccimg/78700/78644-89-0.png)
![(4-AMINO-1,2,5-OXADIAZOL-3-YL)-[(4-AMINO-1,2,5-OXADIAZOL-3-YL)IMINO]-OXIDOAZANIUM](http://img.cochemist.com/ccimg/78700/78644-89-0_b.png)




























![Pentacyclo[4.2.0.02,5.03,8.04,7]octane, octanitro-](http://img.cochemist.com/ccimg/99400/99393-63-2.png)
![Pentacyclo[4.2.0.02,5.03,8.04,7]octane, octanitro-](http://img.cochemist.com/ccimg/99400/99393-63-2_b.png)






![5,2,6-(Iminomethenimino)-1H-imidazo[4,5-b]pyrazine,octahydro-1,3,4,7,8,10-hexanitro-](http://img.cochemist.com/ccimg/135300/135285-90-4.png)
![5,2,6-(Iminomethenimino)-1H-imidazo[4,5-b]pyrazine,octahydro-1,3,4,7,8,10-hexanitro-](http://img.cochemist.com/ccimg/135300/135285-90-4_b.png)

