Co-reporter:Atsushi Kato;Izumi Nakagome;Shinpei Nakagawa;Kyoko Kinami;Isao Adachi;Sarah F. Jenkinson;Jérôme Désiré;Yves Blériot;Robert J. Nash;George W. J. Fleet
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 44) pp:9297-9304
Publication Date(Web):2017/11/15
DOI:10.1039/C7OB02281F
The affinity of a series of iminosugar-based inhibitors exhibiting various ring sizes toward Hex A and their essential interactions with the enzyme active site were investigated. All the Hex A-inhibiting iminosugars tested formed hydrogen bonds with Arg178, Asp322, Tyr421 and Glu462 and had the favorable cation–π interaction with Trp460. Among them, DMDP amide (6) proved to be the most potent competitive inhibitor with a Ki value of 0.041 μM. We analyzed the dynamic properties of both DMDP amide (6) and DNJNAc (1) in aqueous solution using molecular dynamics (MD) calculations; the distance of the interaction between Asp322 and 3-OH and Glu323 and 6-OH was important for stable interactions with Hex A, reducing fluctuations in the plasticity of the active site. DMDP amide (6) dose-dependently increased intracellular Hex A activity in the G269S mutant cells and restored Hex A activity up to approximately 43% of the wild type level; this effect clearly exceeded the border line treatment for Tay–Sachs disease, which is regarded as 10–15% of the wild type level. This is a significantly greater effect than that of pyrimethamine, which is currently in Phase 2 clinical trials. DMDP amide (6), therefore, represents a new promising pharmacological chaperone candidate for the treatment of Tay–Sachs disease.
Co-reporter:Daichi Hayakawa, Noriyuki Yamaotsu, Izumi Nakagome, Shin-ichiro Ozawa, Tomoki Yoshida, Shuichi Hirono
Biophysical Chemistry 2017 Volume 228(Volume 228) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.bpc.2017.06.014
•The structural flexibility of PAH is increased by the point mutation A313T.•The increase in flexibility of the PAH mutant decreases its enzymatic activity.•Binding to a pharmacological chaperone stabilizes the mutant PAH.•The enhanced stabilization contributes to restoring enzymatic activity.Phenylketonuria (PKU) is an inborn error of phenylalanine metabolism due to mutations in phenylalanine hydroxylase (PAH). Recently, small compounds, known as pharmacological chaperones (PhCs), have been identified that restore the enzymatic activity of mutant PAHs. Understanding the mechanism of the reduction in enzymatic activity due to a point mutation in PAH and its restoration by PhC binding is important for the design of more effective PhC drugs. Thermal fluctuations of an enzyme can alter its activity. Here, molecular dynamics simulation show the thermal fluctuation of PAH is increased by introduction of the A313T mutation. Moreover, a simulation using the A313T-PhC complex model was also performed. Thermal fluctuation of the mutant was found to be reduced upon PhC binding, which contributes to restoring its enzymatic activity.Download high-res image (408KB)Download full-size image
Co-reporter:Harutoshi Kato, Noriyuki Yamaotsu, Norihiko Iwazaki, Shigeaki Okamura, ... Shuichi Hirono
Drug Metabolism and Pharmacokinetics 2017 Volume 32, Issue 3(Volume 32, Issue 3) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.dmpk.2017.02.001
The constitutive androstane receptor (CAR, NR1I3) regulates the expression of numerous drug-metabolizing enzymes and transporters. The upregulation of various enzymes, including CYP2B6, by CAR activators is a critical problem leading to clinically severe drug–drug interactions (DDIs). To date, however, few effective computational approaches for identifying CAR activators exist. In this study, we aimed to develop three-dimensional quantitative structure–activity relationship (3D-QSAR) models to predict the CAR activating potency of compounds emerging in the drug-discovery process. Models were constructed using comparative molecular field analysis (CoMFA) based on the molecular alignments of ligands binding to CAR, which were obtained from ensemble ligand-docking using 28 compounds as a training set. The CoMFA model, modified by adding a lipophilic parameter with calculated logD7.4 (S+logD7.4), demonstrated statistically good predictive ability (r2 = 0.99, q2 = 0.74). We also confirmed the excellent predictability of the 3D-QSAR model for CAR activation (r2pred = 0.71) using seven compounds as a test set for external validation. Collectively, our results indicate that the 3D-QSAR model developed in this study provides precise prediction of CAR activating potency and, thus, should be useful for selecting drug candidates with minimized DDI risk related to enzyme-induction in the early drug-discovery stage.Download high-res image (257KB)Download full-size image
Co-reporter:Atsushi Kato, Izumi Nakagome, Kasumi Sato, Arisa Yamamoto, Isao Adachi, Robert J. Nash, George W. J. Fleet, Yoshihiro Natori, Yasuka Watanabe, Tatsushi Imahori, Yuichi Yoshimura, Hiroki Takahata and Shuichi Hirono
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 3) pp:1039-1048
Publication Date(Web):27 Nov 2015
DOI:10.1039/C5OB02223A
We report on the synthesis and biological evaluation of a series of α-1-C-alkylated 1,4-dideoxy-1,4-imino-D-arabinitol (DAB) derivatives as pharmacological chaperones for Gaucher disease. The parent compound, DAB, did not show inhibition of human β-glucocerebrosidase but showed moderate intestinal α-glucosidase inhibition; in contrast, extension of α-1-C-alkyl chain length gave a series of highly potent and selective inhibitors of the β-glucocerebrosidase. Our design of α-1-C-tridecyl-DAB (5j) produced a potent inhibitor of the β-glucocerebrosidase, with IC50 value of 0.77 μM. A molecular docking study revealed that the α-1-C-tridecyl group has a favorable interaction with the hydrophobic pocket and the sugar analogue part (DAB) interacted with essential hydrogen bonds formed to Asp127, Glu235 and Glu340. Furthermore, α-1-C-tridecyl-DAB (5j) displayed enhancement of activity at an effective concentration 10-times lower than isofagomine. α-1-C-Tridecyl-DAB therefore provides the first example of a pyrrolidine iminosugar as a new class of promising pharmacological chaperones with the potential for treatment of Gaucher disease.
Co-reporter:Koichi Ha;Izumi Nakagome;Noriyuki Yamaotsu;Hiroaki Gouda
Journal of Pharmaceutical Sciences 2015 Volume 104( Issue 1) pp:223-232
Publication Date(Web):
DOI:10.1002/jps.24235
The pregnane X receptor [PXR (NR1I2)] induces the expression of xenobiotic metabolic genes and transporter genes. In this study, we aimed to establish a computational method for quantifying the enzyme-inducing potencies of different compounds via their ability to activate PXR, for the application in drug discovery and development. To achieve this purpose, we developed a three-dimensional quantitative structure–activity relationship (3D-QSAR) model using comparative molecular field analysis (CoMFA) for predicting enzyme-inducing potencies, based on computer-ligand docking to multiple PXR protein structures sampled from the trajectory of a molecular dynamics simulation. Molecular mechanics-generalized born/surface area scores representing the ligand–protein-binding free energies were calculated for each ligand. As a result, the predicted enzyme-inducing potencies for compounds generated by the CoMFA model were in good agreement with the experimental values. Finally, we concluded that this 3D-QSAR model has the potential to predict the enzyme-inducing potencies of novel compounds with high precision and therefore has valuable applications in the early stages of the drug discovery process. © 2014 Wiley Periodicals, Inc. and the American Pharmacists Association J Pharm Sci 104:223–232, 2015
Co-reporter:Atsushi Kato, Zhao-Lan Zhang, Hong-Yao Wang, Yue-Mei Jia, Chu-Yi Yu, Kyoko Kinami, Yuki Hirokami, Yutaro Tsuji, Isao Adachi, Robert J. Nash, George W. J. Fleet, Jun Koseki, Izumi Nakagome, and Shuichi Hirono
The Journal of Organic Chemistry 2015 Volume 80(Issue 9) pp:4501-4515
Publication Date(Web):April 3, 2015
DOI:10.1021/acs.joc.5b00342
This paper identifies the required configuration and orientation of α-glucosidase inhibitors, miglitol, α-1-C-butyl-DNJ, and α-1-C-butyl-LAB for binding to ntSI (isomaltase). Molecular dynamics (MD) calculations suggested that the flexibility around the keyhole of ntSI is lower than that of ctSI (sucrase). Furthermore, a molecular-docking study revealed that a specific binding orientation with a CH−π interaction (Trp370 and Phe648) is a requirement for achieving a strong affinity with ntSI. On the basis of these results, a new class of nortropane-type iminosugars, labystegines, hybrid iminosugars of LAB and calystegine, have been designed and synthesized efficiently from sugar-derived cyclic nitrones with intramolecular 1,3-dipolar cycloaddition or samarium iodide catalyzed reductive coupling reaction as the key step. Biological evaluation showed that our newly designed 3(S)-hydroxy labystegine (6a) inherited the selectivity against intestinal α-glucosidases from LAB, and its inhibition potency was 10 times better than that of miglitol. Labystegine, therefore, represents a promising new class of nortropane-type iminosugar for improving postprandial hyperglycemia.
Co-reporter:Masaki Wakasugi, Hiroaki Gouda, Tomoyasu Hirose, Akihiro Sugawara, Tsuyoshi Yamamoto, Kazuro Shiomi, Toshiaki Sunazuka, Satoshi Ōmura, Shuichi Hirono
Bioorganic & Medicinal Chemistry 2013 21(11) pp: 3214-3220
Publication Date(Web):
DOI:10.1016/j.bmc.2013.03.047
Co-reporter:Hiroaki Gouda, Toshiaki Sunazuka, Kanami Iguchi, Akihiro Sugawara, Tomoyasu Hirose, Yoshihiko Noguchi, Yoshifumi Saito, Yuichi Yanai, Tsuyoshi Yamamoto, Takeshi Watanabe, Kazuro Shiomi, Satoshi Ōmura, Shuichi Hirono
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 10) pp:2630-2633
Publication Date(Web):15 May 2009
DOI:10.1016/j.bmcl.2009.04.013
Argifin, a novel pentapeptide chitinase inhibitor isolated from Gliocladium fungal culture, is a promising candidate for the development of new fungicides, insecticides, and anti-asthma medications. In this study, we undertook rational molecular design of argifin-derivatives and tested them against chitinase B from Serratia marcescens (SmChiB). The work involved molecular dynamics simulation with explicit water molecules, the molecular docking calculation, and free-energy analysis using the molecular mechanics Poisson–Boltzmann surface area method. The custom-designed derivatives were synthesized via effective solid phase synthesis, developed recently in our laboratory, and their inhibitory activities were measured against SmChiB. Finally, we identified and obtained a derivative which exhibited 28-fold more inhibition than argifin itself, a compound in which the d-Ala(5) of argifin was replaced with d-Leu and the 4-benzylpiperdine was attached to l-Asp(4).
Co-reporter:Hiroaki Gouda;Toshiaki Sunazuka;Hideaki Ui;Masaki Handa;Yusuke Sakoh;Yuzuru Iwai;Satoshi Ōmura
PNAS 2005 102 (51 ) pp:18286-18291
Publication Date(Web):2005-12-20
DOI:10.1073/pnas.0508425102
The absolute stereostructure of luminamicin, an anaerobic antibiotic, has been determined by using conformational analysis
via high-temperature molecular dynamics, NMR spectroscopy, and the modified Mosher method. It was found that luminamicin has
the S, S, R, R, R, R, S, S, S, R, and S configurations at C2, C4, C7, C9, C10, C11, C12, C13, C16, C28, and C29, respectively. This configuration is the same as
that found in nodusmicin, which has a chemical structure quite similar to luminamicin. The structure of luminamicin consists
of three different rings, i.e., a decalin ring, a 10-membered macrolactone ring, and a 14-membered macrolactone ring. The
resulting three-dimensional structure of luminamicin shows an interesting feature in that the maleic anhydride functionality
in conjugation with the enol ether group of the 14-membered macrolactone is nearly perpendicular to the plane of the other
two rings.
Co-reporter:Noriyuki Yamaotsu;Masaki Suga
Biopolymers 2001 Volume 58(Issue 4) pp:
Publication Date(Web):12 JAN 2001
DOI:10.1002/1097-0282(20010405)58:4<410::AID-BIP1017>3.0.CO;2-0
Trifluoperazine (TFP) has been widely studied in relation to its mode of binding and its inactivation of calmodulin (CaM). Most studies in solution have indicated that CaM has two high-affinity binding sites for TFP. The crystal structure of the 1:4 CaM-TFP complex (CaM-4TFP) shows that three TFP molecules bind to the C-domain of CaM, and that one TFP molecule binds to the N-domain. In contrast, the crystal structure of the 1:1 CaM-TFP complex (CaM-1TFP) shows that one TFP molecule binds to the C-domain. It has been thought that the binding of one TFP molecule to the C-domain is followed by binding to the N-domain. The crystal structure of the 1:2 CaM-TFP complex (CaM-2TFP), moreover, has recently been determined, showing that two TFP molecules bind to the C-domain. In order to determine the structure of the CaM-TFP complex and to clarify the interaction between CaM and TFP in solution, we performed a molecular dynamics simulation of the CaM-TFP complex in aqueous solution starting from the CaM-4TFP crystal structure. The obtained solution structure is very similar to the CaM-2TFP crystal structure. The computer simulation showed that the binding ability of the secondary binding site of the C-domain is higher than that of the primary binding site of the N-domain. © 2001 John Wiley & Sons, Inc. Biopolymers 58: 410–421, 2001
Co-reporter:Tomoko Koyama, Noriyuki Yamaotsu, Izumi Nakagome, Shin-ichiro Ozawa, Tomoki Yoshida, Daichi Hayakawa, Shuichi Hirono
Journal of Molecular Graphics and Modelling (March 2017) Volume 72() pp:229-239
Publication Date(Web):March 2017
DOI:10.1016/j.jmgm.2017.01.014
•Multi-step virtual screening for selective DYRK1A inhibitors was performed.•Our regression analysis revealed the key residues for potency against DYRK1A.•Of the 10 virtual hits, five compounds showed selectivity for DYRK1A over CDK5.•Two hits showed IC50 values of several μM for DYRK1A with >20-fold selectivity.Developing selective inhibitors for a particular kinase remains a major challenge in kinase-targeted drug discovery. Here we performed a multi-step virtual screening for dual-specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A) inhibitors by focusing on the selectivity for DYRK1A over cyclin-dependent kinase 5 (CDK5). To examine the key factors contributing to the selectivity, we constructed logistic regression models to discriminate between actives and inactives for DYRK1A and CDK5, respectively, using residue-based binding free energies. The residue-based parameters were calculated by molecular mechanics-generalized Born surface area (MM-GBSA) decomposition methods for kinase–ligand complexes modeled by computer ligand docking. Based on the findings from the logistic regression models, we built a three-dimensional (3D) pharmacophore model and chose filter criteria for the multi-step virtual screening. The virtual hit compounds obtained from the screening were assessed for their inhibitory activities against DYRK1A and CDK5 by in vitro assay. Our screening identified two novel selective DYRK1A inhibitors with IC50 values of several μM for DYRK1A and >100 μM for CDK5, which can be further optimized to develop more potent selective DYRK1A inhibitors.
Co-reporter:Koichi Handa, Izumi Nakagome, Noriyuki Yamaotsu, Hiroaki Gouda, Shuichi Hirono
Journal of Pharmaceutical Sciences (January 2015) Volume 104(Issue 1) pp:223-232
Publication Date(Web):1 January 2015
DOI:10.1002/jps.24235
The pregnane X receptor [PXR (NR1I2)] induces the expression of xenobiotic metabolic genes and transporter genes. In this study, we aimed to establish a computational method for quantifying the enzyme-inducing potencies of different compounds via their ability to activate PXR, for the application in drug discovery and development. To achieve this purpose, we developed a three-dimensional quantitative structure–activity relationship (3D-QSAR) model using comparative molecular field analysis (CoMFA) for predicting enzyme-inducing potencies, based on computer-ligand docking to multiple PXR protein structures sampled from the trajectory of a molecular dynamics simulation. Molecular mechanics-generalized born/surface area scores representing the ligand–protein-binding free energies were calculated for each ligand. As a result, the predicted enzyme-inducing potencies for compounds generated by the CoMFA model were in good agreement with the experimental values. Finally, we concluded that this 3D-QSAR model has the potential to predict the enzyme-inducing potencies of novel compounds with high precision and therefore has valuable applications in the early stages of the drug discovery process.
Co-reporter:Atsushi Kato, Izumi Nakagome, Kasumi Sato, Arisa Yamamoto, Isao Adachi, Robert J. Nash, George W. J. Fleet, Yoshihiro Natori, Yasuka Watanabe, Tatsushi Imahori, Yuichi Yoshimura, Hiroki Takahata and Shuichi Hirono
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 3) pp:NaN1048-1048
Publication Date(Web):2015/11/27
DOI:10.1039/C5OB02223A
We report on the synthesis and biological evaluation of a series of α-1-C-alkylated 1,4-dideoxy-1,4-imino-D-arabinitol (DAB) derivatives as pharmacological chaperones for Gaucher disease. The parent compound, DAB, did not show inhibition of human β-glucocerebrosidase but showed moderate intestinal α-glucosidase inhibition; in contrast, extension of α-1-C-alkyl chain length gave a series of highly potent and selective inhibitors of the β-glucocerebrosidase. Our design of α-1-C-tridecyl-DAB (5j) produced a potent inhibitor of the β-glucocerebrosidase, with IC50 value of 0.77 μM. A molecular docking study revealed that the α-1-C-tridecyl group has a favorable interaction with the hydrophobic pocket and the sugar analogue part (DAB) interacted with essential hydrogen bonds formed to Asp127, Glu235 and Glu340. Furthermore, α-1-C-tridecyl-DAB (5j) displayed enhancement of activity at an effective concentration 10-times lower than isofagomine. α-1-C-Tridecyl-DAB therefore provides the first example of a pyrrolidine iminosugar as a new class of promising pharmacological chaperones with the potential for treatment of Gaucher disease.

![Cyclo[(aS)-a-[(3S,5R)-3-amino-5-hydroxy-2-oxo-1-pyrrolidinyl]-1H-imidazole-5-propanoyl-5-carboxy-L-norvalyl-N5-[(acetylamino)iminomethyl]-L-ornithyl-D-prolyl]](http://img.cochemist.com/ccimg/289700/289665-92-5.png)
![Cyclo[(aS)-a-[(3S,5R)-3-amino-5-hydroxy-2-oxo-1-pyrrolidinyl]-1H-imidazole-5-propanoyl-5-carboxy-L-norvalyl-N5-[(acetylamino)iminomethyl]-L-ornithyl-D-prolyl]](http://img.cochemist.com/ccimg/289700/289665-92-5_b.png)
![Cyclo[D-alanyl-N5-[imino[[(methylamino)carbonyl]amino]methyl]-L-ornithyl-N-methyl-L-phenylalanyl-L-b-aspartyl-L-b-aspartyl]](http://img.cochemist.com/ccimg/244000/243975-37-3.png)
![Cyclo[D-alanyl-N5-[imino[[(methylamino)carbonyl]amino]methyl]-L-ornithyl-N-methyl-L-phenylalanyl-L-b-aspartyl-L-b-aspartyl]](http://img.cochemist.com/ccimg/244000/243975-37-3_b.png)
![(2E)-N-[(5alpha,6beta)-17-(cyclopropylmethyl)-3,14-dihydroxy-4,5-epoxymorphinan-6-yl]-3-furan-3-yl-N-methylprop-2-enamide](http://img.cochemist.com/ccimg/152700/152657-84-6.png)
![(2E)-N-[(5alpha,6beta)-17-(cyclopropylmethyl)-3,14-dihydroxy-4,5-epoxymorphinan-6-yl]-3-furan-3-yl-N-methylprop-2-enamide](http://img.cochemist.com/ccimg/152700/152657-84-6_b.png)
![7-Hydroxy-[1,2,4]triazolo[4,3-a]pyrimidine-6-carboxylic acid](http://img.cochemist.com/ccimg/937800/937700-95-3.png)
![7-Hydroxy-[1,2,4]triazolo[4,3-a]pyrimidine-6-carboxylic acid](http://img.cochemist.com/ccimg/937800/937700-95-3_b.png)




![Propanal, 2-[(4-methoxyphenyl)methoxy]-, (2R)-](http://img.cochemist.com/ccimg/218500/218435-12-2.png)
![Propanal, 2-[(4-methoxyphenyl)methoxy]-, (2R)-](http://img.cochemist.com/ccimg/218500/218435-12-2_b.png)






![Benzenepropanamine, g-[4-(trifluoromethyl)phenoxy]-, (gS)-](http://img.cochemist.com/ccimg/127000/126924-38-7.png)
![Benzenepropanamine, g-[4-(trifluoromethyl)phenoxy]-, (gS)-](http://img.cochemist.com/ccimg/127000/126924-38-7_b.png)




![b-D-Allopyranoside,(3aR,4R,5R,6S,6aS)-2-(dimethylamino)-3a,5,6,6a-tetrahydro-4-hydroxy-6-(hydroxymethyl)-4H-cyclopentoxazol-5-yl2-(acetylamino)-4-O-[2-(acetylamino)-2-deoxy-b-D-allopyranosyl]-2-deoxy-](http://img.cochemist.com/ccimg/103800/103782-08-7.png)
![b-D-Allopyranoside,(3aR,4R,5R,6S,6aS)-2-(dimethylamino)-3a,5,6,6a-tetrahydro-4-hydroxy-6-(hydroxymethyl)-4H-cyclopentoxazol-5-yl2-(acetylamino)-4-O-[2-(acetylamino)-2-deoxy-b-D-allopyranosyl]-2-deoxy-](http://img.cochemist.com/ccimg/103800/103782-08-7_b.png)
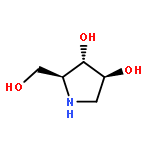
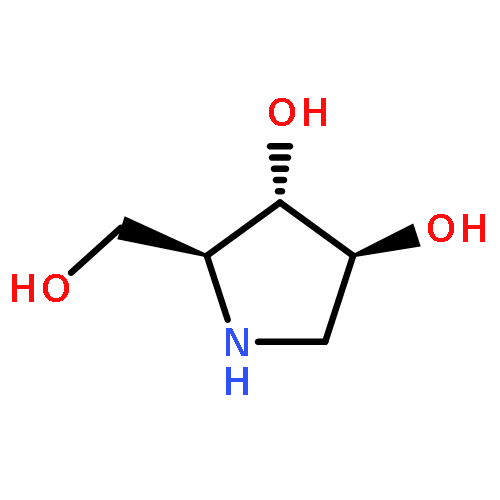
![(S)-N-Methyl-gamma-[4-(trifluoromethyl)phenoxy]benzenepropanamine](http://img.cochemist.com/ccimg/100600/100568-02-3.png)
![(S)-N-Methyl-gamma-[4-(trifluoromethyl)phenoxy]benzenepropanamine](http://img.cochemist.com/ccimg/100600/100568-02-3_b.png)


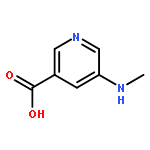
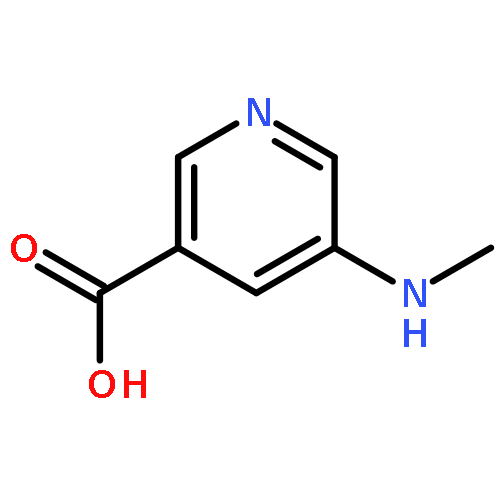
![4-(2-CHLORO-11H-DIBENZO[B,E][1,4]OXATHIEPIN-11-YL)-1-METHYLPIPERI<WBR />DINE (2E)-2-BUTENEDIOATE (1:1)](http://img.cochemist.com/ccimg/72800/72782-05-9.png)
![4-(2-CHLORO-11H-DIBENZO[B,E][1,4]OXATHIEPIN-11-YL)-1-METHYLPIPERI<WBR />DINE (2E)-2-BUTENEDIOATE (1:1)](http://img.cochemist.com/ccimg/72800/72782-05-9_b.png)


![4,5-epoxy-3-methoxy-17-methyl-[9,16-14C]morphin-7-en-6-ol](http://img.cochemist.com/ccimg/16300/16206-70-5.png)
![4,5-epoxy-3-methoxy-17-methyl-[9,16-14C]morphin-7-en-6-ol](http://img.cochemist.com/ccimg/16300/16206-70-5_b.png)


![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2_b.png)