Co-reporter:Jiefeng Hu;Xiaowei Han; Dr. Yu Yuan; Dr. Zhuangzhi Shi
Angewandte Chemie 2017 Volume 129(Issue 43) pp:13527-13531
Publication Date(Web):2017/10/16
DOI:10.1002/ange.201708224
AbstractA copper catalytic system was established for the stereoselective hydrodefluorination of gem-difluoroalkenes through C−F activation to synthesize various Z fluoroalkenes. H2O is used as the hydrogen source for the fluorine acceptor moiety. This mild catalytic system shows good-functional group compatibility, accepting a range of carbonyls as precursors to the gem-difluoroalkenes, including aliphatic, aromatic, and α,β-unsaturated aldehydes and even ketones. It serves as a powerful synthetic method for the late-stage modification of complex compounds.
Co-reporter:Binlin Zhao; Dr. Zhuangzhi Shi
Angewandte Chemie International Edition 2017 Volume 56(Issue 41) pp:12727-12731
Publication Date(Web):2017/10/02
DOI:10.1002/anie.201707181
AbstractThe first example of intermolecular olefination of cyclobutanone oximes with alkenes via selective C−C bond cleavage leading to the synthesis of nitriles in the presence of a cheap copper catalyst is reported. The procedure is distinguished by mild and safe reaction conditions that avoid ligand, oxidant, base, or toxic cyanide salt. A wide scope of cyclobutanones and olefin coupling components can be used without compromising efficiency and scalability. The alternative visible-light-driven photoredox process for this coupling reaction was also uncovered.
Co-reporter:Arun Jyoti Borah
Chemical Communications 2017 vol. 53(Issue 28) pp:3945-3948
Publication Date(Web):2017/04/04
DOI:10.1039/C7CC01274H
An exclusive catalytic C4-selective fluoroalkylation of indoles with highly active (1H, 1H-perfluoroalkyl)mesityliodonium triflate has been described. The key to its high regioselectivity is the appropriate choice of an easily accessible, cheap and removable directing group at the C3 position in the presence of a Pd(OAc)2 catalyst. Besides indole fluoroalkylation, the application of this strategy in other heteroarenes such as benzo[b]thiophene is also described.
Co-reporter:Xiaodong Qiu;Dr. Minyan Wang;Yue Zhao; Dr. Zhuangzhi Shi
Angewandte Chemie 2017 Volume 129(Issue 25) pp:7339-7343
Publication Date(Web):2017/06/12
DOI:10.1002/ange.201703354
AbstractModification of commercially available monophosphine ligands with either aryl bromides or chlorides by rhodium(I)-catalyzed, tertiary phosphine directed C−H activation is described. A series of ligand libraries containing mono- and diaryl-substituted groups, having different steric and electronic properties, were obtained in high yields. Based on the outstanding properties of their parent scaffolds, the modified ligands have been found to be powerful in organic reactions.
Co-reporter:Xiaodong Qiu;Dr. Minyan Wang;Yue Zhao; Dr. Zhuangzhi Shi
Angewandte Chemie International Edition 2017 Volume 56(Issue 25) pp:7233-7237
Publication Date(Web):2017/06/12
DOI:10.1002/anie.201703354
AbstractModification of commercially available monophosphine ligands with either aryl bromides or chlorides by rhodium(I)-catalyzed, tertiary phosphine directed C−H activation is described. A series of ligand libraries containing mono- and diaryl-substituted groups, having different steric and electronic properties, were obtained in high yields. Based on the outstanding properties of their parent scaffolds, the modified ligands have been found to be powerful in organic reactions.
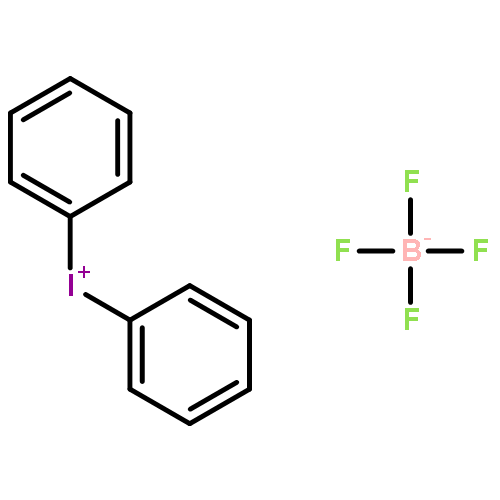
Co-reporter:Youqing Yang; Ruirui Li; Yue Zhao; Dongbing Zhao
Journal of the American Chemical Society 2016 Volume 138(Issue 28) pp:8734-8737
Publication Date(Web):June 30, 2016
DOI:10.1021/jacs.6b05777
The first example of direct and site-selective arylation of indoles at the C6 position has been reported. The key to this high regioselectivity is the appropriate choice of the N–P(O)tBu2 directing group and the use of diaryliodonium triflate salts as the coupling partners in the presence of catalytic CuO. The protocol is distinguished by mild reaction system that avoids ligand and additives, exhibiting wide scope of indole and arene coupling components without compromising its efficiency and scalability, thus representing a significant advancement in the implementation of regioselective direct arylation of indoles.
Co-reporter:Xinghui Pu, Jiefeng Hu, Yue Zhao, and Zhuangzhi Shi
ACS Catalysis 2016 Volume 6(Issue 10) pp:6692
Publication Date(Web):August 30, 2016
DOI:10.1021/acscatal.6b01956
The nickel-catalyzed direct borylation and silylation of phenolic esters has been established. The key to this highly efficient acyl C–O bond borylative and silylative cleavage depends on the appropriate choice of different ligands and additives in the presence of nickel catalyst. Both transformations exhibit good functional group compatibility and can serve as powerful synthetic tools for late-stage functionalization of complex compounds. The elucidation of key mechanistic features of this newly developed acyl C–O bond activation in esters was confirmed by two well-characterized organonickel(II) intermediates.Keywords: borylation; decarbonylation; esters; nickel; silylation
Co-reporter:Pan Gao, Li Liu, Zhuangzhi Shi and Yu Yuan
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 29) pp:7109-7113
Publication Date(Web):29 Jun 2016
DOI:10.1039/C6OB01145D
A regioselective direct arylation of arenes and olefins at the ortho position is reported. The key to the high selectivity is the appropriate choice of diaryliodonium salts as the arylating reagent in the presence of a cationic iridium(III) catalyst. The coordination of the metal with an oxygen atom or a nitrogen atom and subsequent C–H activation allows for direct arylation with coupling partners. This reaction proceeds under mild reaction conditions and with a high tolerance of various functional groups including many halide functional groups.
Co-reporter:Binlin Zhao;Yu Yuan
The Chemical Record 2016 Volume 16( Issue 2) pp:886-896
Publication Date(Web):
DOI:10.1002/tcr.201500241
Abstract
In the past decade, transition-metal-catalyzed C–H activations have been very popular in the research field of organometallic chemistry, and have been considered as efficient and convenient strategies to afford complex natural products, functional advanced materials, fluorescent compounds, and pharmaceutical compounds. In this account, we begin with a brief introduction to the development of transition-metal-catalyzed C–H activation, especially the development of transition-metal-catalyzed chelation-assisted C–H activation. Then, a more detailed discussion is directed towards our recent studies on the transition-metal-catalyzed chelation-assisted oxidative C–H/C–H functionalization of aromatic substrates bearing directing functional groups.
Co-reporter:Jiefeng Hu, Heqing Sun, Wangshui Cai, Xinghui Pu, Yemin Zhang, and Zhuangzhi Shi
The Journal of Organic Chemistry 2016 Volume 81(Issue 1) pp:14-24
Publication Date(Web):December 2, 2015
DOI:10.1021/acs.joc.5b02557
By developing a mild Ni-catalyzed system, a method for direct borylation of sp2 and sp3 C–N bonds has been established. The key to this hightly efficient C–N bond borylative cleavage depends on the appropriate choice of the nickel catalyst Ni(COD)2, ICy·HCl as a ligand, and the use of 2-ethoxyethanol as the cosolvent. This transformation shows good functional group compatibility and can serve as a powerful synthetic tool for gram-scale synthesis and late-stage C–N borylation of complex compounds.
Co-reporter:Pan Gao; Wei Guo; Jingjing Xue; Yue Zhao; Yu Yuan; Yuanzhi Xia
Journal of the American Chemical Society 2015 Volume 137(Issue 38) pp:12231-12240
Publication Date(Web):September 8, 2015
DOI:10.1021/jacs.5b06758
By developing a new Ir(III)-catalyzed C–C cross-coupling, a versatile method for direct arylation of sp2 and sp3 C–H bonds in ketoximes, nitrogen-containing heterocycles, various arenes, and olefins has been established. The key to this arylation depends on the appropriate choice of catalyst and the use of diaryliodonium triflate salts as the coupling partners. This transformation has good functional group compatibility and can serve as a powerful synthetic tool for late-stage C–H arylation of complex compounds. Mechanistic studies by density functional theory calculations suggested that the sp3 C–H activation was realized by a triflate-involved concerted metalation–deprotonation process, and the following oxidation of Ir(III) to Ir(V) is the most favorable when a bistriflimide is contained in the diaryliodonium salt. Calculations indicated that both steps are enabled by initial anion exchange between the reactant complexes.
Co-reporter:Youqing Yang; Xiaodong Qiu; Yue Zhao; Yucheng Mu
Journal of the American Chemical Society 2015 Volume 138(Issue 2) pp:495-498
Publication Date(Web):December 28, 2015
DOI:10.1021/jacs.5b11569
In the past decade, direct C–H arylation of indoles has been developed with high selectivity at the C2 and C3 positions via transition-metal-catalyzed cross-coupling reactions. Here we show that C–H activation can be directed to the C7 position with high selectivity in Pd-catalyzed coupling of indoles with arylboronic acids. The key to this high regioselectivity is the appropriate choice of a phosphinoyl directing group and a pyridine-type ligand in the presence of Pd(OAc)2 catalyst. This previously elusive transformation should provide insight for the design of other cross-couplings as well.
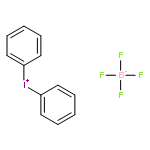
Co-reporter:Chendan Zhu; Yong Liang; Xin Hong; Heqing Sun; Wei-Yin Sun; K. N. Houk
Journal of the American Chemical Society 2015 Volume 137(Issue 24) pp:7564-7567
Publication Date(Web):June 2, 2015
DOI:10.1021/jacs.5b03488
A new strategy is reported for intramolecular sp3 C–H amination under mild reaction conditions using iodoarene as catalyst and m-CPBA as oxidant. This C–H functionalization involving iodine(III) reagents generated in situ occurs readily at sterically hindered tertiary C–H bonds. DFT (M06-2X) calculations show that the preferred pathway involves an iodonium cation intermediate and proceeds via an energetically concerted transition state, through hydride transfer followed by the spontaneous C–N bond formation. This leads to the experimentally observed amination at a chiral center without loss of stereochemical information.
Co-reporter:Pan Gao, Li Liu, Zhuangzhi Shi and Yu Yuan
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 29) pp:NaN7113-7113
Publication Date(Web):2016/06/29
DOI:10.1039/C6OB01145D
A regioselective direct arylation of arenes and olefins at the ortho position is reported. The key to the high selectivity is the appropriate choice of diaryliodonium salts as the arylating reagent in the presence of a cationic iridium(III) catalyst. The coordination of the metal with an oxygen atom or a nitrogen atom and subsequent C–H activation allows for direct arylation with coupling partners. This reaction proceeds under mild reaction conditions and with a high tolerance of various functional groups including many halide functional groups.
Co-reporter:Yucheng Mu, Xiaodong Tan, Yemin Zhang, Xiaobi Jing and Zhuangzhi Shi
Inorganic Chemistry Frontiers 2016 - vol. 3(Issue 3) pp:NaN384-384
Publication Date(Web):2016/01/21
DOI:10.1039/C5QO00438A
A Pd(II)-catalyzed selective β-arylation of O-methyl ketoximes was developed using iodoarenes as the coupling partners. This transformation has good functional group compatibility and can serve as a powerful synthetic tool for late-stage C–H arylation of complex compounds. Moreover, when employing 2-iodobenzoic acids as the substrates in this developed catalytic system, a type of unexpected five-membered lactones could be formed by tandem sp3 C–H arylation and oxygenation.
Co-reporter:Arun Jyoti Borah and Zhuangzhi Shi
Chemical Communications 2017 - vol. 53(Issue 28) pp:NaN3948-3948
Publication Date(Web):2017/03/06
DOI:10.1039/C7CC01274H
An exclusive catalytic C4-selective fluoroalkylation of indoles with highly active (1H, 1H-perfluoroalkyl)mesityliodonium triflate has been described. The key to its high regioselectivity is the appropriate choice of an easily accessible, cheap and removable directing group at the C3 position in the presence of a Pd(OAc)2 catalyst. Besides indole fluoroalkylation, the application of this strategy in other heteroarenes such as benzo[b]thiophene is also described.

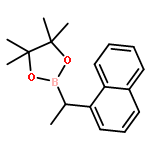
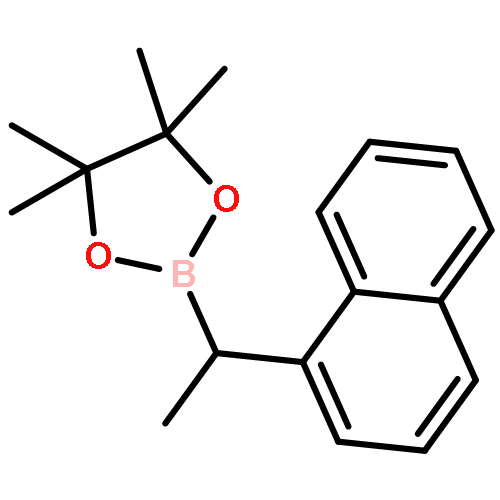




![9-Ethyl-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-9H-carbazole](http://img.cochemist.com/ccimg/1020700/1020657-86-6.png)
![9-Ethyl-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-9H-carbazole](http://img.cochemist.com/ccimg/1020700/1020657-86-6_b.png)






















![DIBENZ[B,F]AZOCIN-6(5H)-ONE, 11,12-DIHYDRO-5-METHOXY-](http://img.cochemist.com/ccimg/517900/517873-48-2.png)
![DIBENZ[B,F]AZOCIN-6(5H)-ONE, 11,12-DIHYDRO-5-METHOXY-](http://img.cochemist.com/ccimg/517900/517873-48-2_b.png)
![6H-Dibenz[b,e]azepin-6-one, 5,11-dihydro-5-methoxy-](http://img.cochemist.com/ccimg/517900/517873-47-1.png)
![6H-Dibenz[b,e]azepin-6-one, 5,11-dihydro-5-methoxy-](http://img.cochemist.com/ccimg/517900/517873-47-1_b.png)




![[1,1'-Biphenyl]-4,4'-diamine, 2-iodo-](http://img.cochemist.com/ccimg/400800/400763-39-5.png)
![[1,1'-Biphenyl]-4,4'-diamine, 2-iodo-](http://img.cochemist.com/ccimg/400800/400763-39-5_b.png)
![1,3,2-Dioxaborolane, 4,4,5,5-tetramethyl-2-[(3-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/365600/365564-12-1.png)
![1,3,2-Dioxaborolane, 4,4,5,5-tetramethyl-2-[(3-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/365600/365564-12-1_b.png)
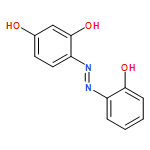
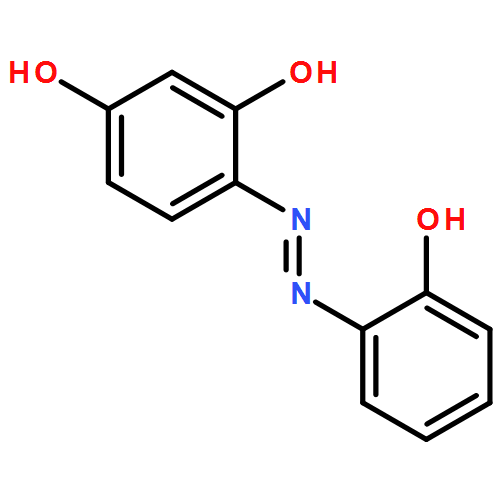
![Carbamic acid,[4-(dimethylamino)phenyl]-, 1,1-dimethylethyl ester (9CI)](http://img.cochemist.com/ccimg/290400/290365-83-2.png)
![Carbamic acid,[4-(dimethylamino)phenyl]-, 1,1-dimethylethyl ester (9CI)](http://img.cochemist.com/ccimg/290400/290365-83-2_b.png)


















![4'-(Methoxycarbonyl)-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/110000/109963-61-3.png)
![4'-(Methoxycarbonyl)-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/110000/109963-61-3_b.png)



























![[1,1'-Biphenyl]-4-carboxylic acid, phenyl ester](http://img.cochemist.com/ccimg/78400/78322-97-1.png)
![[1,1'-Biphenyl]-4-carboxylic acid, phenyl ester](http://img.cochemist.com/ccimg/78400/78322-97-1_b.png)


![Acetamide, N-(4'-methyl[1,1'-biphenyl]-2-yl)-](http://img.cochemist.com/ccimg/76500/76472-82-7.png)
![Acetamide, N-(4'-methyl[1,1'-biphenyl]-2-yl)-](http://img.cochemist.com/ccimg/76500/76472-82-7_b.png)




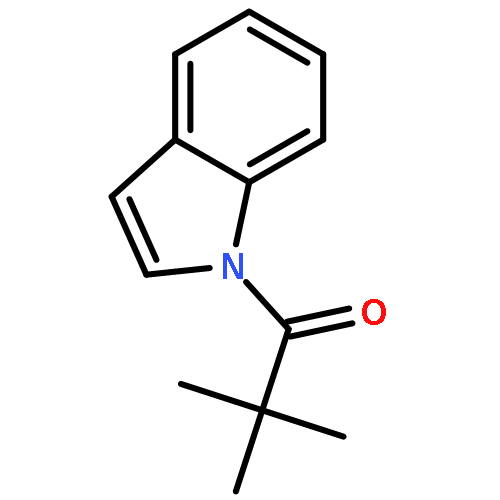


![[1,1'-Biphenyl]-2-carboxamide, N,4'-dimethyl-](http://img.cochemist.com/ccimg/66100/66087-66-9.png)
![[1,1'-Biphenyl]-2-carboxamide, N,4'-dimethyl-](http://img.cochemist.com/ccimg/66100/66087-66-9_b.png)








![1-(Benzo[d][1,3]dioxol-5-yl)-N,N-dimethylmethanamine](http://img.cochemist.com/ccimg/59000/58995-64-5.png)
![1-(Benzo[d][1,3]dioxol-5-yl)-N,N-dimethylmethanamine](http://img.cochemist.com/ccimg/59000/58995-64-5_b.png)


















![[1,1'-Biphenyl]-2-amine,4,4'-dinitro-](http://img.cochemist.com/ccimg/51800/51787-75-8.png)
![[1,1'-Biphenyl]-2-amine,4,4'-dinitro-](http://img.cochemist.com/ccimg/51800/51787-75-8_b.png)


















![Benzoic acid, 4-[(methylamino)carbonyl]-](http://img.cochemist.com/ccimg/23800/23754-45-2.png)
![Benzoic acid, 4-[(methylamino)carbonyl]-](http://img.cochemist.com/ccimg/23800/23754-45-2_b.png)


















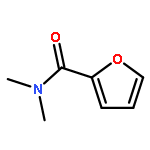
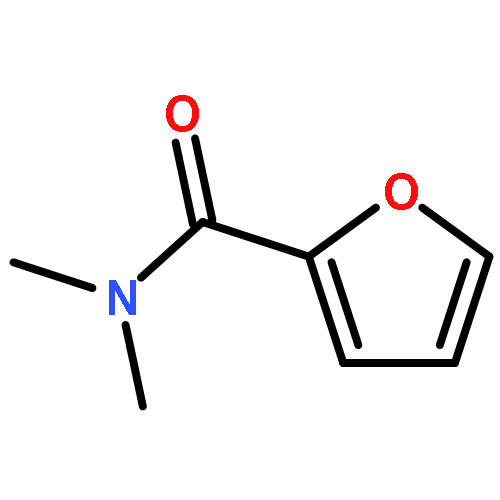


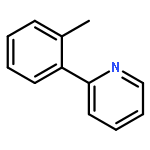




![[1,1'-Biphenyl]-2-amine, 4,4'-dichloro-](http://img.cochemist.com/ccimg/4900/4810-30-4.png)
![[1,1'-Biphenyl]-2-amine, 4,4'-dichloro-](http://img.cochemist.com/ccimg/4900/4810-30-4_b.png)










































![1H-INDOLE, 1-[BIS(1,1-DIMETHYLETHYL)PHOSPHINYL]-](http://img.cochemist.com/ccimg/548800/548775-56-0.png)
![1H-INDOLE, 1-[BIS(1,1-DIMETHYLETHYL)PHOSPHINYL]-](http://img.cochemist.com/ccimg/548800/548775-56-0_b.png)
![1,3,2-Dioxaborinane, 5,5-dimethyl-2-[4-(trifluoromethyl)phenyl]-](/data/chemimg/1128200/501374-30-7.png)
![1,3,2-Dioxaborinane, 5,5-dimethyl-2-[4-(trifluoromethyl)phenyl]-](/data/chemimg/1128200/501374-30-7_b.png)






![1,3,2-Dioxaborinane, 2-[1,1'-biphenyl]-4-yl-5,5-dimethyl-](http://img.cochemist.com/ccimg/5200/5123-05-7.png)
![1,3,2-Dioxaborinane, 2-[1,1'-biphenyl]-4-yl-5,5-dimethyl-](http://img.cochemist.com/ccimg/5200/5123-05-7_b.png)