Co-reporter:Houluo Cong;Sen Xu 郑思珣
Chinese Journal of Polymer Science 2017 Volume 35( Issue 12) pp:1572-1586
Publication Date(Web):29 September 2017
DOI:10.1007/s10118-017-2001-y
A series of mixed, random cylindrical brush copolymers bearing polystyrene (PS) and poly(ε-caprolactone) (PCL) side chains were synthesized via the combination of ring-opening polymerization (ROP) and atom transfer radical polymerization (ATRP). These novel cylindrical brush copolymers have been characterized by means of nuclear magnetic resonance (NMR) spectroscopy, gel permeation chromatography (GPC) and differential scanning calorimetry (DSC). It was found that the mikto-armed cylindrical brush copolymers were microphase-separated in bulks and that the morphologies were dependent on the mass ratios of PS to PCL side chains. One of the cylindrical brush copolymers was employed to incorporate into epoxy thermoset to investigate effect of the mikto-armed cylindrical brush architecture on the reaction-induced microphase separation behavior. Depending on the concentration of the cylindrical brush in epoxy, the thermosets can display the morphologies with the spherical, worm-like and lamellar PS microdomains dispersing in continuous thermosetting matrices.
Co-reporter:Sen Xu, Chongyin Zhang, Lei Li, Sixun Zheng
Polymer 2017 Volume 128(Volume 128) pp:
Publication Date(Web):16 October 2017
DOI:10.1016/j.polymer.2017.09.002
•PS-b-PE-b-PS triblock copolymers were synthesized via sequential polymerizations.•Microphase-separated morphologies depended on the compositions of copolymers.•The triblock copolymers displayed crystallization-driven self-assembly behavior.In this work, we reported the synthesis of a series of novel semicrystalline ABA triblock copolymers with polyethylene midblock and polystyrene endblocks via the combination of ring-opening metathesis polymerization (ROMP), hydrogenation and reversible addition-fragmentation chain transfer (RAFT) polymerization. First, an α,ω-dihydroxyl-terminated polycyclooctadiene (PCOD) was synthesized via the ROMP of cyclooctadiene by the use of the Grubbs second generation catalyst and with cis-2-butene-1,4-diol as the chain transfer agent. The α,ω-dihydroxyl-terminated PCOD was then hydrogenated into an α,ω-dihydroxyl-terminated polyethylene (PE); the latter was allowed to react with 2-methyl-2-[(dodecylsulfanylthiocarbonyl) sulfanyl]propanoic acid to afford a macromolecular chain transfer agent (i.e., Macro-CTA) for the RAFT polymerization of styrene. By controlling the lengths of PS endblocks, a series of polystyrene-block-polyethylene-block-polystyrene (PS-b-PE-b-PS) triblock copolymers were obtained. The successful synthesis of the ABA triblock copolymers were demonstrated by means of nuclear magnetic resonance (NMR) spectroscopy and gel permeation chromatography (GPC). The results of differential scanning calorimetry (DSC) and atomic force microscopy (AFM) indicate that the PS-b-PE-b-PS triblock copolymers were microphase-separated in bulks and their morphologies were quite dependent on the mass ratios of PE to PS blocks. Transmission electron microscopy (TEM), atomic force microscopy (AFM) and dynamic light scattering (DLS) showed that the triblock copolymers displayed crystallization-driven self-assembly (CDSA) behavior in selective solvent (viz. 1,4-dioxane). Depending on the lengths of PS blocks, the CDSA behavior of all the triblock copolymers generated spherical and cylindrical micelles. It was found that the quantity of the cylindrical micelles increased with decreasing the lengths of PS blocks.Download high-res image (215KB)Download full-size image
Co-reporter:Ning Liu, Lei Li, Lei Wang, Sixun Zheng
Polymer 2017 Volume 109() pp:254-265
Publication Date(Web):27 January 2017
DOI:10.1016/j.polymer.2016.12.049
•A well-defined POSS diamine was used for the synthesis of linear polybenzoxazines.•The linear polybenzoxazine copolymers displayed good film-forming properties.•The main-chain POSS significantly affected the curing kinetics of the thermosets.A series of organic-inorganic polybenzoxazine (PBZ) copolymers with double deck silsesquioxane (DDSQ) in the main chains were synthesized via Mannich polycondensation. First, 3,13-dianilino DDSQ, a well-defined polyhedral oligomeric silsesquioxane (POSS) diamine was synthesized. Thereafter, this POSS diamine together with 4,4-diaminodiphenylmethane was utilized to react with 4,4’-dihydroxyldiphenylisopropane and formaldehyde to obtain the PBZ hybrids with DDSQ in the main chains. The high-molecular-weight products were obtained and they displayed excellent film-forming properties. Transmission electron microscopy showed that the PBZ-DDSQ copolymers were microphase-separated in bulks; the spherical POSS microdomains with the diameter of 10–20 nm were dispersed in the continuous PBZ matrix. Compared to plain PBZ, the PBZ-DDSQ copolymers displayed enhanced surface hydrophobicity as evidenced with the static contact angle measurements. These PBZ-DDSQ copolymers can easily undergo the thermally-activated ring-opening polymerization and the organic-inorganic thermosetting nanocomposites were obtained. The curing kinetics showed that the inclusion of DDSQ in the main chains of PBZ enhanced the activation energy of curing reaction. All the organic-inorganic thermosets possessed the enhanced thermal stability as evidenced with thermogravimetric analysis.In this contribution, we reported the synthesis of a series of organic-inorganic polybenzoxazine (PBZ) copolymers with double deck silsesquioxane (DDSQ) in the main chains via Mannich polycondensation. Their thermally activated ring-opening polymerization behavior and thermomechanical properties have been investigated in this work.
Co-reporter:
Journal of Polymer Science Part B: Polymer Physics 2017 Volume 55(Issue 7) pp:587-600
Publication Date(Web):2017/04/01
DOI:10.1002/polb.24303
ABSTRACTIn this work, we synthesized a novel organic–inorganic semitelechelic polymer from polyhedral oligomeric silsesquioxane (POSS) and poly(acrylate amide) (PAA) via reversible addition-fragmentation chain transfer (RAFT) polymerization. The organic–inorganic semitelechelic polymers have been characterized by means of nuclear magnetic resonance spectroscopy, thermal gravimetric analysis, and dynamic mechanical thermal analysis. It was found that capping POSS groups to the single ends of PAA chains caused a series of significant changes in the morphologies and thermomechanical properties of the polymer. The organic–inorganic semitelechelics were microphase-separated; the POSS microdomains were formed via the POSS–POSS interactions. In a selective solvent (e.g., methanol), the organic–inorganic semitelechelics can be self-assembled into the micelle-like nanoobjects. Compared to plain PAA, the POSS-capped PAAs significantly displayed improved surface hydrophobicity as evidenced by the measurements of static contact angles and surface atomic force microscopy. More importantly, the organic–inorganic semitelechelics displayed typical shape memory properties, which was in marked contrast to plain PAA. The shape memory behavior is attributable to the formation of the physically cross-linked networks from the combination of the POSS–POSS interactions with the intermolecular hydrogen-bonding interactions in the organic–inorganic semitelechelics. © 2017 Wiley Periodicals, Inc. J. Polym. Sci., Part B: Polym. Phys. 2017, 55, 587–600
Co-reporter:Ning Liu, Kun Wei, Lei Wang and Sixun Zheng
Polymer Chemistry 2016 vol. 7(Issue 5) pp:1158-1167
Publication Date(Web):06 Jan 2016
DOI:10.1039/C5PY01827G
In this contribution, we report a facile synthesis of a well-defined POSS diamine via the Heck reaction. First, 3,13-divinyloctaphenyl double decker silsesquioxane (3,13-divinyl DDSQ) was synthesized via the silylation reaction of octaphenyldicycloocatasiloxane tetrasodium silanolate with methylvinyldichlorosilane. Thereafter, the 3,13-divinyl DDSQ was reacted with 4-bromoaniline with a palladium catalyst. This POSS diamine (viz. 3,13-dianilino DDSQ) has been employed to synthesize the organic–inorganic polyimides with DDSQ in the main chains. Compared to the plain polyimide, the organic–inorganic hybrids displayed improved thermal stability and surface hydrophobicity. The dielectric analysis indicates that the dielectric constants of the organic–inorganic polyimides were significantly decreased with the inclusion of the DDSQ in the main chains whereas the dielectric loss of the polyimides remained almost unchanged.
Co-reporter:Jingang Li, Lei Li, Yixin Xiang, and Sixun Zheng
Industrial & Engineering Chemistry Research 2016 Volume 55(Issue 3) pp:586-596
Publication Date(Web):January 12, 2016
DOI:10.1021/acs.iecr.5b04057
The nanostructured thermosets involving epoxy and poly(methyl methacrylate)-grafted poly(vinylidene fluoride-co-trifluoroethylene) copolymer (P(VDF-TrFE)-g-PMMA) were successfully prepared in this work. The P(VDF-TrFE)-g-PMMA graft copolymer was synthesized via the atom transfer radical polymerization of methyl methacrylate (MMA) with poly(vinylidene fluoride-co-chlorotrifluoroethylene) copolymer as the macromolecular initiator. It was characterized by means of proton nuclear magnetic resonance spectroscopy, gel permeation chromatography, differential scanning calorimetry, and dynamic laser scattering. Owing to the amphiphilicity, this graft copolymer can be readily incorporated into epoxy to obtain the nanostructured thermosets. Transmission electron microscopy showed that the spherical nanophases with a size of 10–100 nm in diameter were dispersed into the continuous epoxy matrix, depending on the content of P(VDF-TrFE)-g-PMMA. The measurement of dielectric properties showed that the modified epoxy thermosets displayed enhanced dielectric constants and quite low dielectric loss. The surface contact angle measurements showed that the surface hydrophobicity of the nanostructured thermosets was higher than the plain epoxy thermoset.
Co-reporter:Lei Li, Miaolin Huang, and Sixun Zheng
Industrial & Engineering Chemistry Research 2016 Volume 55(Issue 44) pp:11502-11511
Publication Date(Web):October 27, 2016
DOI:10.1021/acs.iecr.6b03048
The nanostructured phenolic thermosets were prepared from the ternary mixtures composed of novolac resin, poly(styrene-alt-maleic anhydride)-block-polystyrene [P(S-alt-MAH)-b-PS] diblock copolymer, and hexamethylenetetramine, which were cured at elevated temperature. The P(S-alt-MAH)-b-PS diblock copolymer was synthesized via a one-pot RAFT polymerization approach. The morphologies of the nanostructured thermosets were modulated by changing the content of the diblock copolymer. The as-obtained nanostructured phenolic thermosets were further used as the precursors to obtain the mesoporous carbons via pyrolysis under highly pure nitrogen atmosphere. The mesoporous carbons were successfully obtained with adjustable porosity. The mesoporous carbons had specific surface areas of 398.9–491.4 m2 g–1 and pore volumes of 0.22–0.31 cm3 g–1.
Co-reporter:Miaolin Huang, Lei Li, Sixun Zheng
Microporous and Mesoporous Materials 2016 Volume 225() pp:9-25
Publication Date(Web):1 May 2016
DOI:10.1016/j.micromeso.2015.12.008
•PS-b-PHEA diblock copolymer was used for preparation of mesoporous silica.•This diblock copolymer was further functionalized with alkyloxysilane.•The inter-component reaction can be used to modulate the porosity of the materials.A polystyrene-block-poly(2-hydroxyethylacrylate) diblock copolymer (PS-b-PHEA) was synthesized via a reversible addition-fragmentation chain transfer (RAFT) polymerization approach. This novel amphiphilic diblock copolymer was successfully used as the template to obtain the mesoporous silica materials. In order to modulate the porosity of mesoporous silica materials, this diblock copolymer was further functionalized via its reaction with 3-isocyanatopropyltriethoxysilane (IPTES) to afford a new diblock copolymer bearing an alkyloxysilane subchain. This derivate diblock copolymer was also employed to prepare the mesoporous silica materials through the inter-component reaction between the block copolymer and silica matrix. The results of small angle X-ray scattering (SAXS), transmission electron microscopy (TEM) and Brunauer–Emmett–Teller (BET) measurements showed that the inter-component reaction can be utilized to modulate the morphologies and porosity of the mesoporous silica materials.In this work, we reported the preparation of mesoporous silica with a novel amphiphilic diblock copolymer, polystyrene-block-poly(2-hydroxylethyl acrylate) (PH-b-PHEA) as the template. In the meantime, PS-b-PHEA was further functionalized via its reaction with 3-isocyanatopropyltriethoxysilane (IPTES) to afford a derivate diblock bearing alkyloxysilane moieties, which was then used for the preparation of mesoporous silica. A comparative investigation was carried out and the effect of the inter-component reaction on the porosity was elucidated.
Co-reporter:Lei Li
Journal of Applied Polymer Science 2016 Volume 133( Issue 16) pp:
Publication Date(Web):
DOI:10.1002/app.43322
ABSTRACT
In this study, we examined a facile approach for achieving a fine dispersion of barium titanate (BT) nanoparticles (NPs) in epoxy thermosets. First, the surfaces of BT NPs were modified with poly(ε-caprolactone) (PCL) via a surface-initiated ring-opening polymerization approach. We found that the PCL-grafted BT NPs were easily dispersed in epoxy thermosets. The fine dispersion of the PCL-grafted BT NPs in the epoxy thermosets was evidenced by transmission electron microscopy and dynamic mechanical thermal analysis. We found that the organic–inorganic nanocomposites displayed significantly enhanced dielectric constants and low dielectric loss compared to the control epoxy. The nanocomposites containing 14.1 wt % BT NPs possessed dielectric constants as high as at a frequency of 103 Hz. The dielectric loss was measured to be 0.002 at a frequency of 103 Hz. The improved dielectric properties are accounted for the fine dispersion of the BT NPs in the epoxy thermosets. © 2015 Wiley Periodicals, Inc. J. Appl. Polym. Sci. 2016, 133, 43322.
Co-reporter:Jingang Li;Yixin Xiang
Journal of Polymer Science Part A: Polymer Chemistry 2016 Volume 54( Issue 3) pp:368-380
Publication Date(Web):
DOI:10.1002/pola.27784
ABSTRACT
In this contribution, we reported the synthesis of a hyperbranched block copolymer composed of poly(ε-caprolactone) (PCL) and polystyrene (PS) subchains. Toward this end, we first synthesized an α-alkynyl- and ω,ω′-diazido-terminated PCL-b-(PS)2 macromonomer via the combination of ring-opening polymerization and atom transfer radical polymerization. By the use of this AB2 macromonomer, the hyperbranched block copolymer (h-[PCL-b-(PS)2]) was synthesized via a copper-catalyzed Huisgen 1,3-dipolar cycloaddition (i.e., click reaction) polymerization. The hyperbranched block copolymer was characterized by means of 1H nuclear magnetic resonance spectroscopy and gel permeation chromatography. Both differential scanning calorimetry and atomic force microscopy showed that the hyperbranched block copolymer was microphase-separated in bulk. While this hyperbranched block copolymer was incorporated into epoxy, the nanostructured thermosets were successfully obtained; the formation of the nanophases in epoxy followed reaction-induced microphase separation mechanism as evidenced by atomic force microscopy, small angle X-ray scattering, and dynamic mechanical thermal analysis. © 2015 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2016, 54, 368–380
Co-reporter:Ning Liu
Journal of Polymer Science Part A: Polymer Chemistry 2016 Volume 54( Issue 18) pp:2949-2961
Publication Date(Web):
DOI:10.1002/pola.28180
ABSTRACT
An organic–inorganic copolymer with polyhedral oligomeric silsesquioxane (POSS) and xanthate moieties in the main chain was synthesized via the polycondensation between 3,13-di(2-bromopropionate)propyl double-decker silsesquioxane (DDSQ) and 1,4-di(xanthate potassium)butane. This hybrid copolymer was used as the macromolecular chain transfer agent to obtain the organic–inorganic poly(N-vinylpyrrolidone) (PVPy) copolymers via a reversible addition fragmentation chain transfer/macromolecular design via the interchange of xanthates (RAFT/MADIX) polymerization approach; the polymerization behavior of N-vinyl pyrrolidone was investigated by means of gel permeation chromatography. It was found that the polymerization was in a living and controlled manner. Transmission electron microscopy (TEM) showed that the organic–inorganic PVPy copolymers with DDSQ in the main chains were microphase-separated in bulks. Compared to plain PVPy, the organic–inorganic PVPy copolymers displayed the decreased glass transition temperatures (Tgs); the decreased Tgs are attributable to the effect of the introduced DDSQ cages on the packing of PVPy chains as evidenced by means of Fourier transform infrared spectroscopy (FTIR). In water, the organic–inorganic PVPy copolymers can self-assemble into the spherical nano-objects with the size of 20–50 nm in diameter. In the self-assembled nano-objects, the aggregates of the hydrophobic DDSQ constituted the cores of the polymeric micelles whereas the PVPy chains between the DDSQ behaved as the coronas of the polymeric micelles. © 2016 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2016, 54, 2949–2961
Co-reporter:Yixin Xiang;Houluo Cong;Lei Li
Journal of Polymer Science Part A: Polymer Chemistry 2016 Volume 54( Issue 12) pp:1852-1863
Publication Date(Web):
DOI:10.1002/pola.28047
ABSTRACT
Poly(N-vinyl pyrrolidone)-block-poly(N-vinyl carbazole)-block-poly(N-vinyl pyrrolidone) (PVP-b-PVK-b-PVP) triblock copolymers were synthesized via sequential reversible addition-fragmentation chain transfer/macromolecular design via the interchange of xanthate (RAFT/MADIX) process. First, 1,4-phenylenebis(methylene)bis(ethyl xanthate) was used as a chain transfer agent to mediate the radical polymerization of N-vinyl carbazole (NVK). It was found that the polymerization was in a controlled and living manner. Second, one of α,ω-dixanthate-terminated PVKs was used as the macromolecular chain transfer agent to mediate the radical polymerization of N-vinyl pyrrolidone (NVP) to obtain the triblock copolymers with various lengths of PVP blocks. Transmission electron microscopy (TEM) showed that the triblock copolymers in bulks were microphase-separated and that PVK blocks were self-organized into cylindrical microdomains, depending on the lengths of PVP blocks. In aqueous solutions, all these triblock copolymers can self-assemble into the spherical micelles. The critical micelle concentrations of the triblock copolymers were determined without external adding fluorescence probe. By analyzing the change in fluorescence intensity as functions of the concentration, it was judged that the onset of micellization occurred at the concentration while the FL intensity began negatively to deviate from the initial linear increase with the concentration. Fluorescence spectroscopy indicates that the self-assembled nanoobjects of the PVP-b-PVK-b-PVP triblock copolymers in water were capable of emitting blue/or purple fluorescence under the irradiation of ultraviolet light. © 2016 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2016, 54, 1852–1863
Co-reporter:Yixin Xiang, Lei Li, Sixun Zheng
Polymer 2016 Volume 98() pp:344-352
Publication Date(Web):19 August 2016
DOI:10.1016/j.polymer.2016.06.037
•PVPy-b-PVK-b-PVPy triblock copolymer was synthesized via RAFT polymerization.•This copolymer was in cooperated into epoxy to obtain the nanostructured thermosets.•The thermosets displayed photoluminescent behavior and enhanced dielectric constant.In this contribution, we reported the preparation of the nanostructured epoxy thermosets containing poly(N-vinylcarbazole) (PVK) microdomains. Toward this end, we first synthesized poly(N-vinylpyrrolidone)-block-poly(N-vinylcarbazole)-block-poly(N-vinyl pyrrolidone) (PVPy-b-PVK-b-PVPy) triblock copolymer via sequential reversible addition-fragmentation chain transfer/macromolecular design via the interchange of xanthate (RAFT/MADIX) process; then this triblock copolymer was incorporated into epoxy to obtain the nanostructured thermosets. It was found that reaction-induced microphase separation occurred in the mixtures of this triblock copolymer with epoxy. The formation of the PVK microdomains in epoxy thermosets were investigated by means of transmission electron microscopy (TEM) and small angle X-ray scattering (SAXS). Owing to the creation of PVK nanophases, the nanostructured epoxy thermosets became light-emissive under ultraviolet irradiation. The photoluminescent behavior is attributable to the formation of the excimers within the PVK microdomains. Compared to control epoxy, the nanostructured thermosets significantly displayed the enhanced dielectric constants due to the formation of PVK nanophases. The enhanced dielectric constants resulted from the dominant contribution of electron polarization and interfacial polarization with the formation of PVK nanophases.
Co-reporter:Wenjun Peng, Sen Xu, Lei Li, Chongyin Zhang, and Sixun Zheng
The Journal of Physical Chemistry B 2016 Volume 120(Issue 46) pp:12003-12014
Publication Date(Web):November 11, 2016
DOI:10.1021/acs.jpcb.6b08026
Organic–inorganic nanocomposites composed of polyhedral oligomeric silsesquioxane (POSS) and epoxy resin were prepared via self-assembly of an amphiphilic triblock copolymer bearing a poly(POSS) midblock in epoxy thermosets. First, this organic–inorganic amphiphilic triblock copolymer was synthesized via hydrosilylation of heptaphenylhydro POSS with an existing triblock copolymer containing a short polybutadiene midblock. It was found that this novel amphiphilic block copolymer can self-assemble into nanophases in epoxy thermosets. In the presence of preformed nanophases, the curing reaction was performed, and the organic–inorganic nanocomposites containing poly(POSS) microdomains were thus obtained. Compared with plain epoxy, the as-obtained thermosets exhibited enhanced surface hydrophobicity; the enhanced surface hydrophobicity is attributed to enrichment of the POSS component at the surface of the materials. Owing to the formation of poly(POSS) microdomains, the dielectric constants of the materials significantly reduced, whereas the dielectric loss remained almost unchanged.
Co-reporter:Kun Wei, Lei Wang, Lei Li and Sixun Zheng
Polymer Chemistry 2015 vol. 6(Issue 2) pp:256-269
Publication Date(Web):22 Sep 2014
DOI:10.1039/C4PY00786G
Bead-like PNIPAAm copolymers with double-decker silsesquioxane (DDSQ) in the main chains were synthesized via a reversible addition–fragmentation chain transfer (RAFT) polymerization approach. The macromolecular chain transfer agent used for the RAFT polymerization was synthesized via the polycondensation of 3,13-dihydroxyproplyl DDSQ with S,S′-bis(α,α′-dimethyl-α′′-propargyl acetate)trithiocarbonate. The organic–inorganic copolymers with variable contents of DDSQ were characterized by means of 1H nuclear magnetic resonance spectroscopy and gel permeation chromatography. Transmission electron microscopy showed that the bead-like PNIPAAm copolymers were microphase-separated in bulk. It was found that the glass transition temperatures (Tg's) of PNIPAAm microdomains of the organic–inorganic copolymers were lower than plain PNIPAAm and decreased with increasing the content of DDSQ. The bead-like PNIPAAm copolymers displayed the self-assembly behavior in aqueous solutions. Depending on the content of DDSQ, the bead-like organic–inorganic copolymers can self-assemble into spherical or vesicular nanoobjects in aqueous solutions. Both micro-differential scanning calorimetry (Micro-DSC) and cloud point analysis with UV-vis spectroscopy showed that the lower critical solution temperature (LCST) behavior of PNIPAAm subchains in the bead-like copolymers was significantly affected by the POSS cages in the main chains.
Co-reporter:Wenjun Peng, Lei Li and Sixun Zheng
RSC Advances 2015 vol. 5(Issue 95) pp:77922-77931
Publication Date(Web):15 Sep 2015
DOI:10.1039/C5RA15804D
In this contribution, we reported the preparation of photoluminescent epoxy microspheres. First, diglycidyl ether of tetraphenylethene (DGETPE), a novel epoxy monomer bearing tetraphenylethene was synthesized and then it was incorporated into a commercial epoxy (viz. diglycidyl ether of bisphenol A) to obtain the epoxy microspheres with sizes of 1–3 μm via a phase-inverted reaction-induced phase separation approach. It was found that the epoxy microspheres were luminescent under UV light radiation. The surfaces of the photoluminescent epoxy microspheres were further functionalized with poly(N-vinyl pyrrolidone) (PVPy) with a grafting-from methodology via reversible addition–fragmentation chain transfer/macromolecular design via the interchange of xanthate (RAFT/MADIX) process. The PVPy-grafted epoxy microspheres can be well dispersed into aqueous solutions. The morphologies and photophysical properties of the epoxy microspheres were investigated by means of scanning electron microscopy, and UV-vis and fluorescence spectroscopy.
Co-reporter:Kun Wei, Ning Liu, Lei Li and Sixun Zheng
RSC Advances 2015 vol. 5(Issue 94) pp:77274-77287
Publication Date(Web):11 Sep 2015
DOI:10.1039/C5RA12843A
In this contribution, we report the synthesis of cis-hexa[(phenyl)(dimethylsiloxypropylglycidylether)cyclohexasiloxane, a novel macrocyclic oligomeric silsesquioxane (MOSS) via the combination of silylation and hydrosilylation reactions. This stereoregular MOSS macromer bearing epoxide groups was then incorporated into polybenzoxazine (PBZ) thermoset and the organic–inorganic nanocomposites were successfully obtained as evidenced by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Dynamic mechanical thermal analysis (DMTA) showed that the organic–inorganic nanocomposites displayed enhanced glass transition temperatures (Tg's) compared to plain PBZ. Thermogravimetric analysis (TGA) indicates that the organic–inorganic PBZ nanocomposites possessed improved thermal stability. The improved thermomechanical properties are attributable to the nanoreinforcement of the stereoregular macrocyclic silsesquioxanes on PBZ matrix as well as the additional crosslinking between PBZ and the MOSS macromer. The results of static contact angle measurement showed that the surface hydrophobicity of the organic–inorganic nanocomposites was significantly improved compared to the plain PBZ thermoset.
Co-reporter:Lei Li and Sixun Zheng
Industrial & Engineering Chemistry Research 2015 Volume 54(Issue 1) pp:171-180
Publication Date(Web):December 15, 2014
DOI:10.1021/ie5038193
Poly(ε-caprolactone)-grafted magnetic iron oxide nanoparticles (Fe3O4 NPs) were prepared via a surface-initiated ring opening polymerization approach. First, the surface of the unmodified Fe3O4 NPs were treated with 3-aminopropyltriethoxysilane to afford the 3-aminopropyl-functionalzed Fe3O4 NPs. Second, the surface-initiated ring-opening polymerization of ε-caprolactone was carried out with the surface-functionalized Fe3O4 NPs as the initiator to afford the poly(ε-caprolactone)(PCL)-grafted Fe3O4 NPs. The PCL-grafted Fe3O4 NPs have been characterized by means of Fourier transform infrared spectroscopy, X-ray diffraction, thermogravimetric analysis, and transmission electron microscopy. It was found that the PCL-grafted Fe3O4 NPs can be well dispersed into epoxy thermosets. The nanocomposites were successfully obtained with the content of the PCL-grafted Fe3O4 NPs up to 40 wt %. The fine dispersion of Fe3O4 nanoparticles in epoxy was demonstrated by transmission electron microscopy and dynamic mechanical thermal analysis. The results of magnetic analysis with vibrating sample measuring technology indicated that the nanocomposites involving epoxy resins and Fe3O4 nanoparticles possessed superparamagnetic properties.
Co-reporter:Rentong Yu and Sixun Zheng
Industrial & Engineering Chemistry Research 2015 Volume 54(Issue 25) pp:6454-6466
Publication Date(Web):June 17, 2015
DOI:10.1021/acs.iecr.5b01435
In this work, we reported a facile preparation of mesoporous silica materials assisted by a commercial polystyrene-block-polybutadiene-block-polystyrene (PS-b-PB-b-PS) triblock copolymer. For this purpose, the PS-b-PB-b-PS triblock copolymer was first functionalized with (3-mercaptopropyl)triethoxysilane via a thiol-ene radical addition approach, and then the functionalized triblock copolymer with the midblock bearing triethoxysilane moieties was employed to perform intercomponent sol–gel reactions with tetraethoxysilane (TEOS) to obtain the organic–inorganic gels. The organic–inorganic gels with variable compositions were then used as precursors to obtain mesoporous silica via pyrolysis at elevated temperatures. The functionalized triblock copolymer was characterized by means of Fourier transform infrared spectroscopy, nuclear magnetic resonance spectroscopy, and small-angle X-ray scattering (SAXS). The results of SAXS, transmission electron microscopy, and Brunauer–Emmett–Teller measurements indicate that the mesoporous silica materials were successfully obtained and the porosity of the materials can be modulated with the mass ratios of the functionalized triblock copolymer to the precursor of silica (viz. TEOS).
Co-reporter:Houluo Cong, Lei Li, Sixun Zheng
Polymer 2015 Volume 79() pp:99-109
Publication Date(Web):19 November 2015
DOI:10.1016/j.polymer.2015.10.014
•A cylindrical brush copolymer with diblock side chains was successfully synthesized.•The cylindrical brush copolymer was microphase-separated in bulks.•Its reaction-induced microphase separation in epoxy afforded fibrillar nanophase.In this contribution, we reported the synthesis of a core-shell cylindrical brush copolymer with poly(2-hydroxyethyl methacrylate) (PHEMA) backbone and polystyrene-block-poly(ε-caprolactone) (PS-b-PCL) diblock side chains [denoted PHEMA-g-(PS-b-PCL)n] with the combination of atom transfer radical polymerization, the Huisgen 1,3-dipolar cycloaddition between azido and alkynyl groups and the ring-opening polymerization via a sequential “grafting from” polymerization approach. The cylindrical brush block copolymer was characterized by means of 1H nuclear magnetic resonance spectroscopy (NMR), gel permeation chromatography (GPC), atomic force microscopy (AFM) and differential scanning calorimetry (DSC). It is found that the fibrillar nanophases were formed with the length of about 400 nm and the diameter of about 15 nm via reaction-induced microphase separation mechanism while this cylindrical block copolymer brush was incorporated into epoxy thermosets. The fibrillar nanophases were characterized by means of transmission electron microscopy (TEM), small-angle X-ray scattering (SAXS) and dynamic mechanical thermal analysis (DMTA). The formation of the fibrillar nanophases was interpreted on the basis of the constraint of cylindrical brush architecture of copolymer on reaction-induced microphase separation behavior.
Co-reporter:Jingang Li, Houluo Cong, Lei Li, Sixun Zheng
Polymer 2015 Volume 69() pp:193-203
Publication Date(Web):9 July 2015
DOI:10.1016/j.polymer.2015.05.057
•PCL-b-P3HT-b-PCL triblock copolymer was synthesized via sequential polymerizations.•Epoxy thermosets containing P3HT nanophases were prepared with the copolymer.•The thermosets displayed enhanced dielectric constant and thermal conductivity.The nanostructured thermosets containing poly(3-hexylthiophene) (P3HT) nanophases were prepared by incorporating poly(ε-caprolactone)-block-poly(3-hexylthiophene)-block-poly(ε-caprolactone) (PCL-b-P3HT-b-PCL) triblock copolymer into epoxy. The PCL-b-P3HT-b-PCL triblock copolymer was synthesized via the combination of the polycondensation of 2-bromo-3-hexyl-5-iodothiophene and the ring-opening polymerization of ε-caprolactone; it was characterized by means of 1H nuclear magnetic resonance spectroscopy (1H NMR), gel permeation chromatography (GPC) and differential scanning calorimetry (DSC). The morphologies of the nanostructured thermosets were investigated by means of transmission electron microscopy (TEM), small angle X-ray scattering (SAXS) and dynamic mechanical thermal analysis (DMTA). The results of small angle X-ray scattering (SAXS) showed that the P3HT nanophases were formed via self-assembly mechanism of the triblock copolymer in epoxy thermosets. Compared to control epoxy, the nanostructured thermosets containing the conjugated nanophases significantly displayed the enhanced dielectric constants. In the meantime, the thermal conductivity of the nanostructured thermosets was also enhanced and increased with increasing the content of P3HT nanophases.
Co-reporter:Jingang Li, Houluo Cong, Lei Li, and Sixun Zheng
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 16) pp:13677
Publication Date(Web):July 18, 2014
DOI:10.1021/am503148v
The block copolymer networks composed of poly(N-isopropylacrylamide) (PNIPAM) and poly(sodium p-styrenesulfonate) were synthesized via sequential reversible addition–fragmentation chain transfer (RAFT) polymerization with α,ω-didithiobenzoate-terminated poly(sodium p-styrenesulfonate) (PSSNa) as the macromolecular chain transfer agent. It was found that the block copolymer networks were microphase-separated as evidenced by means of transmission electron microscopy (TEM) and small-angle X-ray scattering (SAXS). In the block copolymer networks, spherical or cylindrical PSSNa microdomains were finely dispersed into continuous PNIPAM matrixes. In comparison with unmodified PNIPAM hydrogel, the nanostructured hydrogels displayed improved thermoresponsive properties. In addition, the swelling ratios of the PSSNa-modified PNIPAM hydrogels were significantly higher than that of plain PNIPAM hydrogel. The improvement of thermoresponse was attributable to the formation of the PSSNa nanophases, which promoted the transportation of water molecules in the cross-linked networks.Keywords: block copolymer; fast thermoresponsive properties; hydrogels; PNIPAM
Co-reporter:Yulin Yi and Sixun Zheng
RSC Advances 2014 vol. 4(Issue 54) pp:28439-28450
Publication Date(Web):06 Jun 2014
DOI:10.1039/C4RA02624A
The novel organic–inorganic molecular brush composed of macrocyclic oligomeric silsesquioxane (MOSS) and poly(N-isopropylacrylamide) (PNIPAAm) (denoted by PNIPAAm@MOSS) was synthesized via the atom transfer radical polymerization (ATRP) approach. In bulk, the organic–inorganic molecular brushes were microphase-separated; the spherical MOSS microdomains with the diameter of 10–50 nm were dispersed into a continuous PNIPAAm matrix. Depending on the lengths of PNIPAAm chains, the PNIPAAm@MOSS molecular brushes were capable of self-assembling into cylindrical or spherical nano-objects in aqueous solutions as evidenced by transmission election microscopy (TEM) and dynamic light scattering (DLS). Both micro-differential scanning calorimetry (Micro-DSC) and ultraviolet-visible spectroscopy showed that the MOSS backbones exerted significant restriction of coil-to-globule transition of PNIPAAm chains.
Co-reporter:Houluo Cong, Jingang Li, Lei Li, Sixun Zheng
European Polymer Journal 2014 Volume 61() pp:23-32
Publication Date(Web):December 2014
DOI:10.1016/j.eurpolymj.2014.09.018
•Triblock copolymers of PNIPAM and PVP were synthesized via RAFT polymerization.•The ABA triblock copolymers displayed thermoresponsive gelation behavior.•The triblock copolymers could be used as injectable hydrogels.In this contribution, we reported the synthesis of poly(N-isopropylacrylamide)-block-poly(N-vinylpyrrolidone)-block-poly(N-isopropylacrylamide) (PNIPAM-b-PVP-b-PNIPAM) triblock copolymers through sequential reversible addition–fragmentation chain transfer/macromolecular design via the interchange of xanthate (RAFT/MADIX) process. The triblock copolymers have been characterized by means of 1H nuclear magnetic resonance (NMR) spectroscopy, gel permeation chromatography (GPC) and differential scanning calorimetry (DSC). This ABA triblock copolymer displayed a typical thermoresponsive gelation behavior. In this work, the thermoresponsive gelation behavior was investigated by means of rheological measurements, micro-differential scanning calorimetry (Micro-DSC) and small angle X-ray scattering (SAXS). The thermoresponsive gelation behavior could endow the triblock copolymers with the potential application as injectable hydrogels.
Co-reporter:Houluo Cong, Lei Li, Sixun Zheng
Polymer 2014 Volume 55(Issue 5) pp:1190-1201
Publication Date(Web):10 March 2014
DOI:10.1016/j.polymer.2014.01.049
In this work, we investigated the effect of formation mechanisms of nanophases on the morphologies and thermomechanical properties of the nanostructured thermosets containing block copolymers. Toward this end, the nanostructured thermosets involving epoxy and block copolymers were prepared via self-assembly and reaction-induced microphase separation approaches, respectively. Two structurally similar triblock copolymers, poly(ε-caprolactone)-block-poly(butadiene-co-styrene)-block-poly(ε-caprolactone) (PCL-b-PBS-b-PCL) and poly(ε-caprolactone)-block-poly(ethylene-co-ethylethylene-co-styrene)-block-poly(ε-caprolactone) (PCL-b-PEEES-b-PCL) were synthesized via the ring-opening polymerization of ε-caprolactone (CL) with α,ω-dihydroxyl-terminated poly(butadiene-co-styrene) (HO-PBS-OH) and α,ω-dihydroxyl-terminated poly(ethylene-co-ethylethylene-co-styrene) (i.e., HO-PEEES-OH) as the macromolecular initiators, respectively; the latter was obtained via the hydrogenation reduction of the former. Both the triblock copolymers had the same architecture, the identical composition and close molecular weights. In spite of the structural resemblance of both the triblock copolymers, the formation mechanisms of the nanophases in the thermosets were quite different. It was found that the formation of nanophases in the thermosets containing PCL-b-PBS-b-PCL followed a reaction-induced microphase separation mechanism whereas that in the thermosets containing PCL-b-PEEES-b-PCL was in a self-assembly manner. The different formation mechanisms of nanophases resulted in the quite different morphologies, glass transition temperatures (Tg's) and fracture toughness of the nanostructured thermosets.
Co-reporter:Houluo Cong, Jingang Li, Lei Li, and Sixun Zheng
The Journal of Physical Chemistry B 2014 Volume 118(Issue 50) pp:14703-14712
Publication Date(Web):December 8, 2014
DOI:10.1021/jp5089355
Poly(ethylene oxide)-block-poly(sodium p-styrenesulfonate) (PEO-b-PSSNa) diblock copolymer was synthesized and then incorporated into epoxy to obtain the nanostructured epoxy thermosets containing polyelectrolyte nanophases. This PEO-b-PSSNa diblock copolymer was synthesized via the radical polymerization of p-styrenesulfonate mediated with 4-cyano-4-(thiobenzoylthio)valeric ester-terminated poly(ethylene oxide). The formation of polyelectrolyte (i.e., PSSNa) nanophases in epoxy followed a self-assembly mechanism. The precursors of epoxy acted as the selective solvent of the diblock copolymer, and thus, the self-assembled nanostructures were formed. The self-organized nanophases were fixed through the subsequent curing reaction. By means of transmission electron microscopy (TEM) and small-angle X-ray scattering (SAXS), the morphologies of the nanostructured epoxy thermosets containing PSSNa nanophases were investigated. In the glassy state, the epoxy matrixes were significantly reinforced by the spherical PSSNa nanodomains, as evidenced by dynamic mechanical analysis. The measurement of dielectric properties showed that, with the incorporation of PSSNa nanophases, the dielectric constants of the epoxy thermoset were significantly increased. Compared to the control epoxy, the dielectric loss of the nanostructured thermosets still remained at quite a low level, although the values of dielectric loss were slightly increased with inclusion of PSSNa nanophases.
Co-reporter:Chongyin Zhang;Lei Li;Houluo Cong
Journal of Polymer Science Part A: Polymer Chemistry 2014 Volume 52( Issue 7) pp:952-962
Publication Date(Web):
DOI:10.1002/pola.27075
ABSTRACT
In this contribution, we reported a facile synthesis of poly(methyl methacrylate)-block-poly(N-vinyl pyrrolidone) (PMMA-b-PVPy) diblock copolymers via sequential radical polymerizations mediated by isopropylxanthic disulfide (DIP). It was found that the radical polymerization of N-vinyl pyrrolidone (NVP) mediated by DIP was in a controlled and living manner. In contrast, the polymerization of methyl methacrylate mediated by DIP displayed the behavior of telomerization, affording xanthate-terminated PMMA with a good control of molecular weights while the conversion of monomer was not very high. The xanthate-terminated PMMA can be successfully used as the macromolecular chain transfer agent for the polymerization of NVP via RAFT/MADIX process and thus PMMA-b-PVPy diblock copolymers can be successfully synthesized via sequential radical polymerization mediated by isopropylxanthic disulfide. One of these diblock copolymers was incorporated into polybenzoxazine and the nanostructured thermosets were obtained as evidenced by transmission electron microscopy, small angle X-ray scattering, and dynamic mechanical thermal analysis. The formation of nanostructures in polybenzoxazine thermosets was ascribed to a reaction-induced microphase separation mechanism. © 2013 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2014, 52, 952–962
Co-reporter:Kun Wei, Lei Wang and Sixun Zheng
Polymer Chemistry 2013 vol. 4(Issue 5) pp:1491-1501
Publication Date(Web):04 Dec 2012
DOI:10.1039/C2PY20930F
In this contribution, we reported the synthesis of organic–inorganic polyurethanes with polyhedral oligomeric silsesquioxane (POSS) in the main chains. Toward this end, 3,13-dihydroxypropyloctaphenyl double-decker silsesquioxane (DDSQ) was synthesized; this POSS diol was used as a chain extender to obtain hybrid polyurethanes with DDSQ in the main chains. By controlling the molar ratio of 3,13-dihydroxypropyloctaphenyl DDSQ to 1,4-butanediol (BDO), organic–inorganic polyurethanes were obtained with a content of DDSQ up to 48 wt%. The results of 1H nuclear magnetic resonance spectroscopy (NMR) and gel permeation chromatography (GPC) showed that 3,13-dihydroxypropyloctaphenyl, DDSQ, can be successfully used as a chain extender to afford linear organic–inorganic polyurethanes. Differential scanning calorimetry (DSC) showed that the organic–inorganic polyurethanes displayed enhanced glass transition temperatures (Tg's) compared to control polyurethane; the Tg's increased with increasing content of DDSQ in the main chains. Compared to control polyurethane, the organic–inorganic polyurethanes displayed improved thermal stability in terms of thermogravimetric analysis (TGA). With the inclusion of DDSQ in the main chains, the organic–inorganic polyurethanes displayed enhanced surface hydrophobicity.
Co-reporter:Xiangping Yu;Chongyin Zhang;Yong Ni
Journal of Applied Polymer Science 2013 Volume 128( Issue 5) pp:2829-2839
Publication Date(Web):
DOI:10.1002/app.38339
Abstract
In this article, we report the preparation of crosslinked epoxy microspheres with diameters of 5–10 μm prepared via phase-inverted phase separation induced by polymerization in the thermosetting blend of epoxy and poly(ε-caprolactone). The surfaces of the epoxy microspheres were functionalized to bear 2-bromopropionyl groups, which were further used as initiators to obtain poly(glycidyl methacrylate) (PGMA) grafted epoxy microspheres via the surface-initiated atom transfer radical polymerization approach. The PGMA-grafted epoxy microspheres were then employed to react with 3-aminopropyltrimethoxylsilane (APTMS) to obtain the functionalized epoxy microspheres, the surface of which contained a great number of trimethoxysilane groups. A co-sol–gel process between the APTMS-functionalized epoxy microspheres and tetraethoxysilane was performed, and organic–inorganic glassy solids were obtained. The organic–inorganic glasses were used as precursors for accessing macroporous silica materials via pyrolysis at elevated temperatures. The hierarchical porosity of the resulting macroporous silica was investigated by means of field emission scanning electronic microscopy, transmission electronic microscopy, and surface-area Brunauer–Emmett–Teller (BET) measurements. We found that the macroporous silica possessed BET surface areas in the range 183.9–235.2 m2/g, depending on the compositions of their precursors. © 2012 Wiley Periodicals, Inc. J. Appl. Polym. Sci., 2013
Co-reporter:Kun Wei;Lei Wang
Journal of Polymer Science Part A: Polymer Chemistry 2013 Volume 51( Issue 19) pp:4221-4232
Publication Date(Web):
DOI:10.1002/pola.26836
ABSTRACT
A series of novel organic–inorganic copolymers with polyhedral oligomeric silsesquioxane (POSS) in the main chains were synthesized via the copper-catalyzed Huisgen 1,3-dipolar cycloaddition polymerization approach. Toward this end, we synthesized 3,13-azidopropyloctaphenyl double-decked silsesquioxane (DDSQ). This difunctional POSS macromer was used to copolymerize with α,ω-dialkynyl-terminated oligoethylenes with variable number of ethylene units. The organic–inorganic copolymers were obtained with the mass fraction of POSS up to 79%. Gel permeation chromatography showed that the high-molecular-weight copolymers were successfully obtained in all the cases. Differential scanning calorimetry showed that the amplitude of glass transitions for these copolymers was very feeble, suggesting that the segmental motions responsible for the glass transitions was highly restricted with DDSQ cages in the main chains. Thermogravimetric analysis showed that the organic–inorganic hybrid copolymers displayed extremely high thermal stability. Contact angle measurements showed that these organic–inorganic copolymers are highly hydrophobic and possessed very low surface energy. © 2013 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 4221–4232
Co-reporter:Chongyin Zhang, Lei Li, and Sixun Zheng
Macromolecules 2013 Volume 46(Issue 7) pp:2740-2753
Publication Date(Web):April 1, 2013
DOI:10.1021/ma4000682
In this contribution, we reported the investigation of the formation and confined crystallization behavior of polyethylene nanophases in epoxy thermosets. The nanostructured epoxy thermosets were prepared by the use of a poly(ε-caprolactone)-block-polyethylene-block-poly(ε-caprolactone) (PCL-b-PE-b-PCL) triblock copolymer. The crystalline midblock (viz. PE) of the triblock copolymer was prepared from an α,ω-diacetoxy-terminated polycyclooctadiene with the molecular weight as high as Mn = 11,000, which was synthesized via the ring-opening metathesis polymerization (ROMP) of cyclooctadiene catalyzed by Grubbs second generation catalyst. The formation of PE nanophases in epoxy thermosets was evidenced with transmission electronic microscopy (TEM), small-angle X-ray scattering (SAXS) and dynamic mechanical thermal analysis (DMTA). It was found that in the nanostructured thermosets, the spherical nanophases of PE with the size of 20–30 nm in diameter were dispersed into the continuous epoxy matrices. Wide angle X-ray diffraction (XRD) showed that the formation of PE nanophases did not alter the structure of PE crystals. The investigations of isothermal and nonisothermal crystallization kinetics showed that the crystallization of PE in the nanostructured thermosets was in a confined manner and the confinement has been interpreted on the basis of nanoscaled space, interdomain connectivity, and the cross-linked structures of epoxy matrices.
Co-reporter:Lei Wang, Chongyin Zhang, Houluo Cong, Lei Li, and Sixun Zheng, Xiuhong Li and Jie Wang
The Journal of Physical Chemistry B 2013 Volume 117(Issue 27) pp:8256-8268
Publication Date(Web):June 14, 2013
DOI:10.1021/jp402084u
In this work, we investigated the effect of topological structures of block copolymers on the formation of the nanophase in epoxy thermosets containing amphiphilic block copolymers. Two block copolymers composed of poly(ε-caprolactone) (PCL) and poly(2,2,2-trifluoroethyl acrylate) (PTFEA) blocks were synthesized to possess linear and star-shaped topologies. The star-shaped block copolymer composed a polyhedral oligomeric silsesquioxane (POSS) core and eight poly(ε-caprolactone)-block-poly(2,2,2-trifluoroethyl acrylate) (PCL-b-PTFEA) diblock copolymer arms. Both block copolymers were synthesized via the combination of ring-opening polymerization and reversible addition–fragmentation chain transfer/macromolecular design via the interchange of xanthate (RAFT/MADIX) process; they were controlled to have identical compositions of copolymerization and lengths of blocks. Upon incorporating both block copolymers into epoxy thermosets, the spherical PTFEA nanophases were formed in all the cases. However, the sizes of PTFEA nanophases from the star-like block copolymer were significantly lower than those from the linear diblock copolymer. The difference in the nanostructures gave rise to the different glass transition behavior of the nanostructured thermosets. The dependence of PTFEA nanophases on the topologies of block copolymers is interpreted in terms of the conformation of the miscible subchain (viz. PCL) at the surface of PTFEA microdomains and the restriction of POSS cages on the demixing of the thermoset-philic block (viz. PCL).
Co-reporter:Houluo Cong, Lei Li, Sixun Zheng
Polymer 2013 Volume 54(Issue 4) pp:1370-1380
Publication Date(Web):18 February 2013
DOI:10.1016/j.polymer.2012.12.069
In this work, we reported the synthesis of poly(N-isopropyl acrylamide)-block- poly(N-vinylpyrrolidone)-block-poly(N-isopropylacrylamide) triblock copolymer (PNIPAAm-b-PVPy-b-PNIPAAm) via reversible addition-fragmentation chain transfer polymerization/macromolecular design via the interchange of xanthate (RAFT/MADIX) process. This approach was further employed to prepare the PNIPAAm-b-PVPy block copolymer networks with N,N′-methylenebisacrylamide as the crosslinker. The results of small angle X-ray scattering (SAXS) indicate that the PNIPAAm-b-PVPy block copolymer networks were microphase-separated, in which PVPy was dispersed into PNIPAAm matrix as the microdomains. The architecture of block copolymer networks allows investigating the effect of the blocked permanently hydrophobic chains (viz. PVPy) on the deswelling and reswelling behavior of the PNIPAAm hydrogels. It was found that the diffusion of water molecules in PNIPAAm-b-PVPy block copolymer networks was in a non-Fickian and accelerating manner. The swelling ratios of the PNIPAAm-b-PVPy hydrogels were significantly higher than that of control PNIPAAm hydrogel. Compared to control PNIPAAm hydrogel, the PNIPAAm-b-PVPy hydrogels displayed an accelerated response to the external temperature changes in terms of deswelling and reswelling tests. The accelerated thermoresponsive properties is ascribed to the presence of the PVPy blocks in the PNIPAAm-b-PVPy block copolymer networks, which could act as the hydrophilic tunnels to facilitate the diffusion of water molecules in the PNIPAAm networks.
Co-reporter:Liliang Zhu, Chongyin Zhang, Jin Han, Sixun Zheng and Xiuhong Li
Soft Matter 2012 vol. 8(Issue 26) pp:7062-7072
Publication Date(Web):31 May 2012
DOI:10.1039/C2SM25730K
In this contribution, we report the synthesis of organic–inorganic macrocyclic molecular brushes with a 24-membered macrocyclic oligomeric silsesquioxane (MOSS) backbone and with poly(ε-caprolactone) (PCL) (or poly(ε-caprolactone)-block-polystyrene (PCL-b-PS) diblock copolymer) side chains. The macrocyclic molecular brushes were incorporated into epoxy to access the nanostructured thermosets. The nanostructures in the thermosets were investigated by means of atomic force microscopy (AFM), small angle X-ray scattering (SAXS) and dynamic mechanical thermal analysis (DMTA). It is judged that the formation of the nanophases in the thermosets containing the macrocyclic molecular brush with PCL side chains (MOSS[PCL]12) follows the self-assembly mechanism in terms of the immiscibility of the MOSS backbone with epoxy after and before the curing reaction. It was found that the spherical, vesicular and lamellar nanophases were formed in the epoxy thermosets containing the macrocyclic molecular brush with PCL-b-PS side chains (MOSS[PCL-b-PS]12). In contrast to the thermosets containing MOSS[PCL]12 the formation of the nanophases in the thermosets containing MOSS[PCL-b-PS]12 was ascribed to the self-assembly-directed reaction-induced microphase separation mechanism.
Co-reporter:Yaochen Zheng, Lei Wang, Sixun Zheng
European Polymer Journal 2012 Volume 48(Issue 5) pp:945-955
Publication Date(Web):May 2012
DOI:10.1016/j.eurpolymj.2012.03.007
In this work, a novel initiator bearing heptaphenyl polyhedral oligomeric silsesquioxane (POSS) was synthesized via the copper-catalyzed Huisgen 1,3-cycloaddition (i.e., click chemistry). With this initiator, the atom transfer radical polymerization (ATRP) of N-isopropylacrylamide (NIPAAm) was carried out to afford the POSS-capped PNIPAAm. The organic–inorganic amphiphiles were characterized by means of nuclear magnetic resonance spectroscopy (NMR) and gel permeation chromatography (GPC). Atomic force microscopy (AFM) showed that the POSS-capped PNIPAAm amphiphiles in bulk displayed microphase-separated morphologies. In aqueous solutions, the POSS-capped PNIPAAm amphiphiles were self-assembled into micelle-like aggregates as evidenced by dynamic light scattering (DLS) and transmission election microscopy (TEM). It was found that the sizes of the self-organized nanoobjects decreased with increasing the lengths of PNIPAAm chains. By means of UV–vis spectroscopy, the lower critical solution temperature (LCST) behavior of the organic–inorganic amphiphiles in aqueous solution was investigated and the LCSTs of the organic–inorganic amphiphiles decreased with increasing the percentage of POSS termini. It is noted that the self-assembly behavior of the POSS-capped PNIPAAm in aqueous solutions exerted the significant restriction on the macromolecular conformation alteration of PNIPAAm chains while the coil-to-globule collapse occurred.Graphical abstractIn this work, we reported the synthesis of heptaphenyl polyhedral oligomeric silsesquioxane (POSS)-capped poly(N-isopropylacrylamide) via the combination of copper-catalyzed Huisgen 1,3-cycloaddition and atom transfer radical polymerization. The organic–inorganic amphiphiles were characterized by means of nuclear magnetic resonance and gel permeation chromatography. The self-assembly behavior in bulk and in aqueous solution was addressed on the basis of atom force microscopy, dynamic laser scattering and transmission electronic microscopy. It was found that the self-assembly behavior in aqueous solution significantly affect the lower critical solution behavior of the organic–inorganic amphiphiles.Highlights► An initiator bearing POSS was synthesized via click chemistry. ► POSS-capped PNIPAAm was synthesized via ATRP using the initiator. ► The self-assembly behavior of the POSS-capped PNIPAAm was investigated.
Co-reporter:Jin Han, Liliang Zhu, Sixun Zheng
European Polymer Journal 2012 Volume 48(Issue 4) pp:730-742
Publication Date(Web):April 2012
DOI:10.1016/j.eurpolymj.2012.01.020
In this work, we reported the synthesis of a dodecahydroxyl-functionalized macrocyclic oligomeric silsesquioxane (MOSS). The novel 24-membered hydroxyl-functionalized MOSS was employed as a macroinitiator for the ring-opening polymerization of ε-caprolactone (CL) and the organic–inorganic macrocyclic molecular brushes with poly(ε-caprolactone) (PCL) side chains were successfully synthesized. The organic–inorganic macrocyclic molecular brushes were characterized by means of nuclear magnetic resonance spectroscopy (NMR) and gel permeation chromatography (GPC). The results of wide angle X-ray diffraction (XRD) indicate that the architecture of the organic–inorganic macrocyclic molecular brushes did not alter the structure of PCL crystals. Differential scanning calorimetry (DSC) shows that the architecture of organic–inorganic macrocyclic molecular brushes significantly affected the rearrangement of PCL crystals. Compared to linear PCL, the organic–inorganic macrocyclic molecular brushes possessed the improved thermal stability in terms of the temperatures at the maximum of degradation rate and the yields of degradation residues.Graphical abstractIn this contribution, we reported the synthesis of a novel dodecahydroxyl-functionalized macrocyclic oligomeric silsesquioxane (MOSS) and its use as a macroinitiator of ring-opening polymerization for organic–inorganic macrocyclic molecular brushes with poly(ε-caprolactone) side chains.Highlights► A macrocyclic oligomeric silsesquioxane (MOSS) bearing 12 hydroxyls was prepared. ► The MOSS was used to synthesize macrocyclic molecular brushes with PCL side chains. ► The structure and properties of the macrocyclic molecular brushes were studied.
Co-reporter:Lei Wang, Sixun Zheng
Materials Chemistry and Physics 2012 Volume 136(2–3) pp:744-754
Publication Date(Web):15 October 2012
DOI:10.1016/j.matchemphys.2012.07.051
A heptaphenyl polyhedral oligomeric silsesquioxane-capped poly(ethylene oxide) (POSS-capped PEO) telechelics was synthesized via the Huisgen 1,3-dipolar cycloaddition between 3-azidopropylheptaphenyl POSS and α,ω-dialkynyl-terminated poly(ethylene oxide). The organic–inorganic amphiphile was incorporated into epoxy to obtain the organic–inorganic nanocomposites. The morphology of the nanocomposites was investigated by means of atomic force microscopy (AFM) and dynamic mechanical thermal analysis (DMTA). It was found that the epoxy thermosets containing POSS-capped PEO telechelics were microphase-separated. The formation of the nanophases in the thermosets followed a self-assembly mechanism. The static contact angle measurements show that the nanocomposites displayed a significant enhancement in surface hydrophobicity as well as reduction in surface free energy. The improvement in surface dewettability was ascribed to the enrichment of POSS cages at the surface of the nanocomposites and the formation of the specific surface morphology as evidenced by X-ray photoelectron spectroscopy (XPS) and surface atomic force microscopy (AFM).Highlights► POSS-capped PEO telechelics was synthesized via click chemistry approach. ► The organic–inorganic amphiphile can be self-assembled into the nanophases in epoxy. ► The hybrid nanocomposites were successfully prepared via a self-assembly approach. ► The nanocomposites displayed a significant enhancement in surface hydrophobicity.
Co-reporter:Yaochen Zheng;Lei Wang;Rentong Yu
Macromolecular Chemistry and Physics 2012 Volume 213( Issue 4) pp:458-469
Publication Date(Web):
DOI:10.1002/macp.201100506
Abstract
In this work, the synthesis of 3-methacryloxypropylheptaphenyl POSS, a new POSS macromer (denoted MA-POSS) is reported. The POSS macromer is used to synthesize PEO-b-P(MA-POSS)-b-PNIPAAm triblock copolymers via sequential atom transfer radical polymerization (ATRP). The organic-inorganic, amphiphilic and thermoresponsive ABC triblock copolymers are characterized by means of nuclear magnetic resonance spectroscopy (NMR) and gel permeation chromatography (GPC). Differential scanning calorimetry (DSC) and atomic force microscopy (AFM) show that the hybrid ABC triblock copolymers are microphase-separated in bulk. Cloud point measurements show that the effect of the hydrophiphilic block (i.e. PEO) on the LCSTs is more pronounced than the hydrophobic block (i.e. P(MA-POSS)). Both transmission electron microscopy (TEM) and dynamic light scattering (DLS) show that all the triblock copolymers can be self-organized into micellar aggregates in aqueous solutions. The sizes of the micellar aggregates can be modulated by changing the temperature. The temperature-tunable self-assembly behavior is interpreted using a combination of the highly hydrophobicity of P(MA-POSS), the water-solubility of PEO and the thermoresponsive property of PNIPAAm in the triblock copolymers.
Co-reporter:Rentong Yu and Sixun Zheng, Xiuhong Li and Jie Wang
Macromolecules 2012 Volume 45(Issue 22) pp:9155-9168
Publication Date(Web):November 14, 2012
DOI:10.1021/ma3017212
We report an investigation of the influence of block copolymer architectures on formation of nanophases in epoxy thermosets via reaction-induced microphase separation approach. Toward this end, three binary block copolymers composed of polystyrene (PS) and poly(ε-caprolactone) (PCL) were synthesized via the combination of ring-opening polymerization (ROP) and atomic transfer radical polymerization (ATRP). These block copolymers possess PS-b-PCL diblock, PS-b-PCL-b-PS triblock, and PCL-b-PS-b-PCL triblock architectures; they were carefully controlled to have the identical composition and overall molecular weights. It was found that the block copolymers with different architectures in epoxy thermosets displayed quite different reaction-induced microphase separation behavior as evidenced with the results of atomic force microscopy (AFM), small-angle X-ray scattering (SAXS), and dynamic mechanical thermal analysis (DMTA). The morphological transition from spherical to cylindrical to lamellar nanophases occurred with increasing the content of the block copolymer in the thermosets containing PS-b-PCL diblock copolymer. In the thermosets containing PS-b-PCL-b-PS triblock copolymer, unilamellar and multilamellar nanophases were formed depending on the content of the triblock copolymer. In contrast, the macroscopic phase separation occurred in the thermosets containing PCL-b-PS-b-PCL triblock copolymer. The behavior of nanophases in these thermosetting blends have been accounted for the demixing behavior of the miscible blocks (viz. PCL) during the reaction-induced microphase separation and the influence of copolymer architectures on the morphologies of PS microdomains.
Co-reporter:Qinqin Zheng
Journal of Polymer Science Part A: Polymer Chemistry 2012 Volume 50( Issue 9) pp:1717-1727
Publication Date(Web):
DOI:10.1002/pola.25938
Abstract
Poly(N-isopropylacrylamide)-block-poly(ethylene oxide)-block-poly(N-isopropylacrylamide) (PNIPAAm-b-PEO-b-PNIPAAm) triblock copolymer was synthesized via the reversible addition-fragmentation chain transfer/macromolecular design via the interchange of xanthate (RAFT/MADIX) process with xanthate-terminated poly(ethylene oxide) (PEO) as the macromolecular chain transfer agent. The successful synthesis of the ABA triblock copolymer inspired the preparation of poly(N-isopropylacrylamide)-block-poly(ethylene oxide) (PNIPAAm-b-PEO) copolymer networks with N,N′-methylenebisacrylamide as the crosslinking agent with the similar approach. With the RAFT/MADIX process, PEO chains were successfully blocked into poly(N-isopropylacrylamide) (PNIPAAm) networks. The unique architecture of PNIPAAm-b-PEO networks allows investigating the effect of the blocked PEO chains on the deswelling and reswelling behavior of PNIPAAm hydrogels. It was found that with the inclusion of PEO chains into the PNIPAAm networks as midblocks, the swelling ratios of the hydrogels were significantly enhanced. Furthermore, the PNIPAAm-b-PEO hydrogels displayed faster response to the external temperature changes than the control PNIPAAm hydrogel. The accelerated deswelling and reswelling behaviors have been interpreted based on the formation of PEO microdomains in the PNIPAAm networks, which could act as the hydrophilic tunnels to facilitate the diffusion of water molecules in the PNIPAAm networks. © 2012 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2012
Co-reporter:Yaochen Zheng, Sixun Zheng
Reactive and Functional Polymers 2012 72(3) pp: 176-184
Publication Date(Web):March 2012
DOI:10.1016/j.reactfunctpolym.2011.12.006
Co-reporter:Lei Wang, Chongyin Zhang and Sixun Zheng
Journal of Materials Chemistry A 2011 vol. 21(Issue 48) pp:19344-19352
Publication Date(Web):02 Nov 2011
DOI:10.1039/C1JM13596A
In this communication, we reported the synthesis of organic-inorganic poly(hydroxyether of bisphenol A) (PH) copolymers with polyhedral oligomeric silsesquioxane (POSS) in the main chains via direct polymerization between diepoxy and bisphenol A. Toward this end, a novel diepoxy POSS macromer, i.e., 3,13-diglycidyloxypropyloctaphenyl double-decker silsesquioxane (denoted diglycidyloxypropyl DDSQ) was synthesized via the hydrosilylation between 3,13-dihydrooctaphenyl double-decker silsesquioxane (denoted dihydro DDSQ) and allyl glycidyl ether. Both diglycidyloxypropyl DDSQ and diglycidyl ether of bisphenol A (DGEBA) were used as the diepoxy monomers. By adjusting the ratio of diglycidyloxypropyl DDSQ to DGEBA, the PH copolymers with variable content of POSS in the main chains were obtained. It is found that with the direct polymerization the PH copolymers with POSS in the main chains of polymers can be successfully obtained while the content of diglycidyloxypropyl DDSQ was not very high. Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) showed that the glass transition temperatures (Tg) of the organic-inorganic PH copolymers were quite dependent on the contents of POSS in the copolymers. Thermogravimetric analysis (TGA) indicates that the stability of the copolymers was significantly improved in terms of the temperatures at the maximum rate of degradation and the yields of degradation residues. Owing to the presence of POSS in the main chains, the surface hydrophobicity of the organic-inorganic copolymers was significantly improved compared to control PH.
Co-reporter:Lei Wang, Ke Zeng, and Sixun Zheng
ACS Applied Materials & Interfaces 2011 Volume 3(Issue 3) pp:898
Publication Date(Web):March 7, 2011
DOI:10.1021/am101258k
Hepta(3,3,3-trifluoropropyl) polyhedral oligomeric silsesquioxane (POSS)-capped poly(N-isopropylacrylamide) (PNIPAAm) telechelics with variable lengths of PNIPAAm midblocks were synthesized by the combination of reversible addition−fragmentation chain transfer polymerization (RAFT) and the copper-catalyzed Huisgen 1,3-cycloaddition (i.e., click chemistry). The POSS-capped trithiocarbonate was synthesized and used as the chain transfer agent for the RAFT polymerization of N-isopropylacrylamide. The organic−inorganic amphiphilic telechelics were characterized by means of nuclear magnetic resonance spectroscopy (NMR) and gel permeation chromatography (GPC). Atomic force microscopy (AFM) shows that all the POSS-capped PNIPAAm telechelics exhibited microphase-separated morphologies, in which the POSS terminal groups were self-assembled into the microdomains and dispersed into the continuous PNIPAAm matrices. The POSS nanodomains could behave as the physical cross-linking sites and as a result the physical hydrogels were formed while these POSS-capped PNIPAAm telechelics were subjected to the solubility tests with water. These physical hydrogels possessed well-defined volume phase transition phenomena and displayed rapid reswelling and deswelling thermoresponsive behavior compared to control PNIPAAm hydrogel.Keywords: poly(N-isopropylacrylamide); polyhedral oligomeric silsesquioxane; telechelics and physical hydrogels
Co-reporter:Yan Li, Chongyin Zhang, Sixun Zheng
European Polymer Journal 2011 Volume 47(Issue 8) pp:1550-1562
Publication Date(Web):August 2011
DOI:10.1016/j.eurpolymj.2011.05.003
Benzoxazine-terminated poly(ethylene oxide) telechelics (Ba-terminated PEO) was synthesized and incorporated into polybenzoxazine to obtain the nanostructured thermosets. The morphology of the thermosets was investigated by means of atomic force microscopy (AFM), small angle X-ray scattering (SAXS) and dynamic mechanical analysis (DMA). The formation of the nanophase structures in the thermosetting composites was addressed on the basis of the mechanism of reaction-induced microphase separation (RIMPS), which was in marked contrast to the case of the binary thermosetting blends of polybenzoxazine with hydroxyl-terminated poly(ethylene oxide). The occurrence of RIMPS resulted from the copolymerization reaction of the end groups of Ba-terminated PEO telechelics with the precursor of thermosetting matrix (i.e., benzoxazine), which suppressed the occurrence of the macroscopic phase separation. It was found that the formation of the nanostructures has a significant effect on the melting behavior of PEO in the thermosets, thermal transition properties of the PBZ thermosets.Benzoxazine-terminated poly(ethylene oxide) telechelics (Ba-terminated PEO) was synthesized and incorporated into polybenzoxazine (PBa) to obtain the nanostructured thermosets, which is in marked contrast to the case that the macroscopic phase separation occurred in thermosetting blends of polybenzoxazine with poly(ethylene oxide) (PEO).
Co-reporter:Di Hu
Journal of Applied Polymer Science 2011 Volume 119( Issue 5) pp:2933-2944
Publication Date(Web):
DOI:10.1002/app.32977
Abstract
Polysulfone-block-polydimethylsiloxane (PSF-b-PDMS) multiblock copolymer was synthesized via the Mannich polycondensation between phenolic hydroxyl-terminated polysulfone and aminopropyl-terminated polydimethylsiloxane in the presence of formaldehyde. The multiblock copolymer was characterized by means of nuclear magnetic resonance spectroscopy (NMR) and gel permeation chromatography (GPC) and used as a modifier to improve the thermomechanical properties of epoxy thermosets. Transmission electron microscopy (TEM) showed that the epoxy thermosets containing PSF-b-PDMS multiblock copolymer possesses the microphase-separated morphological structures. Depending on the content of the PSF-b-PDMS multiblock copolymer, the spherical particles with the size of 50–200 nm in diameter were dispersed into the continuous epoxy matrices. The measurement of static contact angles showed that with the inclusion of PSF-b-PDMS multiblock copolymer, the epoxy thermosets displayed the improved surface hydrophobicity. It is noted that the epoxy resin was significantly toughened in terms of the measurement of critical stress field intensity factor (K1C). © 2010 Wiley Periodicals, Inc. J Appl Polym Sci, 2011
Co-reporter:Fangping Yi, Rentong Yu, Sixun Zheng, Xiuhong Li
Polymer 2011 Volume 52(Issue 24) pp:5669-5680
Publication Date(Web):10 November 2011
DOI:10.1016/j.polymer.2011.09.055
Poly(2,2,2-trifluoroethyl acrylate)-block-poly(glycidyl methacrylate) (PTFEA-b-PGMA) diblock copolymer was synthesized via sequential reversible addition-fragmentation chain transfer (RAFT) polymerization. The reactive diblock copolymer was incorporated into epoxy to obtain the nanostructured thermosets. The morphology of the thermosets was investigated by means of atomic force microscopy (AFM) and small-angle X-ray scattering (SAXS). It is identified that the demixing of the reactive subchain (viz. PGMA) out of epoxy matrix occurred in the process of curing reaction, which exerted a profound impact on the glass transition temperatures of the nanostructured thermosets. The static contact angle measurements showed that the nanostructured thermosets displayed a significant enhancement in surface hydrophobicity as well as a reduction in surface free energy. The improvement in surface properties was attributed to the enrichment of the fluorine-containing block (i.e., PTFEA) of amphiphilic diblock copolymer on the surface of the thermosets, which was further evidenced by surface atomic force microscopy (AFM). The measurement of critical stress intensity factor (K1C) showed that the fracture toughness of the materials was significantly enhanced by the inclusion of a small amount of PTFEA-b-PGMA diblock copolymer.
Co-reporter:Junzhang Song, Rentong Yu, Lei Wang, Sixun Zheng, Xiuhong Li
Polymer 2011 Volume 52(Issue 10) pp:2340-2350
Publication Date(Web):4 May 2011
DOI:10.1016/j.polymer.2011.03.038
Poly(N-isopropylacrylamide)-block-poly(N-vinylpyrrolidone) diblock copolymer (PNIPAAm-b-PVPy) was successfully synthesized via sequential reversible addition-fragmentation chain transfer/macromolecular design via the interchange of xanthate (RAFT/MADIX) process, in which the chain transfer agent of xanthate was in situ afforded via the reaction of isopropylxanthic disulfide (DIP) with 2,2-azobisisobutylnitrile (AIBN). The RAFT/MADIX technique was employed to prepare the poly(N-vinylpyrrolidone)-grafted poly(N-isopropylacrylamide) copolymers (PNIPAAm-g-PVPy) with N,N-methylenebisacrylamide as the crosslinking agent. The comb-like PNIPAAm-g-PVPy copolymer networks with PVPy as the pendent chains were characterized by means of Fourier transform infrared spectroscopy (FTIR), differential scanning calorimetry (DSC) and small angle X-ray scattering (SAXS). The hydrogel behavior of PNIPAAm-g-PVPy networks was investigated in terms of swelling, deswelling and reswelling tests. With the inclusion of PVPy chains, the swelling ratios of the hydrogels were significantly enhanced compared to the control PNIPAAm hydrogel. It is found that the PVPy-modified PNIPAAm hydrogels displayed faster response to the external temperature changes than the control PNIPAAm hydrogel. The improved thermoresponsive properties of hydrogels are ascribed to the formation of the comb-like architectures in the copolymer networks.
Co-reporter:Rentong Yu and Sixun Zheng
Macromolecules 2011 Volume 44(Issue 21) pp:8546-8557
Publication Date(Web):October 4, 2011
DOI:10.1021/ma201456x
In this study, we synthesized poly(ethylene oxide)-block-poly(ε-caprolactone)-block-polystyrene (PEO-b-PCL-b-PS) triblock copolymer via the combination of ring-opening polymerization (ROP) and atomic transfer radical polymerization (ATRP). The ABC triblock copolymer was incorporated into epoxy to access the nanostructured thermosets. It is found that the nanophases of the epoxy thermosets can be modulated by using different hardeners. While cured with 4,4′-methylenebis(2-chloroaniline), the thermosets displayed the long-ranged ordered nanostructures in which the spherical nanophases were arranged into body-centered cubic (bcc) lattice at the compositions investigated. While 4,4′-diaminodiphenylsulfone was used as the hardener, the thermosets displayed the lamellar nanostructure. The formation of nanostructures in the thermosets has been evidenced by atomic force microscopy and small-angle X-ray scattering. The morphological transition from spherical to lamellar nanophases has been interpreted in terms of the microphase separation of different subchains of the ABC triblock copolymer out of the epoxy–amine matrix during the curing reactions owing to the dependence of miscibility of epoxy networks with PCL subchain of the triblock copolymer on types of hardeners. The kinetics of curing and microphase separation shows the tandem reaction-induced microphase separation occurred while DDS was used as the hardener, which gave rise to the formation of lamellar nanostructures in the epoxy thermosets containing the ABC triblock copolymer.
Co-reporter:Di Hu, Sixun Zheng
Polymer 2010 Volume 51(Issue 26) pp:6346-6354
Publication Date(Web):10 December 2010
DOI:10.1016/j.polymer.2010.10.047
Poly(N-vinyl pyrrolidone)-block-polystyrene diblock copolymer (PVPy-b-PS) was synthesized via sequential reversible radical-fragmentation transfer polymerization with S-1-phenylethyl O-ethylxanthate as a chain transfer agent. The block copolymer was incorporated into polybenzoxazine to access the nanostructures in the thermosets. The nanostructures in the thermosets were investigated by means of transmission electron microscopy (TEM) and small-angle X-ray scattering (SAXS). It was found that disordered and/or ordered PS nanophases were formed in the PBa thermosets. It is judged that the formation of nanophases followed the mechanism of reaction-induced microphase separation in terms of the miscibility of the subchains of the diblock copolymer (viz. PVPy and PS) with polybenzoxazine after and before curing reaction.
Co-reporter:Lei Wang, Sixun Zheng
Polymer 2010 Volume 51(Issue 5) pp:1124-1132
Publication Date(Web):2 March 2010
DOI:10.1016/j.polymer.2010.01.008
The main-chain polybenzoxazine-block-polydimethylsiloxane multiblock copolymers were synthesized via the Mannich polycondensation among 4,4′-dihydroxyldiphenylisopropane, 4,4′-diaminodiphenylmethane, aminopropyl-terminated polydimethylsiloxane and paraformaldehyde. The multiblock copolymers were characterized by means of Fourier transform infrared spectroscopy (FTIR), nuclear magnetic resonance spectroscopy (NMR) and size exclusion chromatography (SEC). Atomic force microscopy (AFM) and small-angle X-ray scattering (SAXS) showed that the multiblock copolymers displayed microphase-separated morphology. Owing to the presence of the main-chain polybenzoxazine blocks, the multiblock copolymers are thermally-crosslinkable. The curing behavior of the multiblock copolymers was investigated according to the analysis of non-isothermal curing kinetics. The measurement of static contact angles showed that with the inclusion of polydimethylsiloxane blocks, the polybenzoxazine thermosets resulting from the multiblock copolymers displayed the improved surface hydrophobicity.
Co-reporter:Di Hu, Zhiguang Xu, Ke Zeng and Sixun Zheng
Macromolecules 2010 Volume 43(Issue 6) pp:2960-2969
Publication Date(Web):February 24, 2010
DOI:10.1021/ma902770r
Polystyrene-block-poly(ethylene oxide) (PS-b-PEO) diblock copolymer was synthesized and incorporated into novolac resin to obtain the nanostructured phenolic thermosets with hexamethylenetetramine (HMTA) as the curing agent. The morphology of the thermosets were investigated by means of atomic force microscopy (AFM) and small-angle X-ray scattering (SAXS). It was found that long-ranged ordered nanostructures were formed after and before curing reaction. In views of the miscibility of the subchains of the diblock copolymer with the phenol−formaldehyde resins after and before curing reaction, it was judged that the formation of the nanostructures followed the mechanism of self-assembly. In the thermosetting blends, the PEO subchain of the diblock copolymer was miscible with phenolic thermosets after and before cuing reaction. Fourier transform infrared (FTIR) spectroscopy showed that the curing reaction significantly weakened the intermolecular hydrogen-bonding interactions between phenolic matrix and PEO subchains of the diblock copolymer. The nanostructured thermosets were subjected to pyrolysis (and/or carbonization) at elevated temperatures to obtain the nanoporous carbons. The hierarchical nanoporosity of the resulting carbons was confirmed by means of transmission electronic microscopy (TEM), field-emission scanning electronic microscopy (FESEM), and surface-area Brunauer−Emmett−Teller (BET) measurements.
Co-reporter:Wenchun Fan, Lei Wang, and Sixun Zheng
Macromolecules 2010 Volume 43(Issue 24) pp:10600-10611
Publication Date(Web):December 1, 2010
DOI:10.1021/ma101945f
Polystyrene-block-poly(ε-caprolactone)-block-poly(n-butyl acrylate) (PS-b-PCL-b-PBA) triblock copolymer was synthesized through the combination of atom transfer radical polymerization, copper-catalyzed Huisgen 1,3-dipolar cycloaddition and ring-opening polymerization. The PS-b-PCL-b-PBA ABC triblock copolymer was incorporated into epoxy to access the nanostructures in the thermoset. The microphase-separated morphology was investigated by means of atomic force microscopy (AFM), small-angle X-ray scattering (SAXS) and dynamic mechanical thermal analysis (DMTA). It was found that depending on the concentration of the triblock copolymer in the thermosets several kinds of nanodomains were formed and they were arranged in lamellar lattice. The formation of the nanostructures was ascribed to the tandem reaction-induced microphase separation of PBA and PS blocks in the thermosetting blends. The investigation of the model binary thermosetting blends showed that the phase separation of PBA occurred at the conversion much lower than that of PS. It is proposed that the PBA nanophases were formed prior to the PS nanophases in the thermosetting blends and the microdomains of PBA subchains could behave as the template for the demixing of PS blocks. The coupling of the two-stage reaction-induced microphase separation exerted a profound impact on the formation of nanostructures in the epoxy thermosets containing the ABC triblock copolymer. Thermal analysis shows that with the formation of the nanostructures in the thermosets a part of poly(ε-caprolactone) subchains were demixed from epoxy matrix; the fractions of demixed PCL blocks have been estimated according to the Tg-composition relation of the model binary blends of epoxy and PCL.
Co-reporter:Yan Li
Journal of Polymer Science Part B: Polymer Physics 2010 Volume 48( Issue 11) pp:1148-1159
Publication Date(Web):
DOI:10.1002/polb.22004
Abstract
A multiblock copolymer consisting of main-chain polybenzoxazine and poly(propylene oxide) blocks was synthesized via Mannich polycondensation among 4,4′-dihydroxyldiphenylisopropane, 4,4′-diaminodiphenylmethane, amino-terminated poly(propylene oxide), and paraformaldehyde, which was evidenced by Fourier transform infrared spectroscopy, nuclear magnetic resonance spectroscopy, and gel permeation chromatography. The multiblock copolymer was incorporated into polybenzoxazine to access the nanostructured polybenzoxazine thermosets. The morphology of the thermosets was investigated by means of atomic force microscopy and small angle X-ray scattering. It was judged that the formation of the nanostructures in the thermosetting composites follows the mechanism of reaction-induced microphase separation. Owing to the big difference in thermal stability between polybenzoxazine thermosets and poly(propylene oxide), the nanostructured thermosets were subjected to the pyrolysis at moderate elevated temperatures to remove poly(propylene oxide) microdomains, to access the nanoporous polybenzoxazine thermosets. The nanoporosity of the resulting polybenzoxazine thermosets was investigated by means of Fourier transform infrared spectroscopy and field-emission scanning electronic microscopy. © 2010 Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys 48: 1148–1159, 2010
Co-reporter:Di Hu, Chongyin Zhang, Rentong Yu, Lei Wang, Sixun Zheng
Polymer 2010 Volume 51(Issue 25) pp:6047-6057
Publication Date(Web):26 November 2010
DOI:10.1016/j.polymer.2010.10.016
In this work, we investigated the self-assembly behavior of poly(ɛ-caprolactone)-block-poly(ethylene-co-ethylethylene)-block-poly(ɛ-caprolactone) (PCL-b-PEEE-b-PCL) triblock copolymer in epoxy thermosets. The PCL-b-PEEE-b-PCL triblock copolymer was synthesized via the ring-opening polymerization of ɛ-caprolactone with a hydroxyl-terminated poly(ethylene-co-ethylethylene) as the macromolecular initiator. The hydroxyl-terminated poly(ethylene-co-ethylethylene) was prepared with the hydrogenation reaction of a hydroxyl-terminated polybutadiene. The triblock copolymer was incorporated into the precursors of epoxy to obtain the nanostructured thermosets. It was found that the self-organized nanophases were formed in the mixture before curing reaction and the nanostructures can be further fixed via curing reaction. The self-assembly behavior of the triblock copolymer in epoxy thermosets was investigated by means of atomic force microscopy (AFM), small-angle X-ray scattering (SAXS) and dynamic mechanical thermal analysis (DMTA). Differential scanning calorimetry (DSC) shows that the formation of the self-organized nanophase in the thermosets caused that a part of poly(ɛ-caprolactone) subchains were demixed from epoxy matrix with the occurrence of curing reaction; the fractions of demixed PCL blocks were estimated according to the Tg-composition relation of the model binary blends of epoxy and PCL.
Co-reporter:Di Hu, Sixun Zheng
European Polymer Journal 2009 Volume 45(Issue 12) pp:3326-3338
Publication Date(Web):December 2009
DOI:10.1016/j.eurpolymj.2009.10.001
Polystyrene-block-poly(ethylene oxide) alternating multiblock copolymer (PS-alt-PEO) was synthesized with the combination of atom transfer radical polymerization (ATRP) and Huisgen 1,3-dipolar cycloaddition (i.e., click chemistry). The copolymer has been characterized by means of Fourier transform infrared spectroscopy (FTIR), 1H nuclear magnetic resonance spectroscopy (NMR), gel permeation chromatography (GPC) and differential scanning calorimetry (DSC). The PS-alt-PEO alternating multiblock copolymer was incorporated into epoxy resin to investigate the behavior of reaction-induced microphase separation, which has been compared to the case of the thermosets containing PS-b-PEO diblock copolymer. The morphology of epoxy thermosets containing PS-alt-PEO alternating multiblock copolymer were investigated by means of atomic force microscopy (AFM), and small-angle X-ray scattering (SAXS) and the nanostructures were detected in all the thermosetting blends investigated. In marked contrast to the case of the thermosets containing PS-b-PEO diblock copolymer, the thermosets containing PS-alt-PEO multiblock copolymer displayed disordered nanostructures, which have been interpreted on the basis of the restriction of the alternating multiblock topology of the block on the formation of the nanostructures via reaction-induced microphase separation.
Co-reporter:Lei Wang;Wei Gong
Polymer International 2009 Volume 58( Issue 2) pp:124-132
Publication Date(Web):
DOI:10.1002/pi.2501
Abstract
BACKGROUND: An important strategy for making polymer materials with combined properties is to prepare block copolymers consisting of well-defined blocks via facile approaches.
RESULTS: Poly(hydroxyether of bisphenol A)-block-polydimethylsiloxane alternating block copolymers (PH-alt-PDMS) were synthesized via Mannich polycondensation involving phenolic hydroxyl-terminated poly(hydroxyether of bisphenol A), diaminopropyl-terminated polydimethylsiloxane and formaldehyde. The polymerization was carried out via the formation of benzoxazine ring linkages between poly(hydroxyether of bisphenol A) and polydimethylsiloxane blocks. Differential scanning calorimetry and small-angle X-ray scattering show that the alternating block copolymers are microphase-separated. Compared to poly(hydroxyether of bisphenol A), the copolymers displayed enhanced surface hydrophobicity (dewettability). In addition, subsequent crosslinking can occur upon heating the copolymers to elevated temperatures owing to the existence of benzoxazine linkages in the microdomains of hard segments.
CONCLUSION: PH-alt-PDMS alternating block copolymers were successfully obtained. The subsequent self-crosslinking of the PH-alt-PDMS alternating block copolymers could lead to these polymer materials having potential applications. Copyright © 2008 Society of Chemical Industry
Co-reporter:Ke Zeng
Macromolecular Chemistry and Physics 2009 Volume 210( Issue 9) pp:783-791
Publication Date(Web):
DOI:10.1002/macp.200800605
Co-reporter:Ke Zeng;Yuan Fang
Journal of Polymer Science Part B: Polymer Physics 2009 Volume 47( Issue 5) pp:504-516
Publication Date(Web):
DOI:10.1002/polb.21655
Abstract
3-Acryloxypropylhepta(3,3,3-trifluoropropyl) polyhedral oligomeric silsesquioxane (POSS) was synthesized and used as a modifier to improve the thermal response rates of poly(N-isopropylacrylamide) (PNIPAM) hydrogel. The radical copolymerization among N-isopropylacrylamide (NIPAM), the POSS macromer and N,N′-methylenebisacrylamide was performed to prepare the POSS-containing PNIPAM cross-linked networks. Differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA) showed that the POSS-containing PNIPAM networks displayed the enhanced glass transition temperatures (Tg's) and improved thermal stability when compared with plain PNIPAM network. The POSS-containing PNIPAM hydrogels exhibited temperature-responsive behavior as the plain PNIPAM hydrogels. It is noted that with the moderate contents of POSS, the POSS-containing PNIPAM hydrogels displayed much faster response rates in terms of swelling, deswelling, and re-swelling experiments than plain PNIPAM hydrogel. The improved thermoresponsive properties of hydrogels have been interpreted on the basis of the formation of the specific microphase-separated morphology in the hydrogels, that is, the POSS structural units in the hybrid hydrogels were self-assembled into the highly hydrophobic nanodomains, which behave as the microporogens and promote the contact of PNIPAM chains and water. © 2009 Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys 47: 504–516, 2009
Co-reporter:Fangping Yi and Sixun Zheng, Tianxi Liu
The Journal of Physical Chemistry B 2009 Volume 113(Issue 7) pp:1857-1868
Publication Date(Web):January 26, 2009
DOI:10.1021/jp8082198
Poly(2,2,2-trifluoroethyl acrylate)-block-poly(ethylene oxide) (PTFEA-b-PEO) amphiphilic diblock copolymer was synthesized via the reversible addition-fragmentation transfer polymerization of 2,2,2-triffluroethyl acrylate with dithiobenzoyl-terminated poly(ethylene oxide) as a chain-transfer agent. The amphiphilic diblock copolymer was incorporated into epoxy resin to prepare the nanostructured epoxy thermosets. The nanostructures were investigated by means of atomic force microscopy, small-angle X-ray scattering, and dynamic mechanical analysis. In terms of the miscibility of the subchains of the block copolymer with epoxy after and before curing reaction, it is judged that the formation of the nanostructures follows the mechanism of self-assembly. The static contact angle measurements indicate that the nanostructured thermosets containing PTFEA-b-PEO diblock copolymer displayed a significant enhancement in surface hydrophobicity as well as a reduction in surface free energy. The improvement in surface properties was ascribed to the enrichment of the fluorine-containing subchain (i.e., PTFEA block) of the amphiphilic diblock copolymer on the surface of the nanostructured thermosets, which was evidenced by surface atomic force microscopy and energy-dispersive X-ray spectroscopy.
Co-reporter:Wenchun Fan, Lei Wang and Sixun Zheng
Macromolecules 2009 Volume 42(Issue 1) pp:327-336
Publication Date(Web):December 4, 2008
DOI:10.1021/ma8018014
Polydimethylsiloxane-block-poly(ε-caprolactone)-block-polystyrene ABC-type triblock copolymer (PDMS-b-PCL-b-PS) was synthesized via the sequential ring-opening polymerization and atom transfer radical polymerization. The ABC triblock copolymer was incorporated into epoxy to prepare the nanostructured thermosets. In terms of the difference in miscibility of epoxy with the subchains of the ABC triblock copolymer after and before curing reaction, it is proposed that the formation of the nanostructures follows the combined mechanisms of self-assembly and reaction-induced microphase separation. The self-organized nanophases could be formed before the curing reaction since the PDMS subchains of the triblock polymer is immiscible with the precursors of epoxy before curing reaction. The reaction-induced microphase separation of PS subchains occurred in the presence of the PDMS nanophases whereas the PCL subchains remain miscible with epoxy after and before curing reaction. By means of atomic force microscopy and small-angle X-ray scattering, the morphological structures of the thermosets containing the ABC triblock copolymer were examined, and the formation of the nanostructures was addressed on the basis of the combination of self-assembly and reaction-induced microphase separation mechanisms.
Co-reporter:Jin Han
Journal of Polymer Science Part A: Polymer Chemistry 2009 Volume 47( Issue 24) pp:6894-6907
Publication Date(Web):
DOI:10.1002/pola.23729
Abstract
Organic–inorganic hybrid brushes comprised of macrocyclic oligomeric silsesquioxane (MOSS) and poly(ε-caprolactone) (PCL) were synthesized via the ring-opening polymerization of ε-caprolactone (CL) with cis-hexa[(phenyl) (2-hydroxyethylthioethyldimethylsiloxy)]cyclohexasiloxane as the initiator. The MOSS macromer bearing hydroxyl groups was synthesized via the thiol-ene radical addition reaction between cis-hexa[(phenyl)(vinyldimethylsiloxy)]cyclohexasiloxane and β-mercaptoethanol. The organic–inorganic PCL cyclic brushes were characterized by means of nuclear magnetic resonance spectroscopy (NMR) and gel permeation chromatography (GPC). These MOSS–PCL brushes were then used to prepare the supramolecular inclusion complexes with α-cyclodextrin (α-CD). The X-ray diffraction (XRD) indicates that the organic–inorganic inclusion complexes (ICs) have a channel-type crystalline structure. It is noted that the molar ratios of CL unit to α-CD for the organic–inorganic ICs are quite dependent on the lengths of the PCL chains bonded to the silsesquioxane macrocycle. While the PCL chains were short, the efficiency of inclusion complexation was significantly decreased. The decreased efficiency could be attributed to the repulsion of the adjacent PCL chains bonded to the silsesquioxane macrocycle and the restriction of the bulky silsesquioxane macrocycle on the motion of PCL chains; this effect is pronounced with decreasing the length of the PCL chains. © 2009 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2009
Co-reporter:Fangping Yi, Sixun Zheng
Polymer 2009 50(2) pp: 670-678
Publication Date(Web):
DOI:10.1016/j.polymer.2008.11.038
Co-reporter:Ke Zeng, Lei Wang, Sixun Zheng, Xuefeng Qian
Polymer 2009 50(2) pp: 685-695
Publication Date(Web):
DOI:10.1016/j.polymer.2008.11.024
Co-reporter:Xingtian Yang, Fangping Yi, Zhirong Xin, Sixun Zheng
Polymer 2009 50(16) pp: 4089-4100
Publication Date(Web):
DOI:10.1016/j.polymer.2009.06.030
Co-reporter:Ke Zeng, Lei Wang and Sixun Zheng
The Journal of Physical Chemistry B 2009 Volume 113(Issue 35) pp:11831-11840
Publication Date(Web):August 11, 2009
DOI:10.1021/jp9043623
Hepta(3,3,3-trifluoropropyl) polyhedral oligomeric silsesquioxane-capped poly(ethylene oxide) telechelics (POSS-capped PEO) were synthesized via click chemistry. The POSS-capped PEO amphiphilic telechelics were incorporated into cross-linked poly(N-isopropylacrylamide) (PNIPAAm) to form the structure of physical interpenetrating polymer networks (IPNs). In the organic−inorganic networks, the POSS terminals of POSS-capped PEO telechelics were self-organized into microdomains to act as the physical cross-linking sites of PEO and the physically cross-linked PEO network was interlocked with the PNIPAAm network. It is identified that the organic−inorganic hydrogels resulting from the PNIPAAm network and the POSS-capped PEO telechelics displayed much faster response rates than the plain PNIPAAm hydrogels in terms of swelling, deswelling, and reswelling tests. The improved thermoresponse of hydrogels has been interpreted on the basis of the formation of the specific supramolecular structures in the hydrogels. The synergism from hydrophobic and hydrophilic components (i.e., POSS domains and PEO chains) is responsible for the improvement of hydrogel properties.
Co-reporter:Lei Li
Journal of Polymer Science Part B: Polymer Physics 2008 Volume 46( Issue 21) pp:2296-2306
Publication Date(Web):
DOI:10.1002/polb.21561
Abstract
Poly(ethylene imine)-graft-poly(ethylene oxide) (PEI-g-PEO) copolymers were synthesized via Michael addition reaction between acryl-terminated poly(ethylene oxide) methyl ether (PEO) and poly(ethylene imine) (PEI). The brush-like copolymers were characterized by means of Fourier transform infrared spectroscopy and nuclear magnetic resonance spectroscopy. It is found that the crystallinity of the PEO side chains in the copolymers remained unaffected by the PEI backbone whereas the crystal structure of PEO side chains was altered to some extent by the PEI backbone. The crystallization behavior of PEO blocks in the copolymers suggests that the bush-shaped copolymers are microphase-separated in the molten state. The PEO side chains of the copolymers were selectively complexed with α-cyclodextrin (α-CD) to afford hydrophobic side chains (i.e., PEO/α-CD inclusion complexes). The X-ray diffraction (XRD) shows that the inclusion complexes (ICs) of the PEO side chains displayed a channel-type crystalline structure. It is identified that the stoichiometry of the inclusion complexation of the PEI-g-PEO with α-CD is close to that of the control PEO with α-CD. © 2008 Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys 46: 2296–2306, 2008
Co-reporter:Jin Han and Sixun Zheng
Macromolecules 2008 Volume 41(Issue 13) pp:4561-4564
Publication Date(Web):June 7, 2008
DOI:10.1021/ma800940t
Co-reporter:Qipeng Guo;Weian Zhang
Journal of Applied Polymer Science 2007 Volume 106(Issue 1) pp:417-424
Publication Date(Web):21 JUN 2007
DOI:10.1002/app.26484
Dendritic star-shaped poly(ε-caprolactone)-block-poly(L-lactide) (PCL-b-PLLA) diblock copolymers were synthesized via sequential ring-opening polymerization. In the first step, an aliphatic dendritic polyester containing 16 terminal hydroxyl groups was used as the core molecule to initiate the ring-opening polymerization of ε-caprolactone, which was catalyzed by stannous(II) octanoate, to obtain dendritic star-shaped poly(ε-caprolactone) (PCL) terminated with hydroxyls, which was used further to initiate the ring-opening polymerization of L-lactide to form the dendritic star-shaped diblock copolymers. The dendritic star-shaped polymers (PCL-b-PLLA) were characterized with nuclear magnetic resonance spectroscopy and gel permeation chromatography. The results showed that the arm length of the dendritic star-shaped polymers could be well controlled in terms of the molar ratios of the initiators (i.e., the aliphatic dendritic polyester and star-shaped PCL) to the monomers (i.e., ε-caprolactone and L-lactide). The crystalline structure and thermal properties of the dendritic star-shaped polymers were investigated with X-ray diffraction and differential scanning calorimetry. The X-ray diffraction indicated that the formation of the dendritic star-shaped topological structure did not affect the structure of the crystals of PCL and poly(L-lactide) (PLLA) blocks. The thermal analyses showed that the crystallization rate of the PCL blocks in the block copolymers was greatly reduced compared to that in the parent dendritic star-shaped PCL. This observation could be attributed to the confinement of the dendritic core and PLLA blocks upon the crystallization of the PCL blocks. © 2007 Wiley Periodicals, Inc. J Appl Polym Sci 2007
Co-reporter:Xingtian Yang
Polymer International 2007 Volume 56(Issue 7) pp:
Publication Date(Web):11 MAY 2007
DOI:10.1002/pi.2231
A novel semi-crystalline polyhydroxyether, poly(hydroxyether ketone) (PHEK), was synthesized via the direct polycondensation between 4,4-dihydroxybenzophenone and epichlorohydrin. By means of Fourier transform infrared and NMR spectroscopy and gel permeation chromatography (GPC), the structure of PHEK was characterized. Differential scanning calorimetry (DSC) and wide-angle X-ray diffraction (WAXRD) show that PHEK is a semi-crystalline polymer with a high rate of crystallization. The polymer possesses a glass transition temperature of 109 °C and a melting temperature of 239 °C. When 4,4′-isopropylidenediphenol was used as a second bisphenol and was added to copolymerize with a stoichiometric amount of epichlorohydin, a series of polyhydroxyether copolymers were obtained. The copolymers with various compositions were characterized by means of NMR, GPC, WAXRD and DSC. It was found that the crystallinity of the copolymers dramatically decreased with increasing content of 4,4′-isopropylidenediphenol. The glass transition temperatures of the copolymers are intermediate between those of PHEK and the poly(hydroxyether of bisphenol A) and decreased with increasing content of 4,4′-isopropylidenediphenol. Copyright © 2007 Society of Chemical Industry
Co-reporter:Sixun Zheng;Yong Ni
Journal of Polymer Science Part B: Polymer Physics 2007 Volume 45(Issue 16) pp:2201-2214
Publication Date(Web):2 JUL 2007
DOI:10.1002/polb.21222
In this study, we investigated the melting and crystallization behavior of polyhedral oligomeric silsesquioxane (POSS)-capped poly(ε-caprolactone) PCL with various lengths of PCL chains by means of X-ray diffraction and differential scanning calorimetry. This organic–inorganic macromolecule possesses a tadpole-like structure in which the bulky POSS cage is the “head” whereas PCL chain the “tail”. The novel organic–inorganic association result in the significant alterations in the melting and crystallization behavior of PCL. The POSS-terminated PCL displayed the enhanced equilibrium melting points compared to the control PCL. Both the overall crystallization rate and the spherulitic growth rate of the POSS-terminated PCLs increased with increasing the concentration of POSS (or with decreasing length of PCL chain in the hybrids). The analysis of Avrami equation shows that the crystallization of the POSS-terminated PCL preferentially followed the mechanism of spherulitic growth with instantaneous nuclei. It is found that the folding free energy of surface for the POSS-terminated PCLs decreased with increasing the concentration of POSS. It is found that the folding free energy of surface for the POSS-terminated PCLs decreased with increasing the concentration of POSS. © 2007 Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys 45: 2201–2214, 2007
Co-reporter:Wenchun Fan
Journal of Polymer Science Part B: Polymer Physics 2007 Volume 45(Issue 18) pp:2580-2593
Publication Date(Web):6 AUG 2007
DOI:10.1002/polb.21264
A tetraarmed star-shaped poly(methyl methacrylate) (s-PMMA) was synthesized via atom transfer radical polymerization with 2-bromoisobutyryl pentaerythritol as the initiator. For comparison, a linear PMMA with the identical molecular weight (l-PMMA) was also prepared. The blends of the two PMMA samples with poly (vinylidene fluoride) (PVDF) were prepared to investigate the effect of macromolecular topological structure on miscibility and crystallization behavior of the binary blends. The behavior of single and composition-dependent glass transition temperatures was found for the blends of s-PMMA with PVDF, indicating that the s-PMMA is miscible with PVDF in the amorphous state just like l-PMMA. The miscibility was further evidenced by the depression of equilibrium melting points. It is found that the blends of s-PMMA and PVDF displayed the larger k value of Gordon–Taylor equation than the blends of l-PMMA and PVDF blends. According to the depression of equilibrium melting points, the intermolecular parameters for the two blends were estimated. It is noted that the s-PMMA/PVDF blends displayed the lower interaction parameter than l-PMMA/PVDF blends. The isothermal crystallization kinetics shows that the crystallization of PVDF in the blends containing s-PMMA is faster than that in the blends containing the linear PMMA. The surface-folding free energy of PVDF chains in the blends containing s-PMMA is significantly lower than those in the blends containing l-PMMA. © 2007 Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys 45: 2580–2593, 2007
Co-reporter:Yong Ni
Journal of Polymer Science Part A: Polymer Chemistry 2007 Volume 45(Issue 7) pp:1247-1259
Publication Date(Web):15 FEB 2007
DOI:10.1002/pola.21893
Supramolecular inclusion complexes (ICs) involving polyhedral oligomeric silsesquioxane (POSS) capped poly(ϵ-caprolactone) (PCL) and α-cyclodextrin (α-CD) were investigated. POSS-terminated PCLs with various molecular weights were prepared via the ring-opening polymerization of ϵ-caprolactone (CL) with 3-hydroxypropylheptaphenyl POSS as an initiator. Because of the presence of the bulky silsesquioxane terminal group, the inclusion complexation between α-CD and the POSS-capped PCL was carried out only with a single end of a PCL chain threading inside the cavity of α-CD, which allowed the evaluation of the effect of the POSS terminal groups on the efficiency of the inclusion complexation. The X-ray diffraction results indicated that the organic–inorganic ICs had a channel-type crystalline structure. The stoichiometry of the organic–inorganic ICs was quite dependent on the molecular weights of the POSS-capped PCLs. With moderate molecular weights of the POSS-capped PCLs (e.g., Mn =3860 or 9880), the stoichiometry was 1:1 mol/mol (CL unit/α-CD), which was close to the literature value based on the inclusion complexation of α-CD with normal linear PCL chains with comparable molecular weights. When the PCL chains were shorter (e.g., for the POSS-capped PCL of Mn = 1720 or 2490), the efficiency of the inclusion complexation decreased. The decreased efficiency of the inclusion complexation could be attributed to the lower mobility of the bulky POSS group, which restricted the motion of the PCL chain attached to the silsesquioxane cage. This effect was pronounced with the decreasing length of the PCL chains. © 2007 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 45: 1247–1259, 2007
Co-reporter:L. Zhu;J. Xi;W. Yuan;Y. Xu;X. Huang;S. Zheng;X. Tang
Advanced Materials 2006 Volume 18(Issue 22) pp:2997-3000
Publication Date(Web):23 OCT 2006
DOI:10.1002/adma.200600562
Templated synthesis of poly(cyclotriphosphaze-co-4,4′-sulfonyldiphenol) nanotubes (see figure) via a one-pot approach was achieved by the in situ formation of triethylammonium chloride nanocrystals during the polymerization. The triethylammonium chloride templates can eventually be removed by washing with water. The dimensions of the nanotubes can be controlled by modification of the experimental conditions.
Co-reporter:Weian Zhang;Yan Chen;Yonghong Liu
Journal of Applied Polymer Science 2006 Volume 99(Issue 3) pp:927-936
Publication Date(Web):18 NOV 2005
DOI:10.1002/app.22270
Benzoxazine bearing trimethoxylsilane (BA-b) was successfully synthesized via Mannich condensation among phenol, paraformaldehyde, and γ-aminopropyltrimethoxysilane. The hydrolysis and condensation of the BA-b catalyzed by hydrochloric acid can afford a soluble polysilsesquioxane bearing benzoxazine groups (denoted PSSQ-b). By initiating the cocuring reaction of PSSQ-b with difunctional benzoxazine of bisphenol A (BA-a), the inorganic–organic hybrids of polybenzoxazine with polysilsesquioxane were prepared. The hybrids displayed enhanced Tg's in comparison with the control polybenzoxazine. In terms of initial decomposition temperature (Td) and char and ceramic yields, the hybrids exhibited improved thermal stability as shown by thermogravimetric analysis (TGA). © 2005 Wiley Periodicals, Inc. J Appl Polym Sci 99: 927–936, 2006
Co-reporter:Yonghong Liu;Yong Ni
Macromolecular Chemistry and Physics 2006 Volume 207(Issue 20) pp:
Publication Date(Web):23 OCT 2006
DOI:10.1002/macp.200600241
Summary: OpePOSS was incorporated into polyurethane to make organic-inorganic hybrid composites and nano-composites containing up to 20 wt.-% POSS were prepared. The formation of the hybrid polyurethane networks is ascribed to two principal cross-linking reactions: i) the cross-linking reaction between MOCA and the polyurethane prepolymer and ii) the inter-component reaction between the PU networks and OpePOSS. The latter was confirmed by model compound reactions. TEM indicates that the POSS was homogeneously dispersed in the polymer matrix at the nanometer scale. DSC showed that the nanocomposites displayed increased glass transition temperatures compared to the control polyurethane. In terms of TGA, the nanocomposites displayed improved thermal stability. Tensile tests indicate that the organic-inorganic hybrid networks were significantly reinforced with the inclusion of POSS. Contact angle measurements show that the organic-inorganic nanocomposites displayed a significant enhancement in surface hydrophobicity as well as a reduction in the surface free energy. The improvement in surface properties was ascribed to the presence of the POSS moiety in place of the polar component of polyurethane. XPS shows enrichment with Si-containing moieties on the surfaces.
Co-reporter:Yonghong Liu;Yong Ni
Macromolecular Chemistry and Physics 2006 Volume 207(Issue 20) pp:
Publication Date(Web):23 OCT 2006
DOI:10.1002/macp.200690038
Co-reporter:Yong Ni
Journal of Polymer Science Part A: Polymer Chemistry 2006 Volume 44(Issue 3) pp:1093-1105
Publication Date(Web):13 DEC 2005
DOI:10.1002/pola.21222
Polyphenylsilsesquioxane (PPSQ) was incorporated into an epoxy resin to prepare organic–inorganic composites, and two strategies were adopted to afford composites with different morphologies. Phase separation induced by polymerization occurred in the physical blending system. However, nanostructured composites were obtained when a catalytic amount of aluminum triacetylacetonate was added to mediate the reaction between PPSQ and diglycidyl ether of bisphenol A (DGEBA). The intercomponent reaction significantly suppressed the phase separation on the micrometer scale. Organic–inorganic composites with different morphologies displayed quite different thermomechanical properties. Both differential scanning calorimetry and dynamic mechanical analysis showed that the nanostructured composites possessed higher glass-transition temperatures than the phase-separated composites with the same loading of PPSQ, although the intercomponent reaction between PPSQ and DGEBA reduced the crosslinking density of the epoxy matrix. This result was ascribed to the presence of nanosized PPSQ domains in the nanostructured composites, which acted as physical crosslinking sites and thus reinforced the epoxy networks. The nanoreinforcement of the PPSQ domains afforded the enhanced dynamic storage modulus for the nanostructured composites in comparison with the phase-separated composites with a PPSQ concentration less than 15 wt %. In terms of thermogravimetric analysis, the organic–inorganic composites displayed improved thermal stability and flame retardancy. © 2005 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 44: 1093–1105, 2006
Co-reporter:Jia Shen
Journal of Polymer Science Part B: Polymer Physics 2006 Volume 44(Issue 6) pp:942-952
Publication Date(Web):1 FEB 2006
DOI:10.1002/polb.20748
The comparative studies on the miscibility and phase behavior between the blends of linear and star-shaped poly(2-methyl-2-oxazoline) with poly(vinylidene fluoride) (PVDF) were carried out in this work. The linear poly(2-methyl-2-oxazoline) was synthesized by the ring opening polymerization of 2-methyl-2-oxazoline in the presence of methyl p-toluenesulfonate (MeOTs) whereas the star-shaped poly(2-methyl-2-oxazoline) was synthesized with octa(3-iodopropyl) polyhedral oligomeric silsesquioxane [(IC3H6)8Si8O12, OipPOSS] as an octafunctional initiator. The polymers with different topological structures were characterized by means of Fourier transform infrared spectroscopy and nuclear magnetic resonance spectroscopy. It is found that the star-shaped poly(2-methyl-2-oxazoline) was miscible with poly(vinylidene fluoride) (PVDF), which was evidenced by single glass-transition temperature behavior and the equilibrium melting-point depression. Nonetheless, the blends of linear poly(2-methyl-2-oxazoline) with PVDF were phase-separated. The difference in miscibility was ascribed to the topological effect of PMOx macromolecules on the miscibility. © 2006 Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys 44: 942–952, 2006
Co-reporter:Bingquan Liu;Weian Zhang;Qipeng Guo
Journal of Polymer Science Part B: Polymer Physics 2006 Volume 44(Issue 11) pp:1618-1626
Publication Date(Web):24 APR 2006
DOI:10.1002/polb.20824
The miscibility of poly(hydroxyether terephthalate ester) (PHETE) with poly(4-vinyl pyridine) (P4VP) was established on the basis of thermal analysis. Differential scanning calorimetry showed that each blend displayed a single glass-transition temperature (Tg), which is intermediate between those of the pure polymers and varies with the composition of blend. The Tg-composition relationship can be well described with Kwei equation with k = 1 and q = −30.8 (K), suggesting the presence of the intermolecular specific interactions in the blend system. To investigate the intermolecular specific interactions in the blends, the model compounds such as 1,3-diphenoxy-2-propanol, 4-methyl pyridine, and ethyl benzoate were used to determine the equilibrium constants, according to Coleman and Painter model, to account for the association equilibriums of several structural moieties, using liquid Fourier transform infrared difference spectroscopy. In terms of the difference in the association equilibrium constant, it is proposed that there are the competitive specific interactions in the blends, which were confirmed by means of Fourier transform infrared spectroscopy of the blends. It is observed that upon adding P4VP to the system, the ester carbonyls of PHETE that were H-bonded with the hydroxyl groups were released because of the formation of the stronger interchain association via the hydrogen bonding between the hydroxyls of PHETE and tertiary nitrogen atoms of P4VP. © 2006 Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys 44: 1618–1626, 2006
Co-reporter:Hongzhi Liu
Macromolecular Rapid Communications 2005 Volume 26(Issue 3) pp:196-200
Publication Date(Web):20 JAN 2005
DOI:10.1002/marc.200400465
Summary: Octaaminophenyl polyhedral oligomeric silsesquioxane (OapPOSS) was used as a crosslinking agent together with 4,4′-methylenebis-(2-chloroaniline) to prepare polyurethane networks containing POSS. Fourier transform infrared spectroscopy (FT-IR), dynamic mechanical analysis (DMA) and thermogravimetric analysis (TGA) were employed to characterize the POSS-reinforced polyurethane. The POSS-containing PU networks displayed enhanced glass transition temperatures (Tgs) and the storage moduli of the networks of the glassy state and rubber plateaus were also observed to be significantly higher than that of the control polyurethane although only a small amount of POSS was incorporated into the systems. The results can be ascribed to the significant nanoscale reinforcement effect of POSS cages on the polyurethane matrix. TGA results showed the thermal stability was also improved with incorporation of POSS into the system.
Co-reporter:Yonghong Liu;Fanliang Meng;Fanliang Meng;Yonghong Liu
Macromolecular Rapid Communications 2005 Volume 26(Issue 11) pp:920-925
Publication Date(Web):2 JUN 2005
DOI:10.1002/marc.200500062
Summary: Octa(propylglycidyl ether) polyhedral oligomeric silsesquioxane (OpePOSS) was used as the crosslinking agent to prepare the nanocrosslinked poly(4-vinylpyridine) (P4VP) with POSS content up to 55.2 wt.-%. The formation of the crosslinked structure is ascribed to the macromolecular reaction between pyridine rings of P4VP and epoxide groups of OpePOSS. The POSS-crosslinked P4VP displayed enhanced glass transition temperatures (Tgs) and an improved thermal stability in terms of the results of thermal analysis.
Co-reporter:Minghua Yin
Macromolecular Chemistry and Physics 2005 Volume 206(Issue 9) pp:929-937
Publication Date(Web):28 APR 2005
DOI:10.1002/macp.200400512
Summary: The ternary thermosetting blends composed of epoxy resin, poly(ethylene oxide) (PEO) and poly(ε-caprolactone) (PCL) were prepared via in situ polymerization of epoxy monomers in the presence of the two crystalline polymers, PEO and PCL. DSC results showed that the binary blends of epoxy with PEO (and/or PCL) are fully miscible in the entire composition in the amorphous state. FTIR indicates that there were interchain specific interactions between the crosslinked epoxy and the linear polymers in the binary blends and the hydrogen bonding interactions between epoxy and PCL are much weaker than those between epoxy and PEO. The difference in the strength of interchain specific interactions gives rise to the competitive hydrogen bonding interactions in the ternary blends of epoxy, PEO and PCL, which were evidenced by the results of FTIR. The results of optical microscopy and DSC showed that in the ternary blends PCL component separated out with inclusion of PEO. The formation of the specific phase structures is ascribed to the competitive interchain specific interactions among the crosslinked epoxy, PEO and PCL.
Co-reporter:Yong Ni
Macromolecular Chemistry and Physics 2005 Volume 206(Issue 20) pp:2075-2083
Publication Date(Web):17 OCT 2005
DOI:10.1002/macp.200500267
Summary: Octamaleimidophenyl polyhedral oligomeric silsesquioxane (OmipPOSS) was synthesized via the imidization reaction between octaaminophenyl polyhedral oligomeric silsesquioxane (OapPOSS) and maleic anhydride, and it was characterized by means of Fourier transform infrared (FTIR) and NMR spectroscopies. OmipPOSS was further employed to prepare epoxy hybrids. The thermosetting hybrids containing OmipPOSS up to 10 wt.-% were obtained via in situ polymerization of diglycidyl ether of bisphenol A (DGEBA) and 4,4′-diaminodiphenylmethane (DDM) in the presence of OmipPOSS. High-resolution transmission electronic microscopy (TEM) indicates that the nanometer-scaled dispersion of POSS molecules was obtained, suggesting that the nanocomposites were successfully prepared. The results of DSC showed that the glass transition temperatures (Tg's) of the POSS-containing nanocomposites are dependent on the content of POSS in the nanocomposites. When the contents of POSS are less than 5 wt.-%, the nanocomposites displayed the enhanced glass transition temperatures (Tg's) in comparison with control epoxy. Thermogravimetric analysis (TGA) showed that all the nanocomposites containing POSS displayed improved char yield, suggesting the flame retardance of the materials is improved.
Co-reporter:Sixun Zheng;Han Lü
Journal of Polymer Science Part B: Polymer Physics 2005 Volume 43(Issue 4) pp:359-367
Publication Date(Web):4 JAN 2005
DOI:10.1002/polb.20338
Thermosetting blends composed of phloroglucinol-cured bisphenol S epoxy resin and poly(ethylene oxide) (PEO) were prepared via the in situ curing reaction of epoxy in the presence of PEO, which started from initially homogeneous mixtures of diglycidyl ether of bisphenol S, phloroglucinol, and PEO. The miscibility of the blends after and before the curing reaction was established on the basis of thermal analysis (differential scanning calorimetry). Single and composition-dependent glass-transition temperatures (Tg's) were observed for all the blend compositions after and before curing. The experimental Tg's could be explained well by the Gordon–Taylor equation. Fourier transform infrared spectroscopy indicated that there were competitive hydrogen-bonding interactions in the binary thermosetting blends upon the addition of PEO to the system, which was involved with the intramolecular and intermolecular hydrogen-bonding interactions, that is, OH···OS, OH···OH, and OH, versus ether oxygen atoms of PEO between crosslinked epoxy and PEO. On the basis of infrared spectroscopy results, it was judged that from weak to strong the strength of the hydrogen-bonding interactions was in the following order: OH···OS, OH···OH, and OH versus ether oxygen atoms of PEO. © 2005 Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys 43: 359–367, 2005
Co-reporter:Sixun Zheng, Kangming Nie, Qipeng Guo
Thermochimica Acta 2004 Volume 419(1–2) pp:267-274
Publication Date(Web):10 September 2004
DOI:10.1016/j.tca.2004.03.002
Miscibility and phase separation in the blends of phenolphthalein poly(aryl ether ketone) (PPAEK) and poly(ethylene oxide) (PEO) were investigated by means of differential scanning calorimetry (DSC). The PPAEK/PEO blends prepared by solution casting from N,N-dimethylformamide (DMF) displayed single composition-dependent glass transition temperatures (Tg), intermediate between those of the pure components, suggesting that the blend system is miscible in the amorphous state at all compositions. All the blends underwent phase separation at higher temperatures and the system exhibited a lower critical solution temperature (LCST) behavior. A step-heating thermal analysis was designed to determine the phase boundaries with DSC. The significant changes in the thermal properties of blends were utilized to judge the mixing status for the blends and the phase diagram was thus established.
Co-reporter:Han Lü;Bing Zhang;Xiaozhen Tang
Macromolecular Chemistry and Physics 2004 Volume 205(Issue 6) pp:
Publication Date(Web):14 APR 2004
DOI:10.1002/macp.200300164
Summary: The blends of poly(hydroxyether sulfone) (PHES) with poly(N-vinylpyrrolidone) (PVPy) were investigated by means of differential scanning calorimetry (DSC) and FTIR spectroscopy. The miscibility of the blend system was established on the basis of the thermal analysis results. DSC showed that the PHES/PVPy blends prepared by casting from N,N-dimethylformamide (DMF) possessed single, composition-dependent glass transition temperatures, indicating that the blends are miscible in the entire composition. The experimental glass transition temperatures have higher values than those calculated on the basis of additive behavior; the variation of the glass transition temperatures of the blends was accounted for by the Kwei equation. FTIR studies indicate that competitive hydrogen bonding interactions exist upon addition of PVPy to the system, which were involved in the self- and cross-association, i.e., OH···OS, OH···OH of PHES and OH···OC< of PVPy. The FTIR spectra in the range of the sulfonyl stretching vibrations showed that the hydroxyl-associated sulfonyl groups are partially “set free” upon addition of PVPy to the system. The IR spectroscopic investigation of both the model compounds and the PHES/PVPy blends suggests that the strength of the hydrogen bonding interactions in the blend system increases in the following order: OH···OS, OH···OH and OH···OC<.
Co-reporter:Sixun Zheng;Han Lü;Qipeng Guo
Macromolecular Chemistry and Physics 2004 Volume 205(Issue 11) pp:1547-1558
Publication Date(Web):20 JUL 2004
DOI:10.1002/macp.200400054
Summary: Polybenzoxazine (PBA-a)/poly(ε-caprolactone) (PCL) blends were prepared by an in situ curing reaction of benzoxazine (BA-a) in the presence of PCL. Before curing, the benzoxazine (BA-a)/PCL blends are miscible, which was evidenced by the behaviors of single and composition-dependant glass transition temperature and equilibrium melting point depression. However, the phase separation induced by polymerization was observed after curing at elevated temperature. It was expected that after curing, the PBA-a/PCL blends would be miscible since the phenolic hydroxyls in the PBA-a molecular backbone have the potential to form intermolecular hydrogen-bonding interactions with the carbonyls of PCL and thus would fulfil the miscibility of the blends. The resulting morphology of the blends prompted an investigation of the status of association between PBA-a and PCL under the curing conditions. Although Fourier-transform infrared spectroscopy (FT-IR) showed that there were intermolecular hydrogen-bonding interactions between PBA-a and PCL at room temperature, especially for the PCL-rich blends, the results of variable temperature FT-IR spectroscopy by the model compound indicate that the phenolic hydroxyl groups could not form efficient intermolecular hydrogen-bonding interactions at elevated temperatures, i.e., the phenolic hydroxyl groups existed mainly in the non-associated form in the system during curing. The results are valuable to understand the effect of curing temperature on the resulting morphology of the thermosetting blends.
Co-reporter:Sixun Zheng;Rong Gao;Jianhua Li;Qipeng Guo
Journal of Applied Polymer Science 2003 Volume 89(Issue 2) pp:505-512
Publication Date(Web):24 APR 2003
DOI:10.1002/app.12272
The poly(sily ether) with pendant chloromethyl groups (PSE) was synthesized by the polyaddition of dichloromethylsilane (DCM) and diglycidylether of bisphenol A (DGEBA) with tetrabutylammonium chloride (TBAC) as a catalyst. This polymer was miscible with diglycidyl ether of bisphenol A (DGEBA), the precursor of epoxy resin. The miscibility is considered to be due mainly to entropy contribution because the molecular weight of DGEBA is quite low. The blends of epoxy resin with PSE were prepared through in situ curing reaction of diglycidyl ether of bisphenol A (DGEBA) and 4,4′-diaminodiphenylmethane (DDM) in the presence of PSE. The DDM-cured epoxy resin/PSE blends with PSE content up to 40 wt % were obtained. The reaction started from the initial homogeneous ternary mixture of DGEBA/DDM/PSE. With curing proceeding, phase separation induced by polymerization occurred. PSE was immiscible with the 4,4′-diaminodiphenylmethane-cured epoxy resin (ER) because the blends exhibited two separate glass transition temperatures (Tgs) as revealed by the means of differential scanning calorimetry (DSC) and dynamic mechanical analysis (DMA). SEM showed that all the ER/PSE blends are heterogeneous. Depending on blend composition, the blends can display PSE- or epoxy-dispersed morphologies, respectively. The mechanical test showed that the DDM-cured ER/PSE blend containing 25 wt % PSE displayed a substantial improvement in Izod impact strength, i.e., epoxy resin was significantly toughened. The improvement in impact toughness corresponded to the formation of PSE-dispersed phase structure. © 2003 Wiley Periodicals, Inc. J Appl Polym Sci 89: 505–512, 2003
Co-reporter:Sixun Zheng;Shuping Ai;Qipeng Guo
Journal of Polymer Science Part B: Polymer Physics 2003 Volume 41(Issue 5) pp:466-475
Publication Date(Web):17 JAN 2003
DOI:10.1002/polb.10392
Poly(hydroxyether of phenolphthalein) (PPH) was synthesized through the polycondensation of phenolphthalein with epichlorohydrin. It was characterized by Fourier transform infrared (FTIR) spectroscopy, NMR spectroscopy, and differential scanning calorimetry (DSC). The miscibility of the blends of PPH with poly(ethylene oxide) (PEO) was established on the basis of the thermal analysis results. DSC showed that the PPH/PEO blends prepared via casting from N,N-dimethylformamide possessed single, composition-dependent glass-transition temperatures. Therefore, the blends were miscible in the amorphous state for all compositions. FTIR studies indicated that there were competitive hydrogen-bonding interactions with the addition of PEO to the system, which were involved with OH…OC〈, OH…OH, and OH vs ether oxygen atoms of PEO hydrogen bonding, that is both intramolecular and intermolecular, between PPH and PEO). Some of the hydroxyl stretching vibration bands significantly shifted to higher frequencies, whereas others shifted to lower frequencies, and this suggested the formation of hydrogen bonds between the pendant hydroxyls of PPH and ether oxygen atoms of PEO, which were stronger than the intramolecular hydrogen bonding between hydroxyls and carbonyls of PPH. The FTIR spectra in the range of carbonyl stretching vibrations showed that the hydroxyl-associated carbonyl groups were partially set free because of the presence of the competitive hydrogen-bonding interactions. © 2003 Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys 41: 466–475, 2003
Co-reporter:Qipeng Guo;Chi-Ming Chan
Journal of Polymer Science Part B: Polymer Physics 2003 Volume 41(Issue 10) pp:1099-1111
Publication Date(Web):31 MAR 2003
DOI:10.1002/polb.10436
Crystalline thermosetting blends composed of 2,2′-bis[4-(4-aminophenoxy)phenyl]propane-crosslinked epoxy resin (ER) and poly(ϵ-caprolactone) (PCL) were investigated by means of Fourier transform infrared (FTIR) spectroscopy and high-resolution solid-state NMR spectroscopy. FTIR investigations indicated that there were specific intermolecular interactions between ER and PCL and that the intermolecular hydrogen-bonding interactions were weaker than the self-association in pure epoxy. The intermolecular hydrogen bonding was considered to be the driving force for the miscibility of the thermosetting blends. For the examination of the miscibility of the thermosetting blends at the molecular level, high-resolution solid-state 13C cross-polarity/magic-angle spinning (CP-MAS) NMR spectroscopy was employed. The line width of 13C CP-MAS spectra decreased with increasing PCL contents, and the chemical shift of the carbonyl carbon resonance of PCL shifted to a low field with an increasing epoxy content in the blends. The proton spin–lattice relaxation experiments in the laboratory frame showed that all the blends possessed identical, composition-dependent relaxation times (i.e., the proton spin–lattice relaxation times in the laboratory frame), suggesting that the thermosetting blends were homogeneous on the scale of 20–30 nm in terms of the spin-diffusion mechanism, and this was in a good agreement with the results of differential scanning calorimetry and dynamic mechanical analysis. For the examination of the miscibility of the blends at the molecular level, the behavior of the proton lattice relaxation in the rotating frame was investigated. The homogeneity of the thermosetting blends at the molecular level was quite dependent on the blend composition. The PCL-lean ER/PCL blends (e.g., 70/30) displayed a single homogeneous amorphous phase, and the molecular chains were intimately mixed on the segmental scale. The PCL-rich blends displayed biexponential decay in experiments concerning the proton spin–lattice relaxation times in the rotating frame, which was ascribed to amorphous and crystalline phases. In the amorphous region, the molecular chains of epoxy and PCL were intimately mixed at the molecular level. © 2003 Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys 41: 1099–1111, 2003
Co-reporter:Sixun Zheng;Qipeng Guo;Haifeng Zheng
Journal of Polymer Science Part B: Polymer Physics 2003 Volume 41(Issue 10) pp:1085-1098
Publication Date(Web):31 MAR 2003
DOI:10.1002/polb.10435
Crystalline thermosetting blends composed of 2,2′-bis[4-(4-aminophenoxy)phenyl]propane (BAPP)-cured epoxy resin (ER) and poly(ϵ-caprolactone) (PCL) were prepared via the in situ curing reaction of epoxy monomers in the presence of PCL, which started from initially homogeneous mixtures of diglycidyl ether of bisphenol A (DGEBA), BAPP, and PCL. The miscibility of the blends after and before the curing reaction was established with differential scanning calorimetry and dynamic mechanical analysis. Single and composition-dependent glass-transition temperatures (Tg's) were observed in the entire blend composition after and before the crosslinking reaction. The experimental Tg's were in good agreement with the prediction by the Fox and Gordon–Taylor equations. The curing reaction caused a considerable increase in the overall crystallization rate and dramatically influenced the mechanism of nucleation and the growth of the PCL crystals. The equilibrium melting point depression was observed for the blends. An analysis of the kinetic data according to the Hoffman–Lauritzen crystallization kinetic theory showed that with an increasing amorphous content, the surface energy of the extremity surfaces increased dramatically for DGEBA/PCL blends but decreased for ER/PCL blends. © 2003 Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys 41: 1085–1098, 2003
Co-reporter:Yonghong Liu, Ke Zeng, Sixun Zheng
Reactive and Functional Polymers (July 2007) Volume 67(Issue 7) pp:627-635
Publication Date(Web):July 2007
DOI:10.1016/j.reactfunctpolym.2007.04.002
Co-reporter:Lei Wang, Chongyin Zhang and Sixun Zheng
Journal of Materials Chemistry A 2011 - vol. 21(Issue 48) pp:NaN19352-19352
Publication Date(Web):2011/11/02
DOI:10.1039/C1JM13596A
In this communication, we reported the synthesis of organic-inorganic poly(hydroxyether of bisphenol A) (PH) copolymers with polyhedral oligomeric silsesquioxane (POSS) in the main chains via direct polymerization between diepoxy and bisphenol A. Toward this end, a novel diepoxy POSS macromer, i.e., 3,13-diglycidyloxypropyloctaphenyl double-decker silsesquioxane (denoted diglycidyloxypropyl DDSQ) was synthesized via the hydrosilylation between 3,13-dihydrooctaphenyl double-decker silsesquioxane (denoted dihydro DDSQ) and allyl glycidyl ether. Both diglycidyloxypropyl DDSQ and diglycidyl ether of bisphenol A (DGEBA) were used as the diepoxy monomers. By adjusting the ratio of diglycidyloxypropyl DDSQ to DGEBA, the PH copolymers with variable content of POSS in the main chains were obtained. It is found that with the direct polymerization the PH copolymers with POSS in the main chains of polymers can be successfully obtained while the content of diglycidyloxypropyl DDSQ was not very high. Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) showed that the glass transition temperatures (Tg) of the organic-inorganic PH copolymers were quite dependent on the contents of POSS in the copolymers. Thermogravimetric analysis (TGA) indicates that the stability of the copolymers was significantly improved in terms of the temperatures at the maximum rate of degradation and the yields of degradation residues. Owing to the presence of POSS in the main chains, the surface hydrophobicity of the organic-inorganic copolymers was significantly improved compared to control PH.
























![Cyclohexasiloxane, 2,4,6,8,10,12-hexakis[(ethenyldimethylsilyl)oxy]-2,4,6,8,10,12-hexaphenyl-](/data/chemimg/3780600/1880497-90-4.png)
![Cyclohexasiloxane, 2,4,6,8,10,12-hexakis[(ethenyldimethylsilyl)oxy]-2,4,6,8,10,12-hexaphenyl-](/data/chemimg/3780600/1880497-90-4_b.png)
![Benzenamine, 3,3',3'',3''',3'''',3'''''-[(2,4,6,8,10,12-hexaphenylcyclohexasiloxane-2,4,6,8,10,12-hexayl)hexakis[oxy(diphenylsilylene)-2,1-ethenediyl]]hexakis-](/data/chemimg/3780600/1880497-96-0.png)
![Benzenamine, 3,3',3'',3''',3'''',3'''''-[(2,4,6,8,10,12-hexaphenylcyclohexasiloxane-2,4,6,8,10,12-hexayl)hexakis[oxy(diphenylsilylene)-2,1-ethenediyl]]hexakis-](/data/chemimg/3780600/1880497-96-0_b.png)
![1,2-Benzenediamine, 4,4',4'',4''',4'''',4'''''-[(2,4,6,8,10,12-hexaphenylcyclohexasiloxane-2,4,6,8,10,12-hexayl)hexakis[oxy(dimethylsilylene)-2,1-ethenediyl]]hexakis-](/data/chemimg/3780600/1880497-98-2.png)
![1,2-Benzenediamine, 4,4',4'',4''',4'''',4'''''-[(2,4,6,8,10,12-hexaphenylcyclohexasiloxane-2,4,6,8,10,12-hexayl)hexakis[oxy(dimethylsilylene)-2,1-ethenediyl]]hexakis-](/data/chemimg/3780600/1880497-98-2_b.png)


















![Propanoic acid, 2,2'-[carbonothioylbis(thio)]bis[2-methyl-](/data/chemimg/104200/355120-40-0.png)
![Propanoic acid, 2,2'-[carbonothioylbis(thio)]bis[2-methyl-](/data/chemimg/104200/355120-40-0_b.png)








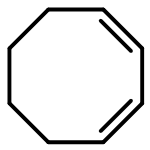




![Poly[(1,3-dihydro-1,3-dioxo-2H-isoindole-2,5-diyl)carbonyl(1,3-dihydro-1,3-dioxo-2H-isoindole-5,2-diyl)-1,4-phenyleneoxy-1,4-phenylene]](http://img.cochemist.com/ccimg/25000/24991-11-5.png)
![Poly[(1,3-dihydro-1,3-dioxo-2H-isoindole-2,5-diyl)carbonyl(1,3-dihydro-1,3-dioxo-2H-isoindole-5,2-diyl)-1,4-phenyleneoxy-1,4-phenylene]](http://img.cochemist.com/ccimg/25000/24991-11-5_b.png)





![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4.png)
![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4_b.png)