Co-reporter:Joseph B. Greer;Richard D. LeDuc;Paul M. Thomas;Bryan P. Early;Ryan T. Fellers
Journal of Proteome Research July 3, 2014 Volume 13(Issue 7) pp:3231-3240
Publication Date(Web):Publication Date (Web): June 12, 2014
DOI:10.1021/pr401277r
The automated processing of data generated by top down proteomics would benefit from improved scoring for protein identification and characterization of highly related protein forms (proteoforms). Here we propose the “C-score” (short for Characterization Score), a Bayesian approach to the proteoform identification and characterization problem, implemented within a framework to allow the infusion of expert knowledge into generative models that take advantage of known properties of proteins and top down analytical systems (e.g., fragmentation propensities, “off-by-1 Da” discontinuous errors, and intelligent weighting for site-specific modifications). The performance of the scoring system based on the initial generative models was compared to the current probability-based scoring system used within both ProSightPC and ProSightPTM on a manually curated set of 295 human proteoforms. The current implementation of the C-score framework generated a marked improvement over the existing scoring system as measured by the area under the curve on the resulting ROC chart (AUC of 0.99 versus 0.78).Keywords: Bayesian scoring; proteoform characterization; top down proteomics;
Co-reporter:Dorothy R. Ahlf;Philip D. Compton;John C. Tran;Bryan P. Early;Paul M. Thomas
Journal of Proteome Research August 3, 2012 Volume 11(Issue 8) pp:4308-4314
Publication Date(Web):2017-2-22
DOI:10.1021/pr3004216
Mass spectrometry based proteomics generally seeks to identify and fully characterize protein species with high accuracy and throughput. Recent improvements in protein separation have greatly expanded the capacity of top-down proteomics (TDP) to identify a large number of intact proteins. To date, TDP has been most tightly associated with Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometry. Here, we couple the improved separations to a Fourier-transform instrument based not on ICR but using the Orbitrap Elite mass analyzer. Application of this platform to H1299 human lung cancer cells resulted in the unambiguous identification of 690 unique proteins and over 2000 proteoforms identified from proteins with intact masses <50 kDa. This is an early demonstration of high throughput TDP (>500 identifications) in an Orbitrap mass spectrometer and exemplifies an accessible platform for whole protein mass spectrometry.Keywords: high-resolution mass spectrometry; Orbitrap Elite; top-down proteomics;
Co-reporter:Nicole A. Haverland;Owen S. Skinner
Journal of The American Society for Mass Spectrometry 2017 Volume 28( Issue 6) pp:1203-1215
Publication Date(Web):03 April 2017
DOI:10.1007/s13361-017-1635-x
Fragmentation of intact proteins in the gas phase is influenced by amino acid composition, the mass and charge of precursor ions, higher order structure, and the dissociation technique used. The likelihood of fragmentation occurring between a pair of residues is referred to as the fragmentation propensity and is calculated by dividing the total number of assigned fragmentation events by the total number of possible fragmentation events for each residue pair. Here, we describe general fragmentation propensities when performing top-down mass spectrometry (TDMS) using denaturing or native electrospray ionization. A total of 5311 matched fragmentation sites were collected for 131 proteoforms that were analyzed over 165 experiments using native top-down mass spectrometry (nTDMS). These data were used to determine the fragmentation propensities for 399 residue pairs. In comparison to denatured top-down mass spectrometry (dTDMS), the fragmentation pathways occurring either N-terminal to proline or C-terminal to aspartic acid were even more enhanced in nTDMS compared with other residues. More generally, 257/399 (64%) of the fragmentation propensities were significantly altered (P ≤ 0.05) when using nTDMS compared with dTDMS, and of these, 123 were altered by 2-fold or greater. The most notable enhancements of fragmentation propensities for TDMS in native versus denatured mode occurred (1) C-terminal to aspartic acid, (2) between phenylalanine and tryptophan (F|W), and (3) between tryptophan and alanine (W|A). The fragmentation propensities presented here will be of high value in the development of tailored scoring systems used in nTDMS of both intact proteins and protein complexes.
Co-reporter:Anthony W. Goering, Jian Li, Ryan A. McClure, Regan J. Thomson, Michael C. Jewett, and Neil L. Kelleher
ACS Synthetic Biology 2017 Volume 6(Issue 1) pp:
Publication Date(Web):August 1, 2016
DOI:10.1021/acssynbio.6b00160
Genome sequencing has revealed that a far greater number of natural product biosynthetic pathways exist than there are known natural products. To access these molecules directly and deterministically, a new generation of heterologous expression methods is needed. Cell-free protein synthesis has not previously been used to study nonribosomal peptide biosynthesis, and provides a tunable platform with advantages over conventional methods for protein expression. Here, we demonstrate the use of cell-free protein synthesis to biosynthesize a cyclic dipeptide with correct absolute stereochemistry. From a single-pot reaction, we measured the expression of two nonribosomal peptide synthetases larger than 100 kDa, and detected high-level production of a diketopiperazine. Using quantitative LC–MS and synthetically prepared standard, we observed production of this metabolite at levels higher than previously reported from cell-based recombinant expression, approximately 12 mg/L. Overall, this work represents a first step to apply cell-free protein synthesis to discover and characterize new natural products.Keywords: biosynthesis; cell-free protein synthesis; cyclic dipeptide; diketopiperazine; natural products; synthetic biology;
Co-reporter:Luca Fornelli, Kenneth R. Durbin, Ryan T. Fellers, Bryan P. Early, Joseph B. Greer, Richard D. LeDuc, Philip D. Compton, and Neil L. Kelleher
Journal of Proteome Research 2017 Volume 16(Issue 2) pp:
Publication Date(Web):November 16, 2016
DOI:10.1021/acs.jproteome.6b00698
Over the past decade, developments in high resolution mass spectrometry have enabled the high throughput analysis of intact proteins from complex proteomes, leading to the identification of thousands of proteoforms. Several previous reports on top-down proteomics (TDP) relied on hybrid ion trap–Fourier transform mass spectrometers combined with data-dependent acquisition strategies. To further reduce TDP to practice, we use a quadrupole-Orbitrap instrument coupled with software for proteoform-dependent data acquisition to identify and characterize nearly 2000 proteoforms at a 1% false discovery rate from human fibroblasts. By combining a 3 m/z isolation window with short transients to improve specificity and signal-to-noise for proteoforms >30 kDa, we demonstrate improving proteome coverage by capturing 439 proteoforms in the 30–60 kDa range. Three different data acquisition strategies were compared and resulted in the identification of many proteoforms not observed in replicate data-dependent experiments. Notably, the data set is reported with updated metrics and tools including a new viewer and assignment of permanent proteoform record identifiers for inclusion of highly characterized proteoforms (i.e., those with C-scores >40) in a repository curated by the Consortium for Top-Down Proteomics.Keywords: AUTOPILOT; data-dependent acquisition; false-discovery rate; gas-phase fractionation; mass spectrometry; medium/high; Orbitrap; proteoform; quadrupole; top-down proteomics;
Co-reporter:Matthew T. Henke and Neil L. Kelleher
Natural Product Reports 2016 vol. 33(Issue 8) pp:942-950
Publication Date(Web):04 Jul 2016
DOI:10.1039/C6NP00024J
Covering: up to 2016
In this highlight, we describe the current landscape for dereplication and discovery of natural products based on the measurement of the intact mass by LC-MS. Often it is assumed that because better mass accuracy (provided by higher resolution mass spectrometers) is necessary for absolute chemical formula determination (≤1 part-per-million), that it is also necessary for dereplication of natural products. However, the average ability to dereplicate tapers off at ∼10 ppm, with modest improvement gained from better mass accuracy when querying focused databases of natural products. We also highlight some recent examples of how these platforms are applied to synthetic biology, and recent methods for dereplication and correlation of substructures using tandem MS data. We also offer this highlight to serve as a brief primer for those entering the field of mass spectrometry-based natural products discovery.
Co-reporter:Yupeng Zheng, Xiaoxiao Huang, Neil L Kelleher
Current Opinion in Chemical Biology 2016 Volume 33() pp:142-150
Publication Date(Web):August 2016
DOI:10.1016/j.cbpa.2016.06.007
•A useful reference for quantitative analysis of histone modifications.•Three general approaches to analyze histone modifications by mass spectrometry.•Analytical challenges of separation, instrumentation, and informatics for histones.•Strategies to implement LC–MS workflows for analysis of histone marks and codes.Histones are a group of proteins with a high number of post-translational modifications, including methylation, acetylation, phosphorylation, and monoubiquitination, which play critical roles in every chromatin-templated activity. The quantitative analysis of these modifications using mass spectrometry (MS) has seen significant improvements over the last decade. It is now possible to perform large-scale surveys of dozens of histone marks and hundreds of their combinations on global chromatin. Here, we review the development of three MS strategies for analyzing histone modifications that have come to be known as Bottom Up, Middle Down, and Top Down. We also discuss challenges and innovative solutions for characterizing and quantifying complicated isobaric species arising from multiple modifications on the same histone molecule.
Co-reporter:Matthew T. Henke, Alexandra A. Soukup, Anthony W. Goering, Ryan A. McClure, Regan J. Thomson, Nancy P. Keller, and Neil L. Kelleher
ACS Chemical Biology 2016 Volume 11(Issue 8) pp:2117
Publication Date(Web):June 16, 2016
DOI:10.1021/acschembio.6b00398
Unlocking the biochemical stores of fungi is key for developing future pharmaceuticals. Through reduced expression of a critical histone deacetylase in Aspergillus nidulans, increases of up to 100-fold were observed in the levels of 15 new aspercryptins, recently described lipopeptides with two noncanonical amino acids derived from octanoic and dodecanoic acids. In addition to two NMR-verified structures, MS/MS networking helped uncover an additional 13 aspercryptins. The aspercryptins break the conventional structural orientation of lipopeptides and appear “backward” when compared to known compounds of this class. We have also confirmed the 14-gene aspercryptin biosynthetic gene cluster, which encodes two fatty acid synthases and several enzymes to convert saturated octanoic and dodecanoic acid to α-amino acids.
Co-reporter:Kenneth R. Durbin; Luca Fornelli; Ryan T. Fellers; Peter F. Doubleday; Masashi Narita
Journal of Proteome Research 2016 Volume 15(Issue 3) pp:976-982
Publication Date(Web):January 22, 2016
DOI:10.1021/acs.jproteome.5b00997
Top-down proteomics is capable of identifying and quantitating unique proteoforms through the analysis of intact proteins. We extended the coverage of the label-free technique, achieving differential analysis of whole proteins <30 kDa from the proteomes of growing and senescent human fibroblasts. By integrating improved control software with more instrument time allocated for quantitation of intact ions, we were able to collect protein data between the two cell states, confidently comparing 1577 proteoform levels. To then identify and characterize proteoforms, our advanced acquisition software, named Autopilot, employed enhanced identification efficiency in identifying 1180 unique Swiss-Prot accession numbers at 1% false-discovery rate. This coverage of the low mass proteome is equivalent to the largest previously reported but was accomplished in 23% of the total acquisition time. By maximizing both the number of quantified proteoforms and their identification rate in an integrated software environment, this work significantly advances proteoform-resolved analyses of complex systems.
Co-reporter:John Paul Savaryn, Owen S. Skinner, Luca Fornelli, Ryan T. Fellers, Philip D. Compton, Scott S. Terhune, Mike M. Abecassis, Neil L. Kelleher
Journal of Proteomics 2016 Volume 134() pp:76-84
Publication Date(Web):16 February 2016
DOI:10.1016/j.jprot.2015.04.025
•Measuring transcription factors by top down mass spectrometry is challenging.•Using HaloTag, we purified sufficient amounts of NF-κB p65 for top down MS.•We resolved isotopes and detected a phosphorylated p65 protein species by FTMS.•We show p65 fragments internally during HCD, increasing protein coverage.•HaloTag purification is amenable to native MS; p65 and p65 dimer were detected.Measuring post-translational modifications on transcription factors by targeted mass spectrometry is hampered by low protein abundance and inefficient isolation. Here, we utilized HaloTag technology to overcome these limitations and evaluate various top down mass spectrometry approaches for measuring NF-κB p65 proteoforms isolated from human cells. We show isotopic resolution of N-terminally acetylated p65 and determined it is the most abundant proteoform expressed following transfection in 293 T cells. We also show MS1 evidence for monophosphorylation of p65 under similar culture conditions and describe a high propensity for p65 proteoforms to fragment internally during beam-style MS2 fragmentation; up to 71% of the fragment ions could be matched as internals in some fragmentation spectra. Finally, we used native spray mass spectrometry to measure proteins copurifying with p65 and present evidence for the native detection of p65, 71 kDa heat shock protein, and p65 homodimer. Collectively, our work demonstrates the efficient isolation and top down mass spectrometry analysis of p65 from human cells, and we discuss the perturbations of overexpressing tagged proteins to study their biochemistry. This article is part of a Special Issue entitled: Protein Species.Biological significanceCharacterizing transcription factor proteoforms in human cells is of high value to the field of molecular biology; many agree that post-translational modifications and combinations thereof play a critical role in modulating transcription factor activity. Thus, measuring these modifications promises increased understanding of molecular mechanisms governing the regulation of complex gene expression outcomes. To date, comprehensive characterization of transcription factor proteoforms within human cells has eluded measurement, owing primarily—with regard to top down mass spectrometry—to large protein size and low relative abundance. Here, we utilized HaloTag technology and recombinant protein expression to overcome these limitations and show top down mass spectrometry characterization of proteoforms of the 60 kDa NF-kB protein, p65. By optimizing the analytical procedure (i.e. purification, MS1, and MS2), our results make important progress toward the ultimate goal of targeted transcription factor characterization from endogenous loci.
Co-reporter:Anthony W. Goering, Ryan A. McClure, James R. Doroghazi, Jessica C. Albright, Nicole A. Haverland, Yongbo Zhang, Kou-San Ju, Regan J. Thomson, William W. Metcalf, and Neil L. Kelleher
ACS Central Science 2016 Volume 2(Issue 2) pp:99
Publication Date(Web):January 20, 2016
DOI:10.1021/acscentsci.5b00331
For more than half a century the pharmaceutical industry has sifted through natural products produced by microbes, uncovering new scaffolds and fashioning them into a broad range of vital drugs. We sought a strategy to reinvigorate the discovery of natural products with distinctive structures using bacterial genome sequencing combined with metabolomics. By correlating genetic content from 178 actinomycete genomes with mass spectrometry-enabled analyses of their exported metabolomes, we paired new secondary metabolites with their biosynthetic gene clusters. We report the use of this new approach to isolate and characterize tambromycin, a new chlorinated natural product, composed of several nonstandard amino acid monomeric units, including a unique pyrrolidine-containing amino acid we name tambroline. Tambromycin shows antiproliferative activity against cancerous human B- and T-cell lines. The discovery of tambromycin via large-scale correlation of gene clusters with metabolites (a.k.a. metabologenomics) illuminates a path for structure-based discovery of natural products at a sharply increased rate.
Co-reporter:Ryan A. McClureAnthony W. Goering, Kou-San Ju, Joshua A. Baccile, Frank C. Schroeder, William W. Metcalf, Regan J. Thomson, Neil. L Kelleher
ACS Chemical Biology 2016 Volume 11(Issue 12) pp:
Publication Date(Web):November 4, 2016
DOI:10.1021/acschembio.6b00779
As microbial genome sequencing becomes more widespread, the capacity of microorganisms to produce an immense number of metabolites has come into better view. Utilizing a metabolite/gene cluster correlation platform, the biosynthetic origins of a new family of natural products, the rimosamides, were discovered. The rimosamides were identified in Streptomyces rimosus and associated with their NRPS/PKS-type gene cluster based upon their high frequency of co-occurrence across 179 strains of actinobacteria. This also led to the discovery of the related detoxin gene cluster. The core of each of these families of natural products contains a depsipeptide bond at the point of bifurcation in their unusual branched structures, the origins of which are definitively assigned to nonlinear biosynthetic pathways via heterologous expression in Streptomyces lividans. The rimosamides were found to antagonize the antibiotic activity of blasticidin S against Bacillus cereus.
Co-reporter:Wei Tang; Huiying Li; Emma H. Doud; Yunqiu Chen; Stephanie Choing; Carla Plaza; Neil L. Kelleher; Thomas L. Poulos;Richard B. Silverman
Journal of the American Chemical Society 2015 Volume 137(Issue 18) pp:5980-5989
Publication Date(Web):April 15, 2015
DOI:10.1021/jacs.5b01202
Nitric oxide synthase (NOS) catalyzes the conversion of l-arginine to l-citrulline and the second messenger nitric oxide. Three mechanistic pathways are proposed for the inactivation of neuronal NOS (nNOS) by (S)-2-amino-5-(2-(methylthio)acetimidamido)pentanoic acid (1): sulfide oxidation, oxidative dethiolation, and oxidative demethylation. Four possible intermediates were synthesized. All compounds were assayed with nNOS, their IC50, KI, and kinact values were obtained, and their crystal structures were determined. The identification and characterization of the products formed during inactivation provide evidence for the details of the inactivation mechanism. On the basis of these studies, the most probable mechanism for the inactivation of nNOS involves oxidative demethylation with the resulting thiol coordinating to the cofactor heme iron. Although nNOS is a heme-containing enzyme, this is the first example of a NOS that catalyzes an S-demethylation reaction; the novel mechanism of inactivation described here could be applied to the design of inactivators of other heme-dependent enzymes.
Co-reporter:Owen S. Skinner, Luis H. F. Do Vale, Adam D. Catherman, Pierre C. Havugimana, Marcelo Valle de Sousa, Philip D. Compton, and Neil L. Kelleher
Analytical Chemistry 2015 Volume 87(Issue 5) pp:3032
Publication Date(Web):February 9, 2015
DOI:10.1021/ac504678d
The cadre of protein complexes in cells performs an array of functions necessary for life. Their varied structures are foundational to their ability to perform biological functions, lending great import to the elucidation of complex composition and dynamics. Native separation techniques that are operative on low sample amounts and provide high resolution are necessary to gain valuable data on endogenous complexes. Here, we detail and optimize the use of tube gel separations to produce samples proven compatible with native, multistage mass spectrometry (nMS/MS). We find that a continuous system (i.e., no stacking gel) with a gradient in its extent of cross-linking and use of the clear native buffer system performs well for both fractionation and native mass spectrometry of heart extracts and a fungal secretome. This integrated advance in separations and nMS/MS offers the prospect of untargeted proteomics at the next hierarchical level of protein organization in biology.
Co-reporter:Jessica C. Albright, Matthew T. Henke, Alexandra A. Soukup, Ryan A. McClure, Regan J. Thomson, Nancy P. Keller, and Neil L. Kelleher
ACS Chemical Biology 2015 Volume 10(Issue 6) pp:1535
Publication Date(Web):March 27, 2015
DOI:10.1021/acschembio.5b00025
The microbial world offers a rich source of bioactive compounds for those able to sift through it. Technologies capable of quantitatively detecting natural products while simultaneously identifying known compounds would expedite the search for new pharmaceutical leads. Prior efforts have targeted histone deacetylases in fungi to globally activate the production of new secondary metabolites, yet no study has directly assessed its effects with minimal bias at the metabolomic level. Using untargeted metabolomics, we monitored changes in >1000 small molecules secreted from the model fungus, Aspergillus nidulans, following genetic or chemical reductions in histone deacetylase activity (HDACi). Through quantitative, differential analyses, we found that nearly equal numbers of compounds were up- and down-regulated by >100 fold. We detected products from both known and unknown biosynthetic pathways and discovered that A. nidulans is capable of producing fellutamides, proteasome inhibitors whose expression was induced by ∼100 fold or greater upon HDACi. This work adds momentum to an “omics”-driven resurgence in natural products research, where direct detection replaces bioactivity as the primary screen for new pharmacophores.
Co-reporter:Ki Hun Kim; Philip D. Compton; John C. Tran
Journal of Proteome Research 2015 Volume 14(Issue 5) pp:2199-2206
Publication Date(Web):April 3, 2015
DOI:10.1021/pr501331q
A fractionation method called gel-eluted liquid fraction entrapment electrophoresis (GELFrEE) has been used to dramatically increase the number of proteins identified in top-down proteomic workflows; however, the technique involves the use of sodium dodecyl sulfate (SDS), a surfactant that interferes with electrospray ionization. Therefore, an efficient removal of SDS is absolutely required prior to mass analysis. Traditionally, methanol/chloroform precipitation and spin columns have been used, but they lack reproducibility and are difficult to automate. Therefore, we developed an in-line matrix removal platform to enable the direct analysis of samples containing SDS and salts. Only small molecules like SDS permeate a porous membrane and are removed in a manner similar to cross-flow filtration. With this device, near-complete removal of SDS is accomplished within 5 min and proteins are subsequently mobilized into a mass spectrometer. The new platform was optimized for the analysis of GELFrEE fractions enriched for histones extracted from human HeLa cells. All four core histones and their proteoforms were detected in a single spectrum by high-resolution mass spectrometry. The new method versus protein precipitation/resuspension showed 2- to 10-fold improved signal intensities, offering a clear path forward to improve proteome coverage and the efficiency of top-down proteomics.
Co-reporter:Kenneth R. Durbin;Owen S. Skinner
Journal of The American Society for Mass Spectrometry 2015 Volume 26( Issue 5) pp:782-787
Publication Date(Web):2015 May
DOI:10.1007/s13361-015-1078-1
Gaseous fragmentation of intact proteins is multifaceted and can be unpredictable by current theories in the field. Contributing to the complexity is the multitude of precursor ion states and fragmentation channels. Terminal fragment ions can be re-fragmented, yielding product ions containing neither terminus, termed internal fragment ions. In an effort to better understand and capitalize upon this fragmentation process, we collisionally dissociated the high (13+), middle (10+), and low (7+) charge states of electrosprayed ubiquitin ions. Both terminal and internal fragmentation processes were quantified through step-wise increases of voltage potential in the collision cell. An isotope fitting algorithm matched observed product ions to theoretical terminal and internal fragment ions. At optimal energies for internal fragmentation of the 10+, nearly 200 internal fragments were observed; on average each of the 76 residues in ubiquitin was covered by 24.1 internal fragments. A pertinent finding was that formation of internal ions occurs at similar energy thresholds as terminal b- and y-ion types in beam-type activation. This large amount of internal fragmentation is frequently overlooked during top-down mass spectrometry. As such, we present several new approaches to visualize internal fragments through modified graphical fragment maps. With the presented advances of internal fragment ion accounting and visualization, the total percentage of matched fragment ions increased from approximately 40% to over 75% in a typical beam-type MS/MS spectrum. These sequence coverage improvements offer greater characterization potential for whole proteins with no needed experimental changes and could be of large benefit for future high-throughput intact protein analysis.
Co-reporter:Kyunggon Kim, Philip D. Compton, Timothy K. Toby, Paul M. Thomas, John T. Wilkins, R.Kannan Mutharasan, Neil L. Kelleher
EuPA Open Proteomics 2015 Volume 8() pp:40-47
Publication Date(Web):September 2015
DOI:10.1016/j.euprot.2015.05.005
•Narrow ID columns yield increased sensitivity for intact protein analysis.•Artificial protein oxidation complicates low-flow top down mass spectrometry.•Low Protein Oxidation (LPOx) mode nanoLC reduces artificial protein oxidation.•LPOx was applied to the characterization of ApoA1 proteoforms.Enabling the implementation of top down proteomic techniques within clinical workflows requires a dramatic increase in sensitivity. It has been previously demonstrated that electrospray ionization (ESI) becomes more efficient with decreasing volumetric flow rates at the emitter. Therefore, narrow inner diameter (I.D.) columns used in front-end chromatographic separations yield increased sensitivity. However, the smaller cross-sectional area of a narrow I.D. column places a larger fraction of the eluent in fluid communication with the electrode within the high voltage union that facilitates electrospray ionization (ESI), leading to increased oxidation of solution-phase proteins. Oxidation of proteins alters their chemical state of the protein, complicates data analysis, and reduces the depth of proteome coverage attained in a typical top-down proteomics experiment. Excessive protein oxidation results in poor deconvolution and exact mass calculations from MS1 spectra, interferes with peak isolation for MS/MS fragmentation, and effectively reduces sensitivity by splitting ion current. All of these factors deteriorate top down mass spectral data quality, an effect that becomes more pronounced as column diameter decreases. Artificial protein oxidation can also mislead investigations of in vivo protein oxidation. All of these effects are accentuated in comparison to bottom up proteomics due to the increased probability of having oxidizable residues within a particular species with increasing mass. Herein, we describe a configuration (which we term “Low Protein Oxidation (LPOx)”) for proteomics experiments created by re-arranging liquid chromatography (LC) plumbing and present its application to artificial protein oxidation and show a marked improvement in detection sensitivity. Using a standard mixture of five intact proteins, we demonstrate that the LPOx configuration reduces protein oxidation up to 90% using 50 μm I.D. columns when compared to a conventional LC plumbing configuration with 50 μm I.D. column. As a proof-of-concept study, at least 11 distinct proteoforms of serum Apolipoprotein A1 were detected with the LPOx configuration. This innovative LC configuration can be applied to the top down identification and characterization of proteoforms obscured by abundant artificial protein oxidation at low flowrates, all while using reduced amounts of valuable protein samples.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Kenneth R. Durbin, Ryan T. Fellers, Ioanna Ntai, Neil L. Kelleher, and Philip D. Compton
Analytical Chemistry 2014 Volume 86(Issue 3) pp:1485
Publication Date(Web):January 8, 2014
DOI:10.1021/ac402904h
The ability to study organisms by direct analysis of their proteomes without digestion via mass spectrometry has benefited greatly from recent advances in separation techniques, instrumentation, and bioinformatics. However, improvements to data acquisition logic have lagged in comparison. Past workflows for Top Down Proteomics (TDPs) have focused on high throughput at the expense of maximal protein coverage and characterization. This mode of data acquisition has led to enormous overlap in the identification of highly abundant proteins in subsequent LC-MS injections. Furthermore, a wealth of data is left underutilized by analyzing each newly targeted species as unique, rather than as part of a collection of fragmentation events on a distinct proteoform. Here, we present a major advance in software for acquisition of TDP data that incorporates a fully automated workflow able to detect intact masses, guide fragmentation to achieve maximal identification and characterization of intact protein species, and perform database search online to yield real-time protein identifications. On Pseudomonas aeruginosa, the software combines fragmentation events of the same precursor with previously obtained fragments to achieve improved characterization of the target form by an average of 42 orders of magnitude in confidence. When HCD fragmentation optimization was applied to intact proteins ions, there was an 18.5 order of magnitude gain in confidence. These improved metrics set the stage for increased proteome coverage and characterization of higher order organisms in the future for sharply improved control over MS instruments in a project- and lab-wide context.
Co-reporter:Owen S. Skinner, Adam D. Catherman, Bryan P. Early, Paul M. Thomas, Philip D. Compton, and Neil L. Kelleher
Analytical Chemistry 2014 Volume 86(Issue 9) pp:4627
Publication Date(Web):April 1, 2014
DOI:10.1021/ac500864w
Integral membrane proteins (IMPs) are of great biophysical and clinical interest because of the key role they play in many cellular processes. Here, a comprehensive top down study of 152 IMPs and 277 soluble proteins from human H1299 cells including 11 087 fragments obtained from collisionally activated dissociation (CAD), 6452 from higher-energy collisional dissociation (HCD), and 2981 from electron transfer dissociation (ETD) shows their great utility and complementarity for the identification and characterization of IMPs. A central finding is that ETD is ∼2-fold more likely to cleave in soluble regions than threshold fragmentation methods, whereas the reverse is observed in transmembrane domains with an observed ∼4-fold bias toward CAD and HCD. The location of charges just prior to dissociation is consistent with this directed fragmentation: protons remain localized on basic residues during ETD but easily mobilize along the backbone during collisional activation. The fragmentation driven by these protons, which is most often observed in transmembrane domains, both is of higher yield and occurs over a greater number of backbone cleavage sites. Further, while threshold dissociation events in transmembrane domains are on average 10.1 (CAD) and 9.2 (HCD) residues distant from the nearest charge site (R, K, H, N-terminus), fragmentation is strongly influenced by the N- or C-terminal position relative to that site: the ratio of observed b- to y-fragments is ∼1:3 if the cleavage occurs >7 residues N-terminal and ∼3:1 if it occurs >7 residues C-terminal to the nearest basic site. Threshold dissociation products driven by a mobilized proton appear to be strongly dependent on not only relative position of a charge site but also N- or C-terminal directionality of proton movement.
Co-reporter:Ioanna Ntai, Kyunggon Kim, Ryan T. Fellers, Owen S. Skinner, Archer D. Smith IV, Bryan P. Early, John P. Savaryn, Richard D. LeDuc, Paul M. Thomas, and Neil L. Kelleher
Analytical Chemistry 2014 Volume 86(Issue 10) pp:4961
Publication Date(Web):May 7, 2014
DOI:10.1021/ac500395k
With the prospect of resolving whole protein molecules into their myriad proteoforms on a proteomic scale, the question of their quantitative analysis in discovery mode comes to the fore. Here, we demonstrate a robust pipeline for the identification and stringent scoring of abundance changes of whole protein forms <30 kDa in a complex system. The input is ∼100–400 μg of total protein for each biological replicate, and the outputs are graphical displays depicting statistical confidence metrics for each proteoform (i.e., a volcano plot and representations of the technical and biological variation). A key part of the pipeline is the hierarchical linear model that is tailored to the original design of the study. Here, we apply this new pipeline to measure the proteoform-level effects of deleting a histone deacetylase (rpd3) in S. cerevisiae. Over 100 proteoform changes were detected above a 5% false positive threshold in WT vs the Δrpd3 mutant, including the validating observation of hyperacetylation of histone H4 and both H2B isoforms. Ultimately, this approach to label-free top down proteomics in discovery mode is a critical technical advance for testing the hypothesis that whole proteoforms can link more tightly to complex phenotypes in cell and disease biology than do peptides created in shotgun proteomics.
Co-reporter:Yunqiu Chen ; Ryan A. McClure ; Yupeng Zheng ; Regan J. Thomson
Journal of the American Chemical Society 2013 Volume 135(Issue 28) pp:10449-10456
Publication Date(Web):June 13, 2013
DOI:10.1021/ja4031193
Due to the importance of proteases in regulating cellular processes, the development of protease inhibitors has garnered great attention. Peptide-based aldehydes are a class of compounds that exhibit inhibitory activities against various proteases and proteasomes in the context of anti-proliferative treatments for cancer and other diseases. More than a dozen peptide-based natural products containing aldehydes have been discovered such as chymostatin, leupeptin, and fellutamide; however, the biosynthetic origin of the aldehyde functionality has yet to be elucidated. Herein we describe the discovery of a new group of lipopeptide aldehydes, the flavopeptins, and the corresponding biosynthetic pathway arising from an orphan gene cluster in Streptomyces sp. NRRL-F6652, a close relative of Streptomyces flavogriseus ATCC 33331. This research was initiated using a proteomics approach that screens for expressed enzymes involved in secondary metabolism in microorganisms. Flavopeptins are synthesized through a non-ribosomal peptide synthetase containing a terminal NAD(P)H-dependent reductase domain likely for the reductive release of the peptide with a C-terminal aldehyde. Solid-phase peptide synthesis of several flavopeptin species and derivatives enabled structural verification and subsequent screening of biological activity. Flavopeptins exhibit sub-micromolar inhibition activities against cysteine proteases such as papain and calpain as well as the human 20S proteasome. They also show anti-proliferative activities against multiple myeloma and lymphoma cell lines.
Co-reporter:Dorothy R Ahlf, Paul M Thomas, Neil L Kelleher
Current Opinion in Chemical Biology 2013 Volume 17(Issue 5) pp:787-794
Publication Date(Web):October 2013
DOI:10.1016/j.cbpa.2013.07.028
•We highlight recent improvements to individual parts of the top down proteomics platforms.•Increased quality and number of identifications to over 1000 ids and ∼3–4 more proteoforms.•Improvements highlighted include separations, mass analyzers, data processing and informatics.Mass spectrometry based proteomics generally seeks to identify and characterize protein molecules with high accuracy and throughput. Recent speed and quality improvements to the independent steps of integrated platforms have removed many limitations to the robust implementation of top down proteomics (TDP) for proteins below 70 kDa. Improved intact protein separations coupled to high-performance instruments have increased the quality and number of protein and proteoform identifications. To date, TDP applications have shown >1000 protein identifications, expanding to an average of ∼3–4 more proteoforms for each protein detected. In the near future, increased fractionation power, new mass spectrometers and improvements in proteoform scoring will combine to accelerate the application and impact of TDP to this century's biomedical problems.
Co-reporter:Adam D. Catherman, Mingxi Li, John C. Tran, Kenneth R. Durbin, Philip D. Compton, Bryan P. Early, Paul M. Thomas, and Neil L. Kelleher
Analytical Chemistry 2013 Volume 85(Issue 3) pp:1880
Publication Date(Web):January 10, 2013
DOI:10.1021/ac3031527
The interrogation of intact integral membrane proteins has long been a challenge for biological mass spectrometry. Here, we demonstrate the application of top down mass spectrometry to whole membrane proteins below 60 kDa with up to 8 transmembrane helices. Analysis of enriched mitochondrial membrane preparations from human cells yielded identification of 83 integral membrane proteins, along with 163 membrane-associated or soluble proteins, with a median q value of 3 × 10–10. An analysis of matching fragment ions demonstrated that significantly more fragment ions were found within transmembrane domains than would be expected based upon the observed protein sequence. In total, 46 proteins from the complexes of oxidative phosphorylation were identified which exemplifies the increasing ability of top down proteomics to provide extensive coverage in a biological network.
Co-reporter:Mikhail E. Belov, Eugen Damoc, Eduard Denisov, Philip D. Compton, Stevan Horning, Alexander A. Makarov, and Neil L. Kelleher
Analytical Chemistry 2013 Volume 85(Issue 23) pp:11163
Publication Date(Web):November 15, 2013
DOI:10.1021/ac4029328
Native mass spectrometry (MS) is becoming an important integral part of structural proteomics and system biology research. The approach holds great promise for elucidating higher levels of protein structure: from primary to quaternary. This requires the most efficient use of tandem MS, which is the cornerstone of MS-based approaches. In this work, we advance a two-step fragmentation approach, or (pseudo)-MS3, from native protein complexes to a set of constituent fragment ions. Using an efficient desolvation approach and quadrupole selection in the extended mass-to-charge (m/z) range, we have accomplished sequential dissociation of large protein complexes, such as phosporylase B (194 kDa), pyruvate kinase (232 kDa), and GroEL (801 kDa), to highly charged monomers which were then dissociated to a set of multiply charged fragmentation products. Fragment ion signals were acquired with a high resolution, high mass accuracy Orbitrap instrument that enabled highly confident identifications of the precursor monomer subunits. The developed approach is expected to enable characterization of stoichiometry and composition of endogenous native protein complexes at an unprecedented level of detail.
Co-reporter:Neil L. Kelleher
Biochemistry 2013 Volume 52(Issue 22) pp:
Publication Date(Web):April 24, 2013
DOI:10.1021/bi400466p
Co-reporter:Yunqiu Chen, Michelle Unger, Ioanna Ntai, Ryan A. McClure, Jessica C. Albright, Regan J. Thomson and Neil L. Kelleher
MedChemComm 2013 vol. 4(Issue 1) pp:233-238
Publication Date(Web):24 Sep 2012
DOI:10.1039/C2MD20232H
“Omic” strategies have been increasingly applied to natural product discovery processes, with (meta-)genome sequencing and mining implemented in many laboratories to date. Using the proteomics-based discovery platform called PrISM (Proteomic Investigation of Secondary Metabolism), we discovered two new siderophores gobichelin A and B from Streptomyces sp. NRRL F-4415, a strain without a sequenced genome. Using the proteomics information as a guide, the 37 kb gene cluster responsible for production of gobichelins was sequenced and its 20 open reading frames interpreted into a biosynthetic scheme. This led to the targeted detection and structure elucidation of the new compounds produced by nonribosomal peptide (NRP) synthesis.
Co-reporter:Neil L. Kelleher;Ljiljana Paša-Tolić
Journal of The American Society for Mass Spectrometry 2013 Volume 24( Issue 7) pp:983-985
Publication Date(Web):2013 July
DOI:10.1007/s13361-013-0640-y
Co-reporter:John F. Kellie, Adam D. Catherman, Kenneth R. Durbin, John C. Tran, Jeremiah D. Tipton, Jeremy L. Norris, Charles E. Witkowski II, Paul M. Thomas, and Neil L. Kelleher
Analytical Chemistry 2012 Volume 84(Issue 1) pp:209
Publication Date(Web):November 21, 2011
DOI:10.1021/ac202384v
As the process of top-down mass spectrometry continues to mature, we benchmark the next installment of an improving methodology that incorporates a tube-gel electrophoresis (TGE) device to separate intact proteins by molecular mass. Top-down proteomics is accomplished in a robust fashion to yield the identification of hundreds of unique proteins, many of which correspond to multiple protein forms. The TGE platform separates 0–50 kDa proteins extracted from the yeast proteome into 12 fractions prior to automated nanocapillary LC–MS/MS in technical triplicate. The process may be completed in less than 72 h. From this study, 530 unique proteins and 1103 distinct protein species were identified and characterized, thus representing the highest coverage to date of the Saccharomyces cerevisiae proteome using top-down proteomics. The work signifies a significant step in the maturation of proteomics based on direct measurement and fragmentation of intact proteins.
Co-reporter:Yunqiu Chen, Ioanna Ntai, Kou-San Ju, Michelle Unger, Leonid Zamdborg, Sarah J. Robinson, James R. Doroghazi, David P. Labeda, William W. Metcalf, and Neil L. Kelleher
Journal of Proteome Research 2012 Volume 11(Issue 1) pp:85-94
Publication Date(Web):2017-2-22
DOI:10.1021/pr2009115
Actinobacteria such as streptomycetes are renowned for their ability to produce bioactive natural products including nonribosomal peptides (NRPs) and polyketides (PKs). The advent of genome sequencing has revealed an even larger genetic repertoire for secondary metabolism with most of the small molecule products of these gene clusters still unknown. Here, we employed a “protein-first” method called PrISM (Proteomic Investigation of Secondary Metabolism) to screen 26 unsequenced actinomycetes using mass spectrometry-based proteomics for the targeted detection of expressed nonribosomal peptide synthetases or polyketide synthases. Improvements to the original PrISM screening approach ( Nat. Biotechnol. 2009, 27, 951−956), for example, improved de novo peptide sequencing, have enabled the discovery of 10 NRPS/PKS gene clusters from 6 strains. Taking advantage of the concurrence of biosynthetic enzymes and the secondary metabolites they generate, two natural products were associated with their previously “orphan” gene clusters. This work has demonstrated the feasibility of a proteomics-based strategy for use in screening for NRP/PK production in actinomycetes (often >8 Mbp, high GC genomes) versus the bacilli (2–4 Mbp genomes) used previously.
Co-reporter:Neil L. Kelleher
Journal of The American Society for Mass Spectrometry 2012 Volume 23( Issue 10) pp:1617-1624
Publication Date(Web):2012 October
DOI:10.1007/s13361-012-0469-9
The general scope of a project to determine the protein molecules that comprise the cells within the human body is framed. By focusing on protein primary structure as expressed in specific cell types, this concept for a cell-based version of the Human Proteome Project (CB-HPP) is crafted in a manner analogous to the Human Genome Project while recognizing that cells provide a primary context in which to define a proteome. Several activities flow from this articulation of the HPP, which enables the definition of clear milestones and deliverables. The CB-HPP highlights major gaps in our knowledge regarding cell heterogeneity and protein isoforms, and calls for development of technology that is capable of defining all human cell types and their proteomes. The main activities will involve mapping and sorting cell types combined with the application of beyond the state-of-the art in protein mass spectrometry.
Co-reporter:Yupeng Zheng;Steve M. M. Sweet;Relja Popovic;Eva Martinez-Garcia;Jeremiah D. Tipton;Paul M. Thomas;Jonathan D. Licht
PNAS 2012 109 (34 ) pp:13549-13554
Publication Date(Web):2012-08-21
DOI:10.1073/pnas.1205707109
We have developed a targeted method to quantify all combinations of methylation on an H3 peptide containing lysines 27 and
36 (H3K27-K36). By using stable isotopes that separately label the histone backbone and its methylations, we tracked the rates
of methylation and demethylation in myeloma cells expressing high vs. low levels of the methyltransferase MMSET/WHSC1/NSD2.
Following quantification of 99 labeled H3K27-K36 methylation states across time, a kinetic model converged to yield 44 effective
rate constants qualifying each methylation and demethylation step as a function of the methylation state on the neighboring
lysine. We call this approach MS-based measurement and modeling of histone methylation kinetics (M4K). M4K revealed that,
when dimethylation states are reached on H3K27 or H3K36, rates of further methylation on the other site are reduced as much
as 100-fold. Overall, cells with high MMSET have as much as 33-fold increases in the effective rate constants for formation
of H3K36 mono- and dimethylation. At H3K27, cells with high MMSET have elevated formation of K27me1, but even higher increases
in the effective rate constants for its reversal by demethylation. These quantitative studies lay bare a bidirectional antagonism
between H3K27 and H3K36 that controls the writing and erasing of these methylation marks. Additionally, the integrated kinetic
model was used to correctly predict observed abundances of H3K27-K36 methylation states within 5% of that actually established
in perturbed cells. Such predictive power for how histone methylations are established should have major value as this family
of methyltransferases matures as drug targets.
Co-reporter:Bradley S. Evans ; Ioanna Ntai ; Yunqiu Chen ; Sarah J. Robinson
Journal of the American Chemical Society 2011 Volume 133(Issue 19) pp:7316-7319
Publication Date(Web):April 26, 2011
DOI:10.1021/ja2015795
Nonribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs) are large enzymes responsible for the biosynthesis of medically and ecologically important secondary metabolites. In a previous report, we described a proteomics approach to screen for expressed NRPSs or PKSs from bacteria with or without sequenced genomes. Here we used this proteome mining approach to discover a novel natural product arising from rare adenylation (A) and reductase (Red) domains in its biosynthetic machinery. We also cloned the entire gene cluster and elucidated the biosynthesis of the new compound, which is produced by an unsequenced Bacillus sp. isolated from soil collected in Koran, Louisiana.
Co-reporter:Bradley S. Evans, Yunqiu Chen, William W. Metcalf, Huimin Zhao, Neil L. Kelleher
Chemistry & Biology 2011 Volume 18(Issue 5) pp:601-607
Publication Date(Web):27 May 2011
DOI:10.1016/j.chembiol.2011.03.008
Many lead compounds in the search for new drugs derive from peptides and polyketides whose similar biosynthetic enzymes have been difficult to engineer for production of new derivatives. Problems with generating multiple analogs in a single experiment along with lack of high-throughput methods for structure-based screening have slowed progress in this area. Here, we use directed evolution and a multiplexed assay to screen a library of >14,000 members to generate three derivatives of the antibacterial compound, andrimid. Another limiting factor in reengineering these mega-enzymes of secondary metabolism has been that commonly used hosts such as Escherichia coli often give lower product titers, so our reengineering was performed in the native producer, Pantoea agglomerans. This integrated in vivo approach can be extended to larger enzymes to create analogs of natural products for bioactivity testing.Graphical AbstractFigure optionsDownload full-size imageDownload high-quality image (149 K)Download as PowerPoint slideHighlights► Site-saturation mutagenesis of an NRPS was performed in the native host, P. agglomerans ► A mass spectrometric screen was developed to sift through thousands of mutant strains ► Three new andrimid derivatives were generated, isolated, and tested for bioactivity
Co-reporter:Philip D. Compton, Leonid Zamdborg, Paul M. Thomas, and Neil L. Kelleher
Analytical Chemistry 2011 Volume 83(Issue 17) pp:6868
Publication Date(Web):July 11, 2011
DOI:10.1021/ac2010795
Top-down proteomics has improved over the past decade despite the significant challenges presented by the analysis of large protein ions. Here, the detection of these high mass species by electrospray-based mass spectrometry (MS) is examined from a theoretical perspective to understand the mass-dependent increases in the number of charge states, isotopic peaks, and interfering species present in typical protein mass spectra. Integrating these effects into a quantitative model captures the reduced ability to detect species over 25 kDa with the speed and sensitivity characteristic of proteomics based on <3 kDa peptide ions. The model quantifies the challenge that top-down proteomics faces with respect to current MS instrumentation and projects that depletion of 13C and 15N isotopes can improve detection at high mass by only <2-fold at 100 kDa whereas the effect is up to 5-fold at 10 kDa. Further, we find that supercharging electrosprayed proteins to the point of producing <5 charge states at high mass would improve detection by more than 20-fold.










![(1R,4S)-2-Azabicyclo[2.2.1]heptan-3-one](http://img.cochemist.com/ccimg/134100/134003-02-4.png)
![(1R,4S)-2-Azabicyclo[2.2.1]heptan-3-one](http://img.cochemist.com/ccimg/134100/134003-02-4_b.png)




![3-Pyrrolidinesulfonic acid,1-[[4-[(iodoacetyl)amino]benzoyl]oxy]-2,5-dioxo-](http://img.cochemist.com/ccimg/106200/106145-13-5.png)
![3-Pyrrolidinesulfonic acid,1-[[4-[(iodoacetyl)amino]benzoyl]oxy]-2,5-dioxo-](http://img.cochemist.com/ccimg/106200/106145-13-5_b.png)


![2-Butenedioic acid(2E)-,1-[(2R,4R,5S,6R)-tetrahydro-2-hydroxy-2-[(1S,2R,3S)-2-hydroxy-3-[(2R,3S,4E,6E,9S,10S,11R,12E,14Z)-10-hydroxy-3,15-dimethoxy-7,9,11,13-tetramethyl-16-oxooxacyclohexadeca-4,6,12,14-tetraen-2-yl]-1-methylbutyl]-5-methyl-6-(1-methylethyl)-2H-pyran-4-yl]ester](http://img.cochemist.com/ccimg/89000/88979-61-7.png)
![2-Butenedioic acid(2E)-,1-[(2R,4R,5S,6R)-tetrahydro-2-hydroxy-2-[(1S,2R,3S)-2-hydroxy-3-[(2R,3S,4E,6E,9S,10S,11R,12E,14Z)-10-hydroxy-3,15-dimethoxy-7,9,11,13-tetramethyl-16-oxooxacyclohexadeca-4,6,12,14-tetraen-2-yl]-1-methylbutyl]-5-methyl-6-(1-methylethyl)-2H-pyran-4-yl]ester](http://img.cochemist.com/ccimg/89000/88979-61-7_b.png)
![2-Butenoicacid, 4-[(2-hydroxy-5-oxo-1-cyclopenten-1-yl)amino]-4-oxo-,(2R,4R,5S,6R)-tetrahydro-2-hydroxy-2-[(1S,2R,3S)-2-hydroxy-3-[(2R,3S,4E,6E,9S,10S,11R,12E,14Z)-10-hydroxy-3,15-dimethoxy-7,9,11,13-tetramethyl-16-oxooxacyclohexadeca-4,6,12,14-tetraen-2-yl]-1-methylbutyl]-5-methyl-6-(1-methylethyl)-2H-pyran-4-ylester, (2E)-](http://img.cochemist.com/ccimg/88900/88899-56-3.png)
![2-Butenoicacid, 4-[(2-hydroxy-5-oxo-1-cyclopenten-1-yl)amino]-4-oxo-,(2R,4R,5S,6R)-tetrahydro-2-hydroxy-2-[(1S,2R,3S)-2-hydroxy-3-[(2R,3S,4E,6E,9S,10S,11R,12E,14Z)-10-hydroxy-3,15-dimethoxy-7,9,11,13-tetramethyl-16-oxooxacyclohexadeca-4,6,12,14-tetraen-2-yl]-1-methylbutyl]-5-methyl-6-(1-methylethyl)-2H-pyran-4-ylester, (2E)-](http://img.cochemist.com/ccimg/88900/88899-56-3_b.png)


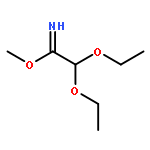
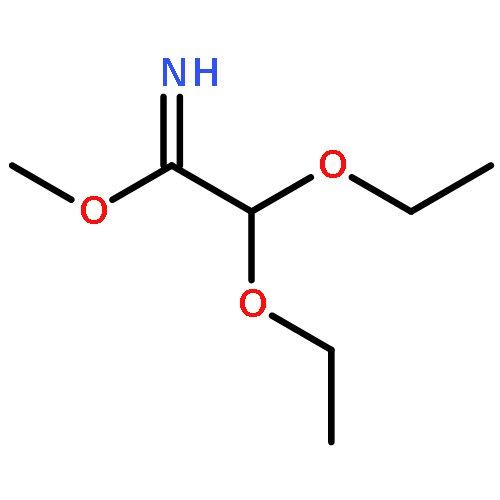
![(2,5-DIOXOPYRROLIDIN-1-YL) 4-[(2-IODOACETYL)AMINO]BENZOATE](http://img.cochemist.com/ccimg/72300/72252-96-1.png)
![(2,5-DIOXOPYRROLIDIN-1-YL) 4-[(2-IODOACETYL)AMINO]BENZOATE](http://img.cochemist.com/ccimg/72300/72252-96-1_b.png)




![Benzoic acid,4-[(2,5-dihydro-2,5-dioxo-1H-pyrrol-1-yl)methyl]-, 2,5-dioxo-1-pyrrolidinylester](http://img.cochemist.com/ccimg/65000/64987-84-4.png)
![Benzoic acid,4-[(2,5-dihydro-2,5-dioxo-1H-pyrrol-1-yl)methyl]-, 2,5-dioxo-1-pyrrolidinylester](http://img.cochemist.com/ccimg/65000/64987-84-4_b.png)


![L-ORNITHINE, N2-[(1,1-DIMETHYLETHOXY)CARBONYL]-, 1,1-DIMETHYLETHYL ESTER](http://img.cochemist.com/ccimg/53100/53054-03-8.png)
![L-ORNITHINE, N2-[(1,1-DIMETHYLETHOXY)CARBONYL]-, 1,1-DIMETHYLETHYL ESTER](http://img.cochemist.com/ccimg/53100/53054-03-8_b.png)
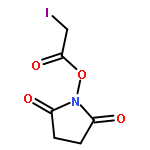
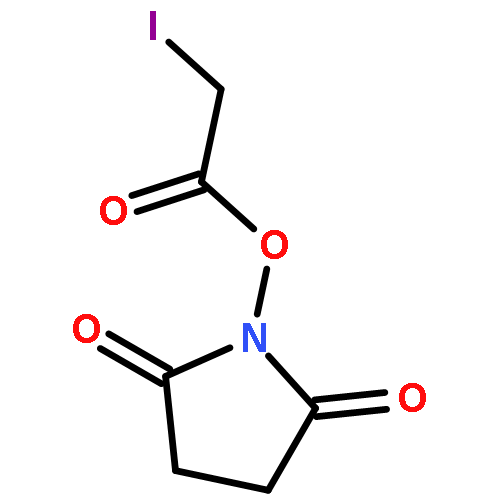
![(3S)-3-[(2S,3R)-3-(acetyloxy)-1-L-valylpyrrolidin-2-yl]-3-{[N-(2-methylbutanoyl)-L-phenylalanyl]oxy}propanoic acid](http://img.cochemist.com/ccimg/37900/37878-19-6.png)
![(3S)-3-[(2S,3R)-3-(acetyloxy)-1-L-valylpyrrolidin-2-yl]-3-{[N-(2-methylbutanoyl)-L-phenylalanyl]oxy}propanoic acid](http://img.cochemist.com/ccimg/37900/37878-19-6_b.png)


![Benzamide,N,N',N''-[(3S,7S,11S)-2,6,10-trioxo-1,5,9-trioxacyclododecane-3,7,11-triyl]tris[2,3-dihydroxy-](http://img.cochemist.com/ccimg/28400/28384-96-5.png)
![Benzamide,N,N',N''-[(3S,7S,11S)-2,6,10-trioxo-1,5,9-trioxacyclododecane-3,7,11-triyl]tris[2,3-dihydroxy-](http://img.cochemist.com/ccimg/28400/28384-96-5_b.png)


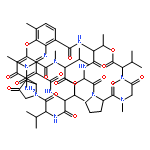
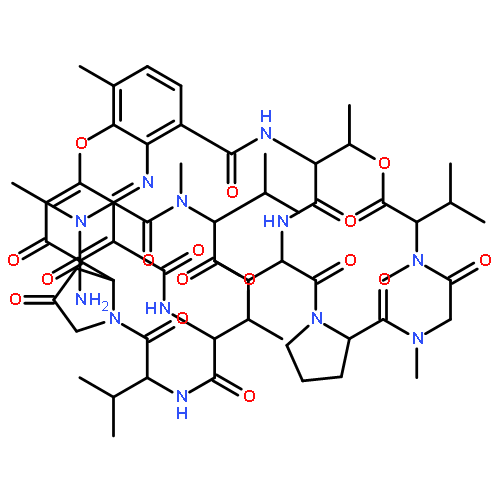
![Actinomycin D,3A-[(4R)-4-hydroxy-L-proline]- (9CI)](http://img.cochemist.com/ccimg/18900/18865-46-8.png)
![Actinomycin D,3A-[(4R)-4-hydroxy-L-proline]- (9CI)](http://img.cochemist.com/ccimg/18900/18865-46-8_b.png)




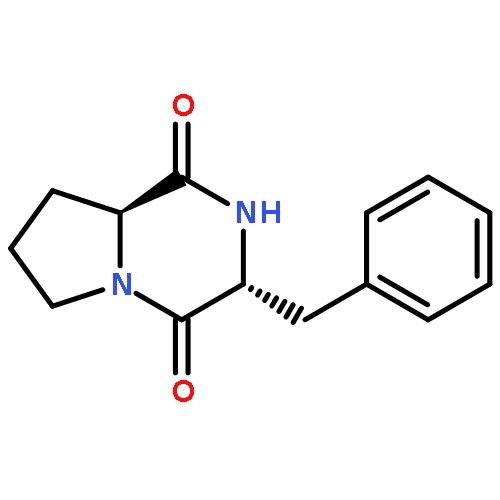
![Pyrrolo[1,2-a]pyrazine-1,4-dione,hexahydro-3-(phenylmethyl)-, (3S,8aS)-](http://img.cochemist.com/ccimg/3800/3705-26-8.png)
![Pyrrolo[1,2-a]pyrazine-1,4-dione,hexahydro-3-(phenylmethyl)-, (3S,8aS)-](http://img.cochemist.com/ccimg/3800/3705-26-8_b.png)






![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9_b.png)
![10H-3,10a-Epidithiopyrazino[1,2-a]indole-1,4-dione,2,3,5a,6-tetrahydro-6-hydroxy-3-(hydroxymethyl)-2-methyl-, (3R,5aS,6S,10aR)-](http://img.cochemist.com/ccimg/100/67-99-2.png)
![10H-3,10a-Epidithiopyrazino[1,2-a]indole-1,4-dione,2,3,5a,6-tetrahydro-6-hydroxy-3-(hydroxymethyl)-2-methyl-, (3R,5aS,6S,10aR)-](http://img.cochemist.com/ccimg/100/67-99-2_b.png)