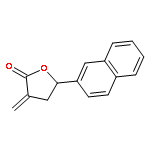
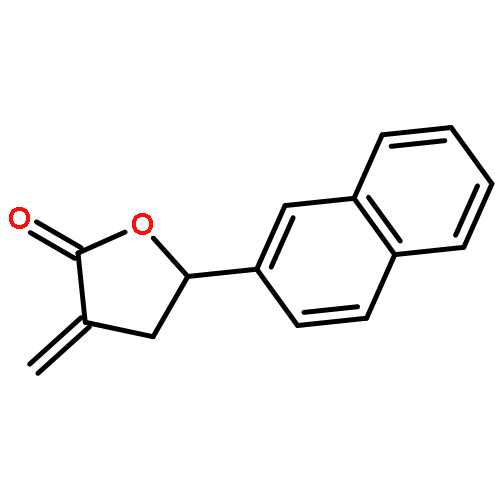
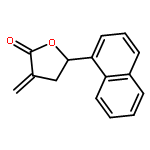
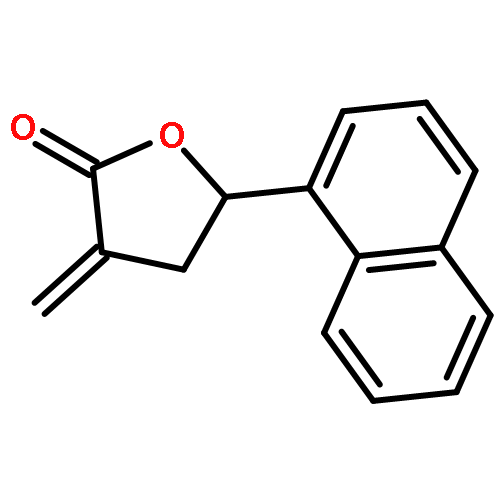
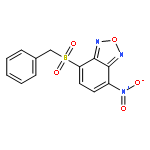
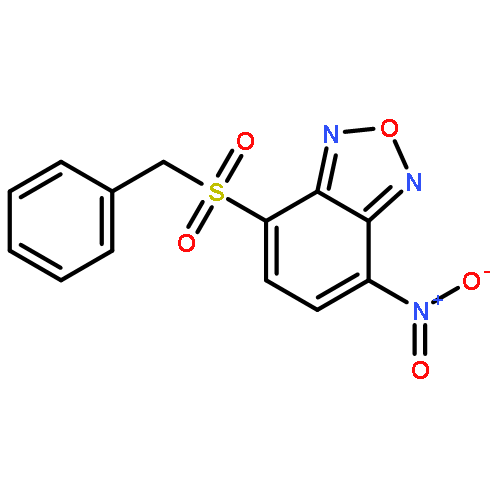
Co-reporter:Caroline M. Robb;Jacob I. Contreras;Smit Kour;Margaret A. Taylor;Mohammad Abid;Yogesh A. Sonawane;Muhammad Zahid;Daryl J. Murry;Sandeep Rana
Chemical Communications 2017 vol. 53(Issue 54) pp:7577-7580
Publication Date(Web):2017/07/04
DOI:10.1039/C7CC03879H
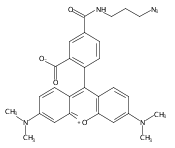
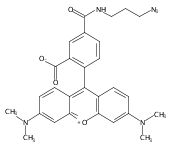
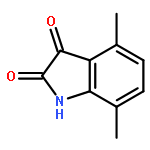
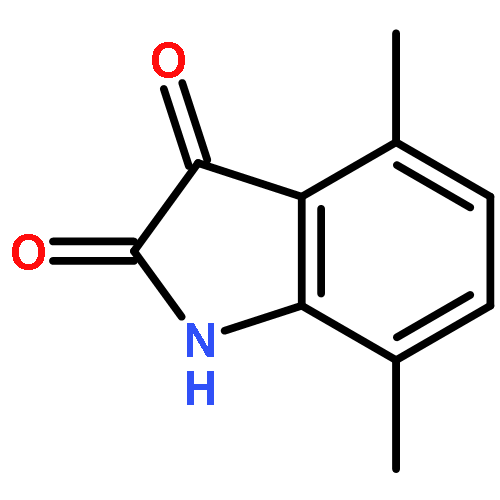
Cyclin-dependent kinase 9 (CDK9), a member of the cyclin-dependent protein kinase (CDK) family, is involved in transcriptional elongation of several target genes. CDK9 is ubiquitously expressed and has been shown to contribute to a variety of malignancies such as pancreatic, prostate and breast cancers. Here we report the development of a heterobifunctional small molecule proteolysis targeting chimera (PROTAC) capable of cereblon (CRBN) mediated proteasomal degradation of CDK9. In HCT116 cells, it selectively degrades CDK9 while sparing other CDK family members. This is the first example of a PROTAC that selectively degrades CDK9.
Co-reporter:Yogesh A. Sonawane, Margaret A. Taylor, John Victor Napoleon, Sandeep Rana, Jacob I. Contreras, and Amarnath Natarajan
Journal of Medicinal Chemistry 2016 Volume 59(Issue 19) pp:8667-8684
Publication Date(Web):May 12, 2016
DOI:10.1021/acs.jmedchem.6b00150
Cyclin dependent kinase (CDK) inhibitors have been the topic of intense research for nearly 2 decades due to their widely varied and critical functions within the cell. Recently CDK9 has emerged as a druggable target for the development of cancer therapeutics. CDK9 plays a crucial role in transcription regulation; specifically, CDK9 mediated transcriptional regulation of short-lived antiapoptotic proteins is critical for the survival of transformed cells. Focused chemical libraries based on a plethora of scaffolds have resulted in mixed success with regard to the development of selective CDK9 inhibitors. Here we review the regulation of CDK9, its cellular functions, and common core structures used to target CDK9, along with their selectivity profile and efficacy in vitro and in vivo.
Co-reporter:Sandeep Rana; Elizabeth C. Blowers; Calvin Tebbe; Jacob I. Contreras; Prakash Radhakrishnan; Smitha Kizhake; Tian Zhou; Rajkumar N. Rajule; Jamie L. Arnst; Adnan R. Munkarah; Ramandeep Rattan
Journal of Medicinal Chemistry 2016 Volume 59(Issue 10) pp:5121-5127
Publication Date(Web):April 14, 2016
DOI:10.1021/acs.jmedchem.6b00400
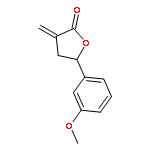
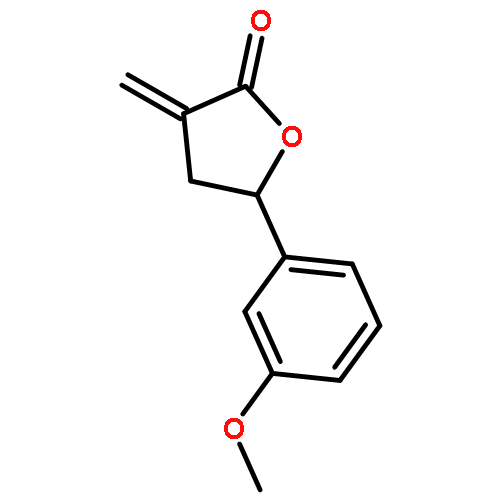
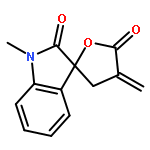
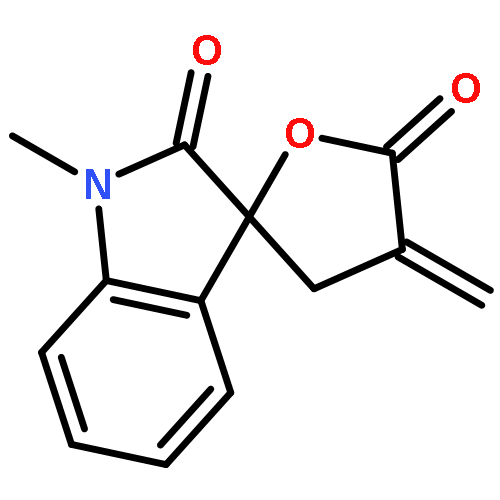
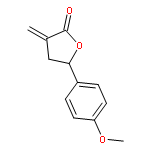
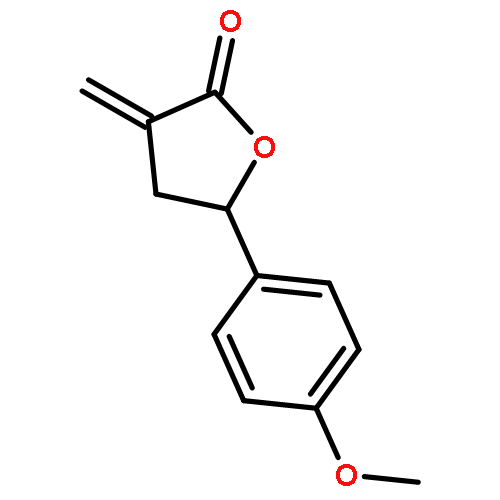
Design, synthesis, and evaluation of α-methylene-γ-butyrolactone analogues and their evaluation as anticancer agents is described. SAR identified a spirocyclic analogue 19 that inhibited TNFα-induced NF-κB activity, cancer cell growth and tumor growth in an ovarian cancer model. A second iteration of synthesis and screening identified 29 which inhibited cancer cell growth with low-μM potency. Our data suggest that an isatin-derived spirocyclic α-methylene-γ-butyrolactone is a suitable core for optimization to identify novel anticancer agents.
Co-reporter:Sandeep Rana; Elizabeth C. Blowers
Journal of Medicinal Chemistry 2015 Volume 58(Issue 1) pp:2-29
Publication Date(Web):August 14, 2014
DOI:10.1021/jm401994c
Adenosine 5′-monophosphate activated protein kinase (AMPK) is a master sensor of cellular energy status that plays a key role in the regulation of whole-body energy homeostasis. AMPK is a serine/threonine kinase that is activated by upstream kinases LKB1, CaMKKβ, and Tak1, among others. AMPK exists as αβγ trimeric complexes that are allosterically regulated by AMP, ADP, and ATP. Dysregulation of AMPK has been implicated in a number of metabolic diseases including type 2 diabetes mellitus and obesity. Recent studies have associated roles of AMPK with the development of cancer and neurological disorders, making it a potential therapeutic target to treat human diseases. This review focuses on the structure and function of AMPK, its role in human diseases, and its direct substrates and provides a brief synopsis of key AMPK modulators and their relevance in human diseases.
Co-reporter:Sandeep Rana and Amarnath Natarajan
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 2) pp:244-247
Publication Date(Web):14 Nov 2012
DOI:10.1039/C2OB27008K
We report an unusual face selective reduction of the exocyclic double bond in the α-methylene-γ-butyrolactone motif of spiro-oxindole systems. The spiro-oxindoles were assembled by an indium metal mediated Barbier-type reaction followed by an acid catalyzed lactonization.
Co-reporter:Jamie L. Arnst, Christopher W. Davies, Srikumar M. Raja, Chittaranjan Das, Amarnath Natarajan
Analytical Biochemistry 2013 Volume 440(Issue 1) pp:71-77
Publication Date(Web):1 September 2013
DOI:10.1016/j.ab.2013.05.017
Abstract
Deubiquitinases (DUBs) play an important role in regulating the ubiquitin landscape of proteins. The DUB AMSH (associated molecule with the SH3 domain of STAM) has been shown to be involved in regulating the ubiquitin-dependent down-regulation of activated cell surface receptors via the endolysosomal degradative pathway. Therefore, small molecule AMSH inhibitors will be useful chemical probes to study the effect of AMSH DUB activity on cell surface receptor degradation. Currently, there are no known selective inhibitors of AMSH or high-throughput compatible assays for their identification. We report the development and optimization of a novel fluorescence resonance energy transfer (FRET)-based add-and-read AMSH DUB assay in a 384-well format. In this format, the optimal temperature for a high-throughput screen (HTS) was determined to be 30 °C, the assay tolerates 5% dimethyl sulfoxide (DMSO), and it has a Z-score of 0.71, indicating HTS compatibility. The assay was used to show that AMSH selectively cleaves Lys63-linked diubiquitin over Lys48- and Lys11-linked diubiquitin. The IC50 value of the nonspecific small molecule DUB inhibitor N-ethylmaleimide was 16.2 ± 3.2 μM and can be used as a qualitative positive control for the screen. We conclude that this assay is high-throughput compatible and can be used to identify novel small molecule inhibitors of AMSH.
Co-reporter:Eric A. Kumar ●; Ziyan Yuan ●; Nicholas Y. Palermo ; Lin Dong ; Gulzar Ahmad ; G. L. Lokesh ●; Carol Kolar ; Smitha Kizhake ●; Gloria E. O. Borgstahl ; Hamid Band ∞ ●
Journal of Medicinal Chemistry 2012 Volume 55(Issue 7) pp:3583-3587
Publication Date(Web):March 6, 2012
DOI:10.1021/jm300078z
We describe truncation and SAR studies to identify a pentapeptide that binds Cbl tyrosine kinase binding domain with a higher affinity than the parental peptide. The pentapeptide has an alternative binding mode that allows occupancy of a previously uncharacterized groove. A peptide library was used to map the binding site and define the interface landscape. Our results suggest that the pentapeptide is an ideal starting point for the development of inhibitors against Cbl driven diseases.
Co-reporter:Rajkumar Rajule, Vashti C. Bryant, Hernando Lopez, Xu Luo, Amarnath Natarajan
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 7) pp:2227-2234
Publication Date(Web):1 April 2012
DOI:10.1016/j.bmc.2012.02.022
In HeLa cells the combinatorial knockdown of Bcl-xL and Mcl-1 is sufficient to induce spontaneous apoptosis. Quinoxaline derivatives were screened for the induction of Mcl-1 dependent apoptosis using a cell line without functional Bcl-xL. Quinoxaline urea analog 1h was able to specifically induce apoptosis in an Mcl-1 dependent manner. We demonstrate that even small changes to 1h results in dramatic loss of activity. In addition, 1h and ABT-737 synergistically inhibit cell growth and induce apoptosis. Our results also suggest that 1h could have therapeutic potential against ABT-737 refractory cancer.
Co-reporter:Vashti C. Bryant, G.D. Kishore Kumar, Abijah M. Nyong, Amarnath Natarajan
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 1) pp:245-248
Publication Date(Web):1 January 2012
DOI:10.1016/j.bmcl.2011.11.025
The macrocyclic diarylether heptanoid (MDEH) natural products have been used in folk medicine for centuries. MDEHs are reported to exert anti-tumor properties by inhibiting the activation of NF-κB. Here we report the synthesis of a small MDEH library (first reported synthesis of racemic platycarynol) using a Grubbs cross metathesis/Ullmann cyclization strategy. Evaluation of the library led to the identification of MDEH 9b which sensitizes pancreatic cancer cells to gemcitabine mediated growth inhibition and apoptosis.
Co-reporter:Ziyan Yuan Pessetto;Ying Yan;Tadayoshi Bessho;Amarnath Natarajan
Breast Cancer Research and Treatment 2012 Volume 134( Issue 2) pp:511-517
Publication Date(Web):2012/07/01
DOI:10.1007/s10549-012-2079-4
Synthetic lethal therapeutic strategy using poly(adenosine diphosphate [ADP]-ribose) polymerase (PARP) inhibitor olaparib in carriers of BRCA1 or BRCA2 mutation has shown promise in clinical settings. Since <5 % of patients are BRCA1 or BRCA2 mutation carriers, small molecules that functionally mimic BRCA1 or BRCA2 mutations will extend the synthetic lethal therapeutic option for non-mutation carriers. Here we provide proof of principle for this strategy using a BRCA1 inhibitor peptide 2 that targets the BRCT(BRCA1)-phosphoprotein interaction and mimics the M177R/K BRCA1 mutation. Reciprocal immunoprecipitation and immunoblotting of BRCA1 and Abraxas was used to demonstrate inhibitor 2 targets BRCT(BRCA1)-Abraxas interface. Immunostaining of γH2AX, cell cycle analysis and homologous recombination (HR) assays were conducted to confirm that inhibitor 2 functionally mimics a chemosensitizing BRCA1 mutation. The concept of synthetic lethal therapeutic strategy with the BRCA1 inhibitor 2 and the PARP inhibitor Olaparib was explored in HeLa, MDA-MB-231, and HCC1937 cell lines. The results show that inhibition of BRCA1 by 2 sensitizes HeLa and MDA-MB-231 cells but not HCC1937 to Olaparib mediated growth inhibition and apoptosis. These results provide the basis for developing high affinity BRCT(BRCA1) inhibitors as adjuvants to treat sporadic breast and ovarian cancers.
Co-reporter:Ziyan Yuan ; Eric A. Kumar ; Smitha Kizhake
Journal of Medicinal Chemistry 2011 Volume 54(Issue 12) pp:4264-4268
Publication Date(Web):May 16, 2011
DOI:10.1021/jm1016413
Carboxy terminal BRCT domains of the breast cancer susceptibility gene 1 (BRCA1) bind to phosphorylated proteins through a pSXXF consensus recognition motif. We report a systematic structure–activity relationship study that maps the BRCT(BRCA1)–pSXXF binding interface, leading to identification of peptides with nanomolar binding affinities comparable to those of the previously reported 13-mer peptides and providing a clear description of the pSXXF–BRCT interface, which is essential for developing small molecule inhibitors via the peptidomimetic approach.
Co-reporter:Ziyan Yuan, Eric A. Kumar, Stephen J. Campbell, Nicholas Y. Palermo, Smitha Kizhake, J. N. Mark Glover, and Amarnath Natarajan
ACS Medicinal Chemistry Letters 2011 Volume 2(Issue 10) pp:764
Publication Date(Web):August 17, 2011
DOI:10.1021/ml200147a
Breast cancer gene 1 carboxy terminus (BRCT) domains are found in a number of proteins that are important for DNA damage response (DDR). The BRCT domains bind phosphorylated proteins, and these protein–protein interactions are essential for DDR and DNA repair. High affinity domain specific inhibitors are needed to facilitate the dissection of the protein–protein interactions in the DDR signaling. The BRCT domains of BRCA1 bind phosphorylated protein through a pSXXF consensus recognition motif. We identified a hydrophobic pocket at the P-1 position of the pSXXF binding site. Here we conducted a structure-guided synthesis of peptide analogues with hydrophobic functional groups at the P-1 position. Evaluation of these led to the identification of a peptide mimic 15 with a inhibitory constant (Ki) of 40 nM for BRCT(BRCA1). Analysis of the TopBP1 and MDC1 BRCT domains suggests a similar approach is viable to design high affinity inhibitors.Keywords: BRCT inhibitors and breast cancer; peptide mimics; Protein−protein interface
Co-reporter:Nicholas Y. Palermo, Amarnath Natarajan
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 20) pp:6081-6084
Publication Date(Web):15 October 2011
DOI:10.1016/j.bmcl.2011.08.051
Nuclear Factor κ B is implicated in tumor progression and chronic inflammatory diseases and is regulated by IκB kinase β (IKKβ). The crystal structure of IKKβ has been recently solved for Xenopus laevis. Homology models of human IKKβ have been developed prior to and after the crystal structure was solved. Here, we compare four models of human IKKβ and evaluate their performance in both broad and focused library docking studies.
Co-reporter:Qianyi Chen, Vashti C. Bryant, Hernando Lopez, David L. Kelly, Xu Luo, Amarnath Natarajan
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 7) pp:1929-1932
Publication Date(Web):1 April 2011
DOI:10.1016/j.bmcl.2011.02.055
The quinoxaline core is considered a privileged scaffold as it is found in a variety of biologically relevant molecules. Here we report the synthesis of a quinoxalin-6-amine library, screening against a panel of cancer cell lines and a structure–activity relationship (SAR). This resulted in the identification of a bisfuranylquinoxalineurea analog (7c) that has low micromolar potency against the panel of cancer cell lines. We also show that cells treated with quinoxalineurea 7c results in caspase 3/7 activation, PARP cleavage and Mcl-1 dependent apoptosis.
Co-reporter:Eric A. Kumar, Casey D. Charvet, G.L. Lokesh, Amarnath Natarajan
Analytical Biochemistry 2011 Volume 411(Issue 2) pp:254-260
Publication Date(Web):15 April 2011
DOI:10.1016/j.ab.2010.11.038
The casitas B-lineage lymphoma (Cbl) proteins play an important role in regulating signal transduction pathways by functioning as E3 ubiquitin ligases. The Cbl proteins contain a conserved tyrosine kinase binding (TKB) domain that binds more than a dozen proteins, including protein tyrosine kinases (PTKs), in a phosphorylation-dependent manner. The cell surface expression levels of the PTKs are regulated by Cbl-mediated ubiquitination, internalization, and degradation. Dysfunction in this signaling cascade has resulted in prolonged activation of the PTKs and, therefore, has been implicated in inflammatory diseases and various cancers. Due to this negative regulatory function, Cbl has been largely ignored as a therapeutic target. However, recent studies, such as the identification of (i) gain of function c-Cbl mutations in subsets of myeloid cancer and (ii) c-Cbl as a prostate basal cell marker that correlates with poor clinical outcome, suggest otherwise. Here we report the development of a competitive high-throughput fluorescence polarization assay in a 384-well format to identify inhibitors of Cbl(TKB). The high-throughput screen readiness of the assay was demonstrated by screening the Prestwick Chemical Library.
Co-reporter:Victor M. Anisimov;Arturas Ziemys
Journal of Computer-Aided Molecular Design 2011 Volume 25( Issue 11) pp:1071-1084
Publication Date(Web):2011 November
DOI:10.1007/s10822-011-9484-3
The C-terminal domain of BRCA1(BRCT) is involved in the DNA repair pathway by recognizing the pSXXF motif in interacting proteins. It has been reported that short peptides containing this motif bind to BRCA1(BRCT) in the micromolar range with high specificity. In this work, the binding of pSXXF peptides has been studied computationally and experimentally in order to characterize their interaction with BRCA1(BRCT). Elucidation of the contacts that drive the protein–ligand interaction is critical for the development of high affinity small-molecule BRCA1 inhibitors. Molecular dynamics (MD) simulations revealed the key role of threonine at the peptide P+2 position in providing structural rigidity to the ligand in the bound state. The mutation at P+1 had minor effects. Peptide extension at the N-terminal position with the naphthyl amino acid exhibited a modest increase in binding affinity, what could be explained by the dispersion interaction of the naphthyl side-chain with a hydrophobic patch. Three in silico end-point methods were considered for the calculation of binding free energy. The Molecular Mechanics Poisson–Boltzmann Surface Area and the Solvated Interaction Energy methods gave reasonable agreement with experimental data, exhibiting a Pearlman predictive index of 0.71 and 0.78, respectively. The MM-quantum mechanics-surface area method yielded improved results, which was characterized by a Pearlman index of 0.78. The correlation coefficients were 0.59, 0.61 and 0.69, respectively. The ability to apply a QM level of theory within an end-point binding free energy protocol may provide a way for a consistent improvement of accuracy in computer-aided drug design.
Co-reporter:Devalina Ray, Abijah M. Nyong, Amarnath Natarajan
Tetrahedron Letters 2010 Volume 51(Issue 19) pp:2655-2656
Publication Date(Web):12 May 2010
DOI:10.1016/j.tetlet.2010.03.034
Aryl and alkenyl amino acid derivatives were synthesized by a palladium-catalyzed 1,4-addition of the corresponding boronic acids to 2-acetamidoacrylate.
Co-reporter:Sandeep Rana and Amarnath Natarajan
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 2) pp:NaN247-247
Publication Date(Web):2012/11/14
DOI:10.1039/C2OB27008K
We report an unusual face selective reduction of the exocyclic double bond in the α-methylene-γ-butyrolactone motif of spiro-oxindole systems. The spiro-oxindoles were assembled by an indium metal mediated Barbier-type reaction followed by an acid catalyzed lactonization.
Co-reporter:Caroline M. Robb, Jacob I. Contreras, Smit Kour, Margaret A. Taylor, Mohammad Abid, Yogesh A. Sonawane, Muhammad Zahid, Daryl J. Murry, Amarnath Natarajan and Sandeep Rana
Chemical Communications 2017 - vol. 53(Issue 54) pp:NaN7580-7580
Publication Date(Web):2017/06/14
DOI:10.1039/C7CC03879H
Cyclin-dependent kinase 9 (CDK9), a member of the cyclin-dependent protein kinase (CDK) family, is involved in transcriptional elongation of several target genes. CDK9 is ubiquitously expressed and has been shown to contribute to a variety of malignancies such as pancreatic, prostate and breast cancers. Here we report the development of a heterobifunctional small molecule proteolysis targeting chimera (PROTAC) capable of cereblon (CRBN) mediated proteasomal degradation of CDK9. In HCT116 cells, it selectively degrades CDK9 while sparing other CDK family members. This is the first example of a PROTAC that selectively degrades CDK9.

![12H-Indolo[2,3-a]pyrrolo[3,4-c]carbazole-12-propanenitrile,5,6,7,13-tetrahydro-13-methyl-5-oxo-](http://img.cochemist.com/ccimg/136200/136194-77-9.png)
![12H-Indolo[2,3-a]pyrrolo[3,4-c]carbazole-12-propanenitrile,5,6,7,13-tetrahydro-13-methyl-5-oxo-](http://img.cochemist.com/ccimg/136200/136194-77-9_b.png)
![Carbamimidothioic acid,3-[3-[2,5-dihydro-4-(1-methyl-1H-indol-3-yl)-2,5-dioxo-1H-pyrrol-3-yl]-1H-indol-1-yl]propylester](http://img.cochemist.com/ccimg/125400/125314-64-9.png)
![Carbamimidothioic acid,3-[3-[2,5-dihydro-4-(1-methyl-1H-indol-3-yl)-2,5-dioxo-1H-pyrrol-3-yl]-1H-indol-1-yl]propylester](http://img.cochemist.com/ccimg/125400/125314-64-9_b.png)