The cyanosporasides A–F are a collection of monochlorinated benzenoid derivatives isolated from the marine actinomycetes Salinispora and Streptomyces sp. All derivatives feature one of two types of cyanocyclopenta[a]indene frameworks, which are regioisomeric in the position of a single chlorine atom. It is proposed that these chloro-substituted benzenoids are formed biosynthetically through the cycloaromatization of a bicyclic nine-membered enediyne precursor. Herein, we report the synthesis of such a bicyclic precursor, its spontaneous transannulation into a p-benzyne, and its differential 1,4 hydrochlorination reactivity under either organochlorine or chloride-salt conditions. Our bioinspired approach culminated in the first regiodivergent total synthesis of the aglycons A/F and B/C, as well as cyanosporasides D and E. In addition, empirical insights into the site selectivity of a natural-like p-benzyne, calculated to be a ground-state triplet diradical, to hydrogen, chlorine, and chloride sources are revealed.
The cyanosporasides A–F are a collection of monochlorinated benzenoid derivatives isolated from the marine actinomycetes Salinispora and Streptomyces sp. All derivatives feature one of two types of cyanocyclopenta[a]indene frameworks, which are regioisomeric in the position of a single chlorine atom. It is proposed that these chloro-substituted benzenoids are formed biosynthetically through the cycloaromatization of a bicyclic nine-membered enediyne precursor. Herein, we report the synthesis of such a bicyclic precursor, its spontaneous transannulation into a p-benzyne, and its differential 1,4 hydrochlorination reactivity under either organochlorine or chloride-salt conditions. Our bioinspired approach culminated in the first regiodivergent total synthesis of the aglycons A/F and B/C, as well as cyanosporasides D and E. In addition, empirical insights into the site selectivity of a natural-like p-benzyne, calculated to be a ground-state triplet diradical, to hydrogen, chlorine, and chloride sources are revealed.
(−)-Platensimycin is a potent inhibitor of fatty acid synthase that holds promise in the treatment of metabolic disorders (e.g., diabetes and “fatty liver”) and pathogenic infections (e.g., those caused by drug-resistant bacteria). Herein, we describe its total synthesis through a four-step preparation of the aromatic amine fragment and an improved stereocontrolled assembly of the ketolide fragment, (−)-platensic acid. Key synthetic advances include 1) a modified Lieben haloform reaction to directly convert an aryl methyl ketone into its methyl ester within 30 seconds, 2) an experimentally improved dialkylation protocol to form platensic acid, 3) a sterically controlled chemo- and diastereoselective organocatalytic conjugate reduction of a spiro-cyclized cyclohexadienone by using the trifluoroacetic acid salt of α-amino di-tert-butyl malonate, 4) a tetrabutylammonium fluoride promoted spiro-alkylative para dearomatization of a free phenol to assemble the cagelike ketolide core with the moderate leaving-group ability of an early tosylate intermediate, and 5) a bismuth(III)-catalyzed Friedel–Crafts cyclization of a free lactol, with LiClO4 as an additive to liberate a more active oxocarbenium perchlorate species and suppress the Lewis basicity of the sulfonyloxy group. The longest linear sequence is 21 steps with an overall yield of 3.8 % from commercially available eugenol.
![Cyclopent[a]indene-5-acetonitrile, 7-chloro-3,3a-dihydro-3,3a-dihydroxy-, (3S,3aR)-](/data/chemimg/3740100/1643768-17-5.png)
![Cyclopent[a]indene-5-acetonitrile, 7-chloro-3,3a-dihydro-3,3a-dihydroxy-, (3S,3aR)-](/data/chemimg/3740100/1643768-17-5_b.png)
![Cyclopent[a]indene-5-acetonitrile, 7-chloro-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, (3S,3aR)-](/data/chemimg/3740100/1643768-14-2.png)
![Cyclopent[a]indene-5-acetonitrile, 7-chloro-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, (3S,3aR)-](/data/chemimg/3740100/1643768-14-2_b.png)
![Cyclopent[a]indene, 7-chloro-5-(chloromethyl)-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, (3S,3aR)-](/data/chemimg/3740100/1643768-13-1.png)
![Cyclopent[a]indene, 7-chloro-5-(chloromethyl)-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, (3S,3aR)-](/data/chemimg/3740100/1643768-13-1_b.png)
![Cyclopent[a]indene-5-methanol, 7-chloro-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, 5-methanesulfonate, (3S,3aR)-](/data/chemimg/3740100/1643768-12-0.png)
![Cyclopent[a]indene-5-methanol, 7-chloro-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, 5-methanesulfonate, (3S,3aR)-](/data/chemimg/3740100/1643768-12-0_b.png)
![Cyclopent[a]indene-5-acetonitrile, 4-chloro-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, (3S,3aR)-](/data/chemimg/3740100/1643768-11-9.png)
![Cyclopent[a]indene-5-acetonitrile, 4-chloro-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, (3S,3aR)-](/data/chemimg/3740100/1643768-11-9_b.png)
![Cyclopent[a]indene, 4-chloro-5-(chloromethyl)-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, (3S,3aR)-](/data/chemimg/3740100/1643768-10-8.png)
![Cyclopent[a]indene, 4-chloro-5-(chloromethyl)-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, (3S,3aR)-](/data/chemimg/3740100/1643768-10-8_b.png)
![Cyclopent[a]indene-5-methanol, 4-chloro-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, 5-methanesulfonate, (3S,3aR)-](/data/chemimg/3740100/1643768-09-5.png)
![Cyclopent[a]indene-5-methanol, 4-chloro-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, 5-methanesulfonate, (3S,3aR)-](/data/chemimg/3740100/1643768-09-5_b.png)
![Cyclopent[a]indene, 7-chloro-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, (3S,3aR)-](/data/chemimg/3740100/1643768-07-3.png)
![Cyclopent[a]indene, 7-chloro-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, (3S,3aR)-](/data/chemimg/3740100/1643768-07-3_b.png)
![Cyclopent[a]indene, 4-chloro-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, (3S,3aR)-](/data/chemimg/3740100/1643768-05-1.png)
![Cyclopent[a]indene, 4-chloro-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, (3S,3aR)-](/data/chemimg/3740100/1643768-05-1_b.png)
![Cyclopent[a]indene, 4,7-dichloro-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, (3S,3aR)-](/data/chemimg/3740100/1643768-03-9.png)
![Cyclopent[a]indene, 4,7-dichloro-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, (3S,3aR)-](/data/chemimg/3740100/1643768-03-9_b.png)
![Cyclopent[a]indene, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, (3S,3aR)-](/data/chemimg/3740100/1643768-00-6.png)
![Cyclopent[a]indene, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-3,3a-dihydro-3a-[(triethylsilyl)oxy]-, (3S,3aR)-](/data/chemimg/3740100/1643768-00-6_b.png)
![1-Cyclopentene-1-carboxaldehyde, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3-[(4-methoxyphenyl)methoxy]-5-[2-[(4R)-2-(4-methoxyphenyl)-4-(2-propyn-1-yl)-1,3-dioxolan-4-yl]ethynyl]-5-[(triethylsilyl)oxy]-, (3R,4S,5S)-](/data/chemimg/3740100/1643767-95-6.png)
![1-Cyclopentene-1-carboxaldehyde, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3-[(4-methoxyphenyl)methoxy]-5-[2-[(4R)-2-(4-methoxyphenyl)-4-(2-propyn-1-yl)-1,3-dioxolan-4-yl]ethynyl]-5-[(triethylsilyl)oxy]-, (3R,4S,5S)-](/data/chemimg/3740100/1643767-95-6_b.png)
![1-Cyclopentene-1-carboxylic acid, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3-[(4-methoxyphenyl)methoxy]-5-[2-[(4R)-2-(4-methoxyphenyl)-4-(2-propyn-1-yl)-1,3-dioxolan-4-yl]ethynyl]-5-[(triethylsilyl)oxy]-, methyl ester, (3R,4S,5S)-](/data/chemimg/3740100/1643767-94-5.png)
![1-Cyclopentene-1-carboxylic acid, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3-[(4-methoxyphenyl)methoxy]-5-[2-[(4R)-2-(4-methoxyphenyl)-4-(2-propyn-1-yl)-1,3-dioxolan-4-yl]ethynyl]-5-[(triethylsilyl)oxy]-, methyl ester, (3R,4S,5S)-](/data/chemimg/3740100/1643767-94-5_b.png)
![1-Cyclopentene-1-carboxylic acid, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3-[(4-methoxyphenyl)methoxy]-5-[2-[(4R)-2-(4-methoxyphenyl)-4-[3-(trimethylsilyl)-2-propyn-1-yl]-1,3-dioxolan-4-yl]ethynyl]-5-[(triethylsilyl)oxy]-, methyl ester, (3R,4S,5S)-](/data/chemimg/3740100/1643767-93-4.png)
![1-Cyclopentene-1-carboxylic acid, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3-[(4-methoxyphenyl)methoxy]-5-[2-[(4R)-2-(4-methoxyphenyl)-4-[3-(trimethylsilyl)-2-propyn-1-yl]-1,3-dioxolan-4-yl]ethynyl]-5-[(triethylsilyl)oxy]-, methyl ester, (3R,4S,5S)-](/data/chemimg/3740100/1643767-93-4_b.png)
![1,3-Dioxolane, 4-[2-[(1R,4R,5S)-5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-iodo-4-[(4-methoxyphenyl)methoxy]-1-[(triethylsilyl)oxy]-2-cyclopenten-1-yl]ethynyl]-2-(4-methoxyphenyl)-4-[3-(trimethylsilyl)-2-propyn-1-yl]-, (4R)-](/data/chemimg/3740100/1643767-92-3.png)
![1,3-Dioxolane, 4-[2-[(1R,4R,5S)-5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-iodo-4-[(4-methoxyphenyl)methoxy]-1-[(triethylsilyl)oxy]-2-cyclopenten-1-yl]ethynyl]-2-(4-methoxyphenyl)-4-[3-(trimethylsilyl)-2-propyn-1-yl]-, (4R)-](/data/chemimg/3740100/1643767-92-3_b.png)
![2-Cyclopenten-1-ol, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-iodo-4-[(4-methoxyphenyl)methoxy]-1-[2-[(4R)-2-(4-methoxyphenyl)-4-[3-(trimethylsilyl)-2-propyn-1-yl]-1,3-dioxolan-4-yl]ethynyl]-, (1R,4R,5S)-](/data/chemimg/3740100/1643767-91-2.png)
![2-Cyclopenten-1-ol, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-iodo-4-[(4-methoxyphenyl)methoxy]-1-[2-[(4R)-2-(4-methoxyphenyl)-4-[3-(trimethylsilyl)-2-propyn-1-yl]-1,3-dioxolan-4-yl]ethynyl]-, (1R,4R,5S)-](/data/chemimg/3740100/1643767-91-2_b.png)
![2-Cyclopenten-1-one, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4-hydroxy-2-iodo-, (4R,5S)-](/data/chemimg/3740100/1643767-90-1.png)
![2-Cyclopenten-1-one, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4-hydroxy-2-iodo-, (4R,5S)-](/data/chemimg/3740100/1643767-90-1_b.png)
![1,5-Hexadiyn-3-ol, 3-[[(4-methoxyphenyl)methoxy]methyl]-6-(trimethylsilyl)-, (3R)-](/data/chemimg/3740100/1643767-89-8.png)
![1,5-Hexadiyn-3-ol, 3-[[(4-methoxyphenyl)methoxy]methyl]-6-(trimethylsilyl)-, (3R)-](/data/chemimg/3740100/1643767-89-8_b.png)
![D-threo-Pent-1-ynitol, 3,4-anhydro-1,2-dideoxy-4-C-ethynyl-5-O-[(4-methoxyphenyl)methyl]-1-(trimethylsilyl)-](/data/chemimg/3740100/1643767-88-7.png)
![D-threo-Pent-1-ynitol, 3,4-anhydro-1,2-dideoxy-4-C-ethynyl-5-O-[(4-methoxyphenyl)methyl]-1-(trimethylsilyl)-](/data/chemimg/3740100/1643767-88-7_b.png)
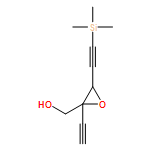
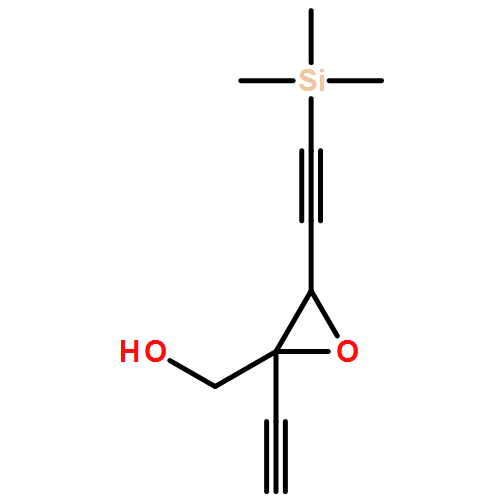
![1,3-Dioxolane, 4-ethynyl-2-(4-methoxyphenyl)-4-[3-(trimethylsilyl)-2-propyn-1-yl]-, (2S,4R)-](/data/chemimg/3740100/1643767-85-4.png)
![1,3-Dioxolane, 4-ethynyl-2-(4-methoxyphenyl)-4-[3-(trimethylsilyl)-2-propyn-1-yl]-, (2S,4R)-](/data/chemimg/3740100/1643767-85-4_b.png)
![1,3-Dioxolane, 4-ethynyl-2-(4-methoxyphenyl)-4-[3-(trimethylsilyl)-2-propyn-1-yl]-, (2R,4R)-](/data/chemimg/3740100/1643767-84-3.png)
![1,3-Dioxolane, 4-ethynyl-2-(4-methoxyphenyl)-4-[3-(trimethylsilyl)-2-propyn-1-yl]-, (2R,4R)-](/data/chemimg/3740100/1643767-84-3_b.png)
![2-Cyclopenten-1-one, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-iodo-4-[(4-methoxyphenyl)methoxy]-, (4R,5S)-](/data/chemimg/3740100/1643767-83-2.png)
![2-Cyclopenten-1-one, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-iodo-4-[(4-methoxyphenyl)methoxy]-, (4R,5S)-](/data/chemimg/3740100/1643767-83-2_b.png)
![Benzene, [[[(1R,2S)-2-(nitromethyl)cyclopropyl]methoxy]methyl]-, rel-](/data/chemimg/3739700/1639844-41-9.png)
![Benzene, [[[(1R,2S)-2-(nitromethyl)cyclopropyl]methoxy]methyl]-, rel-](/data/chemimg/3739700/1639844-41-9_b.png)
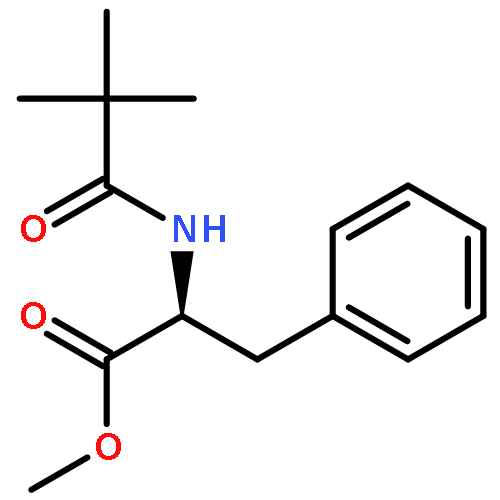
![L-Valine, (2R)-N-[(1,1-dimethylethoxy)carbonyl]-2-phenylglycyl-, methyl ester](/data/chemimg/3740100/1637252-43-7.png)
![L-Valine, (2R)-N-[(1,1-dimethylethoxy)carbonyl]-2-phenylglycyl-, methyl ester](/data/chemimg/3740100/1637252-43-7_b.png)
![Carbamic acid, N-[(S)-phenyl(phenylsulfonyl)methyl]-, 1,1-dimethylethyl ester](/data/chemimg/3739900/1427513-22-1.png)
![Carbamic acid, N-[(S)-phenyl(phenylsulfonyl)methyl]-, 1,1-dimethylethyl ester](/data/chemimg/3739900/1427513-22-1_b.png)
![Cyclopent[a]indene-5-acetonitrile, 3-(acetyloxy)-7-chloro-3,3a-dihydro-3a-hydroxy-, (3S,3aR)-](/data/chemimg/3739900/1423138-35-5.png)
![Cyclopent[a]indene-5-acetonitrile, 3-(acetyloxy)-7-chloro-3,3a-dihydro-3a-hydroxy-, (3S,3aR)-](/data/chemimg/3739900/1423138-35-5_b.png)
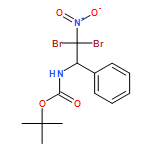
![Pyrrolidine, 2-[diphenyl[(trimethylsilyl)oxy]methyl]-1-[(1Z)-2-phenyl-1-propen-1-yl]-, (2R)-](/data/chemimg/3732100/1380445-17-9.png)
![Pyrrolidine, 2-[diphenyl[(trimethylsilyl)oxy]methyl]-1-[(1Z)-2-phenyl-1-propen-1-yl]-, (2R)-](/data/chemimg/3732100/1380445-17-9_b.png)
![Pyrrolidine, 2-[diphenyl[(trimethylsilyl)oxy]methyl]-1-[(1E)-2-phenyl-1-propen-1-yl]-, (2R)-](/data/chemimg/3739900/1380445-16-8.png)
![Pyrrolidine, 2-[diphenyl[(trimethylsilyl)oxy]methyl]-1-[(1E)-2-phenyl-1-propen-1-yl]-, (2R)-](/data/chemimg/3739900/1380445-16-8_b.png)
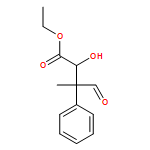
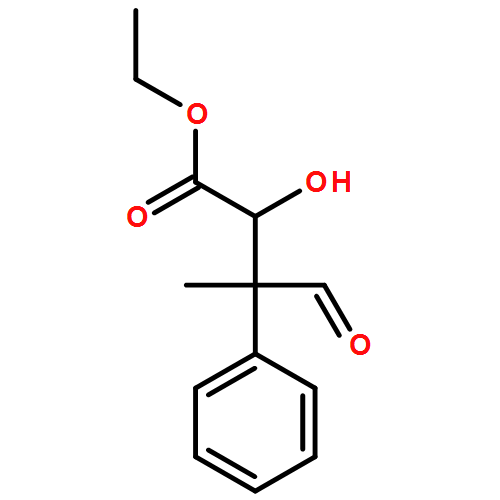
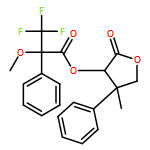
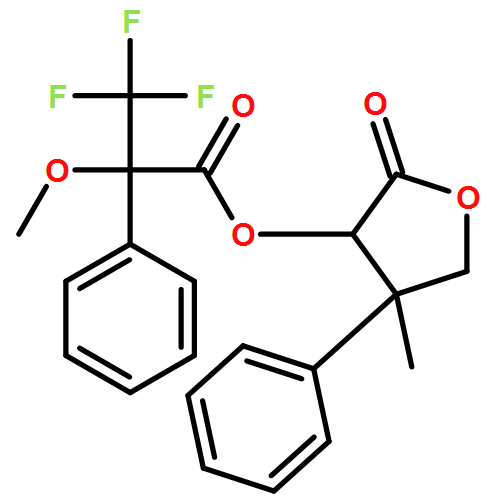
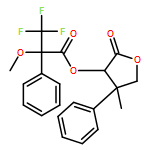
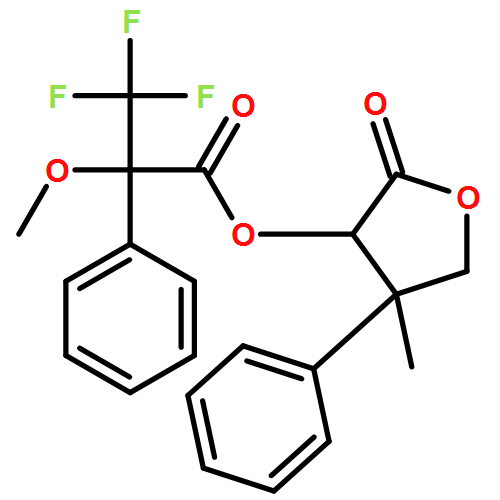
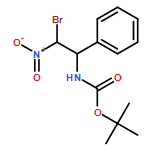
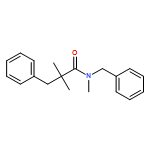
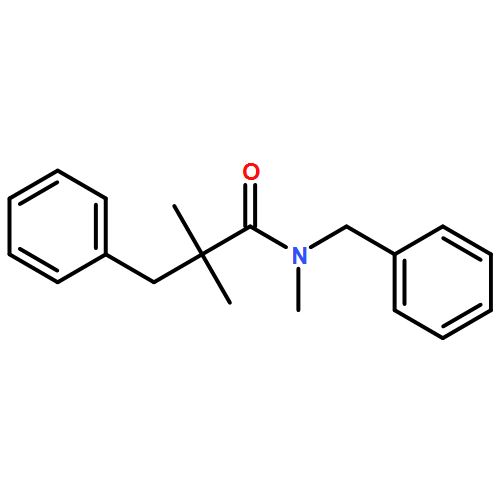
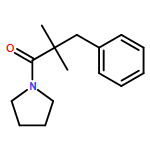
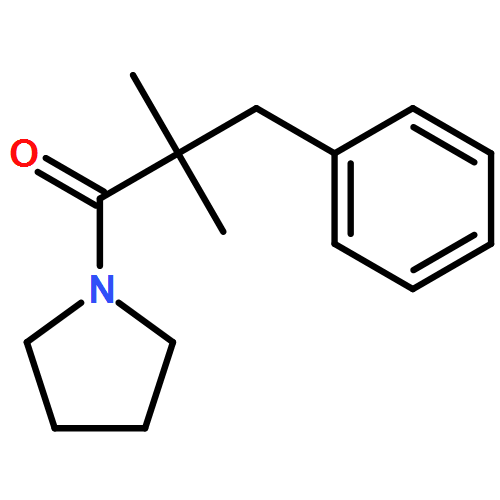
![Carbamic acid, N-[2-oxo-1-phenyl-2-(1-piperidinyl)ethyl]-, 1,1-dimethylethyl ester](/data/chemimg/3739900/1323203-74-2.png)
![Carbamic acid, N-[2-oxo-1-phenyl-2-(1-piperidinyl)ethyl]-, 1,1-dimethylethyl ester](/data/chemimg/3739900/1323203-74-2_b.png)
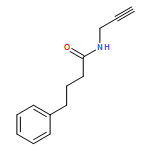
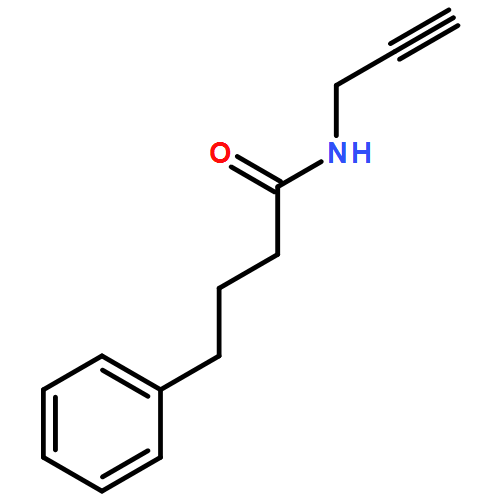
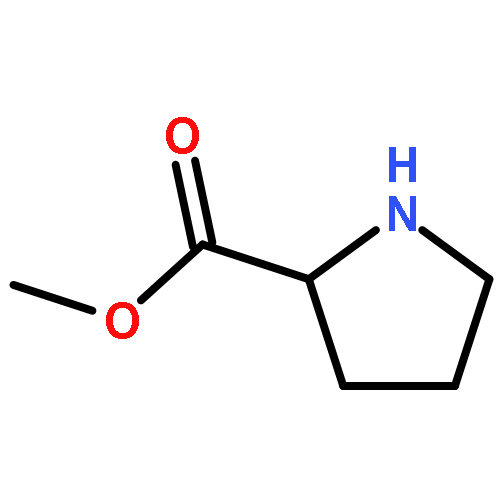
![L-Proline, (2R)-N-[(1,1-dimethylethoxy)carbonyl]-2-phenylglycyl-, methyl ester](/data/chemimg/3739800/1186497-03-9.png)
![L-Proline, (2R)-N-[(1,1-dimethylethoxy)carbonyl]-2-phenylglycyl-, methyl ester](/data/chemimg/3739800/1186497-03-9_b.png)
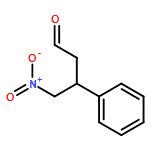

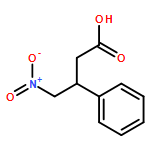
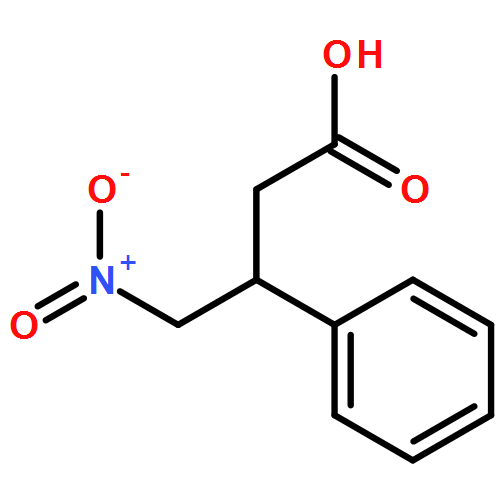


![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-2-phenylglycyl-, methyl ester](/data/chemimg/3739800/535993-38-5.png)
![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-2-phenylglycyl-, methyl ester](/data/chemimg/3739800/535993-38-5_b.png)
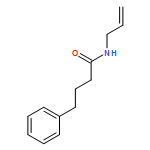
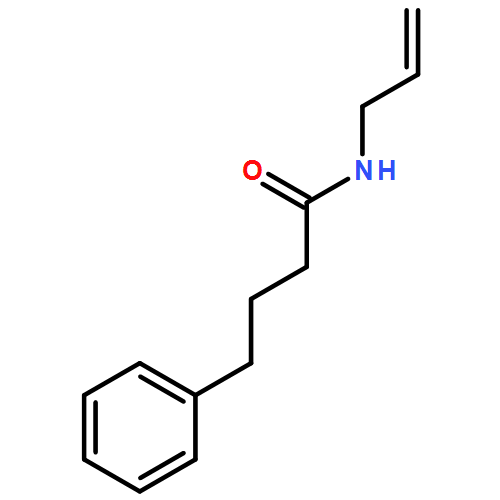
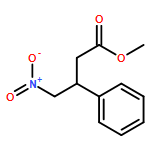
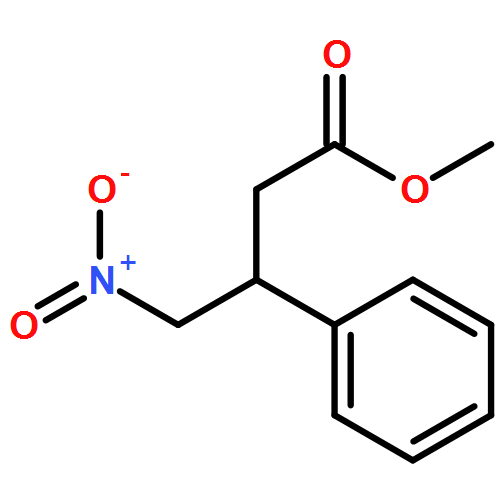
![2-Cyclopenten-1-one, 4-(acetyloxy)-5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-iodo-, (4R,5S)-](/data/chemimg/3739800/361482-73-7.png)
![2-Cyclopenten-1-one, 4-(acetyloxy)-5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-iodo-, (4R,5S)-](/data/chemimg/3739800/361482-73-7_b.png)
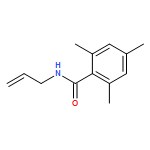
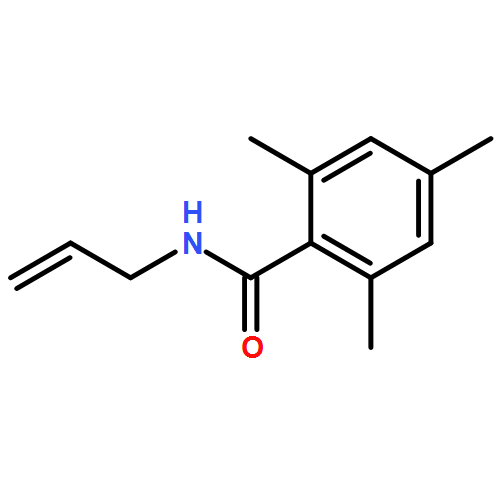
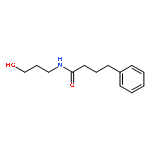
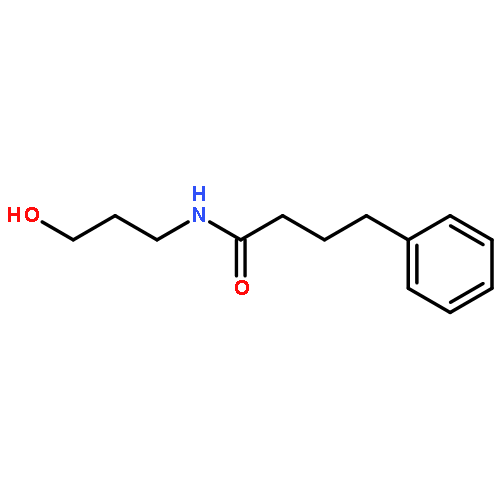
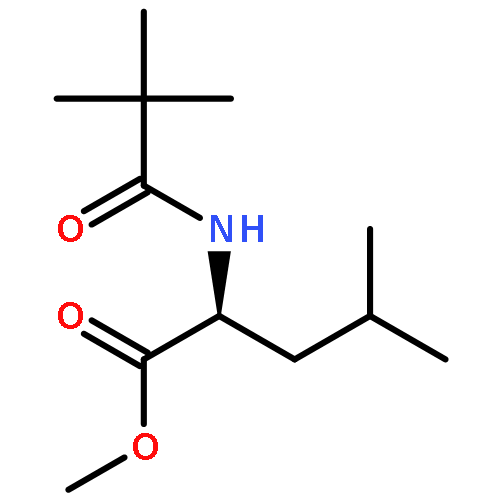
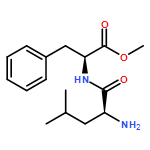
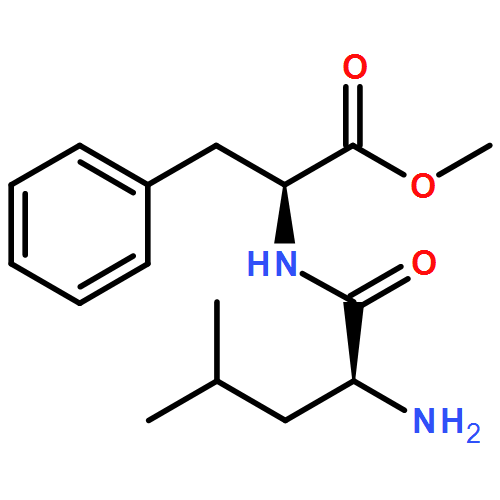
![L-Leucine, (2R)-N-[(1,1-dimethylethoxy)carbonyl]-2-phenylglycyl-, methyl ester](/data/chemimg/3739800/232929-02-1.png)
![L-Leucine, (2R)-N-[(1,1-dimethylethoxy)carbonyl]-2-phenylglycyl-, methyl ester](/data/chemimg/3739800/232929-02-1_b.png)


![Cyclopropanecarboxaldehyde, 2-[(phenylmethoxy)methyl]-, cis- (9CI)](/data/chemimg/3739800/145013-14-5.png)
![Cyclopropanecarboxaldehyde, 2-[(phenylmethoxy)methyl]-, cis- (9CI)](/data/chemimg/3739800/145013-14-5_b.png)
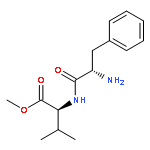
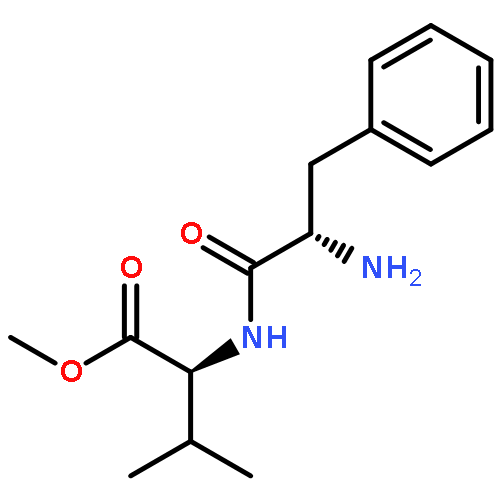
![L-Phenylalanine, N-[N-(2,2-dimethyl-1-oxopropyl)-L-valyl]-, methyl ester (9CI)](/data/chemimg/3739800/133221-39-3.png)
![L-Phenylalanine, N-[N-(2,2-dimethyl-1-oxopropyl)-L-valyl]-, methyl ester (9CI)](/data/chemimg/3739800/133221-39-3_b.png)


![Acetic acid, methoxy-,(1S,2S)-2-[2-[[3-(1H-benzimidazol-2-yl)propyl]methylamino]ethyl]-6-fluoro-1,2,3,4-tetrahydro-1-(1-methylethyl)-2-naphthalenylester](http://img.cochemist.com/ccimg/116700/116644-53-2.png)
![Acetic acid, methoxy-,(1S,2S)-2-[2-[[3-(1H-benzimidazol-2-yl)propyl]methylamino]ethyl]-6-fluoro-1,2,3,4-tetrahydro-1-(1-methylethyl)-2-naphthalenylester](http://img.cochemist.com/ccimg/116700/116644-53-2_b.png)
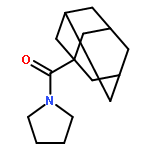
![Tricyclo[3.3.1.13,7]decane-1-carboxamide, N-methyl-N-(phenylmethyl)-](http://img.cochemist.com/ccimg/113500/113435-65-7.png)
![Tricyclo[3.3.1.13,7]decane-1-carboxamide, N-methyl-N-(phenylmethyl)-](http://img.cochemist.com/ccimg/113500/113435-65-7_b.png)
![L-Serine, O-[(1,1-dimethylethyl)dimethylsilyl]-, methyl ester](http://img.cochemist.com/ccimg/98700/98642-61-6.png)
![L-Serine, O-[(1,1-dimethylethyl)dimethylsilyl]-, methyl ester](http://img.cochemist.com/ccimg/98700/98642-61-6_b.png)
![L-Phenylalanine, (2R)-N-[(1,1-dimethylethoxy)carbonyl]-2-phenylglycyl-, methyl ester](/data/chemimg/3739800/94778-64-0.png)
![L-Phenylalanine, (2R)-N-[(1,1-dimethylethoxy)carbonyl]-2-phenylglycyl-, methyl ester](/data/chemimg/3739800/94778-64-0_b.png)
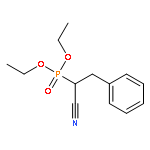
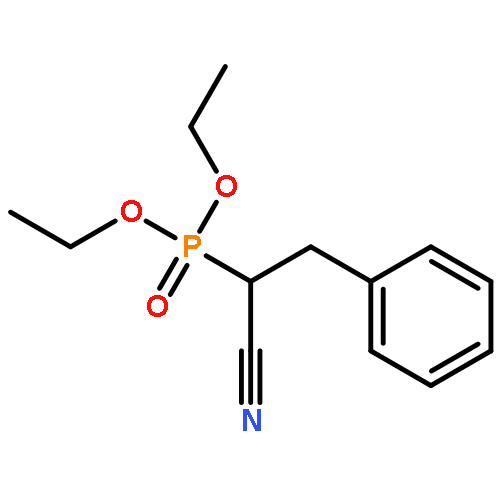
![Benzenepropanenitrile, α-[[(4-methylphenyl)sulfonyl]oxy]-](/data/chemimg/3739800/92963-73-0.png)
![Benzenepropanenitrile, α-[[(4-methylphenyl)sulfonyl]oxy]-](/data/chemimg/3739800/92963-73-0_b.png)
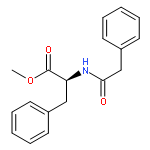
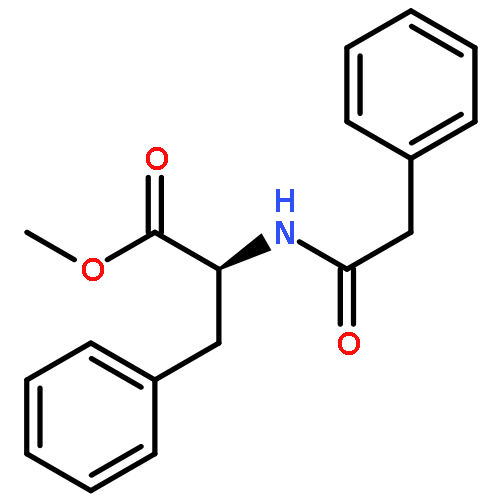


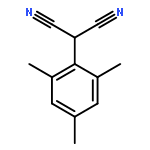
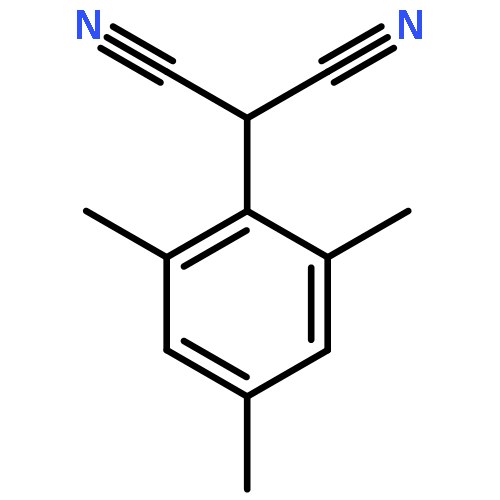
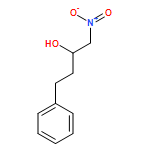
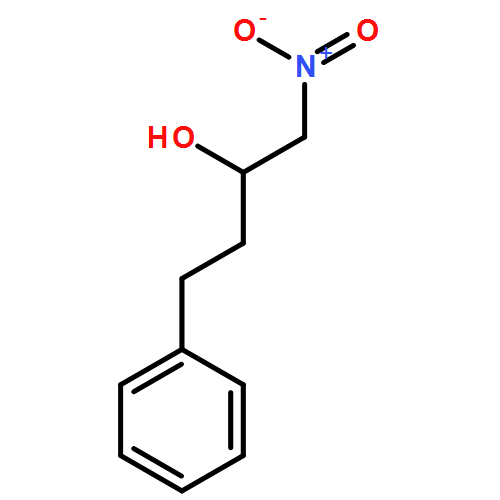


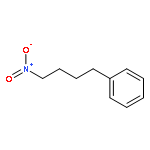
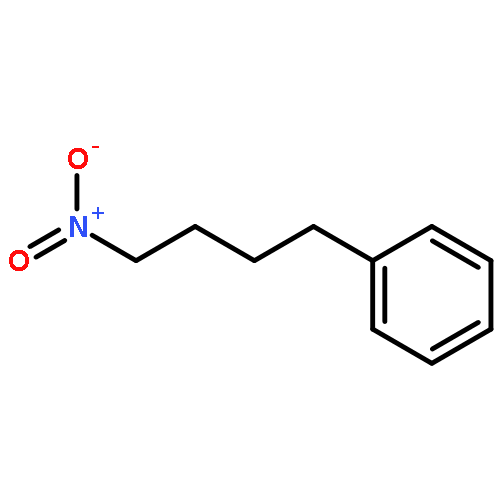



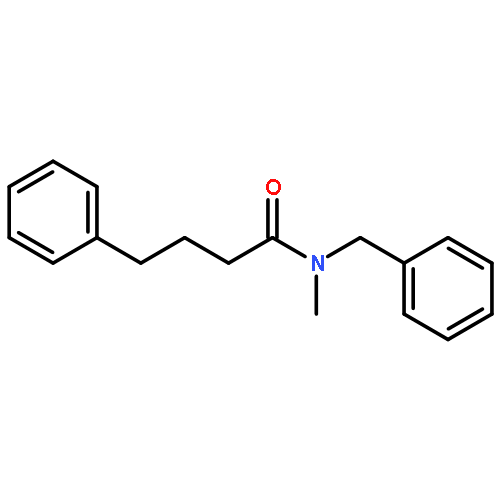
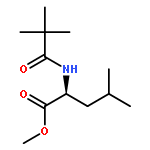
![Benzene, [(1-nitro-2-phenylethyl)sulfonyl]-](http://img.cochemist.com/ccimg/74800/74737-94-3.png)
![Benzene, [(1-nitro-2-phenylethyl)sulfonyl]-](http://img.cochemist.com/ccimg/74800/74737-94-3_b.png)
![L-Phenylalanine, N-(tricyclo[3.3.1.13,7]dec-1-ylcarbonyl)-, methyl ester](/data/chemimg/3739800/74010-50-7.png)
![L-Phenylalanine, N-(tricyclo[3.3.1.13,7]dec-1-ylcarbonyl)-, methyl ester](/data/chemimg/3739800/74010-50-7_b.png)



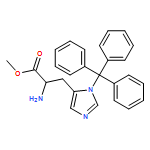
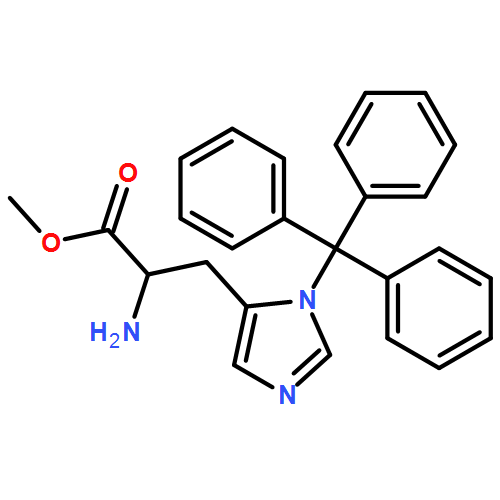
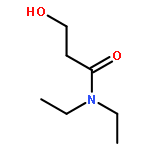

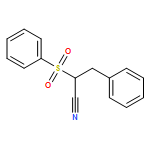
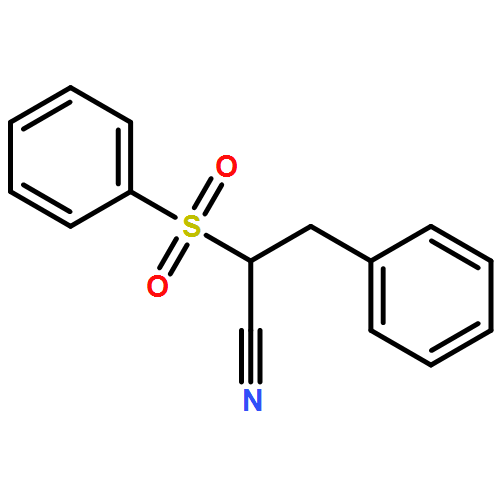


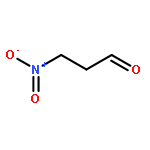


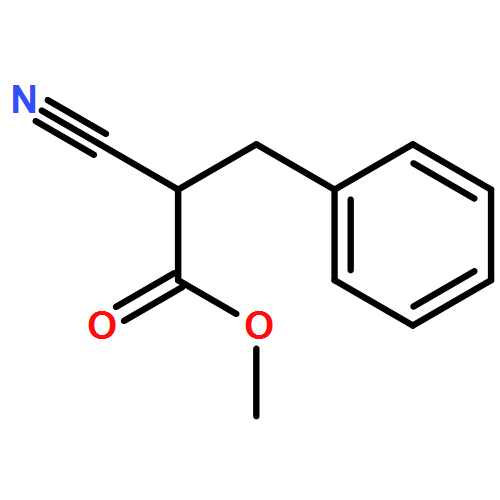
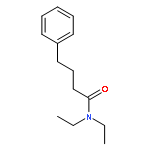










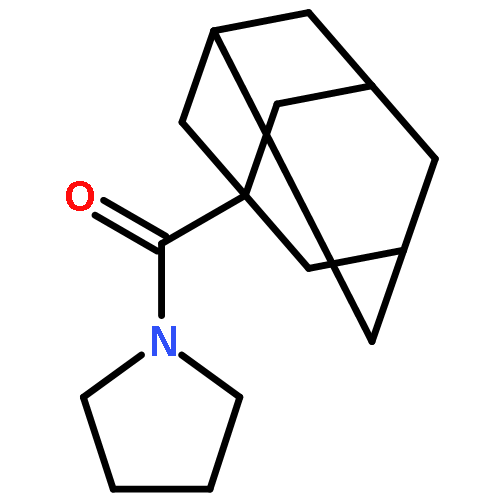
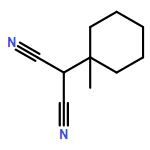
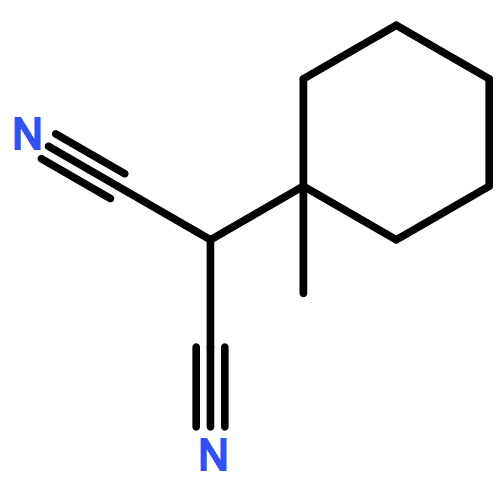
![Propanedinitrile, 2-tricyclo[3.3.1.13,7]dec-1-yl-](/data/chemimg/3739800/21982-89-8.png)
![Propanedinitrile, 2-tricyclo[3.3.1.13,7]dec-1-yl-](/data/chemimg/3739800/21982-89-8_b.png)
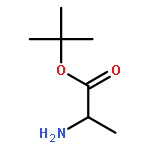
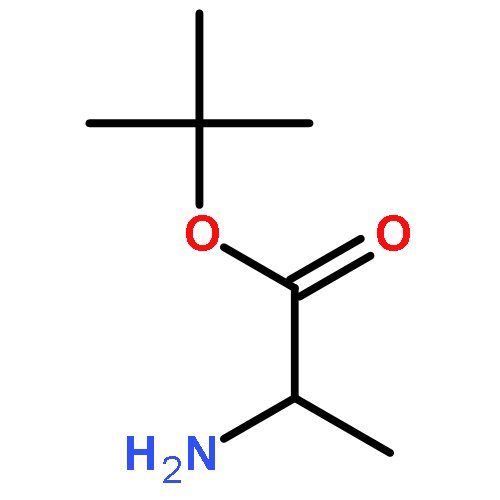








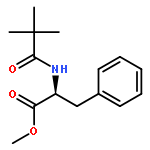


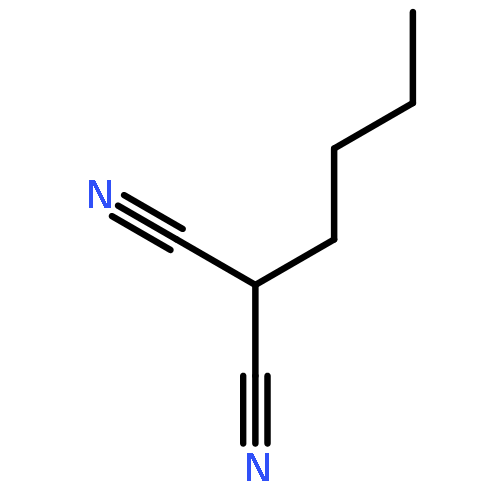
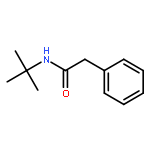
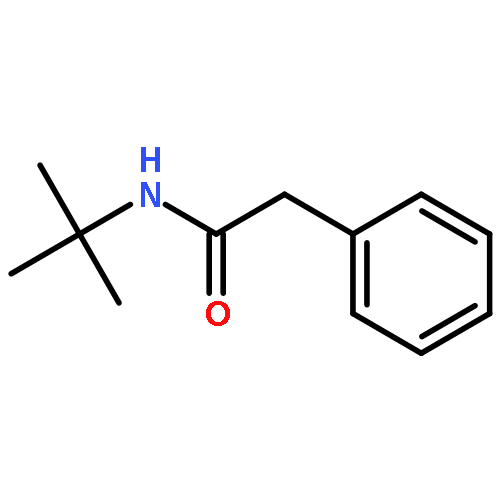




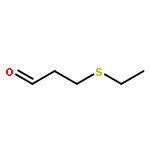
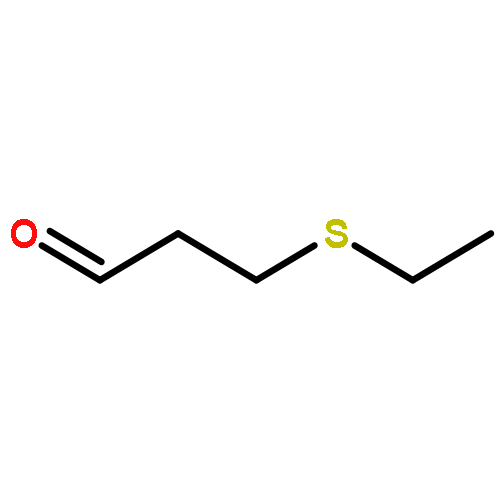


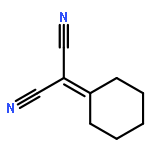
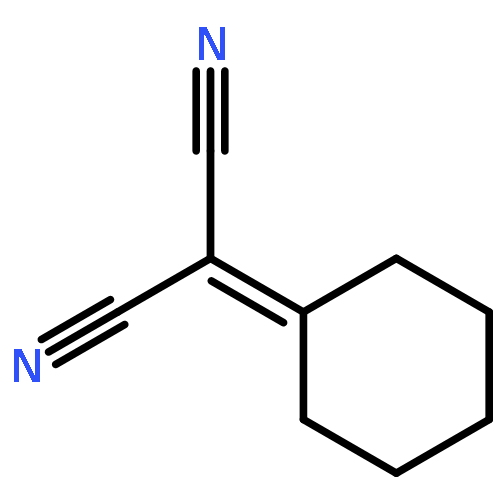
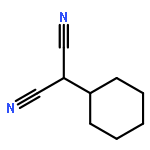
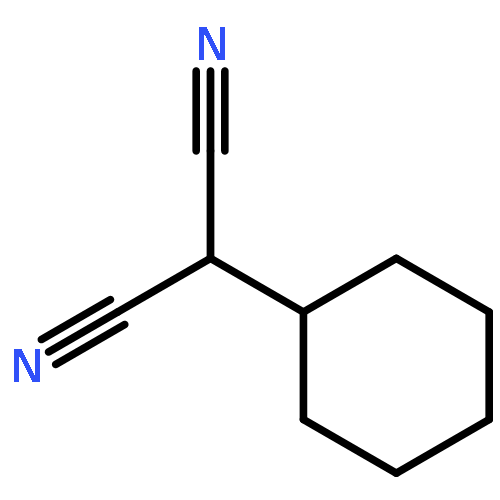
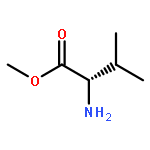
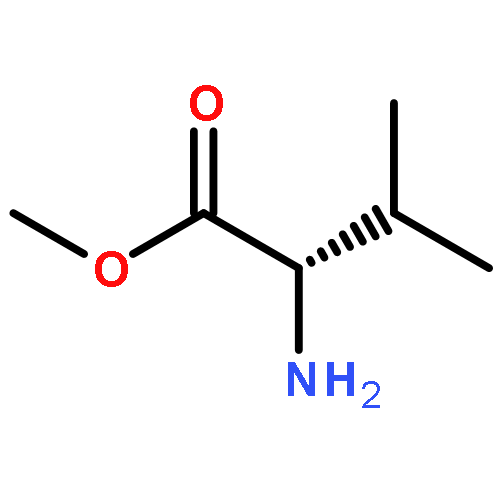
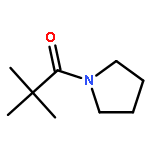
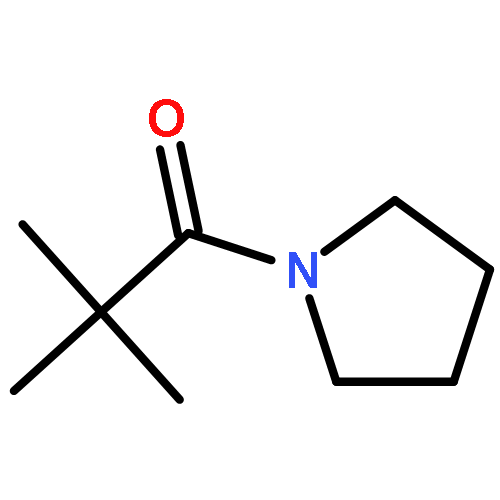




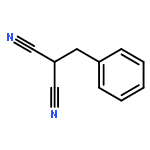

![1,3-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea](/data/chemimg/315700/1060-92-0.png)
![1,3-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea](/data/chemimg/315700/1060-92-0_b.png)




![Cyclopropanecarboxamide, 2-[(phenylmethoxy)methyl]-N-2-propen-1-yl-, (1R,2R)-rel-](/data/chemimg/3739800/1885848-44-1.png)
![Cyclopropanecarboxamide, 2-[(phenylmethoxy)methyl]-N-2-propen-1-yl-, (1R,2R)-rel-](/data/chemimg/3739800/1885848-44-1_b.png)
![Cyclopropanecarboxamide, 2-[(phenylmethoxy)methyl]-N-2-propen-1-yl-, (1R,2S)-rel-](/data/chemimg/3739800/1885848-43-0.png)
![Cyclopropanecarboxamide, 2-[(phenylmethoxy)methyl]-N-2-propen-1-yl-, (1R,2S)-rel-](/data/chemimg/3739800/1885848-43-0_b.png)
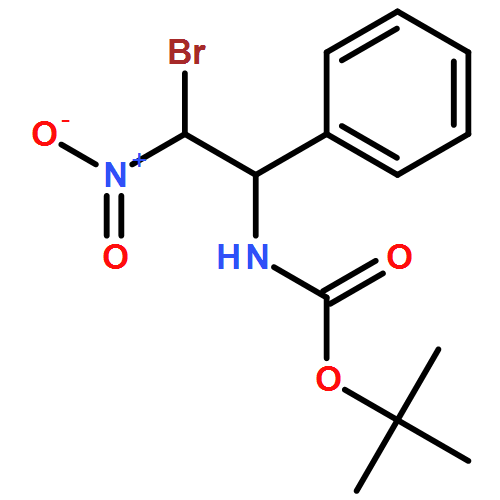
![Carbamic acid, N-[(1R,2S)-2-bromo-2-nitro-1-phenylethyl]-, 1,1-dimethylethyl ester, rel-](/data/chemimg/3739800/1809651-56-6.png)
![Carbamic acid, N-[(1R,2S)-2-bromo-2-nitro-1-phenylethyl]-, 1,1-dimethylethyl ester, rel-](/data/chemimg/3739800/1809651-56-6_b.png)
![Carbamic acid, N-[(1R)-2-iodo-2-nitro-1-phenylethyl]-, 1,1-dimethylethyl ester](/data/chemimg/3739800/1809651-53-3.png)
![Carbamic acid, N-[(1R)-2-iodo-2-nitro-1-phenylethyl]-, 1,1-dimethylethyl ester](/data/chemimg/3739800/1809651-53-3_b.png)
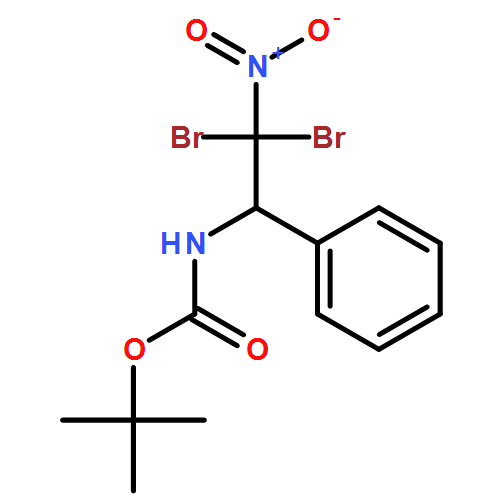
![Carbamic acid, N-[(1R,2R)-2-bromo-2-nitro-1-phenylethyl]-, 1,1-dimethylethyl ester, rel-](/data/chemimg/3739800/1809651-47-5.png)
![Carbamic acid, N-[(1R,2R)-2-bromo-2-nitro-1-phenylethyl]-, 1,1-dimethylethyl ester, rel-](/data/chemimg/3739800/1809651-47-5_b.png)
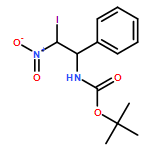
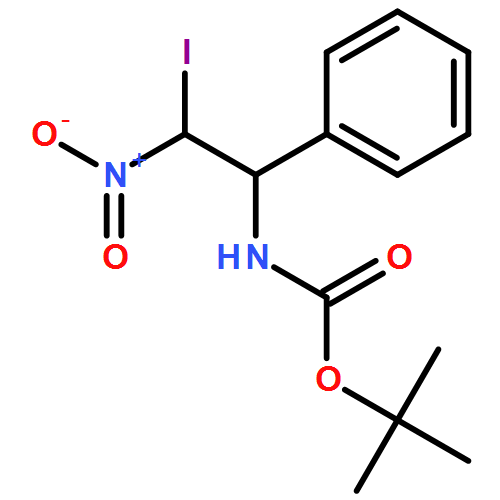
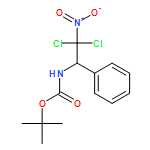
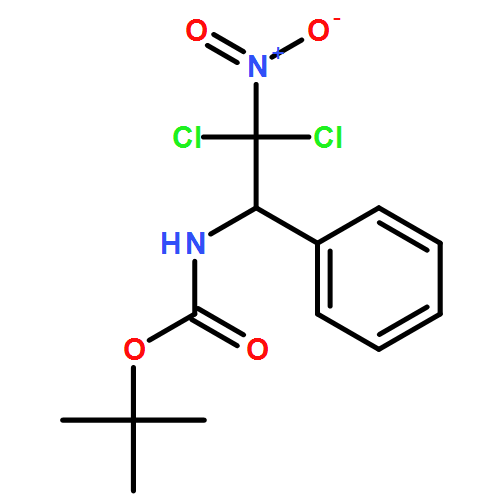
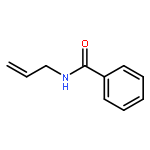
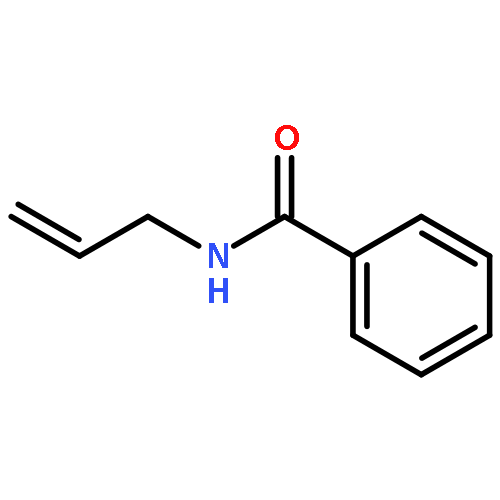
![Carbamic acid, N-[2-oxo-1-phenyl-2-(2-propen-1-ylamino)ethyl]-, 1,1-dimethylethyl ester](/data/chemimg/3739700/1323094-42-3.png)
![Carbamic acid, N-[2-oxo-1-phenyl-2-(2-propen-1-ylamino)ethyl]-, 1,1-dimethylethyl ester](/data/chemimg/3739700/1323094-42-3_b.png)
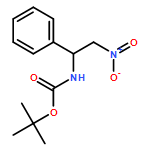
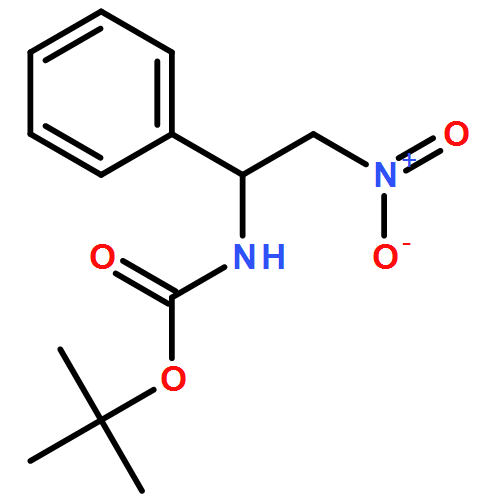
![Carbamic acid, [(1R)-2-nitro-1-phenylethyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/685200/685132-79-0.png)
![Carbamic acid, [(1R)-2-nitro-1-phenylethyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/685200/685132-79-0_b.png)