Co-reporter:Xiaofei Xin;Xue Pei;Xin Yang;Yaqi Lv;Li Zhang;Lifang Yin
Advanced Science 2017 Volume 4(Issue 11) pp:
Publication Date(Web):2017/11/01
DOI:10.1002/advs.201700324
AbstractEfficient microRNAs (miRNA) delivery into cells is a promising strategy for disease therapy, but is a major challenge because the available conventional nonviral vectors have significant drawbacks. In particular, after these vectors are entrapped in lysosomes, the escape efficiency of genes from lysosomes into the cytosol is less than 2%. Here, a novel approach for lethal-7a (let-7a) replacement therapy using rod-shaped active pure drug nanoparticles (≈130 nm in length, PNPs) with a dramatically high drug-loading of ≈300% as vectors is reported. Importantly, unlike other vectors, the developed PNPs/let-7a complexes (≈178 nm, CNPs) can enter cells and bypass the lysosomal route to localize to the cytosol, achieving efficient intracellular delivery of let-7a and a 50% reduction in expression of the target protein (KRAS). Also, CNPs prolong the t1/2 of blood circulation by ≈threefold and increase tumor accumulation by ≈1.5–2-fold, resulting in significantly improved antitumor efficacies. Additionally, no damage to normal organs is observed following systemic injection of CNPs. In conclusion, rod-shaped active PNPs enable efficient and safe delivery of miRNA with synergistic treatment for disease. This nanoplatform would also offer a viable strategy for the potent delivery of proteins and peptides in vitro and in vivo.
Co-reporter:Yongji Li; Zhannan Wu; Wei He; Chao Qin; Jing Yao; Jianping Zhou;Lifang Yin
Molecular Pharmaceutics 2015 Volume 12(Issue 5) pp:1485-1500
Publication Date(Web):March 23, 2015
DOI:10.1021/mp5008037
About 40% of the marketed drugs and 70–90% of new drug candidates are insoluble in water and therefore poorly bioavailable, which significantly compromises their therapeutic effects. A formulation of nanosuspensions achieved by reducing the pure drug particle size down to seb-micron range is one of the most promising approaches to overcome the insolubility. However, the nanosuspension formulations are subject to instability because of nucleation and particle growth. Therefore, a stabilizer is needed to be incorporated into the nanosuspension formulation during the preparation process to suppress the aggregation of drug particles. β-LG, a globular protein, is broken by heat-induced denaturation, and its hydrophobic area is exposed, which allows it to associate with organic particles. PTX, an insoluble drug, is widely used for the clinical treatment of human cancer. However, this drug’s clinical application is greatly limited by intrinsic defects including poor solubility, adverse side effects, and poor tumor penetration. In this study, we prepared β-LG-stabilized PTX nanosuspensions (PTX-NS) by coating the protein onto nanoscaled drug particles, investigating the stabilization effect of β-LG on PTX-NS, and evaluating its in vitro and in vivo performance. PTX-NS with a diameter of approximately 200 nm was easily prepared. β-LG produced significantly stabilized effect on PTX-NS via the interaction between the hydrophobic area of the protein and the hydrophobic surface of the drug particles, which resulted in a conformational change of the protein, the loss of both secondary and tertiary structures, and the transition of Trp residues to a less hydrophobic condition. Importantly, unlike other conventional nanoparticles, PTX-NS could directly translocated across the membrane into the cytosol in an energy-independent manner, without entrapment within the endosomal–lysosomal system. Moreover, compared with Taxol, PTX-NS increased AUC and Cmax by 26- and 16-fold, respectively, and prolonged T1/2 by 314-fold. As expected, PTX-NS had better in vitro and in vivo antitumor activity compared to PTX alone. Additionally, β-LG is cyto- and bio-compatible, and PTX-NS is not toxic to healthy tissues. In conclusion, the present study has suggested the high potency of globular proteins, such as β-LG, as novel biomaterials for nanosuspension platform to improve the drug delivery for disease treatment.
Co-reporter:Lan Wu;Yanli Qiao;Lina Wang;Jiahua Guo;Guocheng Wang
AAPS PharmSciTech 2015 Volume 16( Issue 5) pp:1051-1058
Publication Date(Web):2015 October
DOI:10.1208/s12249-014-0280-y
AJS is the code name of an untitled novel medicative compound synthesized by the Tasly Holding Group Company (Tianjin, China) based on the structure of cinnamamide, which is one of the Biopharmaceutics Classification System (BCS) class II drugs. The drug has better antidepressant effect, achieved by acting on the 5-hydroxytryptamine receptor. However, the therapeutic effects of the drug are compromised due to its poor water solubility and lower bioavailability. Herein, a self-microemulsifying drug delivery system (SMEDDS) was developed to improve its solubility and oral bioavailability. AJS-SMEDDS formulation was optimized in terms of drug solubility in the excipients, droplet size, stability, and drug precipitation using a pseudo-ternary diagram. The pharmacokinetic study was performed in rats, and the drug concentration in plasma samples was assayed using the high-performance liquid chromatography-electrospray tandem mass spectrometry (HPLC-MS/MS) method. The optimized formulation for SMEDDS has a composition of castor oil 24.5%, Labrasol 28.6%, Cremphor EL 40.8%, and Transcutol HP 2.7% (co-surfactant). No drug precipitation or phase separation was observed from the optimized formulation after 3 months of storing at 25°C. The droplet size of microemulsion formed by the optimized formulation was 26.08 ± 1.68 nm, and the zeta potential was −2.76 mV. The oral bioavailability of AJS-SMEDDS was increased by 3.4- and 35.9-fold, respectively, compared with the solid dispersion and cyclodextrin inclusion; meanwhile, the Cmax of AJS-SMEDDS was about 2- and 40-fold as great as the two controls, respectively. In summary, the present SMEDDS enhanced oral bioavailability of AJS and was a promising strategy to orally deliver the drug.
Co-reporter:Li Zhang, Qingqing Xiao, Yiran Wang, Chenshuang Zhang, Wei He, Lifang Yin
International Journal of Pharmaceutics (15 May 2017) Volume 523(Issue 1) pp:1-14
Publication Date(Web):15 May 2017
DOI:10.1016/j.ijpharm.2017.03.026
Many lead compounds have a low solubility in water, which substantially hinders their clinical application. Nanosuspensions have been considered a promising strategy for the delivery of water-insoluble drugs. Here, denatured soy protein isolate (SPI)-coated docetaxel nanosuspensions (DTX-NS) were developed using an anti-solvent precipitation-ultrasonication method to improve the water-solubility of DTX, thus improving its intracellular delivery. DTX-NS, with a diameter of 150–250 nm and drug-loading up to 18.18%, were successfully prepared by coating drug particles with SPI. Interestingly, the drug state of DTX-NS was alterable. Amorphous drug nanoparticles were obtained at low drug-loading, whereas at a high drug-loading, the DTX-NS drug was mainly present in the crystalline state. Moreover, DTX-NS could be internalized at high levels by cancer cells and enter the cytosol by lysosomal escape, enhancing cell cytotoxicity and apoptosis compared with free DTX. Taken together, denatured SPI has a strong stabilization effect on nanosuspensions, and the drug state in SPI-coated nanosuspensions is alterable by changing the drug-loading. Moreover, DTX-NS could achieve cytosolic delivery, generating enhanced cell cytotoxicity against cancer cells.Download high-res image (200KB)Download full-size image

![2H-Naphth[1,2-b]-1,4-oxazin-9-ol,3,4,4a,5,6,10b-hexahydro-4-propyl-, hydrochloride (1:1), (4aR,10bR)-](http://img.cochemist.com/ccimg/99800/99705-65-4.png)
![2H-Naphth[1,2-b]-1,4-oxazin-9-ol,3,4,4a,5,6,10b-hexahydro-4-propyl-, hydrochloride (1:1), (4aR,10bR)-](http://img.cochemist.com/ccimg/99800/99705-65-4_b.png)


![(1aS,1bR,3S,4aS,6R,7aR,7bR,8R,9aR)-1,1,3,6,8-pentamethyltetradecahydro-1H-cyclopropa[3,4]benzo[1,2-e]azulene](http://img.cochemist.com/ccimg/67800/67707-87-3.png)
![(1aS,1bR,3S,4aS,6R,7aR,7bR,8R,9aR)-1,1,3,6,8-pentamethyltetradecahydro-1H-cyclopropa[3,4]benzo[1,2-e]azulene](http://img.cochemist.com/ccimg/67800/67707-87-3_b.png)
![Technetate(1-)-99Tc,[N,N-bis[2-[bis(carboxymethyl)amino]ethyl]glycinato(5-)]-, sodium (9CI)](http://img.cochemist.com/ccimg/65500/65454-61-7.png)
![Technetate(1-)-99Tc,[N,N-bis[2-[bis(carboxymethyl)amino]ethyl]glycinato(5-)]-, sodium (9CI)](http://img.cochemist.com/ccimg/65500/65454-61-7_b.png)
![2-(Hydroxymethyl)-4-{(1R)-1-hydroxy-2-[(2-methyl-2-propanyl)amino ]ethyl}phenol hydrochloride (1:1)](http://img.cochemist.com/ccimg/50300/50293-90-8.png)
![2-(Hydroxymethyl)-4-{(1R)-1-hydroxy-2-[(2-methyl-2-propanyl)amino ]ethyl}phenol hydrochloride (1:1)](http://img.cochemist.com/ccimg/50300/50293-90-8_b.png)
![5H-Cyclopropa[3,4]benz[1,2-e]azulen-5-one,1,1a,1b,4,4a,7a,7b,8,9,9a-decahydro-4a,7b,9,9a-tetrahydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-,(1aR,1bS,4aR,7aS,7bS,8R,9R,9aS)-](http://img.cochemist.com/ccimg/17700/17673-25-5.png)
![5H-Cyclopropa[3,4]benz[1,2-e]azulen-5-one,1,1a,1b,4,4a,7a,7b,8,9,9a-decahydro-4a,7b,9,9a-tetrahydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-,(1aR,1bS,4aR,7aS,7bS,8R,9R,9aS)-](http://img.cochemist.com/ccimg/17700/17673-25-5_b.png)
![Tungstate(3-),tetracosa-m-oxododecaoxo[m12-[phosphato(3-)-kO:kO:kO:kO':kO':kO':kO'':kO'':kO'':kO''':kO''':kO''']]dodeca-,hydrogen (1:3)](http://img.cochemist.com/ccimg/1400/1343-93-7.png)
![Tungstate(3-),tetracosa-m-oxododecaoxo[m12-[phosphato(3-)-kO:kO:kO:kO':kO':kO':kO'':kO'':kO'':kO''':kO''':kO''']]dodeca-,hydrogen (1:3)](http://img.cochemist.com/ccimg/1400/1343-93-7_b.png)
![5-Heptenoic acid,7-[(2R,3S,4S)-tetrahydro-4,6-dihydroxy-2-[(1E,3S)-3-hydroxy-1-octen-1-yl]-2H-pyran-3-yl]-,(5Z)-](http://img.cochemist.com/ccimg/54400/54397-85-2.png)
![5-Heptenoic acid,7-[(2R,3S,4S)-tetrahydro-4,6-dihydroxy-2-[(1E,3S)-3-hydroxy-1-octen-1-yl]-2H-pyran-3-yl]-,(5Z)-](http://img.cochemist.com/ccimg/54400/54397-85-2_b.png)


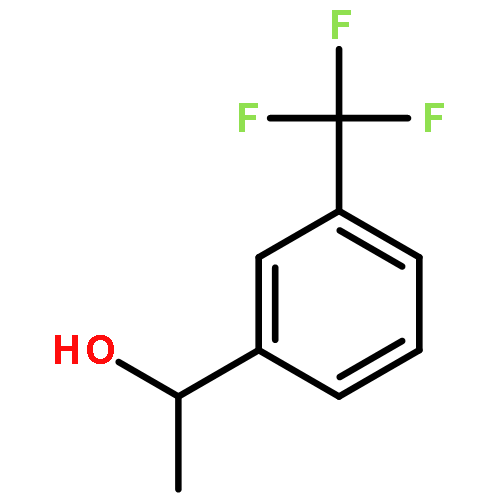
![Propanedioic acid, [(1R,2E)-1,3-diphenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/96500/96482-67-6.png)
![Propanedioic acid, [(1R,2E)-1,3-diphenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/96500/96482-67-6_b.png)
![Propanedioic acid, [(1S,2E)-1,3-diphenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/96500/96482-64-3.png)
![Propanedioic acid, [(1S,2E)-1,3-diphenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/96500/96482-64-3_b.png)




![Formamide, N-[2-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/91600/91533-85-6.png)
![Formamide, N-[2-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/91600/91533-85-6_b.png)
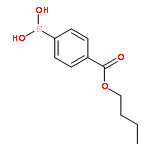







![METHANONE, [(2R,3S)-3-(4-METHOXYPHENYL)OXIRANYL]PHENYL-](http://img.cochemist.com/ccimg/82900/82864-16-2.png)
![METHANONE, [(2R,3S)-3-(4-METHOXYPHENYL)OXIRANYL]PHENYL-](http://img.cochemist.com/ccimg/82900/82864-16-2_b.png)
![2-Propenoyl chloride, 3-[3,4-bis(acetyloxy)phenyl]-](http://img.cochemist.com/ccimg/82600/82592-68-5.png)
![2-Propenoyl chloride, 3-[3,4-bis(acetyloxy)phenyl]-](http://img.cochemist.com/ccimg/82600/82592-68-5_b.png)
![Benzene, 1-methoxy-4-[(2-phenylethoxy)methyl]-](http://img.cochemist.com/ccimg/82600/82529-79-1.png)
![Benzene, 1-methoxy-4-[(2-phenylethoxy)methyl]-](http://img.cochemist.com/ccimg/82600/82529-79-1_b.png)
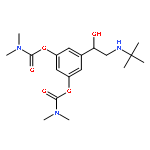
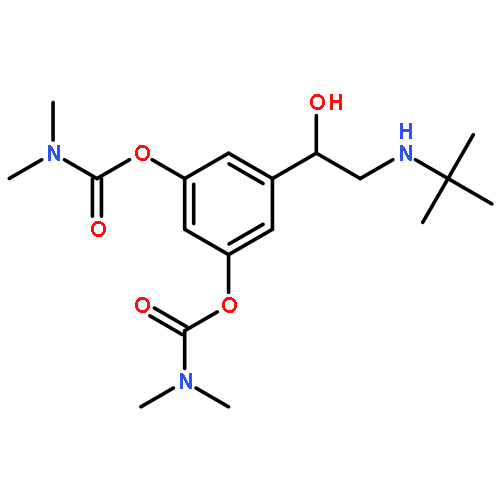
![Bicyclo[4.2.0]octa-1,3,5-trien-7-one, 5-chloro-](http://img.cochemist.com/ccimg/81500/81447-63-4.png)
![Bicyclo[4.2.0]octa-1,3,5-trien-7-one, 5-chloro-](http://img.cochemist.com/ccimg/81500/81447-63-4_b.png)
![BICYCLO[4.2.0]OCTA-1,3,5-TRIEN-7-ONE, 4,5-DIMETHOXY-](http://img.cochemist.com/ccimg/81500/81447-58-7.png)
![BICYCLO[4.2.0]OCTA-1,3,5-TRIEN-7-ONE, 4,5-DIMETHOXY-](http://img.cochemist.com/ccimg/81500/81447-58-7_b.png)












![Threonine, N-[[5-(dimethylamino)-1-naphthalenyl]sulfonyl]-](http://img.cochemist.com/ccimg/77500/77481-11-9.png)
![Threonine, N-[[5-(dimethylamino)-1-naphthalenyl]sulfonyl]-](http://img.cochemist.com/ccimg/77500/77481-11-9_b.png)
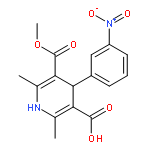



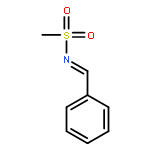
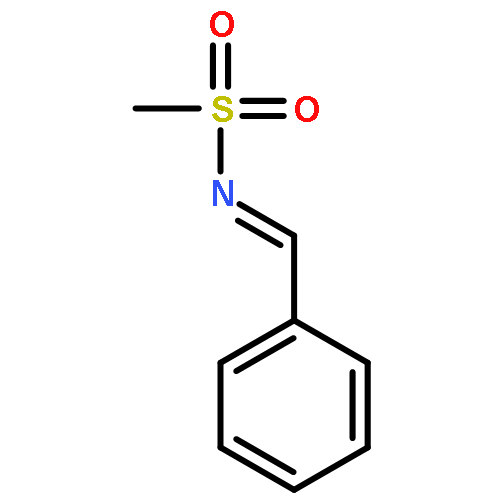
![Benzenesulfonamide, 4-methyl-N-[(2-methylphenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/73900/73845-01-9.png)
![Benzenesulfonamide, 4-methyl-N-[(2-methylphenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/73900/73845-01-9_b.png)









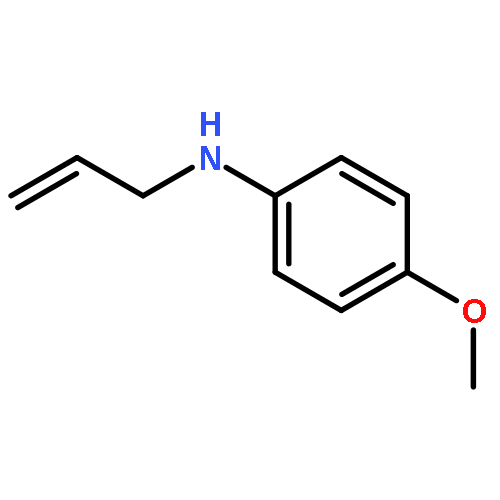
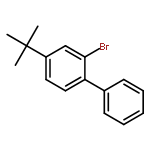
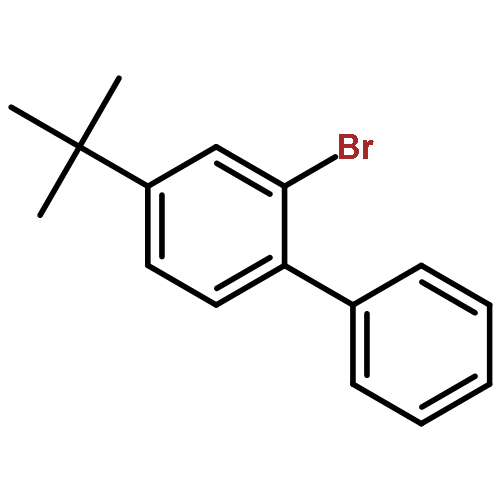






![Bicyclo[4.2.0]octa-1,3,5-trien-7-one, 5-methoxy-](http://img.cochemist.com/ccimg/67000/66947-60-2.png)
![Bicyclo[4.2.0]octa-1,3,5-trien-7-one, 5-methoxy-](http://img.cochemist.com/ccimg/67000/66947-60-2_b.png)
![Leucine, N-[[5-(dimethylamino)-1-naphthalenyl]sulfonyl]-](http://img.cochemist.com/ccimg/65500/65452-14-4.png)
![Leucine, N-[[5-(dimethylamino)-1-naphthalenyl]sulfonyl]-](http://img.cochemist.com/ccimg/65500/65452-14-4_b.png)
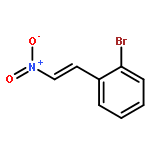
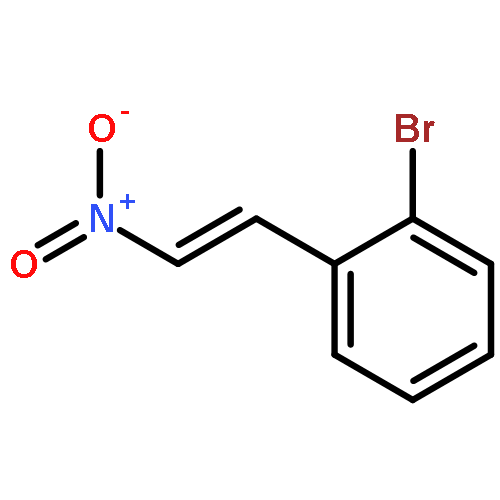










![2-Propen-1-one, 3-phenyl-1-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/62100/62056-10-4.png)
![2-Propen-1-one, 3-phenyl-1-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/62100/62056-10-4_b.png)

![Methanone, [(2R,3S)-3-(4-chlorophenyl)oxiranyl]phenyl-](http://img.cochemist.com/ccimg/61900/61840-96-8.png)
![Methanone, [(2R,3S)-3-(4-chlorophenyl)oxiranyl]phenyl-](http://img.cochemist.com/ccimg/61900/61840-96-8_b.png)
![Methanone, phenyl[(2R,3S)-3-phenyloxiranyl]-](http://img.cochemist.com/ccimg/61900/61840-92-4.png)
![Methanone, phenyl[(2R,3S)-3-phenyloxiranyl]-](http://img.cochemist.com/ccimg/61900/61840-92-4_b.png)
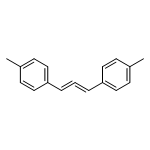
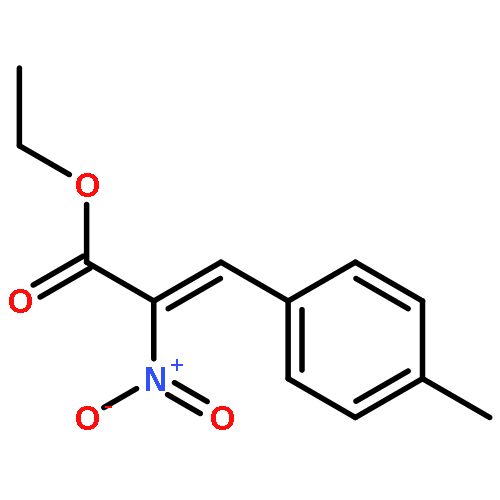
![2-Propen-1-one, 1-phenyl-3-[4-(trifluoromethyl)phenyl]-](/data/chemimg/1516700/61637-11-4.png)
![2-Propen-1-one, 1-phenyl-3-[4-(trifluoromethyl)phenyl]-](/data/chemimg/1516700/61637-11-4_b.png)
![6A-[(2-aminoethyl)amino]-6A-deoxy- beta-Cyclodextrin](http://img.cochemist.com/ccimg/61000/60984-63-6.png)
![6A-[(2-aminoethyl)amino]-6A-deoxy- beta-Cyclodextrin](http://img.cochemist.com/ccimg/61000/60984-63-6_b.png)



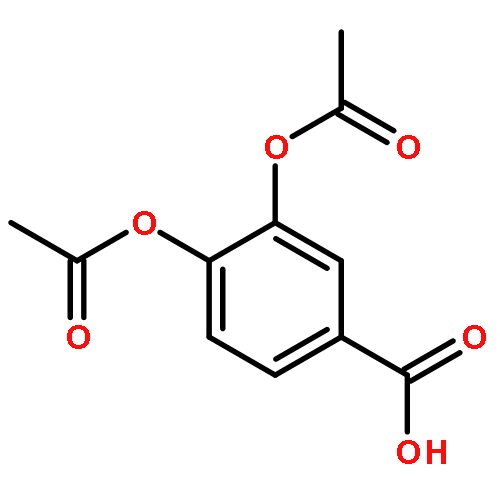


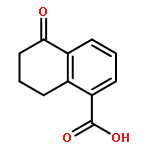
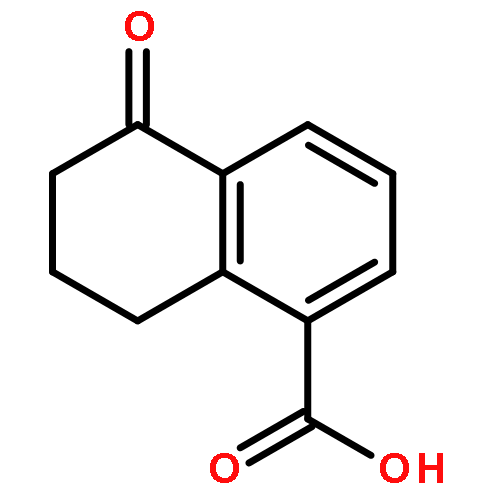





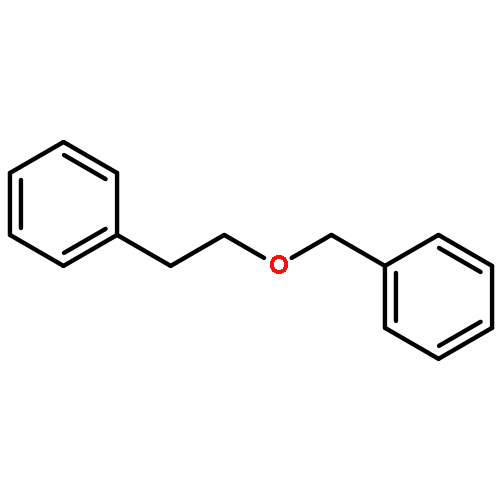
![2-PROPENOYL CHLORIDE, 3-[4-(ACETYLOXY)PHENYL]-](http://img.cochemist.com/ccimg/54000/53901-95-4.png)
![2-PROPENOYL CHLORIDE, 3-[4-(ACETYLOXY)PHENYL]-](http://img.cochemist.com/ccimg/54000/53901-95-4_b.png)
![2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]benzoic acid](http://img.cochemist.com/ccimg/53700/53663-18-6.png)
![2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]benzoic acid](http://img.cochemist.com/ccimg/53700/53663-18-6_b.png)
![2-Propenoyl chloride, 3-[4-(acetyloxy)-3-methoxyphenyl]-](http://img.cochemist.com/ccimg/51000/50906-12-2.png)
![2-Propenoyl chloride, 3-[4-(acetyloxy)-3-methoxyphenyl]-](http://img.cochemist.com/ccimg/51000/50906-12-2_b.png)


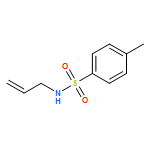

![Methionine, N-[[5-(dimethylamino)-1-naphthalenyl]sulfonyl]-](http://img.cochemist.com/ccimg/48300/48208-47-5.png)
![Methionine, N-[[5-(dimethylamino)-1-naphthalenyl]sulfonyl]-](http://img.cochemist.com/ccimg/48300/48208-47-5_b.png)
![Serine, N-[[5-(dimethylamino)-1-naphthalenyl]sulfonyl]-](http://img.cochemist.com/ccimg/48200/48196-47-0.png)
![Serine, N-[[5-(dimethylamino)-1-naphthalenyl]sulfonyl]-](http://img.cochemist.com/ccimg/48200/48196-47-0_b.png)
![Aspartic acid, N-[[5-(dimethylamino)-1-naphthalenyl]sulfonyl]-](http://img.cochemist.com/ccimg/42900/42808-07-1.png)
![Aspartic acid, N-[[5-(dimethylamino)-1-naphthalenyl]sulfonyl]-](http://img.cochemist.com/ccimg/42900/42808-07-1_b.png)










![Benzene, [(1E)-3-phenoxy-1-propenyl]-](http://img.cochemist.com/ccimg/37500/37464-41-8.png)
![Benzene, [(1E)-3-phenoxy-1-propenyl]-](http://img.cochemist.com/ccimg/37500/37464-41-8_b.png)
![2-METHYL-2-PROPANYL [1-(6-BROMO-2-PYRIDINYL)-3-PIPERIDINYL]CARBAM<WBR />ATE](http://img.cochemist.com/ccimg/37300/37220-82-9.png)
![2-METHYL-2-PROPANYL [1-(6-BROMO-2-PYRIDINYL)-3-PIPERIDINYL]CARBAM<WBR />ATE](http://img.cochemist.com/ccimg/37300/37220-82-9_b.png)






![4-[(1r)-2-(tert-butylamino)-1-hydroxyethyl]-2-(hydroxymethyl)phenol](http://img.cochemist.com/ccimg/34400/34391-04-3.png)
![4-[(1r)-2-(tert-butylamino)-1-hydroxyethyl]-2-(hydroxymethyl)phenol](http://img.cochemist.com/ccimg/34400/34391-04-3_b.png)
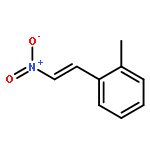
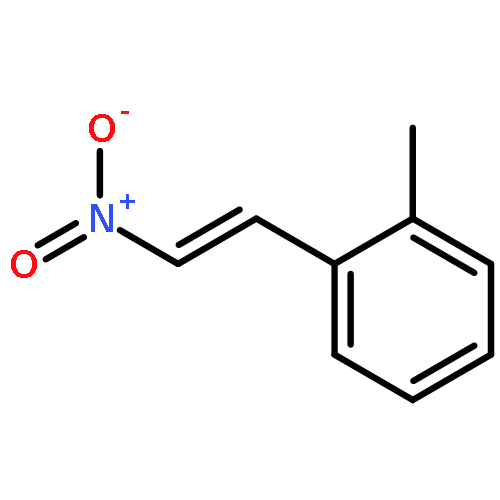
![3aH-Cyclopenta[3,4]cyclobuta[1,2]benzen-3a-ol, 1,2,3,7b-tetrahydro-](http://img.cochemist.com/ccimg/32800/32725-90-9.png)
![3aH-Cyclopenta[3,4]cyclobuta[1,2]benzen-3a-ol, 1,2,3,7b-tetrahydro-](http://img.cochemist.com/ccimg/32800/32725-90-9_b.png)






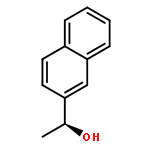

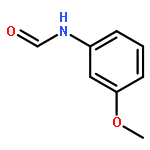





![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2.png)
![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2_b.png)
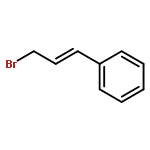
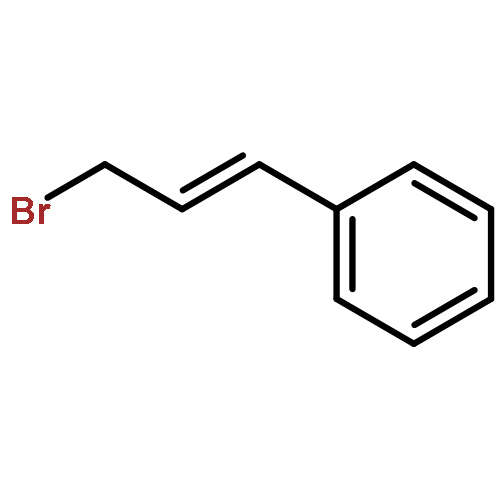
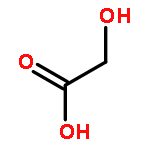
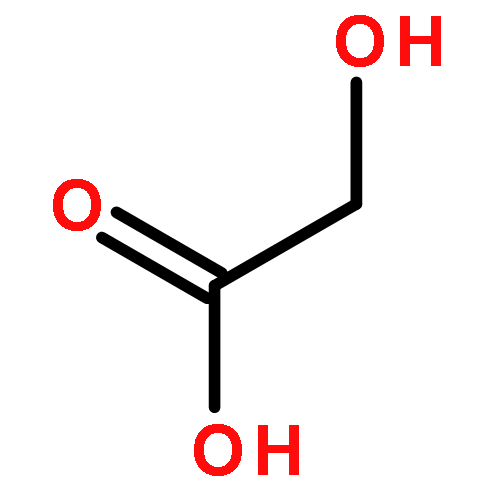



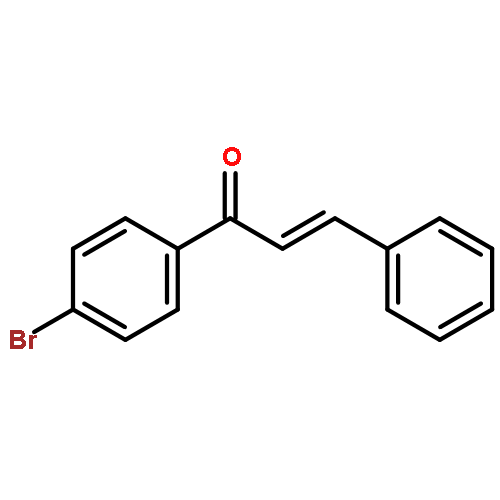
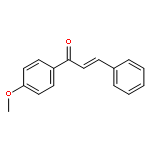
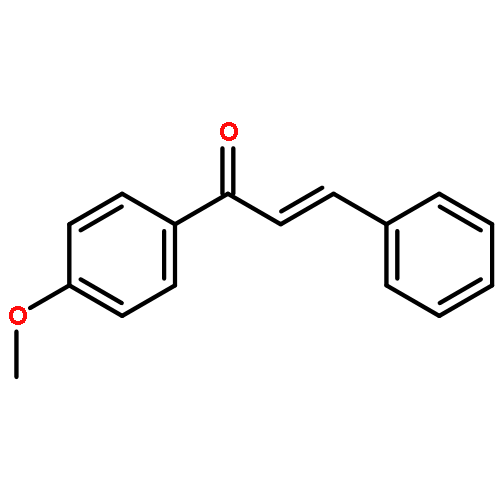
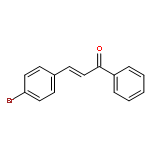
![Benzene, [(1E)-3-methoxy-1-propen-1-yl]-](/data/chemimg/1798100/22688-03-5.png)
![Benzene, [(1E)-3-methoxy-1-propen-1-yl]-](/data/chemimg/1798100/22688-03-5_b.png)



![Benzene, 1-methoxy-2-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/21300/21298-69-1.png)
![Benzene, 1-methoxy-2-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/21300/21298-69-1_b.png)
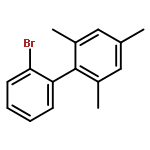


![Dibenzo[a,g]quinolizinium,5,6-dihydro-](http://img.cochemist.com/ccimg/19800/19716-69-9.png)
![Dibenzo[a,g]quinolizinium,5,6-dihydro-](http://img.cochemist.com/ccimg/19800/19716-69-9_b.png)
![Bicyclo[4.2.0]octa-1,3,5-trien-7-ol, 7-phenyl-](http://img.cochemist.com/ccimg/19200/19164-61-5.png)
![Bicyclo[4.2.0]octa-1,3,5-trien-7-ol, 7-phenyl-](http://img.cochemist.com/ccimg/19200/19164-61-5_b.png)
![Bicyclo[4.2.0]octa-1,3,5-trien-7-ol, 7-methyl-](http://img.cochemist.com/ccimg/19200/19164-60-4.png)
![Bicyclo[4.2.0]octa-1,3,5-trien-7-ol, 7-methyl-](http://img.cochemist.com/ccimg/19200/19164-60-4_b.png)

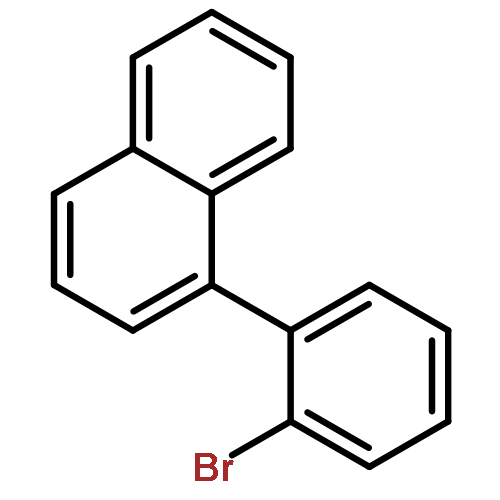


![Tryptophan, N-[[5-(dimethylamino)-1-naphthalenyl]sulfonyl]-](http://img.cochemist.com/ccimg/17100/17039-57-5.png)
![Tryptophan, N-[[5-(dimethylamino)-1-naphthalenyl]sulfonyl]-](http://img.cochemist.com/ccimg/17100/17039-57-5_b.png)




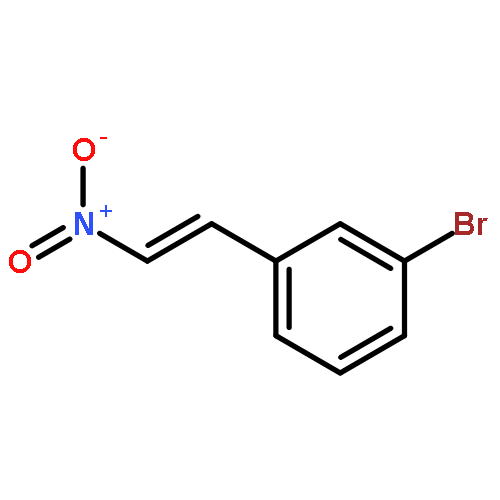


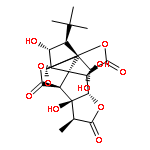

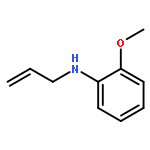

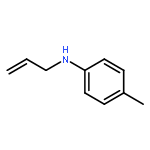

![Benzene, [3-(1-methylethoxy)-1-propenyl]-, (E)-](/data/chemimg/1724800/14289-95-3.png)
![Benzene, [3-(1-methylethoxy)-1-propenyl]-, (E)-](/data/chemimg/1724800/14289-95-3_b.png)
![Benzene,[2-(2-propen-1-yloxy)ethyl]-](http://img.cochemist.com/ccimg/14300/14289-65-7.png)
![Benzene,[2-(2-propen-1-yloxy)ethyl]-](http://img.cochemist.com/ccimg/14300/14289-65-7_b.png)

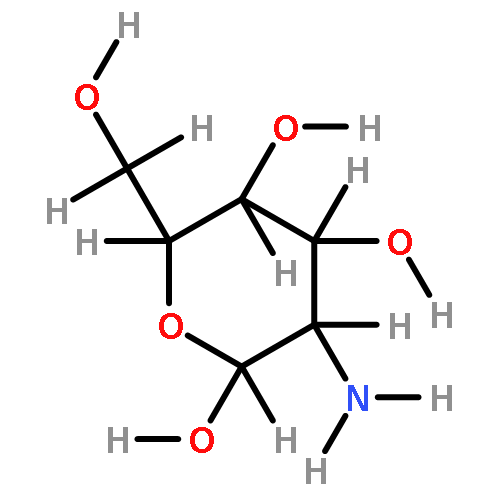
![2-Propenoic acid,3-[3,4-bis(acetyloxy)phenyl]-](http://img.cochemist.com/ccimg/13800/13788-48-2.png)
![2-Propenoic acid,3-[3,4-bis(acetyloxy)phenyl]-](http://img.cochemist.com/ccimg/13800/13788-48-2_b.png)
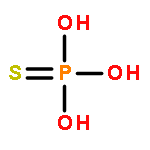
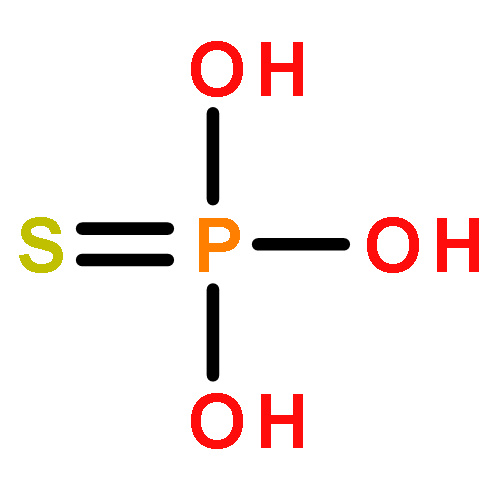




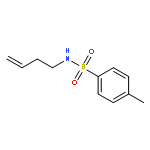



























![BENZENE, [(1E)-3-ETHOXY-1-PROPENYL]-](http://img.cochemist.com/ccimg/5900/5882-84-8.png)
![BENZENE, [(1E)-3-ETHOXY-1-PROPENYL]-](http://img.cochemist.com/ccimg/5900/5882-84-8_b.png)


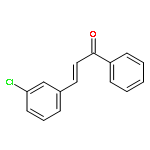

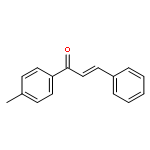
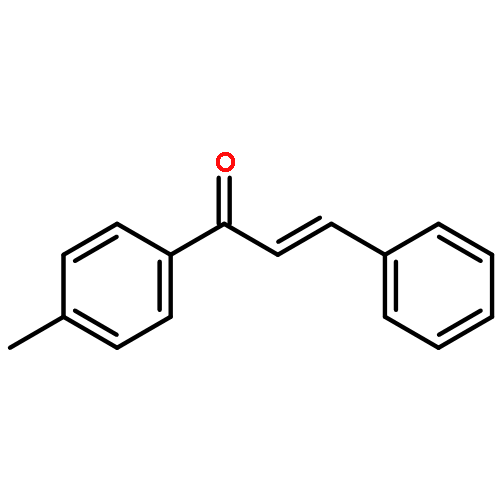
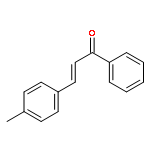

![[1(3H)-ISOINDOLINONE-3-YL]ACETIC ACID](http://img.cochemist.com/ccimg/3900/3849-22-7.png)
![[1(3H)-ISOINDOLINONE-3-YL]ACETIC ACID](http://img.cochemist.com/ccimg/3900/3849-22-7_b.png)
![2-[(3S,4S)-4-(2-BROMO-5-CHLOROPHENYL)-1-METHYL-3-PYRROLIDINYL]PHE<WBR />NOL](http://img.cochemist.com/ccimg/3700/3621-36-1.png)
![2-[(3S,4S)-4-(2-BROMO-5-CHLOROPHENYL)-1-METHYL-3-PYRROLIDINYL]PHE<WBR />NOL](http://img.cochemist.com/ccimg/3700/3621-36-1_b.png)






![Bicyclo[4.2.0]octa-1,3,5-trien-7-one](http://img.cochemist.com/ccimg/3500/3469-06-5.png)
![Bicyclo[4.2.0]octa-1,3,5-trien-7-one](http://img.cochemist.com/ccimg/3500/3469-06-5_b.png)







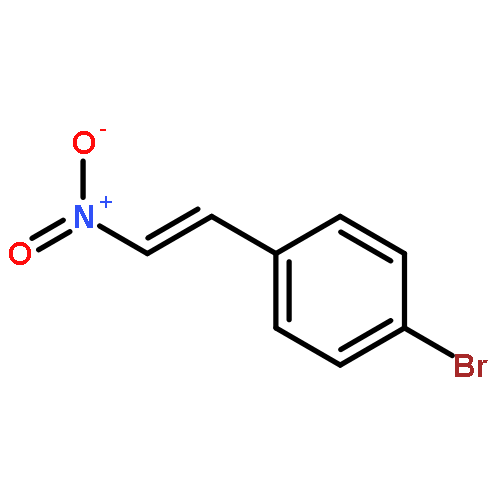


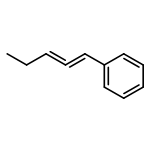
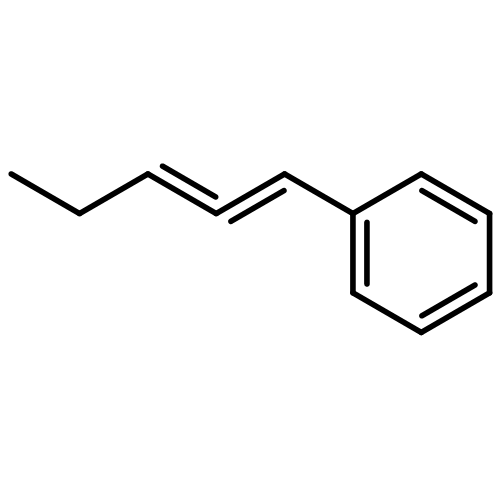


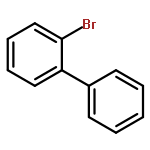
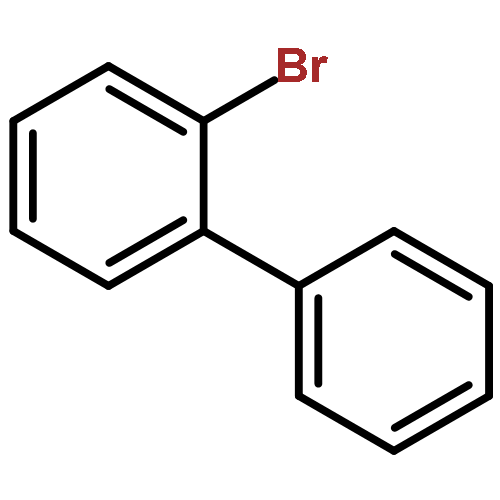

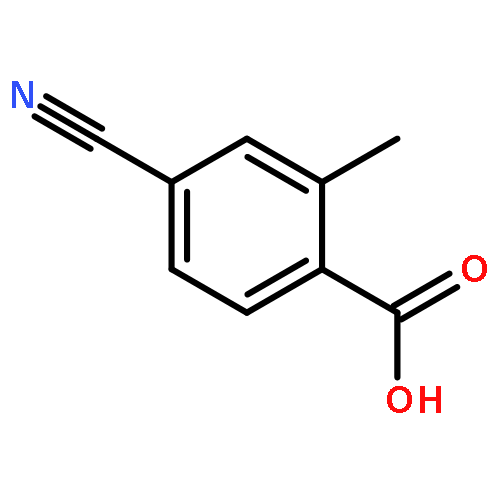








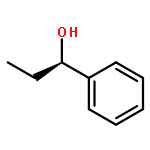
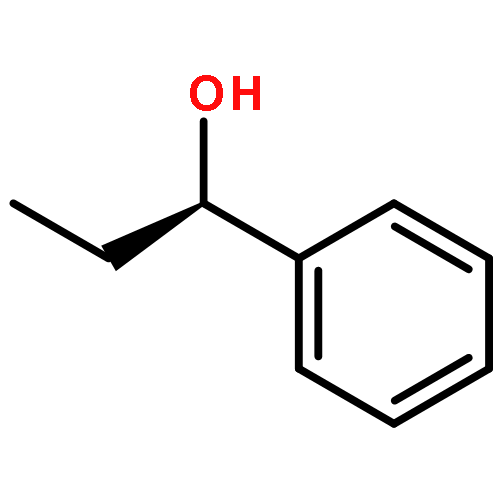
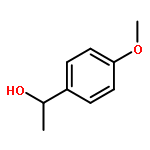
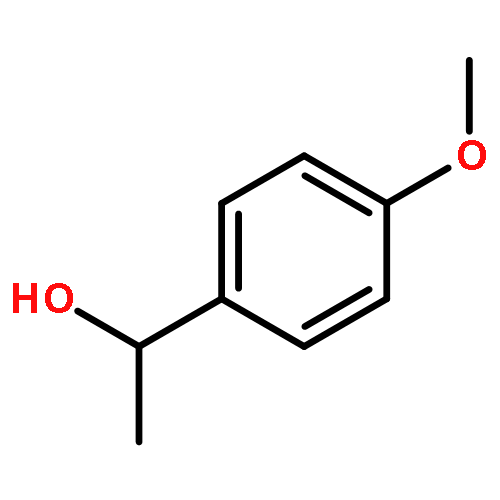
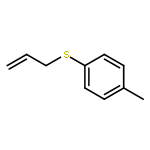
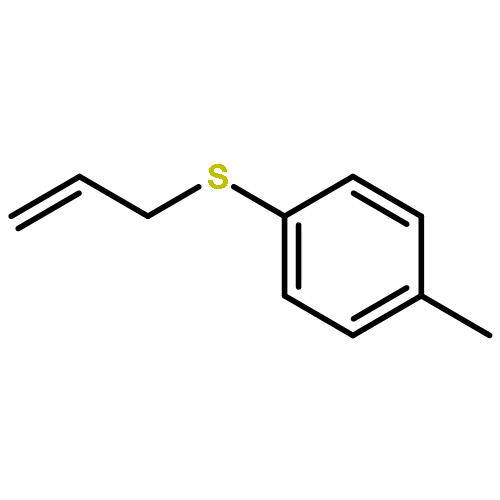


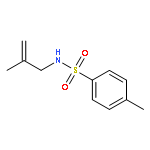
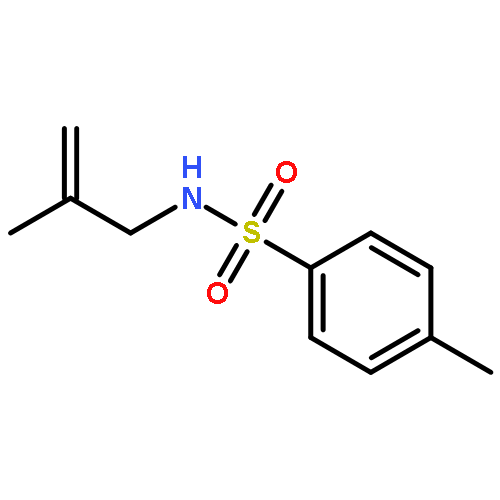






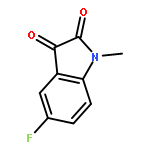
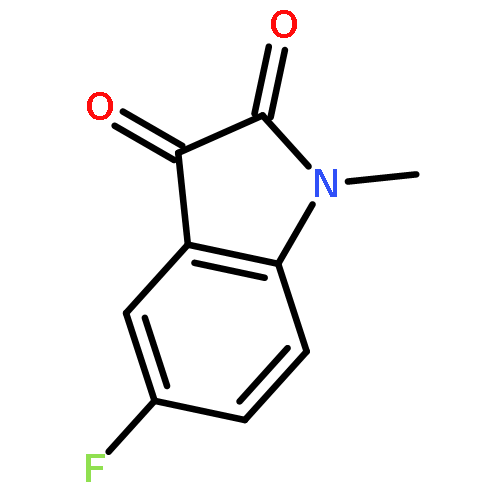




![1-Propanaminium,2-[(aminocarbonyl)oxy]-N,N,N-trimethyl-](http://img.cochemist.com/ccimg/700/674-38-4.png)
![1-Propanaminium,2-[(aminocarbonyl)oxy]-N,N,N-trimethyl-](http://img.cochemist.com/ccimg/700/674-38-4_b.png)
![Formamide,N-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/700/657-78-3.png)
![Formamide,N-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/700/657-78-3_b.png)

![1H,3H-Furo[3,4-c]furan,1,4-bis(3,4-dimethoxyphenyl)tetrahydro-, (1R,3aS,4R,6aS)-](http://img.cochemist.com/ccimg/600/526-06-7.png)
![1H,3H-Furo[3,4-c]furan,1,4-bis(3,4-dimethoxyphenyl)tetrahydro-, (1R,3aS,4R,6aS)-](http://img.cochemist.com/ccimg/600/526-06-7_b.png)
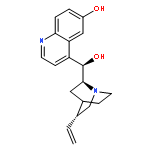
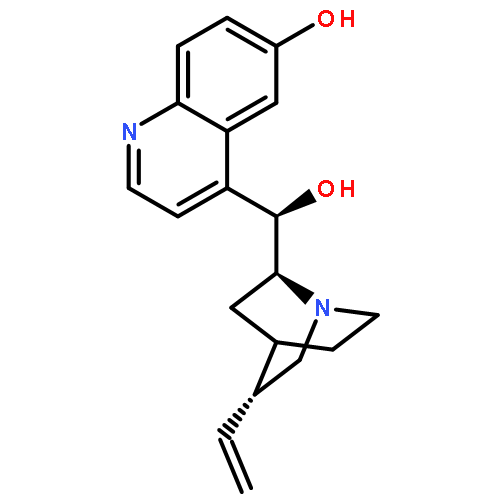




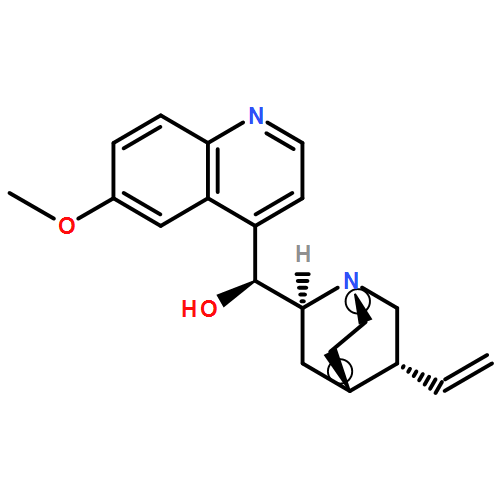
![9-(3-methylbut-2-enyloxy)-7H-furo[3,2-g]chromen-7-one](http://img.cochemist.com/ccimg/500/482-44-0.png)
![9-(3-methylbut-2-enyloxy)-7H-furo[3,2-g]chromen-7-one](http://img.cochemist.com/ccimg/500/482-44-0_b.png)
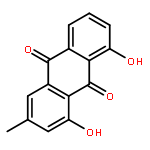
![[2,3'-Biindolinylidene]-2',3-dione](http://img.cochemist.com/ccimg/500/479-41-4.png)
![[2,3'-Biindolinylidene]-2',3-dione](http://img.cochemist.com/ccimg/500/479-41-4_b.png)




















![CARBAMIC ACID, [(1R)-1-CYCLOHEXYL-2-NITROETHYL]-, 1,1-DIMETHYLETHYL ESTER](http://img.cochemist.com/ccimg/875500/875483-12-8.png)
![CARBAMIC ACID, [(1R)-1-CYCLOHEXYL-2-NITROETHYL]-, 1,1-DIMETHYLETHYL ESTER](http://img.cochemist.com/ccimg/875500/875483-12-8_b.png)
![Carbamic acid, [(1R)-1-(1-naphthalenyl)-2-nitroethyl]-, 1,1-dimethylethylester](http://img.cochemist.com/ccimg/875400/875304-09-9.png)
![Carbamic acid, [(1R)-1-(1-naphthalenyl)-2-nitroethyl]-, 1,1-dimethylethylester](http://img.cochemist.com/ccimg/875400/875304-09-9_b.png)











![CARBAMIC ACID, [(3-CHLOROPHENYL)METHYLENE]-, 1,1-DIMETHYLETHYL ESTER](http://img.cochemist.com/ccimg/848200/848194-74-1.png)
![CARBAMIC ACID, [(3-CHLOROPHENYL)METHYLENE]-, 1,1-DIMETHYLETHYL ESTER](http://img.cochemist.com/ccimg/848200/848194-74-1_b.png)


![Carbamic acid, [(1R,2S)-2-nitro-1-phenylpropyl]-, 1,1-dimethylethylester](/data/chemimg/1931300/685132-82-5.png)
![Carbamic acid, [(1R,2S)-2-nitro-1-phenylpropyl]-, 1,1-dimethylethylester](/data/chemimg/1931300/685132-82-5_b.png)
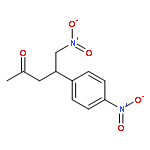





![CARBAMIC ACID, [(3-BROMOPHENYL)METHYLENE]-, 1,1-DIMETHYLETHYL ESTER](http://img.cochemist.com/ccimg/479500/479423-42-2.png)
![CARBAMIC ACID, [(3-BROMOPHENYL)METHYLENE]-, 1,1-DIMETHYLETHYL ESTER](http://img.cochemist.com/ccimg/479500/479423-42-2_b.png)
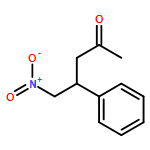

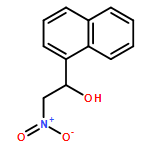
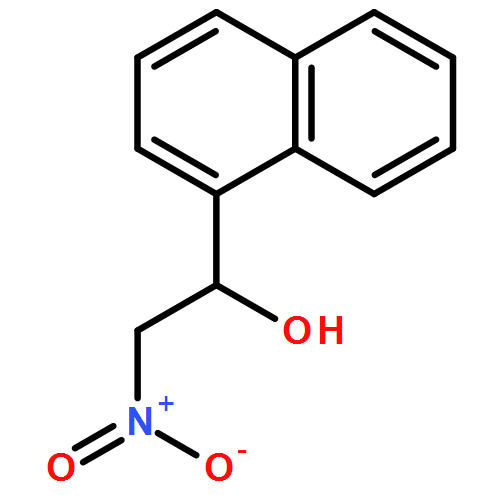
![Carbamic acid, [phenyl(phenylsulfonyl)methyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/155400/155396-71-7.png)
![Carbamic acid, [phenyl(phenylsulfonyl)methyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/155400/155396-71-7_b.png)




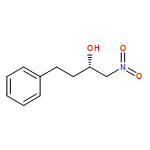
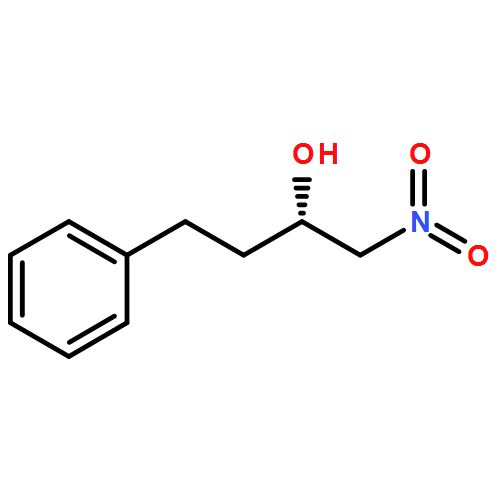
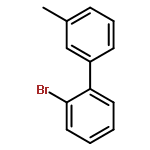





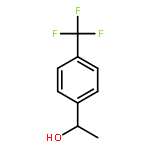
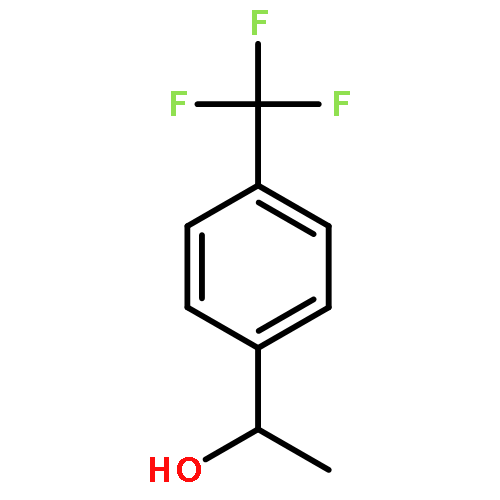




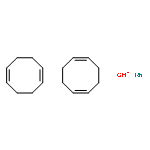
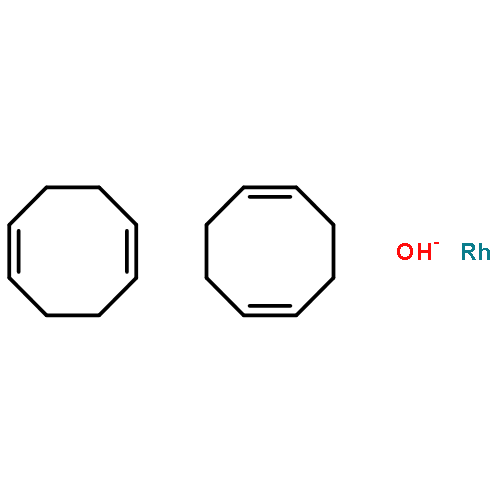




![7-[3-(3-HYDROXYOCT-1-ENYL)-4,6-DIOXABICYCLO[3.1.1]HEPT-2-YL]HEPT-5-ENOIC ACID](http://img.cochemist.com/ccimg/57600/57576-52-0.png)
![7-[3-(3-HYDROXYOCT-1-ENYL)-4,6-DIOXABICYCLO[3.1.1]HEPT-2-YL]HEPT-5-ENOIC ACID](http://img.cochemist.com/ccimg/57600/57576-52-0_b.png)
![Naphthalene, 1-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/37700/37630-26-5.png)
![Naphthalene, 1-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/37700/37630-26-5_b.png)
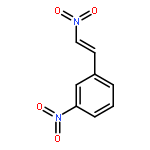
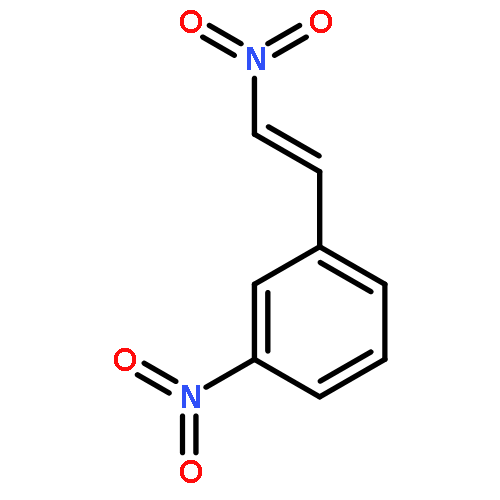




![2,9,10-trimethoxy-5,6-dihydroisoquinolino[2,1-b]isoquinolin-7-ium-3-ol](http://img.cochemist.com/ccimg/3700/3621-38-3.png)
![2,9,10-trimethoxy-5,6-dihydroisoquinolino[2,1-b]isoquinolin-7-ium-3-ol](http://img.cochemist.com/ccimg/3700/3621-38-3_b.png)
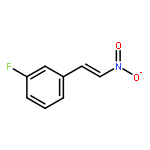
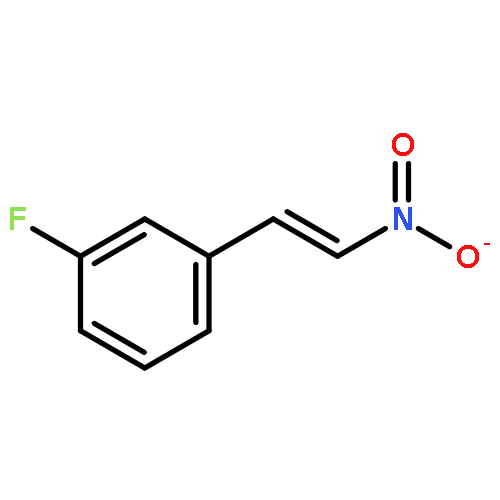




![Carbamic acid, [[4-(trifluoromethyl)phenyl]methylene]-, 1,1-dimethylethylester](http://img.cochemist.com/ccimg/871100/871090-34-5.png)
![Carbamic acid, [[4-(trifluoromethyl)phenyl]methylene]-, 1,1-dimethylethylester](http://img.cochemist.com/ccimg/871100/871090-34-5_b.png)


![Carbamic acid, [(2-chlorophenyl)methylene]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/848200/848194-75-2.png)
![Carbamic acid, [(2-chlorophenyl)methylene]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/848200/848194-75-2_b.png)


![TERT-BUTYL N-[(2-BROMOPHENYL)METHYLIDENE]CARBAMATE](http://img.cochemist.com/ccimg/779400/779342-74-4.png)
![TERT-BUTYL N-[(2-BROMOPHENYL)METHYLIDENE]CARBAMATE](http://img.cochemist.com/ccimg/779400/779342-74-4_b.png)
![Carbamic acid, [(1R)-2-nitro-1-phenylethyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/685200/685132-79-0.png)
![Carbamic acid, [(1R)-2-nitro-1-phenylethyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/685200/685132-79-0_b.png)
![Carbamic acid, [(4-bromophenyl)methylene]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/479500/479423-43-3.png)
![Carbamic acid, [(4-bromophenyl)methylene]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/479500/479423-43-3_b.png)




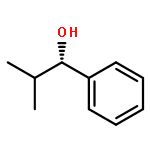
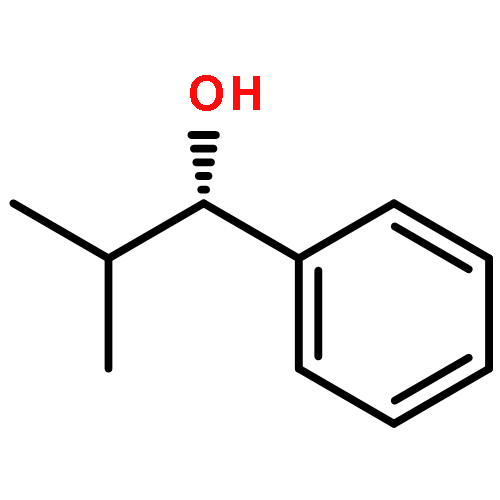
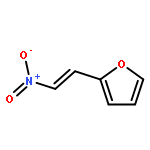
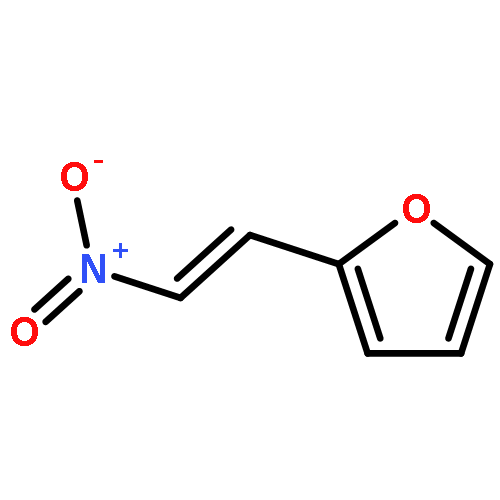
![1-chloro-2-[(e)-2-nitrovinyl]benzene](http://img.cochemist.com/ccimg/22600/22568-07-6.png)
![1-chloro-2-[(e)-2-nitrovinyl]benzene](http://img.cochemist.com/ccimg/22600/22568-07-6_b.png)






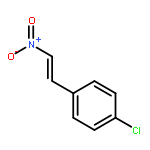
![Bis[1,3]benzodioxolo[5,6-a:4',5'-g]quinolizinium,6,7-dihydro-](http://img.cochemist.com/ccimg/3500/3486-66-6.png)
![Bis[1,3]benzodioxolo[5,6-a:4',5'-g]quinolizinium,6,7-dihydro-](http://img.cochemist.com/ccimg/3500/3486-66-6_b.png)