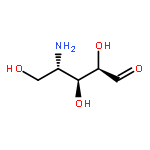
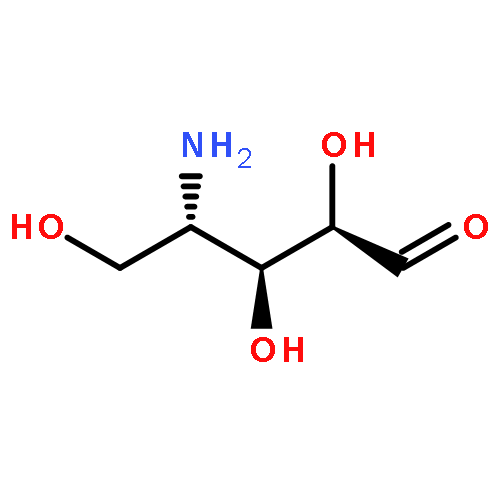
Co-reporter:Pamela Arden Doerner and Marcelo C. Sousa
Biochemistry June 20, 2017 Volume 56(Issue 24) pp:3142-3142
Publication Date(Web):June 1, 2017
DOI:10.1021/acs.biochem.7b00281
BamA is an essential component of the β-barrel assembly machine (BAM) that is responsible for insertion and folding of β-barrel outer membrane proteins (OMPs) in Gram-negative bacteria. BamA is an OMP itself, and its β-barrel transmembrane domain is thought to catalyze OMP insertion and folding, although the molecular mechanism remains poorly understood. Crystal structures of BamA and complementary molecular dynamics simulations have shown that its β-barrel seam (the interface between the first and last barrel strands) is destabilized. This has led to mechanistic models in which the BamA barrel seam functions as a lateral gate that opens and successively accepts β-hairpins from a nascent OMP such that a nascent barrel can bud from BamA. Consistent with this model, disulfide locking of the BamA barrel seam is lethal in Escherichia coli. Here we show that disulfide locking of the BamA barrel has no effect on its ability to catalyze folding of a model OMP into liposomes. However, disulfide trapping experiments indicate that the BamA barrel is highly dynamic in the liposome membranes, with the β-strands at the barrel seam undergoing “register sliding” by more than 14 Å both up and down the membrane. Remarkably, these extreme dynamics were also observed in the BamA barrel in the context of the native E. coli outer membrane. These results are consistent with a model in which the BamA barrel dynamics induce defects in the outer membrane that facilitate insertion of nascent OMPs.
Co-reporter:Robert Walder, Marc-André LeBlanc, William J. Van Patten, Devin T. Edwards, Jacob A. Greenberg, Ayush Adhikari, Stephen R. Okoniewski, Ruby May A. Sullan, David Rabuka, Marcelo C. Sousa, and Thomas T. Perkins
Journal of the American Chemical Society July 26, 2017 Volume 139(Issue 29) pp:9867-9867
Publication Date(Web):July 5, 2017
DOI:10.1021/jacs.7b02958
Atomic force microscopy (AFM)-based single-molecule force spectroscopy (SMFS) is a powerful yet accessible means to characterize mechanical properties of biomolecules. Historically, accessibility relies upon the nonspecific adhesion of biomolecules to a surface and a cantilever and, for proteins, the integration of the target protein into a polyprotein. However, this assay results in a low yield of high-quality data, defined as the complete unfolding of the polyprotein. Additionally, nonspecific surface adhesion hinders studies of α-helical proteins, which unfold at low forces and low extensions. Here, we overcame these limitations by merging two developments: (i) a polyprotein with versatile, genetically encoded short peptide tags functionalized via a mechanically robust Hydrazino-Pictet-Spengler ligation and (ii) the efficient site-specific conjugation of biomolecules to PEG-coated surfaces. Heterobifunctional anchoring of this polyprotein construct and DNA via copper-free click chemistry to PEG-coated substrates and a strong but reversible streptavidin–biotin linkage to PEG-coated AFM tips enhanced data quality and throughput. For example, we achieved a 75-fold increase in the yield of high-quality data and repeatedly probed the same individual polyprotein to deduce its dynamic force spectrum in just 2 h. The broader utility of this polyprotein was demonstrated by measuring three diverse target proteins: an α-helical protein (calmodulin), a protein with internal cysteines (rubredoxin), and a computationally designed three-helix bundle (α3D). Indeed, at low loading rates, α3D represents the most mechanically labile protein yet characterized by AFM. Such efficient SMFS studies on a commercial AFM enable the rapid characterization of macromolecular folding over a broader range of proteins and a wider array of experimental conditions (pH, temperature, denaturants). Further, by integrating these enhancements with optical traps, we demonstrate how efficient bioconjugation to otherwise nonstick surfaces can benefit diverse single-molecule studies.
Co-reporter:Lisa R. Warner, Petia Z. Gatzeva-Topalova, Pamela A. Doerner, Arthur Pardi, Marcelo C. Sousa
Structure 2017 Volume 25, Issue 1(Volume 25, Issue 1) pp:
Publication Date(Web):3 January 2017
DOI:10.1016/j.str.2016.11.013
•BamA with a transmembrane β-barrel and soluble POTRA motifs mediates OMP biogenesis•NMR relaxation studies show POTRA1–2 is flexibly linked to POTRA3–5 in BamA•Residual dipolar couplings indicate POTRA1–2 and POTRA4–5 behave as rigid species•Disulfides that restrict POTRA2–3 flexibility impair in vivo function of BamAThe β-barrel assembly machine (BAM) mediates the biogenesis of outer membrane proteins (OMPs) in Gram-negative bacteria. BamA, the central BAM subunit composed of a transmembrane β-barrel domain linked to five polypeptide transport-associated (POTRA) periplasmic domains, is thought to bind nascent OMPs and undergo conformational cycling to catalyze OMP folding and insertion. One model is that conformational flexibility between POTRA domains is part of this conformational cycling. Nuclear magnetic resonance (NMR) spectroscopy was used here to study the flexibility of the POTRA domains 1–5 in solution. NMR relaxation studies defined effective rotational correlational times and together with residual dipolar coupling data showed that POTRA1–2 is flexibly linked to POTRA3–5. Mutants of BamA that restrict flexibility between POTRA2 and POTRA3 by disulfide crosslinking displayed impaired function in vivo. Together these data strongly support a model in which conformational cycling of hinge motions between POTRA2 and POTRA3 in BamA is required for biological function.Download high-res image (238KB)Download full-size image
Co-reporter:Lisa R. Warner, Petia Z. Gatzeva-Topalova, Pamela A. Doerner, Arthur Pardi, Marcelo C. Sousa
Structure 2017 Volume 25, Issue 1(Volume 25, Issue 1) pp:
Publication Date(Web):3 January 2017
DOI:10.1016/j.str.2016.11.013
•BamA with a transmembrane β-barrel and soluble POTRA motifs mediates OMP biogenesis•NMR relaxation studies show POTRA1–2 is flexibly linked to POTRA3–5 in BamA•Residual dipolar couplings indicate POTRA1–2 and POTRA4–5 behave as rigid species•Disulfides that restrict POTRA2–3 flexibility impair in vivo function of BamAThe β-barrel assembly machine (BAM) mediates the biogenesis of outer membrane proteins (OMPs) in Gram-negative bacteria. BamA, the central BAM subunit composed of a transmembrane β-barrel domain linked to five polypeptide transport-associated (POTRA) periplasmic domains, is thought to bind nascent OMPs and undergo conformational cycling to catalyze OMP folding and insertion. One model is that conformational flexibility between POTRA domains is part of this conformational cycling. Nuclear magnetic resonance (NMR) spectroscopy was used here to study the flexibility of the POTRA domains 1–5 in solution. NMR relaxation studies defined effective rotational correlational times and together with residual dipolar coupling data showed that POTRA1–2 is flexibly linked to POTRA3–5. Mutants of BamA that restrict flexibility between POTRA2 and POTRA3 by disulfide crosslinking displayed impaired function in vivo. Together these data strongly support a model in which conformational cycling of hinge motions between POTRA2 and POTRA3 in BamA is required for biological function.Download high-res image (238KB)Download full-size image
Co-reporter:Myeongseon Lee and Marcelo C. Sousa
Biochemistry 2014 Volume 53(Issue 4) pp:796-805
Publication Date(Web):January 10, 2014
DOI:10.1021/bi4015677
Cationic Antimicrobial Peptides (CAMPs) represent a first line of defense against bacterial colonization. When fighting Gram-negative bacteria, CAMPs initially interact electrostatically with the negatively charged phosphate groups in lipid A and are thought to kill bacteria by disrupting their membrane integrity. However, many human pathogens, including Salmonella and Pseudomonas, have evolved lipid A modification mechanisms that result in resistance to CAMPs and related antibiotics such as Colistin. The addition of 4-amino-4-deoxy-l-Arabinose (Ara4N) to a phosphate group in lipid A is one such modification, frequently found in Pseudomonas isolated from cystic fibrosis patients. The pathway for biosynthesis of Ara4N-lipid A requires conversion of UDP-Glucuronic acid into UDP-Ara4N and subsequent transfer of the amino-sugar to lipid A. ArnB is a pyridoxal-phosphate (PLP) dependent transaminase that catalyzes a crucial step in the pathway: synthesis of UDP-Ara4N from UDP-4-keto-pentose. Here we present the 2.3 Å resolution crystal structure of an active site mutant of ArnB (K188A) in complex with the reaction intermediate aldimine formed by UDP-Ara4N and PLP. The sugar–nucleotide binding site is in a cleft between the subunits of the ArnB dimer with the uracil buried at the interface and the UDP ribose and phosphate groups exposed to the solvent. The Ara4N moiety is found in the 4C1 conformation and its positioning, stabilized by interactions with both the protein and cofactor, is compatible with catalysis. The structure suggests strategies for the development of specific inhibitors that may prove useful in the treatment of resistant bacteria such as Pseudomonas found in cystic fibrosis patients.
Co-reporter:Michelle Marian Turco and Marcelo Carlos Sousa
Biochemistry 2014 Volume 53(Issue 31) pp:5131-5139
Publication Date(Web):July 18, 2014
DOI:10.1021/bi500593e
Many pathogenic bacteria utilize the type III secretion system (T3SS) to translocate effector proteins directly into host cells, facilitating colonization. In enterohemmorhagic Escherichia coli (EHEC), a subset of T3SS effectors is essential for suppression of the inflammatory response in hosts, including humans. Identified as a zinc protease that cleaves NF-κB transcription factors, NleC is one such effector. Here, we investigate NleC substrate specificity, showing that four residues around the cleavage site in the DNA-binding loop of the NF-κB subunit RelA strongly influence the cleavage rate. Class I NF-κB subunit p50 is cleaved at a reduced rate consistent with conservation of only three of these four residues. However, peptides containing 10 residues on each side of the scissile bond were not efficiently cleaved by NleC, indicating that elements distal from the cleavage site are also important for substrate recognition. We present the crystal structure of NleC and show that it mimics DNA structurally and electrostatically. Consistent with this model, mutation of phosphate-mimicking residues in NleC reduces the level of RelA cleavage. We propose that global recognition of NF-κB subunits by DNA mimicry combined with a high sequence selectivity for the cleavage site results in exquisite NleC substrate specificity. The structure also shows that despite undetectable similarity of its sequence to those of other Zn2+ proteases beyond its conserved HExxH Zn2+-binding motif, NleC is a member of the Zincin protease superfamily, albeit divergent from its structural homologues. In particular, NleC displays a modified Ψ-loop motif that may be important for folding and refolding requirements implicit in T3SS translocation.
Co-reporter:Paul D. Templeton, Elizabeth S. Litman, Sandra I. Metzner, Natalie G. Ahn, and Marcelo C. Sousa
Biochemistry 2013 Volume 52(Issue 33) pp:5675-5684
Publication Date(Web):July 16, 2013
DOI:10.1021/bi400556e
Metastatic melanoma is among the most intractable cancers to treat; patients show resistance to therapy and limited survival time. A critical step in the development of metastatic melanoma is the acquisition of invasion and transition from thin to thick tumors on the skin, followed by invasion to lymph nodes. Prior studies have shown that metastatic melanoma is associated with dysregulation of RhoA and enhanced expression of a protein named “mediator of RhoA-dependent invasion (MRDI)”. Importantly, MRDI is a “moonlighting” enzyme, with two distinct functions in melanoma cells. First, MRDI acts as a methylthioribose-1-phosphate (MTR-1-P) isomerase, catalyzing a critical step in methionine salvage. Second, MRDI promotes and is necessary for melanoma cell invasion, independent of its catalytic activity. This paper demonstrates that MtnA, a bacterial MTR-1-P isomerase, rescues the methionine salvage function of MRDI, but is unable to rescue its role in invasion. The crystal structure of MRDI was solved to a resolution of 2.5 Å to identify structural elements important for its invasion activity. This structure and its comparison with other MTR-1-P isomerases are presented, and mutations within a region separate from the MTR-1-P binding site, which interfere with invasion, are identified. Thus, structural elements in MRDI distal from the MTR-1-P catalytic site are responsible for the invasion phenotype.
Co-reporter:Troy A. Walton;Arthur Pardi;Marcelo C. Sousa;Cristina M. Sandoval;C. Andrew Fowler
PNAS 2009 Volume 106 (Issue 6 ) pp:1772-1777
Publication Date(Web):2009-02-10
DOI:10.1073/pnas.0809275106
Outer membrane proteins (OMPs) of Gram-negative bacteria are synthesized in the cytosol and must cross the periplasm before
insertion into the outer membrane. The 17-kDa protein (Skp) is a periplasmic chaperone that assists the folding and insertion
of many OMPs, including OmpA, a model OMP with a membrane embedded β-barrel domain and a periplasmic αβ domain. Structurally,
Skp belongs to a family of cavity-containing chaperones that bind their substrates in the cavity, protecting them from aggregation.
However, some substrates, such as OmpA, exceed the capacity of the chaperone cavity, posing a mechanistic challenge. Here,
we provide direct NMR evidence that, while bound to Skp, the β-barrel domain of OmpA is maintained in an unfolded state, whereas
the periplasmic domain is folded in its native conformation. Complementary cross-linking and NMR relaxation experiments show
that the OmpA β-barrel is bound deep within the Skp cavity, whereas the folded periplasmic domain protrudes outside of the
cavity where it tumbles independently from the rest of the complex. This domain-based chaperoning mechanism allows the transport
of β-barrels across the periplasm in an unfolded state, which may be important for efficient insertion into the outer membrane.
Co-reporter:Ricardo Stephen;S&x142;awomir Filipek;Krzysztof Palczewski
Photochemistry and Photobiology 2008 Volume 84( Issue 4) pp:903-910
Publication Date(Web):
DOI:10.1111/j.1751-1097.2008.00323.x
Abstract
Photon absorption by rhodopsin triggers the phototransduction signaling pathway that culminates in degradation of cGMP, closure of cGMP-gated ion channels and hyperpolarization of the photoreceptor membrane. This process is accompanied by a decrease in free Ca2+ concentration in the photoreceptor cytosol sensed by Ca2+-binding proteins that modulate phototransduction and activate the recovery phase to reestablish the photoreceptor dark potential. Guanylate cyclase-activating proteins (GCAPs) belong to the neuronal calcium sensor (NCS) family and are responsible for activating retinal guanylate cyclases (retGCs) at low Ca2+ concentrations triggering synthesis of cGMP and recovery of the dark potential. Here we review recent structural insight into the role of the N-terminal myristoylation in GCAPs and compare it to other NCS family members. We discuss previous studies identifying regions of GCAPs important for retGC1 regulation in the context of the new structural data available for myristoylated GCAP1. In addition, we present a hypothetical model for the Ca2+-triggered conformational change in GCAPs and retGC1 regulation. Finally, we briefly discuss the involvement of mutant GCAP1 proteins in the etiology of retinal degeneration as well as the importance of other Ca2+ sensors in the modulation of phototransduction.
Co-reporter:Ricardo Stephen, Grzegorz Bereta, Marcin Golczak, Krzysztof Palczewski, Marcelo Carlos Sousa
Structure (13 November 2007) Volume 15(Issue 11) pp:1392-1402
Publication Date(Web):13 November 2007
DOI:10.1016/j.str.2007.09.013
Guanylate cyclase-activating proteins (GCAPs) are Ca2+-binding proteins myristoylated at the N terminus that regulate guanylate cyclases in photoreceptor cells and belong to the family of neuronal calcium sensors (NCS). Many NCS proteins display a recoverin-like “calcium-myristoyl switch” whereby the myristoyl group, buried inside the protein in the Ca2+-free state, becomes fully exposed upon Ca2+ binding. Here we present a 2.0 Å resolution crystal structure of myristoylated GCAP1 with Ca2+ bound. The acyl group is buried inside Ca2+-bound GCAP1. This is in sharp contrast to Ca2+-bound recoverin, where the myristoyl group is solvent exposed. Furthermore, we provide direct evidence that the acyl group in GCAP1 remains buried in the Ca2+-free state and does not undergo switching. A pronounced kink in the C-terminal helix and the presence of the myristoyl group allow clustering of sequence elements crucial for GCAP1 activity.
Co-reporter:Petia Z. Gatzeva-Topalova, Troy A. Walton, Marcelo C. Sousa
Structure (12 December 2008) Volume 16(Issue 12) pp:1873-1881
Publication Date(Web):12 December 2008
DOI:10.1016/j.str.2008.09.014
The envelope of Gram-negative bacteria consists of inner and outer membranes surrounding the peptidoglycan wall. The outer membrane (OM) is rich in integral membrane proteins (OMPs), which have a characteristic β barrel domain embedded in the OM. The Omp85 family of proteins, ubiquitous among Gram-negative bacteria and also present in chloroplasts and mitochondria, is required for folding and insertion of OMPs into the outer membrane. Bacterial Omp85 proteins are characterized by a periplasmic domain containing five repeats of polypeptide transport-associated (POTRA) motifs. Here we report the crystal structure of a periplasmic fragment of YaeT (the Escherichia coli Omp85) containing the first four POTRA domains in an extended conformation consistent with recent solution X-ray scattering data. Analysis of the YaeT structure reveals conformational flexibility around a hinge point between POTRA2 and 3 domains. The structure's implications for substrate binding and folding mechanisms are also discussed.
Co-reporter:Petia Zvezdanova Gatzeva-Topalova, Lisa Rosa Warner, Arthur Pardi, Marcelo Carlos Sousa
Structure (10 November 2010) Volume 18(Issue 11) pp:1492-1501
Publication Date(Web):10 November 2010
DOI:10.1016/j.str.2010.08.012
Folding and insertion of β-barrel outer membrane proteins (OMPs) is essential for Gram-negative bacteria. This process is mediated by the multiprotein complex BAM, composed of the essential β-barrel OMP BamA and four lipoproteins (BamBCDE). The periplasmic domain of BamA is key for its function and contains five “polypeptide transport-associated” (POTRA) repeats. Here, we report the crystal structure of the POTRA4-5 tandem, containing the essential for BAM complex formation and cell viability POTRA5. The domain orientation observed in the crystal is validated by solution NMR and SAXS. Using previously determined structures of BamA POTRA1-4, we present a spliced model of the entire BamA periplasmic domain validated by SAXS. Solution scattering shows that conformational flexibility between POTRA2 and 3 gives rise to compact and extended conformations. The length of BamA in its extended conformation suggests that the protein may bridge the inner and outer membranes across the periplasmic space.Graphical AbstractDownload high-res image (433KB)Download full-size imageHighlights► BamA POTRA4-5 adopts an L-shaped conformation ► A spliced model of BamA POTRA1-5 is validated by SAXS ► Conformational flexibility gives rise to bent and extended conformations ► In the extended conformation, BamA may bridge the inner and outer membranes
Co-reporter:Cristina M. Sandoval, Susan L. Baker, Katarina Jansen, Sandra I. Metzner, Marcelo C. Sousa
Journal of Molecular Biology (10 June 2011) Volume 409(Issue 3) pp:348-357
Publication Date(Web):10 June 2011
DOI:10.1016/j.jmb.2011.03.035
Folding and insertion of integral β-barrel proteins in the outer membrane (OM) is an essential process for Gram-negative bacteria that requires the β-barrel assembly machinery (BAM). Efficient OM protein (OMP) folding and insertion appears to require a consensus C-terminal signal in OMPs characterized by terminal F or W residues. The BAM complex is embedded in the OM and, in Escherichia coli, consists of the β-barrel BamA and four lipoproteins BamBCDE. BamA and BamD are broadly distributed across all species of Gram-negative bacteria, whereas the other components are present in only a subset of species. BamA and BamD are also essential for viability, suggesting that these two proteins constitute the functional core of the bacterial BAM complex. Here, we present the crystal structure of BamD from the thermophilic bacteria Rhodothermus marinus refined to 2.15 Å resolution. The protein contains five tetratricopeptide repeats (TPRs) organized into two offset tandems, each capped by a terminal helix. The N-terminal domain contains three TPRs and displays remarkable structural similarity with proteins that recognize targeting signals in extended conformations. The C-terminal domain harbors the remaining two TPRs and previously described mutations that impair binding to other BAM components map to this domain. Therefore, the structure suggests a model where the C-terminal domain provides a scaffold for interaction with BAM components, while the N-terminal domain participates in interaction with the substrates, either recognizing the C-terminal consensus sequence or binding unfolded OMP intermediates.Research Highlights► BamD is essential for folding and insertion of OMPs.We determined the crystal structure of R. marinus BamD. ► Five TPRs are arranged into two offset tandems in R. marinus BamD. ► Three N-terminal TPRs are similar to proteins that recognize targeting signals. ► Two C-terminal TPRs harbor determinants for interaction with other BAM components.









![L-Glutamic acid,N-[4-[[(2-amino-3,4,5,6,7,8-hexahydro-4-oxo-6-pteridinyl)methyl]formylamino]benzoyl]-](http://img.cochemist.com/ccimg/2900/2800-34-2.png)
![L-Glutamic acid,N-[4-[[(2-amino-3,4,5,6,7,8-hexahydro-4-oxo-6-pteridinyl)methyl]formylamino]benzoyl]-](http://img.cochemist.com/ccimg/2900/2800-34-2_b.png)