Co-reporter:Cheng-Xing Cui;Yu-Ping Zhang;Ling-Bo Qu;Zhao-Pei Zhang
The Journal of Physical Chemistry A January 19, 2017 Volume 121(Issue 2) pp:523-531
Publication Date(Web):December 23, 2016
DOI:10.1021/acs.jpca.6b10231
Bis-adducts of fullerenes are important for both material and biological science. The first added substituent greatly impacts the reactivity and regioselectivity of fullerene. What determines the bond reactivity and how to control the regioselectivity are two crucial questions in synthesizing bis-adduct of C70. Recently, an unexpected 12 o’clock isomer of anthracene bis-adduct of C70 was prepared with high yield by the Diels–Alder (DA) reaction although three possible isomers (12, 2, and 5 o’clock isomers) may be formed. In the current study, the beneath mechanism is systematically investigated by density functional theory methods. Moreover, effects of charges and encapsulated lanthanum atom on the regioselectivity are reported. The computational results successfully rationalize experimental observations by Venkata et al. A possible way to change the regioselectivity of DA reaction is put forward. The kinetical promotion effect of an encapsulated La atom on the 12 o’clock reaction is elucidated.
Co-reporter:Yuanyuan Cheng;Yajun Liu
Chinese Journal of Chemistry 2017 Volume 35(Issue 5) pp:733-741
Publication Date(Web):2017/05/01
DOI:10.1002/cjoc.201600731
AbstractAcryloyl fluoride is an ideal molecule for investigating the phenomenon of hindered internal rotation. In concert with recently acquired high-resolution UV absorption spectrum of acryloyl fluoride, in this study, the absorption spectra of the s-trans and s-cis isomers of acryloyl fluoride were theoretically simulated. The simulated spectra were convoluted by a Gaussian function with displacement, distortion, Franck-Condon, Herzberg-Teller, and Duschinsky effects in the framework of the time-independent model. The statistical vibronic transition analysis reveals the unity of the spectrum transition property, the relevant normal modes, and the primary geometrical variations, enriching the understanding of the experimental observation. The discrepancy between the theoretical and experimental spectra was interpreted clearly.
Co-reporter:
Photochemistry and Photobiology 2017 Volume 93(Issue 2) pp:511-518
Publication Date(Web):2017/03/01
DOI:10.1111/php.12657
AbstractDinoflagellates are the most ubiquitous luminescent protists in the marine environment and have drawn much attention for their crucial roles in marine ecosystems. Dinoflagellate bioluminescence has been applied in underwater target detection. The luminescent system of dinoflagellates is a typical luciferin–luciferase one. However, the excited-state oxyluciferin is not the light emitter of dinoflagellate bioluminescence as in most luciferin–luciferase bioluminescent organisms. The oxyluciferin of bioluminescent dinoflagellates is not fluorescent, whereas its luciferin emits bright fluorescence with similar wavelength of the bioluminescence. What is the light emitter of dinoflagellate bioluminescence and what is the chemical process of the light emission like? These questions have not been answered by the limited experimental evidence so far. In this study, for the first time, the density functional calculation is employed to investigate the geometries and properties of luciferin and oxyluciferin of bioluminescent dinoflagellate. The calculated results agree with the experimental observations and indicate the luciferin or its analogue, rather than oxyluciferin, is the bioluminophore of dinoflagellate bioluminescence. A rough mechanism involving energy transfer is proposed for dinoflagellate bioluminescence.
Co-reporter:Bo-Wen Ding and Ya-Jun Liu
Journal of the American Chemical Society 2016 Volume 139(Issue 3) pp:1106-1119
Publication Date(Web):December 29, 2016
DOI:10.1021/jacs.6b09119
Watasenia scintillans (W. scintillans) is a deep-sea luminescent squid with a popular name of firefly squid. It produces flashes of blue light via a series of complicated luciferin-luciferase reactions involving ATP, Mg2+, and molecular oxygen. Tsuji has proposed a hypothetical scheme for this mysterious bioluminescence (BL) process, but the proposal is short of strong evidence experimentally or theoretically, especially for two key steps. They are the addition of molecular oxygen to luciferin and the formation of light emitter. For the first time, the present study investigates the two steps by reliable density functional theory (DFT) and time-dependent DFT. The results of calculated energetics, charge transfer process, electronic structures, and molecular dynamics give convincing support for Tsuji’s proposal. The oxygenation reaction occurs with a single electron-transfer (SET) mechanism, and the light emitter is produced via the mechanism of gradually reversible charge-transfer-induced luminescence (GRCTIL). The simulation of nonadiabatic molecular dynamics further confirms the GRCTIL mechanisms and evaluates the quantum yield of the light emitter to be 43%. The knowledge obtained in the current study will help to understand a large amount of BL systems in nature, since the core structure of W. scintillans luciferin, imidazopyrazinone, is common in the luciferins of about eight phyla of luminescent organisms.
Co-reporter:Ning Lou, Yanbang Li, Chengxing Cui, Yajun Liu, and Liangbing Gan
Organic Letters 2016 Volume 18(Issue 9) pp:2236-2239
Publication Date(Web):April 19, 2016
DOI:10.1021/acs.orglett.6b00872
[60]Fullerene hexaadducts C60R5Cl (R = OMe or Ar) reacted with hydroxylamine to form C60R5(NHOH) with the hydroxylamino group attached on the central pentagon as in the starting material. Further reactions including treatment with PCl5 and basic alumina led to the insertion of the nitrogen atom into the fullerene cage skeleton and decarbonylation to form azafullerenes C59N(H)(OMe)4 and C59N(OMe)5. The fullerene derivatives C59N(CO)R5 and C60NAr5 with a pyridinone and a pyridine moiety on the cage skeleton, respectively, were also synthesized starting from the hydroxylamine adducts.
Co-reporter:Yuan-Yuan Cheng
Photochemistry and Photobiology 2016 Volume 92( Issue 4) pp:552-560
Publication Date(Web):
DOI:10.1111/php.12601
Abstract
Firefly bioluminescence has been applied in several fields. However, the absorption and fluorescence spectra of the substrate, luciferin, have not been observed at the vibrational level. In this study, the vibrationally resolved absorption and fluorescence spectra of firefly luciferin (neutral form LH2, phenolate ion form LH− and dianion form L2−) are simulated using the density functional method and convoluted by a Gaussian function, with displacement, distortion and Duschinsky effects in the framework of the Franck–Condon approximation. Both neutral and anionic forms of the luciferin are considered in the gas phase and in solution. The simulated spectra have desired band maxima with the experimental ones. The vibronic structure analysis reveals that the features of the most contributive vibrational modes coincide with the key geometry-changing region during transition between the ground state and the first singlet excited state.
Co-reporter:Yan-Li Song, Cheng-Xing Cui, Ya-Jun Liu
Journal of Photochemistry and Photobiology A: Chemistry 2016 Volume 317() pp:68-71
Publication Date(Web):15 February 2016
DOI:10.1016/j.jphotochem.2015.11.014
Co-reporter:Yuan-Yuan Cheng and Ya-Jun Liu
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 11) pp:5360-5370
Publication Date(Web):October 9, 2015
DOI:10.1021/acs.jctc.5b00659
Firefly bioluminescence attracts people by its glaring beauty and fascinating applications, but what is the light emitter of a firefly? The answer to this question has been explored since before the 1960s. The unanimously accepted answer is that excited-state oxyluciferin is the light emitter. The complexity of this question arises from the existence of six chemical forms (keto, enol, keto-1, enol-1, enol-1′, and enol-2) of oxyluciferin. After decades of experimental and theoretical efforts, a consistent conclusion was almost reached in 2011: excited-state keto-1 is the only light emitter in fireflies. However, the debate is raised again by the latest in vitro experimental results. This study will solve this contradiction via hybrid quantum mechanics and molecular mechanics (QM/MM) calculations combined with molecular dynamics (MD). The calculations were performed in the real protein for the six chemical forms of oxyluciferin and their corresponding analogues employed in the latest experiments. By considering the real environment, the pH value, and a possible equilibrium of the chemical forms of oxyluciferin in vivo, the calculated results indicate that the main emitter is still the excited-state keto-1 form.
Co-reporter:Bo-Wen Ding, Panče Naumov, and Ya-Jun Liu
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 2) pp:591-599
Publication Date(Web):January 8, 2015
DOI:10.1021/ct5009203
Cypridina hilgendorfii (sea firefly) is a bioluminescent crustacean whose bioluminescence (BL) reaction is archetypal for a number of marine organisms, notably other bioluminescent crustaceans and coelenterates. Unraveling the mechanism of its BL is paramount for future applications of its strongly emissive lumophore. Cypridina produces light in a three-step reaction: First, the cypridinid luciferin is activated by an enzyme to produce a peroxide intermediate, cypridinid dioxetanone (CDO), which then decomposes to generate excited oxyluciferin (OxyCLnH*). Finally, OxyCLnH* deexcites to its ground state along with emission of bright blue light. Unfortunately, the detailed mechanism of the critical step, the thermolysis of CDO, remains unknown, and it is unclear whether the light emitter is generated from a neutral form (CDOH) or anionic form (CDO–) of the CDO precursor. In this work, we investigated the key step in the process by modeling the thermal decompositions of both CDOH and CDO–. The calculated results indicate that the decomposition of CDO– occurs via the gradually reversible charge transfer (CT)-initiated luminescence (GRCTIL) mechanism, whereas CDOH decomposes through an entropic trapping mechanism without an obvious CT process. The thermolysis of CDO– is sensitive to solvent effects and is energetically favorable in polar environments compared with the thermolysis of CDOH. The thermolysis of CDO– produces the excited oxyluciferin anion (OxyCLn–*), which combines with a proton from the environment to form OxyCLnH*, the actual light emitter for the natural system.
Co-reporter:Cheng-Xing Cui
Journal of Physical Organic Chemistry 2015 Volume 28( Issue 4) pp:281-289
Publication Date(Web):
DOI:10.1002/poc.3408
Investigation of the relative reactivity of bonds in fullerenes will provide fundamental theory for the design of fullerene-based materials. We have theoretically investigated the reactivity of the Diels–Alder (DA) cycloaddition of cis-1,3-butadiene to all types of bonds in C60 and C70 using the M06-2X hybrid density functional theory (DFT) calculations (J. Phys. Org. Chem. 2012, 25 850–855) and have pointed out that the DA cycloadditions of cis and trans forms of 1,3-butadiene to ethylene have a specially intimate relationship (J. Phys. Org. Chem. 2014, 27 652–660). For the aim of telling a whole story of the DA cycloaddition concerning C60 and C70, the DA cycloadditions of trans-1,3-butadiene to all types of bonds in C60 and C70 were explored at the same theoretical level as those of the cis-1,3-butadiene. The calculated results related with the trans- and cis-1,3-butadienes were compared. The potential energy curves of DA cycloadditions of trans- and cis-1,3-butadiene to C60 and C70 were discussed. The distortion–interaction energy model was employed to elucidate the origin of different reactivity of all kinds of CC bonds. The solvent effects were examined using the continuum solvent model. These current results, along with our previous research, will help to obtain an overall view of the DA cycloadditions of 1,3-butadiene to C60 and C70. Copyright © 2014 John Wiley & Sons, Ltd.
Co-reporter:Cheng-Xing Cui and Ya-Jun Liu
The Journal of Physical Chemistry A 2015 Volume 119(Issue 12) pp:3098-3106
Publication Date(Web):February 25, 2015
DOI:10.1021/acs.jpca.5b00194
Endohedral metallofullerene has novel properties because of the interaction between the encapsulated metal atom or cation and fullerene. Experiments have demonstrated that the insertion of Li+ into C60 can greatly promote the reactivity of the Diels–Alder (DA) cycloaddition of cyclopentadiene (CpH) to C60. However, the reaction is sufficiently fast that its quantitative kinetic data cannot be obtained experimentally. In addition, knowledge regarding the effects of other alkali metal cations and metal cations with more charges on the reactivity and regioselectivity of C60 is almost nonexistent. In the current study, DA cycloadditions of CpH to M+@C60 (where M = Li, Na, K, Rb, and Cs) and Ca2+@C60 were investigated via density functional theory in the gas phase and in solvent. Via careful discussion and comparison with the results of C60, we concluded the following for the DA reaction of CpH to C60 and, more generally, for DA reactions of other fullerenes: (1) the encapsulated metal cations enhance the reactivity; (2) among alkali metal cations, Na+ could be the best catalyst; (3) Ca2+ is more favorable in promoting the reactivity than any alkali metal cation; (4) encapsulated metal cations with more positive charges enhance the reactivity of the 6–5 bond in C60, which is significant when the 6–5 adduct is the target product.
Co-reporter:Liang Xu;Hongjiang Ren;Sisi Liang;Jiahao Sun;Dr. Yajun Liu;Dr. Liangbing Gan
Chemistry - A European Journal 2015 Volume 21( Issue 39) pp:13539-13543
Publication Date(Web):
DOI:10.1002/chem.201502306
Abstract
A reversible wetting/dewetting procedure is reported for an open-cage fullerene with an 18-membered orifice. In a homogeneous mixture of H2O/EtOH/CHCl3, water was encapsulated into the cavity of the open-cage compound quantitatively at 80 °C. Addition of aqueous hydrogen fluoride into the water-encapsulated complex removed the encapsulated water completely at room temperature. H-bonding between the trapped water and fluoride is shown to play a key role for the water release process.
Co-reporter:Ling Yue; Zhenggang Lan
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 3) pp:540-548
Publication Date(Web):January 23, 2015
DOI:10.1021/jz502305g
The firefly is famous for its high bioluminescent efficiency, which has attracted both scientific and public attention. The chemical origin of firefly bioluminescence is the thermolysis of the firefly dioxetanone anion (FDO–). Although considerable theoretical research has been conducted, and several mechanisms were proposed to elucidate the high efficiency of the chemi- and bioluminescence of FDO–, there is a lack of direct experimental and theoretical evidence. For the first time, we performed a nonadiabatic molecular dynamics simulation on the chemiluminescent decomposition of FDO– under the framework of the trajectory surface hopping (TSH) method and theoretically estimated the chemiluminescent quantum yield. The TSH simulation reproduced the gradually reversible charge-transfer initiated luminescence mechanism proposed in our previous study. More importantly, the current study, for the first time, predicted the bioluminescence efficiency of the firefly from a theoretical viewpoint, and the theoretical prediction efficiency is in good agreement with experimental measurements.
Co-reporter:Cheng-Xing Cui
Journal of Physical Organic Chemistry 2014 Volume 27( Issue 8) pp:652-660
Publication Date(Web):
DOI:10.1002/poc.3313
The Diels–Alder (DA) reaction is one of the most important reactions in organic chemistry. The controversy surrounding this reaction as to whether it follows a concerted or stepwise mechanism has existed for a long time. The reaction of 1,3-butadiene and ethylene is the paradigmatic example of the DA reaction. We have reinvestigated the mechanism of this reaction using density functional theory. The theoretical study considered all types of possible pathways for the reaction of 1,3-butadiene and ethylene using six functionals at different rungs of Jacob's ladder. Therefore, a complete picture is given for a thorough understanding of the iconic DA reaction, and a new stationary point during the reaction processes has been reported for the first time. The calculated results indicated that three functionals, ωB97X-D, M06-2X, and B2-PLYP, of the fourth and fifth rungs of Jacob's ladder performed well in the investigation of the mechanism of this reaction and that the reliable basis set should be larger than 6-311+G(2d,p). The cis-1,3-butadiene more easily reacted with ethylene compared with 1,3-butadiene in the trans conformation. The concerted mechanism was found to be energetically favorable, whose energy barrier is around 10 kcal/mol lower than that of the stepwise mechanism. Two investigated solvents, toluene and CH3CN, had little impact on this simple DA reaction. Copyright © 2014 John Wiley & Sons, Ltd.
Co-reporter:Yuan-Yuan Cheng, Jia Zhu, Ya-Jun Liu
Chemical Physics Letters 2014 Volume 591() pp:156-160
Publication Date(Web):20 January 2014
DOI:10.1016/j.cplett.2013.11.023
Highlights
- •
Three density functionals were employed to predict the efficiently firefly luminescent analogs.
- •
The reliable functionals for calculating the firefly luminescent analogs were suggested.
- •
Theoretically designed new efficient firefly oxyluciferin analogs.
Co-reporter:Cheng-Xing Cui
Journal of Physical Organic Chemistry 2014 Volume 27( Issue 10) pp:823-832
Publication Date(Web):
DOI:10.1002/poc.3342
The 1,3-dipolar cycloaddition (1,3-DPCA) reaction plays a crucial role during the functionalization of fullerenes, which have broad applications in the materials and pharmaceutical fields. In concert with previous experiments, we theoretically investigated the mechanisms of 1,3-DPCA of diphenyldiazomethane (DDMf) to two fullerenes (C60 and C70) using the M06-2X density functional method under vacuum and in solvents. To understand the influence of the dipolarophile on these reactions, the 1,3-DPCA of DDMf to three common acceptors, specifically tetracyanoethylene (TCNE), 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), and chloranil (CA), was also studied at the same computational level. The substituent effects on the five reactions were investigated by modeling 1,3-DPCA reactions with 12 different substituted DDMf (DDMs) with five dipolarophiles, totaling 60 reactions. Including the five unsubstituted DDMf reactions, 65 1,3-DPCA reactions were studied. The stereoselectivity, relative reactivity, solvent effects, and distortion/interaction energy model were carefully considered and analyzed based on their corresponding electronic structures, electrostatic potential surfaces, interaction models, solvent models, and thermodynamic data. An intermediate was identified for each of the 65 reactions. A possible biradical pathway for the reactions between DDMf and the two fullerenes was also investigated. The calculated results corroborate and enrich the experimental observations. The conclusion and detailed discussion are generally important for understanding the 1,3-DPCA reactions to fullerenes. Copyright © 2014 John Wiley & Sons, Ltd.
Co-reporter:Cong Hou;Dr. Ya-Jun Liu;Dr. Nicolas Ferré;Dr. Wei-Hai Fang
Chemistry - A European Journal 2014 Volume 20( Issue 26) pp:7979-7986
Publication Date(Web):
DOI:10.1002/chem.201400253
Abstract
Bacterial bioluminescence (BL) has been successfully applied in water-quality monitoring and in vivo imaging. The attention of researchers has been attracted for several decades, but the mechanism of bacterial BL is still largely unknown due to the complexity of the multistep reaction process. Debates mainly focus on three key questions: How is the bioluminophore produced? What is the exact chemical form of the bioluminophore? How does the protein environment affect the light emission? Using quantum mechanics (QM), combined QM and molecular mechanics (QM/MM) and molecular dynamic (MD) calculations in gas-phase, solvent and protein environments, the entire process of bacterial BL was investigated, from flavin reduction to light emission. This investigation revealed that: 1) the chemiluminescent decomposition of flavin peroxyhemiacetal does not occur through the intramolecular chemical initiated electron exchange luminescence (CIEEL) or the “dioxirane” mechanism, as suggested in the literature. Instead, the decomposition occurs according to the charge-transfer initiated luminescence (CTIL) mechanism for the thermolysis of dioxetanone. 2) The first excited state of 4a-hydroxy-4a,5-dihydroFMN (HFOH) was affirmed to be the bioluminophore of bacterial BL. This study provides details regarding the mechanism by which bacterial BL is produced and is helpful in understanding bacterial BL in general.
Co-reporter:Dr. Oleg V. Maltsev;Ling Yue;Dr. Mateusz Rebarz; Lukas Hintermann;Dr. Michel Sliwa;Dr. Cyril Ruckebusch; Ljup&x10d;o Pejov; Ya-Jun Liu; Pan&x10d;e Naumov
Chemistry - A European Journal 2014 Volume 20( Issue 34) pp:10782-10790
Publication Date(Web):
DOI:10.1002/chem.201400210
Abstract
The chemical complexity of oxyluciferin (OxyLH2), the light-emitting molecule in the bioluminescence of fireflies, originates from the possibility of keto/enol tautomerism and single or double deprotonation. Herein, we present detailed infrared spectroscopic analysis of OxyLH2 and several of its chemical isomers and isotopomers. To facilitate the future characterization of its biogenic forms, we provide accurate assignments of the solid-state and solution FTIR spectra of OxyLH2 based on comparison to six isotopically labeled variants ([2-13C]-OxyLH2, [3-15N]-OxyLH2, [4-13C]-OxyLH2, [5-13C]-OxyLH2, [2′-13C]-OxyLH2, [3′-15N]-OxyLH2), five closely related structural analogues, and theoretically computed spectra. The computed DFT harmonic vibrational force fields (B3LYP and M06 functionals with basis sets of varying flexibility up to 6-311++G**) reproduce well the observed shifts in the IR spectra of both isotopically labeled and structurally related analogues.
Co-reporter:Shufeng Chen, Isabelle Navizet, Roland Lindh, Yajun Liu, and Nicolas Ferré
The Journal of Physical Chemistry B 2014 Volume 118(Issue 11) pp:2896-2903
Publication Date(Web):March 6, 2014
DOI:10.1021/jp412198w
Obelia longissima, a tiny hydrozoan living in temperate and cold seas, features the Obelin photoprotein, which emits blue light. The Obelin bioluminescence and the Ca2+-discharged Obelin fluorescence spectra show multimodal characteristics that are currently interpreted by the concomitant participation of several light emitters. Up to now, the coelenteramide luminophore is thought to exist in different protonation states, one of them engaged in an ion-pair with the nearby residue, His22. Using hybrid quantum mechanics/molecular mechanics (QM/MM) calculations, we demonstrate that such an ion-pair cannot exist as a stable light emitter. However, when His22 electric neutrality is maintained by means of another proton transfer, the phenolate state of coelenteramide exhibits emission properties in agreement with experiment. Finally, an alternative nonradiative decay pathway, involving the formation of a diradical excited state, is postulated for the first time.
Co-reporter:Ling Yue and Ya-Jun Liu
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 5) pp:2300-2312
Publication Date(Web):April 15, 2013
DOI:10.1021/ct400206k
The chemiluminescence phenomenon of 3-(2′-spiroadamantyl)-4-methoxy-4-(3″-phosphoryloxy)-phenyl-1,2-dioxetane (AMPPD or m-AMPPD) has been routinely applied in clinical diagnostics. Although the AMPDD chemiluminescence immunoassay is one of the most successful immunoassays, the mechanism of AMPPD chemiluminescence remains largely unknown. The AMPPD chemiluminescence process is composed of three steps: AMPPD is enzymatically triggered to produce 3-(2′-spiroadamantyl)-4-methoxy-4-(3″-hydroxyphenyl)-1,2-dioxetane (m-AMPD); m-AMPD decomposes into the excited-state methyl m-oxybenzoate anion (m-MOB–); the excited-state m-MOB– relaxes to its ground state and emits light. Obviously, the middle step is critical for the chemiluminescence and has not been well understood because of both experimental and theoretical difficulties. We performed the first theoretical study on the chemiluminescent decomposition mechanism of m-AMPD and its para isomer, p-AMPD, using the complete active space self-consistent field and the second-order multiconfigurational perturbation methods in addition to the density functional method. This investigation revealed that (1) neither the intramolecular chemical initiated electron exchange luminescence (CIEEL) nor the concerted charge transfer (CT) mechanism can describe the decomposition of m- and p-AMPD well. Instead, their decomposition occurs according to our previously proposed mechanism of gradually reversible CT-initiated luminescence. (2) The different stabilities of the m- and p-AMPD chemiexcited states might be the basis for the large difference in their chemiluminescence efficiencies. (3) The relationship between the chemiluminescence efficiency and the position of the electron donor on the aromatic ring, the so-called “odd/even selection rule,” does not fully explain the chemiluminescence efficiency of dioxetanes. The odd/even selection rule is only correct for partial dioxetanes, because it does not capture the origin of the relationship between the chemiluminescence and the donor. We revealed that the origin consists of a combination of conjugation, induction, and steric effects. On the basis of this combination of effects, we theoretically designed some 1,2-dioxetanes to guide experimentalists in the synthesis of these excellent chemiluminescent molecules.
Co-reporter:Dr. Shu-Feng Chen; Nicolas Ferré; Ya-Jun Liu
Chemistry - A European Journal 2013 Volume 19( Issue 26) pp:8466-8472
Publication Date(Web):
DOI:10.1002/chem.201300678
Abstract
Aequorea victoria is a type of jellyfish that is known by its famous protein, green fluorescent protein (GFP), which has been widely used as a probe in many fields. Aequorea has another important protein, aequorin, which is one of the members of the EF-hand calcium-binding protein family. Aequorin has been used for intracellular calcium measurements for three decades, but its bioluminescence mechanism remains largely unknown. One of the important reasons is the lack of clear and reliable knowledge about the light emitters, which are complex. Several neutral and anionic forms exist in chemiexcited, bioluminescent, and fluorescent states and are connected with the H-bond network of the binding cavity in the protein. We first theoretically investigated aequorin chemiluminescence, bioluminescence, and fluorescence in real proteins by performing hybrid quantum mechanics and molecular mechanics methods combined with a molecular dynamics method. For the first time, this study reported the origin and clear differences in the chemiluminescence, bioluminescence and fluorescence of aequorin, which is important for understanding the bioluminescence not only of jellyfish, but also of many other marine organisms (that have the same coelenterazine caved in different coelenterazine-type luciferases).
Co-reporter:Ling Yue ; Ya-Jun Liu ;Wei-Hai Fang
Journal of the American Chemical Society 2012 Volume 134(Issue 28) pp:11632-11639
Publication Date(Web):June 21, 2012
DOI:10.1021/ja302979t
The peroxide decomposition that generates the excited-state carbonyl compound is the key step in most organic chemiluminescence, and chemically initiated electron exchange luminescence (CIEEL) has been widely accepted for decades as the general mechanism for this decomposition. The firefly dioxetanone, which is a peroxide, is the intermediate in firefly bioluminescence, and its decomposition is the most important step leading to the emission of visible light by a firefly. However, the firefly dioxetanone decomposition mechanism has never been explored at a reliable theoretical level, because the decomposition process includes biradical, charge-transfer (CT) and several nearly degenerate states. Herein, we have investigated the thermolysis of firefly dioxetanone in its neutral (FDOH) and anionic (FDO–) forms using second-order multiconfigurational perturbation theories in combination with the ground-state intrinsic reaction coordinate calculated via the combined hybrid functional with Coulomb attenuated exchange-correlation, and considered the solvent effect on the ground-state reaction path using the combined hybrid functional with Coulomb attenuated exchange-correlation. The calculated results indicate that the chemiluminescent decomposition of FDOH or FDO– does not take place via the CIEEL mechanism. An entropic trap was found to lead to an excited-state carbonyl compound for FDOH, and a gradually reversible CT initiated luminescence (GRCTIL) was proposed as a new mechanism for the decomposition of FDO–.
Co-reporter:Ling Yue, Daniel Roca-Sanjuán, Roland Lindh, Nicolas Ferré, and Ya-Jun Liu
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 11) pp:4359-4363
Publication Date(Web):September 12, 2012
DOI:10.1021/ct3006562
The chemiluminescent decomposition of 1,2-dioxetanone has in the past been studied by state-of-the-art multireference quantum chemical calculations, and a stepwise biradical mechanism was established. Recently, this decomposition has been reinvestigated, and a concerted mechanism has been proposed based on calculations performed at the closed-shell density functional theory (DFT) level of theory. In order to solve this apparent mechanistic contradiction, the present paper presents restricted and unrestricted DFT results obtained using functionals including different amounts of Hartree–Fock (HF) exchange, repeating and complementing the above-mentioned DFT calculations. The calculated results clearly indicate that the closed-shell DFT methods cannot correctly describe the thermolysis of 1,2-dioxetanone. It is found that unrestricted Kohn–Sham reaction energies and barriers are always lower than the ones obtained using a restricted formalism. Hence, from energy principles, the biradical mechanism is found to be prevailing in the understanding of the 1,2-dioxetanone thermolysis.
Co-reporter:Shu-Feng Chen, Isabelle Navizet, Daniel Roca-Sanjuán, Roland Lindh, Ya-Jun Liu, and Nicolas Ferré
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 8) pp:2796-2807
Publication Date(Web):July 10, 2012
DOI:10.1021/ct300356j
A systematic investigation of the structural and spectroscopic properties of coelenteramide has been performed at the TD-CAM-B3LYP/6-31+G(d,p) level of theory, including various fluorescence and chemiluminescence states. The influence of geometric conformations, solvent polarity, protonation state, and the covalent character of the O–H bond of the hydroxyphenyl moiety were carefully studied. Striking differences in geometries and electronic structures among the states responsible for light emission were characterized. All fluorescence states can be described as a limited charge transfer process for a planar amide moiety. However, the chemiluminescence state is characterized by a much larger charge transfer that takes place over a longer distance. Moreover, the chemiluminescent coelenteramide structure exhibits an amide moiety that is no longer planar, in agreement with recent, more accurate ab initio results [Roca-Sanjuán et al. J. Chem. Theory Comput.2011, 7, 4060]. Because the chemiluminescence state appears to be completely dark, a new mechanism is tentatively introduced for this process.
Co-reporter:Xue-Fen Gao;Cheng-Xing Cui
Journal of Physical Organic Chemistry 2012 Volume 25( Issue 10) pp:850-855
Publication Date(Web):
DOI:10.1002/poc.2930
All possible types of Diels–Alder cycloadditions of 1,3-cis-butadiene to C60 (2 in total) and to C70 (8 in total) were theoretically investigated by the M06-2X density functional method in gas phase and solutions. An intermediate between the reactant and the transition state was located for each reaction. These intermediates except one have not been experimentally or theoretically reported before. The reactivities of the 10 reactions in both the gas phase and solutions were systematically compared based on the calculated results. The present conclusion agrees with the experimental observations and partly disagrees with the previously theoretical conclusion. Copyright © 2012 John Wiley & Sons, Ltd.
Co-reporter:Shu-Feng Chen, Ya-Jun Liu, Isabelle Navizet, Nicolas Ferré, Wei-Hai Fang, and Roland Lindh
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 3) pp:798-803
Publication Date(Web):February 16, 2011
DOI:10.1021/ct200045q
This is a systematic theoretical investigation on all the possible light emitters of firefly using a multireference method. Six chemical forms of oxyluciferin (OxyLH2) molecules/anions were studied by a multistate complete active space second-order perturbation (MS-CASPT2) method in vacuum and dimethyl sulfoxide. The calculated results and subsequent analysis excluded enol-OxyLH2, keto-OxyLH2, and enolate-OxyLH− as possible light emitters. The remaining three candidates, phenolate-enol-OxyLH−, phenolate-keto-OxyLH−, and OxyL2−, were further investigated in protein by a MS-CASPT2/molecular mechanics (MM) study to explain the natural bioluminescence of firefly. By comparison of the MS-CASPT2/MM calculated results of phenolate-enol-OxyLH−, phenolate-keto-OxyLH−, and OxyL2− with the experimental observation and detailed analysis, we concluded that the direct decomposition excited-state product of firefly dioxetanone in vivo and the only light emitter of firefly in natural bioluminescence is the first singlet excited state (S1) of phenolate-keto-OxyLH−.
Co-reporter:Qiu Fang and Ya-Jun Liu
The Journal of Physical Chemistry A 2010 Volume 114(Issue 1) pp:680-684
Publication Date(Web):December 18, 2009
DOI:10.1021/jp908567m
In concert with the latest laser-induced fluorescence (LIF) experiment [Wei et al. J. Phys. Chem. A 2008, 112, 4727], we investigated the photodissociation mechanics of the benzoic acid monomer (BAM) with α C−O fission by means of state-of-the-art ab initio calculations. Complete active space self-consistent-field (CASSCF) and multireference CASSCF second-order perturbation theory (MSCASPT2) calculations were performed on the ground and a number of low-lying excited states of BAM. Our calculations indicated that α C−O fission from the S1 state is in competition with the fission from the T2 state upon the 266−284 nm wavelength photon. This differs from the conclusion of the previous theoretical investigation and clarified the vague experimental conclusion made earlier. According to our calculations, α C−O fission mainly occurs at the T2 state upon photoexcitation at 284−294 nm, and the photon with a wavelength longer than 294 nm is unable to present the α C−O fission. This conclusion agrees with the LIF experimental observation.
Co-reporter:Fengyi Liu ; Yajun Liu ; Luca De Vico ;Roland Lindh
Journal of the American Chemical Society 2009 Volume 131(Issue 17) pp:6181-6188
Publication Date(Web):April 9, 2009
DOI:10.1021/ja808511t
The unimolecular chemiluminescent decomposition of unsubstituted dioxetanone was studied at the complete active space self-consistent field level of theory combined with the multistate second-order multiconfigurational perturbation theory energy correction. The calculations revealed interesting features. Two transition states, two conical intersections, and one intermediate stable biradical structure along the lowest energy reaction path were identified. It was noted that the conical intersections are found at or in very close proximity to the transition states. The first and second transition states correspond to O−O and C−C cleavages, respectively. In particular, a planar structure is supported by the 1(σ,σ*) state during the O−O dissociation up to the first transition state and conical intersection. At this point the 1(σ,σ*) state dissociation path bifurcates, corresponding to a torsion of the O−C−C−O angle. Simultaneously, the 1(n,σ*) state becomes lower in energy while still favoring a planar structure. As the lowest-energy reaction path proceeds toward the second transition state and conical intersection, the 1(n,σ*), 3(n,σ*), and 1(σ,σ*) states are close in energy. This work suggests that the vibrational distribution at the first conical intersection and the interactions among the states as the reaction proceeds between the two transition states are the origin of the population of the chemiluminescent (n,σ*) states.
Co-reporter:Isabelle Navizet ; Ya-Jun Liu ; Nicolas Ferré ; Hong-Yan Xiao ; Wei-Hai Fang ;Roland Lindh
Journal of the American Chemical Society 2009 Volume 132(Issue 2) pp:706-712
Publication Date(Web):December 16, 2009
DOI:10.1021/ja908051h
This is the first report on a multiconfigurational reference second-order perturbation theory−molecular mechanics study of the color modulation of the observed bioluminescence of the oxyluciferin−luciferase complex of the Japanese genji-botaru firefly using structures according to recent X-ray data. Our theoretical results do not support the experimentally deduced conclusion that the color modulation of the emitted light primarily depends on the size of the compact luciferase protein cavity embedding the excited oxyluciferin molecule. Rather, we find, in agreement with recent experimental observations, that the wavelength of the emitted light depends on the polarity of the microenvironment at the phenol/phenolate terminal of the benzothiazole fragment in oxyluciferin.
Co-reporter:Fengyi Liu, Yajun Liu, Luca De Vico, Roland Lindh
Chemical Physics Letters 2009 Volume 484(1–3) pp:69-75
Publication Date(Web):8 December 2009
DOI:10.1016/j.cplett.2009.11.009
Abstract
The gas-phase decompositions of a thiazole-substituted dioxetanone, in both the natural and anionic forms, were investigated theoretically in a CASSCF/CASPT2 study. The neutral conjugated thiazole (with or without a hydroxyl group) substitution on the dioxetanone has no evident effect on the dissociation; however, a subsequent deprotonation – invoking charge-transfer excitations from the thiazole to the dioxetanone moiety – will dramatically change the reaction mechanism from stepwise to concerted, and reduce the activation barrier by ∼8 kcal mol−1. These findings are helpful for the better understanding of dioxetanone chemiluminescence and the charge-transfer induced electron excitation in chemi- and bioluminescence processes.
Co-reporter:Ya-Jun Liu Dr.;Wei-Hai Fang ;Luca De Vico Dr.;Rol Lindh and
ChemPhysChem 2007 Volume 8(Issue 6) pp:890-898
Publication Date(Web):12 MAR 2007
DOI:10.1002/cphc.200600737
The UV photodissociation (<5 eV) of diiodomethane (CH2I2) is investigated by spin-orbit ab initio calculations. The experimentally observed photodissociation channels in the gas and condensed phases are clearly assigned by multi-state second-order multiconfigurational perturbation theory in conjunction with spin-orbit interaction through complete active space-state interaction potential energy curves. The calculated results indicate that the fast dissociations of the first two singlet states of CH2I2 and CH2II lead to geminate-radical products, CH2I .+I(2P3/2) or CH2I .+ I*(2P1/2). The recombination process from CH2II to CH2I2 is explained by an isomerization process and a secondary photodissociation reaction of CH2II. Finally, the study reveals that spin-orbits effects are significant in the quantitative analysis of the electronic spectrum of the CH2II species.
Co-reporter:YaJun Liu;WeiHai Fang
Science China Chemistry 2007 Volume 50( Issue 6) pp:725-730
Publication Date(Web):2007 December
DOI:10.1007/s11426-007-0127-4
The ground state (S0) geometry of the firefly luciferin (LH2) was optimized by both DFT B3LYP and CASSCF methods. The vertical excitation energies (Tv) of three low-lying states (S1, S2, and S3) were calculated by TD-DFT B3LYP//CASSCF method. The S1 geometry was optimized by CASSCF method. Its Tv and the transition energy (Te) were calculated by MS-CASPT2//CASSCF method. Both the TD-DFT and MS-CASPT2 calculated S1 state Tv values agree with the experimental one. The IPEA shift greatly affects the MS-CASPT2 calculated Tv values. Some important excited states of LH2 and oxyluciferin (oxyLH2) are charge-transfer states and have more than one dominant configuration, so for deeply researching the firefly bioluminescence, the multireference calculations are desired.
Co-reporter:HongJiang Ren, ChengXing Cui, XiaoJun Li, YaJun Liu
International Journal of Hydrogen Energy (5 January 2017) Volume 42(Issue 1) pp:312-321
Publication Date(Web):5 January 2017
DOI:10.1016/j.ijhydene.2016.10.151











![Butanamide,N-[3-[3-[(aminoiminomethyl)amino]propyl]-5-(1H-indol-3-yl)pyrazinyl]-2-methyl-(9CI)](http://img.cochemist.com/ccimg/17300/17297-78-8.png)
![Butanamide,N-[3-[3-[(aminoiminomethyl)amino]propyl]-5-(1H-indol-3-yl)pyrazinyl]-2-methyl-(9CI)](http://img.cochemist.com/ccimg/17300/17297-78-8_b.png)
![2-[3-[2-[(2S)-BUTAN-2-YL]-6-(1H-INDOL-3-YL)-3-OXO-7H-IMIDAZO[1,2-A]PYRAZIN-8-YL]PROPYL]GUANIDINE](http://img.cochemist.com/ccimg/7300/7273-34-9.png)
![2-[3-[2-[(2S)-BUTAN-2-YL]-6-(1H-INDOL-3-YL)-3-OXO-7H-IMIDAZO[1,2-A]PYRAZIN-8-YL]PROPYL]GUANIDINE](http://img.cochemist.com/ccimg/7300/7273-34-9_b.png)
![(S)-2-(6-Hydroxybenzo[d]thiazol-2-yl)-4,5-dihydrothiazole-4-carboxylic acid](/data/chemimg/473800/2591-17-5.png)
![(S)-2-(6-Hydroxybenzo[d]thiazol-2-yl)-4,5-dihydrothiazole-4-carboxylic acid](/data/chemimg/473800/2591-17-5_b.png)
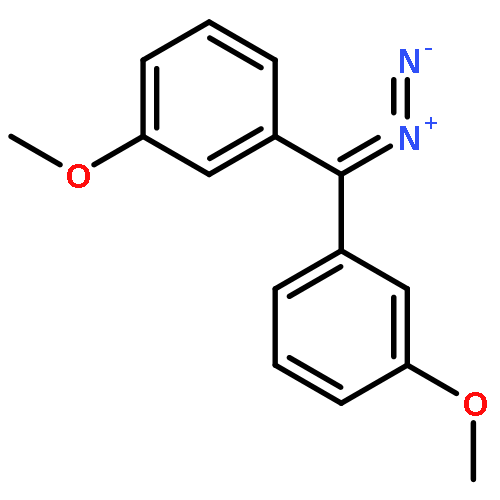
![[bis(4-methoxyphenyl)methylidene]diazenium](http://img.cochemist.com/ccimg/1300/1221-72-3.png)
![[bis(4-methoxyphenyl)methylidene]diazenium](http://img.cochemist.com/ccimg/1300/1221-72-3_b.png)