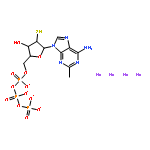
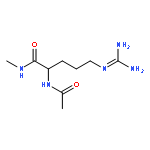
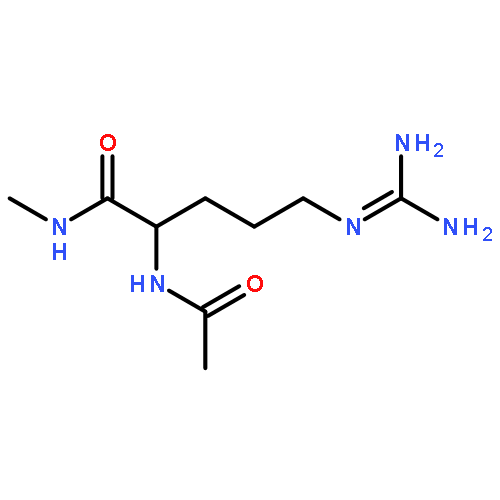
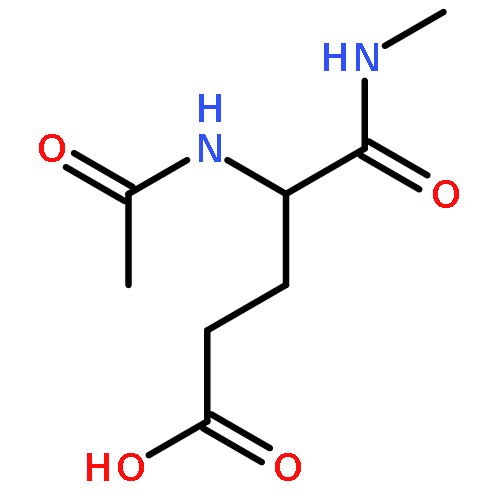
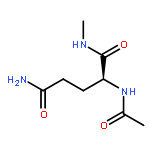
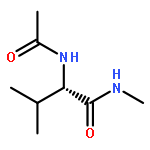
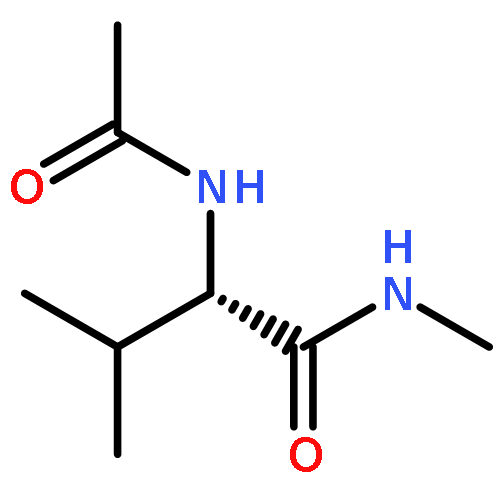
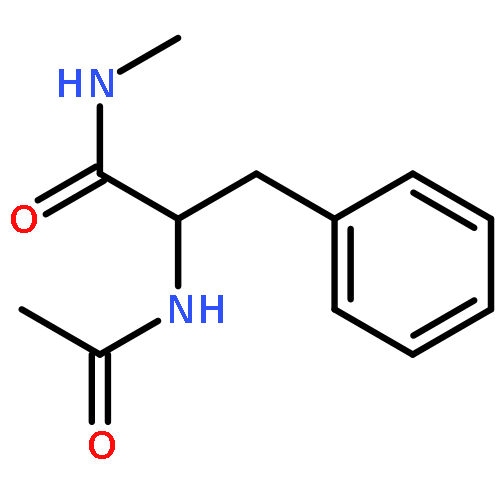
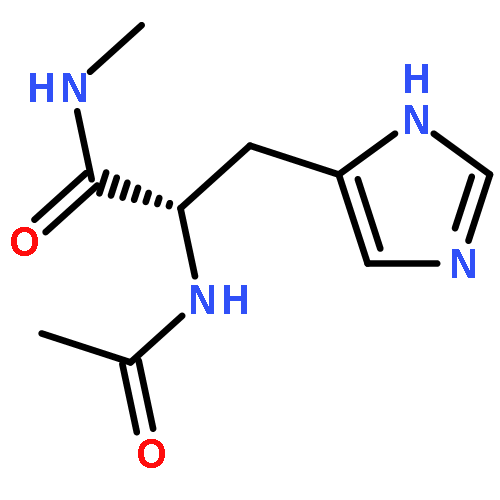
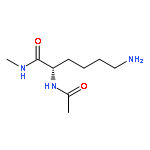
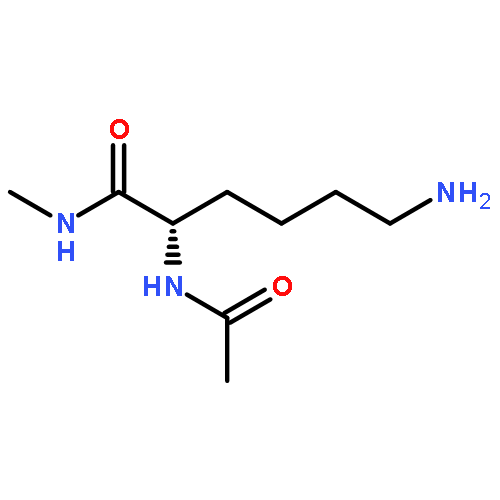
Co-reporter:Ruxi Qi, Wesley M. Botello-Smith, and Ray Luo
Journal of Chemical Theory and Computation July 11, 2017 Volume 13(Issue 7) pp:3378-3378
Publication Date(Web):May 29, 2017
DOI:10.1021/acs.jctc.7b00336
Electrostatic interactions play crucial roles in biophysical processes such as protein folding and molecular recognition. Poisson–Boltzmann equation (PBE)-based models have emerged as widely used in modeling these important processes. Though great efforts have been put into developing efficient PBE numerical models, challenges still remain due to the high dimensionality of typical biomolecular systems. In this study, we implemented and analyzed commonly used linear PBE solvers for the ever-improving graphics processing units (GPU) for biomolecular simulations, including both standard and preconditioned conjugate gradient (CG) solvers with several alternative preconditioners. Our implementation utilizes the standard Nvidia CUDA libraries cuSPARSE, cuBLAS, and CUSP. Extensive tests show that good numerical accuracy can be achieved given that the single precision is often used for numerical applications on GPU platforms. The optimal GPU performance was observed with the Jacobi-preconditioned CG solver, with a significant speedup over standard CG solver on CPU in our diversified test cases. Our analysis further shows that different matrix storage formats also considerably affect the efficiency of different linear PBE solvers on GPU, with the diagonal format best suited for our standard finite-difference linear systems. Further efficiency may be possible with matrix-free operations and integrated grid stencil setup specifically tailored for the banded matrices in PBE-specific linear systems.
Co-reporter:Li Xiao, Changhao Wang, Xiang Ye, and Ray Luo
The Journal of Physical Chemistry B 2016 Volume 120(Issue 33) pp:8707-8721
Publication Date(Web):May 4, 2016
DOI:10.1021/acs.jpcb.6b04439
Continuum solvation modeling based upon the Poisson–Boltzmann equation (PBE) is widely used in structural and functional analysis of biomolecules. In this work, we propose a charge-central interpretation of the full nonlinear PBE electrostatic interactions. The validity of the charge-central view or simply charge view, as formulated as a vacuum Poisson equation with effective charges, was first demonstrated by reproducing both electrostatic potentials and energies from the original solvated full nonlinear PBE. There are at least two benefits when the charge-central framework is applied. First the convergence analyses show that the use of polarization charges allows a much faster converging numerical procedure for electrostatic energy and forces calculation for the full nonlinear PBE. Second, the formulation of the solvated electrostatic interactions as effective charges in vacuum allows scalable algorithms to be deployed for large biomolecular systems. Here, we exploited the charge-view interpretation and developed a particle–particle particle–mesh (P3M) strategy for the full nonlinear PBE systems. We also studied the accuracy and convergence of solvation forces with the charge-view and the P3M methods. It is interesting to note that the convergence of both the charge-view and the P3M methods is more rapid than the original full nonlinear PBE method. Given the developments and validations documented here, we are working to adapt the P3M treatment of the full nonlinear PBE model to molecular dynamics simulations.
Co-reporter:D’Artagnan Greene, Wesley M. Botello-Smith, Alec Follmer, Li Xiao, Eleftherios Lambros, and Ray Luo
The Journal of Physical Chemistry B 2016 Volume 120(Issue 48) pp:12293-12304
Publication Date(Web):November 10, 2016
DOI:10.1021/acs.jpcb.6b09535
Membrane proteins, due to their roles as cell receptors and signaling mediators, make prime candidates for drug targets. The computational analysis of protein–ligand binding affinities has been widely employed as a tool in rational drug design efforts. Although efficient implicit solvent-based methods for modeling globular protein–ligand binding have been around for many years, the extension of such methods to membrane protein–ligand binding is still in its infancy. In this study, we extended the widely used Amber/MMPBSA method to model membrane protein–ligand systems, and we used it to analyze protein–ligand binding for the human purinergic platelet receptor (P2Y12R), a prominent drug target in the inhibition of platelet aggregation for the prevention of myocardial infarction and stroke. The binding affinities, computed by the Amber/MMPBSA method using standard parameters, correlate well with experiment. A detailed investigation of these parameters was conducted to assess their impact on the accuracy of the method. These analyses show the importance of properly treating the nonpolar solvation interactions and the electrostatic polarization in the binding of nucleotide agonists and non-nucleotide antagonists to P2Y12R. On the basis of the crystal structures and the experimental conditions in the binding assay, we further hypothesized that the nucleotide agonists lose their bound magnesium ion upon binding to P2Y12R, and our computational study supports this hypothesis. Ultimately, this work illustrates the value of computational analysis in the interpretation of experimental binding reactions.
Co-reporter:Wei Ye, Dingjue Ji, Wei Wang, Ray Luo, and Hai-Feng Chen
Journal of Chemical Information and Modeling 2015 Volume 55(Issue 5) pp:1021-1029
Publication Date(Web):April 28, 2015
DOI:10.1021/acs.jcim.5b00043
Over 40% of eukaryotic proteomic sequences have been predicted to be intrinsically disordered proteins (IDPs) or intrinsically disordered regions (IDRs) and confirmed to be associated with many diseases. However, widely used force fields cannot well reproduce the conformers of IDPs. Previously the ff99IDPs force field was released to simulate IDPs with CMAP energy corrections for the eight disorder-promoting residues. In order to further confirm the performance of ff99IDPs, three representative IDP systems (arginine-rich HIV-1 Rev, aspartic proteinase inhibitor IA3, and α-synuclein) were used to test and evaluate the simulation results. The results show that for free disordered proteins, the chemical shifts from the ff99IDPs simulations are in quantitative agreement with those from reported NMR measurements and better than those from ff99SBildn. Thus, ff99IDPs can sample more clusters of disordered conformers than ff99SBildn. For structural proteins, both ff99IDPs and ff99SBildn can well reproduce the conformations. In general, ff99IDPs can successfully be used to simulate the conformations of IDPs and IDRs in both bound and free states. However, relative errors could still be found at the boundaries of ordered residues scattered in long disorder-promoting sequences. Therefore, polarizable force fields might be one of the possible ways to further improve the performance on IDPs.
Co-reporter:Wesley M. Botello-Smith
Journal of Chemical Information and Modeling 2015 Volume 55(Issue 10) pp:2187-2199
Publication Date(Web):September 21, 2015
DOI:10.1021/acs.jcim.5b00341
Continuum solvent models have been widely used in biomolecular modeling applications. Recently much attention has been given to inclusion of implicit membranes into existing continuum Poisson–Boltzmann solvent models to extend their applications to membrane systems. Inclusion of an implicit membrane complicates numerical solutions of the underlining Poisson–Boltzmann equation due to the dielectric inhomogeneity on the boundary surfaces of a computation grid. This can be alleviated by the use of the periodic boundary condition, a common practice in electrostatic computations in particle simulations. The conjugate gradient and successive over-relaxation methods are relatively straightforward to be adapted to periodic calculations, but their convergence rates are quite low, limiting their applications to free energy simulations that require a large number of conformations to be processed. To accelerate convergence, the Incomplete Cholesky preconditioning and the geometric multigrid methods have been extended to incorporate periodicity for biomolecular applications. Impressive convergence behaviors were found as in the previous applications of these numerical methods to tested biomolecules and MMPBSA calculations.
Co-reporter:Lishi Xu, Wei Ye, Cheng Jiang, Jingxu Yang, Jinmai Zhang, Yan Feng, Ray Luo, and Hai-Feng Chen
The Journal of Physical Chemistry B 2015 Volume 119(Issue 7) pp:2844-2856
Publication Date(Web):January 29, 2015
DOI:10.1021/jp510940w
Lac repressor is a DNA-binding protein which inhibits the expression of a series of genes involved in lactose metabolism. Lac repressor can bind at a random DNA site via nonspecific interactions; then, it rapidly translocates through the double chain of DNA until it finds the specific binding site. Therefore, the site transform between these two modes is essential for the specific recognition between Lac repressor and DNA. Here, the recognition mechanism between Lac repressor and DNA was illustrated with molecular dynamics simulations and correlation network analyses. We have found that the correlation network of the specific system (2KEI) is more centralized and denser than that of the nonspecific system (1OSL). The significant difference in the networks between the nonspecific and specific systems is apparently due to the different binding modes. Then, different interaction modes were found where electrostatic and hydrogen bonding interactions in the nonspecific system are stronger than those in the specific system. Hydrophobic interactions were found only in specific complexes and mostly focused on the hinge helices. Furthermore, the hinge helix will induce the bending of DNA for the specific system. At the same time, a common specific sequence of DNA was revealed for three specific systems. Then, two design systems (positive and control) were used to evaluate the specific recognition between DNA and Lac repressor. These combined methods can be used to reveal the recognition mechanism between other transcription factors and DNA.
Co-reporter:Wei Wang;Wei Ye;Cheng Jiang;Hai-Feng Chen
Chemical Biology & Drug Design 2014 Volume 84( Issue 3) pp:253-269
Publication Date(Web):
DOI:10.1111/cbdd.12314
Intrinsically disordered proteins or intrinsically disordered protein regions comprise a large portion of eukaryotic proteomes (between 35% and 51%). These intrinsically disordered proteins were found to link with cancer and various other diseases. However, widely used additive force field parameter sets are insufficient in quantifying the structural properties of intrinsically disordered proteins. Therefore, we explored to a systematic correction of a base additive force field parameter set (chosen as Amber ff99SBildn) to correct the biases that was first demonstrated in simulations with the base parameter set. Specifically, the φ/ψ distributions of disorder-promoting residues were systematically corrected with the CMAP method. Our simulations show that the CMAP corrected Amber parameter set, termed ff99IDPs, improves the φ/ψ distributions of the disorder-promoting residues with respect to the benchmark data of intrinsically disordered protein structures, with root mean-squared percentage deviation less than 0.15% between the simulation and the benchmark. Our further validation shows that the chemical shifts from the ff99IDPs simulations are in quantitative agreement with those from reported NMR measurements for two tested IDPs, MeV NTAIL, and p53. The predicted residue dipolar couplings also show high correlation with experimental data. Interestingly, our simulations show that ff99IDPs can still be used to model the ordered state when the intrinsically disordered proteins are in complex, in contrast to ff99SBildn that can be applied well only to the ordered complex structures. These findings confirm that the newly proposed Amber ff99IDPs parameter set provides a reasonable tool in further studies of intrinsically disordered protein structures. In addition, our study also shows the importance of considering intrinsically disordered protein structures in general-purposed force field developments for both additive and non-additive models.
Co-reporter:Qingfen Yu, Wei Ye, Cheng Jiang, Ray Luo, and Hai-Feng Chen
The Journal of Physical Chemistry B 2014 Volume 118(Issue 43) pp:12426-12434
Publication Date(Web):October 9, 2014
DOI:10.1021/jp5079289
The KH3 domain of Nova-2 protein can precisely recognize the sequence-specific target RNA of human glycine receptor α2. However, the recognition mechanism between the protein and its target RNA is still hotly debated. In this study, molecular dynamic simulations in explicit solvent were utilized to understand the recognition mechanism. The structural analysis and the Kolmogorov–Smirnov P-test statistics reveal that the KH3 domain might obey a conformational selection mechanism upon RNA binding. However, the induced fit mechanism could not be completely ruled out. Unfolding kinetics indicates that the folding of RNA and KH3 happens first and then the binding between RNA and KH3 follows. Principle component analysis shows that the invariant Gly-Lys-Gly-Gly loop moves toward to the RNA molecule but the C-terminal domain moves away from the RNA molecule upon binding. These specific dominant motions were hypothesized to stabilize the complex structure. The hydrophobic and hydrogen bonding interactions were found to be the driving forces for the specific recognition, in contrast to the dominant electrostatic interactions for nonspecific recognition.
Co-reporter:Wesley M. Botello-Smith, Xingping Liu, Qin Cai, Zhilin Li, Hongkai Zhao, Ray Luo
Chemical Physics Letters 2013 Volume 555() pp:274-281
Publication Date(Web):3 January 2013
DOI:10.1016/j.cplett.2012.10.081
Abstract
Membrane protein systems are important computational research topics due to their roles in rational drug design. In this study, we developed a continuum membrane model utilizing a level set formulation under the numerical Poisson–Boltzmann framework within the Amber molecular mechanics suite for applications such as protein–ligand binding affinity and docking pose predictions. Two numerical solvers were adapted for periodic systems to alleviate possible edge effects. Validation on systems ranging from organic molecules to membrane proteins up to 200 residues, demonstrated good numerical properties. This lays foundations for sophisticated models with variable dielectric treatments and second-order accurate modeling of solvation interactions.
Co-reporter:Jun Wang, Qin Cai, Ye Xiang, and Ray Luo
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 8) pp:2741-2751
Publication Date(Web):June 18, 2012
DOI:10.1021/ct300341d
Grid dependence in numerical reaction field energies and solvation forces is a well-known limitation in the finite-difference Poisson–Boltzmann methods. In this study, we have investigated several numerical strategies to overcome the limitation. Specifically, we have included trimeric solvent accessible arc dots during analytical molecular surface generation to improve the convergence of numerical reaction field energies and solvation forces. We have also utilized the level set function to trace the molecular surface implicitly to simplify the numerical mapping of the grid-independent molecular surface. We have further explored combining the weighted harmonic averaging of boundary dielectrics with a charge-based approach to improve the convergence and stability of numerical reaction field energies and solvation forces. Our test data show that the convergence and stability in both numerical energies and forces can be improved significantly when the combined strategy is applied to either the Poisson equation or the full Poisson–Boltzmann equation.
Co-reporter:Qin Cai, Xiang Ye and Ray Luo
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 45) pp:15917-15925
Publication Date(Web):08 Oct 2012
DOI:10.1039/C2CP43237D
Continuum solvation representations based on the Poisson–Boltzmann equation have become widely accepted in biomolecular applications after years of basic research and development. Since analytical solution of the differential equation can be achieved only in a few specific cases with simple solute geometry, only numerical solution is possible for biomolecular applications. However, it is conceptually difficult to assign solvation forces in the numerical methods, limiting their applications into direct simulations of energy minimization and molecular dynamics. In this study a dielectric pressure formulation was derived from the general Maxwell stress tensor for continuum solvation of biomolecules modeled with the widely used abrupt-transitioned dielectrics. A charge-central strategy was then proposed to improve the numerical behavior of the computed pressure. An interesting observation is the highly similar charge-central formulations between the smooth-transition dielectric and the abrupt-transition dielectric models utilized in the biomolecular solvation treatments. The connections of the new formulation with both the Davis–McCammon and Gilson et al. approaches were further presented after applying the normal field approximation. The consistency was verified with the numerical tests on a realistic biomolecule. The numerical experiments on the tested biomolecule further indicate that the charge-central strategy combined with the normal field approximation not only improves the accuracy of the dielectric boundary force but also reduces its grid dependence for biomolecular applications.
Co-reporter:Qin Cai, Xiang Ye, Jun Wang, Ray Luo
Chemical Physics Letters 2011 Volume 514(4–6) pp:368-373
Publication Date(Web):6 October 2011
DOI:10.1016/j.cplett.2011.08.067
Abstract
Continuum modeling of electrostatic interactions based upon the numerical solutions of the Poisson–Boltzmann equation has been widely adopted in biomolecular applications. To extend their applications to molecular dynamics and energy minimization, robust and efficient methodologies to compute solvation forces must be developed. In this study, we have first reviewed the theory for the computation of dielectric boundary force based on the definition of the Maxwell stress tensor. This is followed by a new formulation of the dielectric boundary force suitable for the finite-difference Poisson–Boltzmann methods. We have validated the new formulation with idealized analytical systems and realistic molecular systems.
Co-reporter:Xiang Ye, Jun Wang and Ray Luo
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 4) pp:1157-1169
Publication Date(Web):March 1, 2010
DOI:10.1021/ct900318u
A revised density function is developed to define the molecular surface for the numerical Poisson−Boltzmann methods to achieve a better convergence and a higher numerical stability. The new density function does not use any predefined functional form but is numerically optimized to reproduce the reaction field energies computed with the solvent excluded surface definition. An exhaustive search in the parameter space is utilized in the optimization, using a wide-range of training molecules including proteins, nucleic acids, and peptides in both folded and unfolded conformations. A cubic-spline function is introduced to guarantee good numerical behavior of the new density function. Our test results show that the average relative energy errors computed with the revised density function are uniformly lower than 1% for both training and test molecules with different sizes and conformations. Our transferability analysis shows that the performance of the new method is mostly size and conformation independent. A detailed analysis further shows that the numerical forces computed with the revised density function converge better with respect to the grid spacing and are numerically more stable in tested peptides.
Co-reporter:Jun Wang, Qin Cai, Zhi-Lin Li, Hong-Kai Zhao, Ray Luo
Chemical Physics Letters 2009 Volume 468(4–6) pp:112-118
Publication Date(Web):22 January 2009
DOI:10.1016/j.cplett.2008.12.049
Violation of energy conservation in Poisson–Boltzmann molecular dynamics, due to the limited accuracy and precision of numerical methods, is a major bottleneck preventing its wide adoption in biomolecular simulations. We explored the ideas of enforcing interface conditions by the immerse interface method and of removing charge singularity to improve the finite-difference methods. Our analysis of these ideas on an analytical test system shows significant improvement in both energies and forces. Our analysis further indicates the need for more accurate force calculation, especially the boundary force calculation.A new Poisson–Boltzmann method was proposed to achieve energy conservation in molecular dynamics simulations.
Co-reporter:Qin Cai, Xiang Ye and Ray Luo
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 45) pp:NaN15925-15925
Publication Date(Web):2012/10/08
DOI:10.1039/C2CP43237D
Continuum solvation representations based on the Poisson–Boltzmann equation have become widely accepted in biomolecular applications after years of basic research and development. Since analytical solution of the differential equation can be achieved only in a few specific cases with simple solute geometry, only numerical solution is possible for biomolecular applications. However, it is conceptually difficult to assign solvation forces in the numerical methods, limiting their applications into direct simulations of energy minimization and molecular dynamics. In this study a dielectric pressure formulation was derived from the general Maxwell stress tensor for continuum solvation of biomolecules modeled with the widely used abrupt-transitioned dielectrics. A charge-central strategy was then proposed to improve the numerical behavior of the computed pressure. An interesting observation is the highly similar charge-central formulations between the smooth-transition dielectric and the abrupt-transition dielectric models utilized in the biomolecular solvation treatments. The connections of the new formulation with both the Davis–McCammon and Gilson et al. approaches were further presented after applying the normal field approximation. The consistency was verified with the numerical tests on a realistic biomolecule. The numerical experiments on the tested biomolecule further indicate that the charge-central strategy combined with the normal field approximation not only improves the accuracy of the dielectric boundary force but also reduces its grid dependence for biomolecular applications.
.jpg)