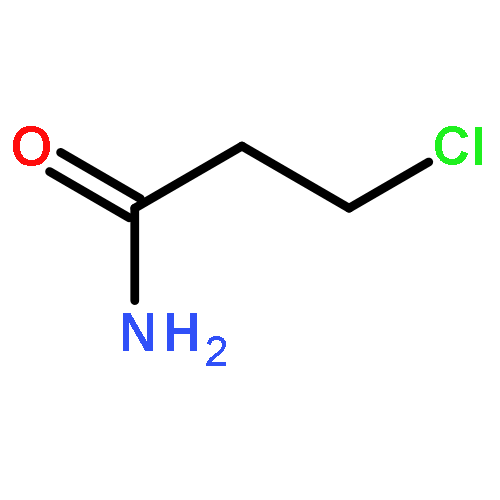
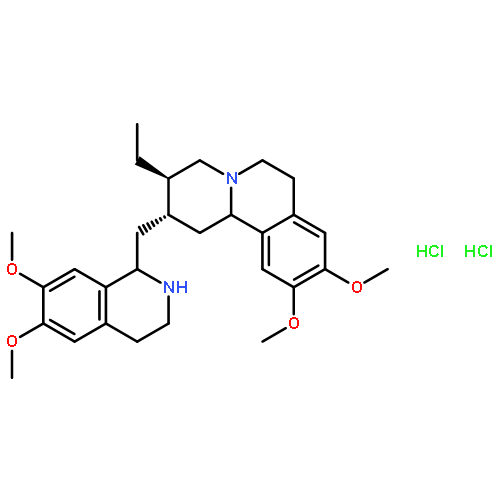
Co-reporter:Emmanuel S. Akinboye, Zebalda D. Bamji, Bernard Kwabi-Addo, David Ejeh, Robert L. Copeland, Samuel R. Denmeade, Oladapo Bakare
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 17) pp:5839-5845
Publication Date(Web):1 September 2015
DOI:10.1016/j.bmc.2015.06.072
A small library of emetine dithiocarbamate ester derivatives were synthesized in 25–86% yield via derivatization of the N2′- position of emetine. Anticancer evaluation of these compounds in androgen receptor positive LNCaP and androgen receptor negative PC3 and DU145 prostate cancer cell lines revealed time dependent and dose-dependent cytotoxicity. With the exception of compound 4c, all the dithiocarbamate ester analogs in this study showed appreciable potency in all the prostate cancer cell lines (regardless of whether it is androgen receptor positive or negative) with a cytotoxicity IC50 value ranging from 1.312 ± 0.032 μM to 5.201 ± 0.125 μM by day 7 of treatment. Compared to the sodium dithiocarbamate salt 1, all the dithiocarbamate ester analogs (2 and 4a–4g) displayed lower cytotoxicity than compound 1 (PC3, IC50 = 0.087 ± 0.005 μM; DU145, IC50 = 0.079 ± 0.003 μM and LNCaP, IC50 = 0.079 ± 0.003 μM) on day 7 of treatment. Consequently, it appears that S-alkylation of compound 1 leads to a more stable dithiocarbamate ester derivative that resulted in lower anticancer activity in the prostate cancer cell lines.Anticancer activities of 4a to 4g in LNCaP, PC3 and DU145 prostate cancer cell lines, IC50 value ranging from 1.312 ± 0.032 μM to 5.201 ± 0.125 μM.
Co-reporter:Emmanuel S. Akinboye ; Marc D. Rosen ; Samuel R. Denmeade ; Bernard Kwabi-Addo
Journal of Medicinal Chemistry 2012 Volume 55(Issue 17) pp:7450-7459
Publication Date(Web):August 6, 2012
DOI:10.1021/jm300426q
The N-2′ position of the natural product emetine has been derivatized to thiourea, urea, sulfonamide, dithiocarbamate, carbamate, and pH responsive hydrolyzable amide analogues. In vitro studies of these analogues in PC3 and LNCaP prostate cancer cell lines showed that the analogues are generally less cytotoxic (average IC50 ranging from 0.079 to 10 μM) than emetine (IC50 ranging from 0.0237 to 0.0329 μM). The pH sensitive sodium dithiocarbamate salt 13 and the amide analogues 21, 22, 26 (obtained from maleic and citraconic anhydrides) showed the most promise as acid-activatable prodrugs under mildly acidic conditions found in the cancer microenvironment. These prodrugs released 12–83% of emetine at pH 6.5 and 41–95% emetine at pH 5.5. Compounds 13 and 26 were further shown to exhibit increased cytotoxicity in PC3 cell culture medium that was already below pH 7.0 at the time of treatment.
Co-reporter:Solomon Berhe, Andrew Slupe, Choice Luster, Henry A. Charlier Jr., Don L. Warner, Leon H. Zalkow, Edward M. Burgess, Nkechi M. Enwerem, Oladapo Bakare
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 1) pp:134-141
Publication Date(Web):1 January 2010
DOI:10.1016/j.bmc.2009.11.011
A series of indazole-dione derivatives were synthesized by the 1,3-dipolar cycloaddition reaction of appropriate substituted benzoquinones or naphthoquinones and N-carboalkoxyamino diazopropane derivatives. These compounds were evaluated for their effects on human carbonyl reductase. Several of the analogs were found to serve as substrates for carbonyl reductase with a wide range of catalytic efficiencies, while four analogs display inhibitory activities with IC50 values ranging from 3–5 μM. Two of the inhibitors were studied in greater detail and were found to be noncompetitive inhibitors against both NADPH and menadione with KI values ranging between 2 and 11 μM. Computational studies suggest that conformation of the compounds may determine whether the indazole-diones bind productively to yield product or nonproductively to inhibit the enzyme.A series of indazole-dione derivatives were synthesized and their biological activities on human carbonyl reductase are studied.
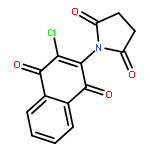
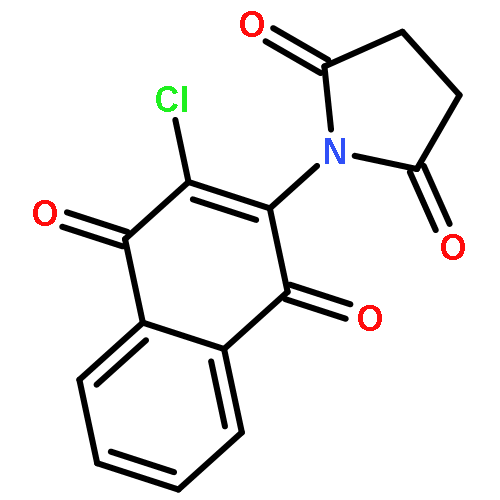
Co-reporter:Oladapo Bakare, Curtis L. Ashendel, Hairuo Peng, Leon H. Zalkow, Edward M. Burgess
Bioorganic & Medicinal Chemistry 2003 Volume 11(Issue 14) pp:3165-3170
Publication Date(Web):17 July 2003
DOI:10.1016/S0968-0896(03)00267-0
Mitogen activated protein kinases are of interest as research tools and as therapeutic target for certain physiological disorders. In this study, we found 2-chloro-3-(N-succinimidyl)-1,4-naphthoquinone 6 to be a selective inhibitor of MEK1 with an IC50 of 0.38 μM. An open-chain homologue, 10, showed selective cytotoxicity against renal cancer in the NCI in vitro tumor screening. Structure–activity relationship study of eight compounds showed the cyclic imido-substituted chloro-1,4-naphthoquinone as more potent and selective MEK1 inhibitors than the open chain homologues. The imido-substituted chloro-1,4-naphthoquinones were synthesized in a straightforward fashion by refluxing 2-amino-3-chloro-1,4-naphthoquinone with the appropriate acid chloride or diacyl dichloride.2-Chloro-3-(N-succinimidyl)-1,4-naphthoquinone 6 was found to be a selective inhibitor of MEK1 with an IC50 of 0.38 μM, while an open-chain homologue 10 showed selective cytotoxicity against renal cancer cell lines in the NCI in vitro tumor screening.





![4-Thiomorpholinamine,3-methyl-N-[(5-nitro-2-furanyl)methylene]-, 1,1-dioxide](http://img.cochemist.com/ccimg/23300/23256-30-6.png)
![4-Thiomorpholinamine,3-methyl-N-[(5-nitro-2-furanyl)methylene]-, 1,1-dioxide](http://img.cochemist.com/ccimg/23300/23256-30-6_b.png)