Co-reporter:Julien Brioche; Sarah J. Pike; Sofja Tshepelevitsh; Ivo Leito; Gareth A. Morris; Simon J. Webb
Journal of the American Chemical Society 2015 Volume 137(Issue 20) pp:6680-6691
Publication Date(Web):April 27, 2015
DOI:10.1021/jacs.5b03284
Biomolecular systems are able to respond to their chemical environment through reversible, selective, noncovalent intermolecular interactions. Typically, these interactions induce conformational changes that initiate a signaling cascade, allowing the regulation of biochemical pathways. In this work, we describe an artificial molecular system that mimics this ability to translate selective noncovalent interactions into reversible conformational changes. An achiral but helical foldamer carrying a basic binding site interacts selectively with the most acidic member of a suite of chiral ligands. As a consequence of this noncovalent interaction, a global absolute screw sense preference, detectable by 13C NMR, is induced in the foldamer. Addition of base, or acid, to the mixture of ligands competitively modulates their interaction with the binding site, and reversibly switches the foldamer chain between its left and right-handed conformations. As a result, the foldamer–ligand mixture behaves as a biomimetic chemical system with emergent properties, functioning as a “proton-counting” molecular device capable of providing a tunable, pH-dependent conformational response to its environment.
Co-reporter:Bryden A. F. Le Bailly, Liam Byrne, Vincent Diemer, Mohammadali Foroozandeh, Gareth A. Morris and Jonathan Clayden
Chemical Science 2015 vol. 6(Issue 4) pp:2313-2322
Publication Date(Web):21 Jan 2015
DOI:10.1039/C4SC03944K
Although foldamers, by definition, are extended molecular structures with a well-defined conformation, minor conformers must be populated at least to some extent in solution. We present a quantitative analysis of these minor conformers for a series of helical oligomers built from achiral but helicogenic α-amino acids. By measuring the chain length dependence or chain position dependence of NMR or CD quantities that measure screw-sense preference in a helical oligomer, we quantify values for the decay constant of a conformational signal as it passes through the molecular structure. This conformational signal is a perturbation of the racemic mixture of M and P helices that such oligomers typically adopt by the inclusion of an N or C terminal chiral inducer. We show that decay constants may be very low (<1% signal loss per residue) in non-polar solvents, and we evaluate the increase in decay constant that results in polar solvents, at higher temperatures, and with more conformationally flexible residues such as Gly. Decay constants are independent of whether the signal originates from the N or the C terminus. By interpreting the decay constant in terms of the probability with which conformations containing a screw-sense reversal are populated, we quantify the populations of these alternative minor conformers within the overall ensemble of secondary structures adopted by the foldamer. We deduce helical persistence lengths for Aib polymers that allow us to show that in a non-polar solvent a peptide helix, even in the absence of chiral residues, may continue with the same screw sense for approximately 200 residues.
Co-reporter:Michael B. Tait, Sam Butterworth, and Jonathan Clayden
Organic Letters 2015 Volume 17(Issue 5) pp:1236-1239
Publication Date(Web):February 18, 2015
DOI:10.1021/acs.orglett.5b00199
2-Aryltetrahydropyridines formed by anionic cyclization or ring-closing metathesis were converted to their N′-aryl urea derivatives. Depending on the position of the unsaturation within the tetrahydropyridine ring, metalation by deprotonative lithiation or carbolithiation led to migration of the N′-aryl substituent to the 2- or 6-position via intramolecular nucleophilic attack of a benzylic organolithium on the aryl ring. The products are a range of 2,2-, 2,2,3-, and 2,6-polysubstituted piperidine derivatives. Related chemistry was observed in pyrroline homologues.
Co-reporter:Fernando Fernández-Nieto, Josep Mas Roselló, Simone Lenoir, Simon Hardy, and Jonathan Clayden
Organic Letters 2015 Volume 17(Issue 15) pp:3838-3841
Publication Date(Web):July 22, 2015
DOI:10.1021/acs.orglett.5b01803
Palladium(II) trifluoroacetate (5 mol %) catalyzes the C-arylation of N,N-disubstituted hydantoins by aryl iodides in good yield. The reaction proceeds through base-promoted enolization of the amino acid derived hydantoins, and the resulting 5,5-disubstituted hydantoins may be deprotected at one or both N atoms to yield biologically active structures or alternatively hydrolyzed to the parent α-aryl α-amino acids. The reaction is successful with a variety of parent amino acids and a range of electron-rich and electron-poor aryl iodides.
Co-reporter:Liam Byrne, Jordi Solà and Jonathan Clayden
Chemical Communications 2015 vol. 51(Issue 54) pp:10965-10968
Publication Date(Web):02 Jun 2015
DOI:10.1039/C5CC01790D
Helical peptides built principally from the achiral quaternary amino acid Aib but with an induced preferred screw-sense exhibit enantioselectivity in their chain-extension reactions when presented with a racemic tertiary amino acid. This is the first demonstration that secondary structure alone, in the absence of local chiral residues, can direct the enantioselectivity of peptide coupling.
Co-reporter:Julien Maury, Bryden A. F. Le Bailly, James Raftery and Jonathan Clayden
Chemical Communications 2015 vol. 51(Issue 59) pp:11802-11805
Publication Date(Web):05 Jun 2015
DOI:10.1039/C5CC02995C
Linking together an oligourea and an oligoamide foldamer gives rise to a conformationally well-defined structure, despite the different hydrogen-bonding patterns in the two domains, provided the oligomers are ligated amide C terminus to urea N terminus. A powerful screw-sense preference induced at the N terminus of the resulting chimeric structure provides evidence for cooperative conformational interactions within the ‘block co-foldamer’.
Co-reporter:Daniele Castagnolo, Leonardo Degennaro, Renzo Luisi and Jonathan Clayden
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 8) pp:2330-2340
Publication Date(Web):16 Dec 2014
DOI:10.1039/C4OB02329C
The addition of n-butyllithium to alkenylthiocarbamates in the presence of (−)-sparteine or the (+)-sparteine surrogate leads to asymmetric carbolithiation, and returns enantiomerically enriched thiocarbamate derivatives of secondary thiols. In THF, with the (+)-sparteine surrogate, in situ aryl migration leads to an enantiomerically enriched tertiary thiol derivative. Remarkably, the two pseudoenantiomeric chiral ligands do not always give enantiomeric products, probably as a result of a complex interplay of kinetic and thermodynamic control. In situ IR and NMR studies of a stable, hindered lithiated thiocarbamate demonstrated its chemical and configurational stability over a period of hours at 0 °C.
Co-reporter:Sarah J. Pike, Thomas Boddaert, James Raftery, Simon J. Webb and Jonathan Clayden
New Journal of Chemistry 2015 vol. 39(Issue 5) pp:3288-3294
Publication Date(Web):17 Dec 2014
DOI:10.1039/C4NJ01547A
The solid state conformational preferences of a series of 2-aminoisobutyric acid (Aib) foldamers bearing a single N-terminal tertiary amino acid (Cbz-L-phenylalanine (Cbz-L-Phe)) have been investigated by X-ray crystallography. The type of β-turn present at the N-terminus and the global screw-sense preferences of the Aib foldamers were determined by analysis of intramolecular hydrogen-bonds and peptide torsion angles. The contrasting influence of a C-terminal ester or amide on the 310 helical conformation of the foldamers was established by identifying the hydrogen-bonding motifs adopted in the solid state. The ability of non-Aib achiral quaternary residues in the middle of the chain to stabilise the 310 helix was similarly confirmed. Combining these structural features, which promote the formation of consecutive i → i + 3 β-turns in Aib foldamers, permitted the formation of long chain oligomers in 310 helical conformations that extend over 21 Å.
Co-reporter:Mark A. Vincent;Julien Maury;Ian H. Hillier
European Journal of Organic Chemistry 2015 Volume 2015( Issue 5) pp:953-959
Publication Date(Web):
DOI:10.1002/ejoc.201403572
Abstract
The intramolecular arylation reactions of lithiated derivatives of ureas, carbamates and thiocarbamates generally proceed stereospecifically, but the reaction is stereochemically retentive with ureas and thiocarbamates, whereas it is stereochemically invertive with carbamates. Using DFT calculations, we have studied the mechanism of the intramolecular attack of a thiocarbamate-stabilised carbanion on an N-aryl substituent and compared the details of the calculated reaction pathway with the corresponding reactions of carbamate- and urea-stabilised carbanions. The different stereochemical outcomes observed in the rearrangements of carbamates and thiocarbamates arise from the sulfur–carbon interaction in the thiocarbamate, which enhances the stabilisation of the anion. As a result, the solvated lithium cation takes different pathways across the potential energy surfaces that lead to stereochemically divergent outcomes. Additionally, we investigated the importance of the intramolecular nature of the aryl migration and compared the pathway for aryl migration within a urea with that of a hypothetical, experimentally unfeasible, intermolecular reaction. The results throw light on the reason why aryl migrations are successful even with much more electron-rich rings than would be tolerated in a typical intermolecular nucleophilic aromatic substitution.
Co-reporter:Rachel C. Atkinson;Dr. Ferno Fernández-Nieto;Josep MasRoselló ; Jonathan Clayden
Angewandte Chemie International Edition 2015 Volume 54( Issue 31) pp:8961-8965
Publication Date(Web):
DOI:10.1002/anie.201502569
Abstract
Available α-amino acids undergo arylation at their α position in an enantioselective manner on treatment with base of N′-aryl urea derivatives ligated to pseudoephedrine as a chiral auxiliary. In situ silylation and enolization induces diastereoselective migration of the N′-aryl group to the α position of the amino acid, followed by ring closure to a hydantoin with concomitant explulsion of the recyclable auxiliary. The hydrolysis of the hydantoin products provides derivatives of quaternary amino acids. The arylation avoids the use of heavy-metal additives, and is successful with a range of amino acids and with aryl rings of varying electronic character.
Co-reporter:Rachel C. Atkinson;Dr. Ferno Fernández-Nieto;Josep MasRoselló ; Jonathan Clayden
Angewandte Chemie 2015 Volume 127( Issue 31) pp:9089-9093
Publication Date(Web):
DOI:10.1002/ange.201502569
Abstract
Available α-amino acids undergo arylation at their α position in an enantioselective manner on treatment with base of N′-aryl urea derivatives ligated to pseudoephedrine as a chiral auxiliary. In situ silylation and enolization induces diastereoselective migration of the N′-aryl group to the α position of the amino acid, followed by ring closure to a hydantoin with concomitant explulsion of the recyclable auxiliary. The hydrolysis of the hydantoin products provides derivatives of quaternary amino acids. The arylation avoids the use of heavy-metal additives, and is successful with a range of amino acids and with aryl rings of varying electronic character.
Co-reporter:Nadia Fleary-Roberts, Gilles Lemière, Jonathan Clayden
Tetrahedron 2015 Volume 71(Issue 39) pp:7204-7208
Publication Date(Web):30 September 2015
DOI:10.1016/j.tet.2015.02.055
Three unsaturated vinylic or allylic halides were made in enantiomerically pure form, ready for coupling with a vinyl metal as part of a divergent strategy for the synthesis of members of the domoic/isodomoic acid family and their analogues. The trisubstituted alkene 3 was made by an E-selective Wittig reaction, while 4 and 5 were made by stereoselective hydrometallation or hydroboration of an alkyne.
Co-reporter:Bryden A. F. Le Bailly and Jonathan Clayden
Chemical Communications 2014 vol. 50(Issue 59) pp:7949-7952
Publication Date(Web):05 Jun 2014
DOI:10.1039/C4CC03261F
The global screw-sense preference of an achiral helical oligomer may be controlled by a single chiral monomer located at one terminus. Remarkably, maximal control is induced in oligomers of the achiral quaternary amino acid Aib by a single C-terminal alaninamide residue, probably because the Ala side chain, though small, is compatible with a 310 helical conformation. The presence or absence of a C-terminal hydrogen bond donor determines the screw sense of the entire oligomer.
Co-reporter:Romina Wechsel, Julien Maury, Juliette Fremaux, Scott P. France, Gilles Guichard and Jonathan Clayden
Chemical Communications 2014 vol. 50(Issue 95) pp:15006-15009
Publication Date(Web):14 Oct 2014
DOI:10.1039/C4CC06754A
The ability of urea-linked oligomers of achiral diamines (achiral analogues of the well-established chiral oligourea foldamers) to adopt helical conformations was explored spectroscopically. Up to four achiral units were ligated either to a well-formed helical trimer or to a single chiral diamine, and the extent to which they adopted a screw-sense preference was determined by NMR and CD. In the best performing cases, a trimeric chiral oligourea and even a single cis-cyclohexanediamine monomer induced folding into a helical conformation.
Co-reporter:Gaëlle Mingat, Joseph J. W. McDouall and Jonathan Clayden
Chemical Communications 2014 vol. 50(Issue 51) pp:6754-6757
Publication Date(Web):06 May 2014
DOI:10.1039/C4CC02596B
Stereospecific [3,3]-sigmatropic rearrangement of O-substituted thiocarbamate derivatives of enantiopure allylic alcohols provides allylic thiocarbamates as single enantiomers. Intramolecular arylation by rearrangement of their allyllithium derivatives provides allylic tertiary thiols. Allylation and ring-closing metathesis gives 2,5-dihydrothiophenes containing sulfur-bearing quaternary centres.
Co-reporter:Gaëlle Mingat, Paul MacLellan, Marju Laars, and Jonathan Clayden
Organic Letters 2014 Volume 16(Issue 4) pp:1252-1255
Publication Date(Web):February 6, 2014
DOI:10.1021/ol5002522
The synthesis of tertiary thiols in enantiomerically enriched form is accomplished by lithiation of enantiomerically enriched N-aryl allylic thiocarbamates. Formation of an allyllithium derivative promotes intramolecular N to C aryl migration to the position α to sulfur, generally with good stereospecificity. The substrates may themselves be obtained by Pd-catalyzed enantioselective [3,3]-sigmatropic rearrangement of N-aryl O-allyl thiocarbamates. Solvolysis of the product thiocarbamates yields tertiary thiols, which may be converted to sulfide derivatives.
Co-reporter:Matteo De Poli and Jonathan Clayden
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 5) pp:836-843
Publication Date(Web):2013/11/25
DOI:10.1039/C3OB42167H
A single thionoglycine (glycine thioamide, –HNCH2C(S)–) residue inserted into a peptide foldamer provides both a pair of germinal protons for use as a 1H NMR stereochemical probe and a chromophore giving rise to a well defined Cotton effect in CD. Comparison of the response of these two features to a local helically chiral environment validates them as independent methods for quantifying the conformational screw-sense preference of a helical oligomer, in this case a peptide made of repeated Aib units. The sign of the Cotton effect provides a measure of the sign of the screw-sense preference, while both the chemical shift separation of the anisochronous signals of the glycine CH2 group and the magnitude of the Cotton effect give an estimate of the helicity excess of the oligomer. The thionoglycine unit is readily introduced synthetically by a thionation of a BocGlyAibOMe dipeptide.
Co-reporter:Sarah J. Pike, James Raftery, Simon J. Webb and Jonathan Clayden
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 24) pp:4124-4131
Publication Date(Web):29 Apr 2014
DOI:10.1039/C4OB00268G
The effect of Schellman motifs on the adoption of stable 310 helical conformations in a series of aminoisobutyric (Aib) oligomers has been studied in the solid state and solution. The destabilising effect of the Schellman motif (a local inversion of helical screw-sense due to a C-terminal ester residue) was quantified in the solid state using X-ray crystallography through analysis of the torsion angles and their deviation from those observed in an ideal 310 helix. Investigation of the intramolecular hydrogen-bonding interactions in the solid state led to the identification of a fully extended C5 conformation in one oligomer, which is a novel folding motif for Aib oligomers. The effect of ester groups with differing steric demands on intermolecular hydrogen-bonding contacts in the solid state was also ascertained. In solution, the adoption of a 310 conformation in Aib oligomers appeared to be more finely tuned, depending on a number of factors, including chain length and the steric demands of the C-terminal destabilising Schellman motif.
Co-reporter:Michael B. Tait, Philipp A. Ottersbach, Daniel J. Tetlow, and Jonathan Clayden
Organic Process Research & Development 2014 Volume 18(Issue 10) pp:1245-1252
Publication Date(Web):June 27, 2014
DOI:10.1021/op500173q
The deprotonation of N′-arylurea derivatives of cyclohexenamines by alkyllithiums leads to migration of the N′-aryl substituent from N′ to the allylic position α to N via rearrangement of a urea-stabilised allyllithium intermediate. The product ureas may be solvolysed to reveal 1-arylcyclohexenamines.
Co-reporter:Matteo De Poli, Liam Byrne, Robert A. Brown, Jordi Solà, Alejandro Castellanos, Thomas Boddaert, Romina Wechsel, Jonathan D. Beadle, and Jonathan Clayden
The Journal of Organic Chemistry 2014 Volume 79(Issue 10) pp:4659-4675
Publication Date(Web):April 7, 2014
DOI:10.1021/jo500714b
Oligomers of α-aminoisobutyric acid (Aib) are achiral peptides that typically adopt 310 helical conformations in which enantiomeric left- and right-handed conformers are, necessarily, equally populated. Incorporating a single protected chiral residue at the N-terminus of the peptide leads to induction of a screw-sense preference in the helical chain, which may be quantified (in the form of “helical excess”) by NMR spectroscopy. Variation of this residue and its N-terminal protecting group leads to the conclusion that maximal levels of screw-sense preference are induced by bulky chiral tertiary amino acids carrying amide protecting groups or by chiral quaternary amino acids carrying carbamate protecting groups. Tertiary l-amino acids at the N-terminus of the oligomer induce a left-handed screw sense, while quaternary l-amino acids induce a right-handed screw sense. A screw-sense preference may also be induced from the second position of the chain, weakly by tertiary amino acids, and much more powerfully by quaternary amino acids. In this position, the l enantiomers of both families induce a right-handed screw sense. Maximal, and essentially quantitative, control is induced by an l-α-methylvaline residue at both positions 1 and 2 of the chain, carrying an N-terminal carbamate protecting group.
Co-reporter:Dr. Sarah J. Pike;Dr. Vincent Diemer;Dr. James Raftery;Dr. Simon J. Webb ; Jonathan Clayden
Chemistry - A European Journal 2014 Volume 20( Issue 48) pp:15981-15990
Publication Date(Web):
DOI:10.1002/chem.201403626
Abstract
The biological activity of antibiotic peptaibols has been linked to their ability to aggregate, but the structure–activity relationship for aggregation is not well understood. Herein, we report a systematic study of a class of synthetic helical oligomer (foldamer) composed of aminoisobutyric acid (Aib) residues, which mimic the folding behavior of peptaibols. NMR spectroscopic analysis was used to quantify the dimerization constants in solution, which showed hydrogen-bond donors at the N terminus promoted aggregation more effectively than similar modifications at the C terminus. Elongation of the peptide chain also favored aggregation. The geometry of aggregation in solution was investigated by means of titrations with [D6]DMSO and 2D NOE NMR spectroscopy, which allowed the NH protons most involved in intermolecular hydrogen bonds in solution to be identified. X-ray crystallography studies of two oligomers allowed a comparison of the inter- and intramolecular hydrogen-bonding interactions in the solid state and in solution and gave further insight into the geometry of foldamer–foldamer interactions. These solution-based and solid-state studies indicated that the preferred geometry for aggregation is through head-to-tail interactions between the N and C termini of adjacent Aib oligomers.
Co-reporter:Samantha Stanil;Bo Yuan;Nelson Giménez-Agulló;Dr. Tommaso Marcelli;Dr. Simon C. Willies;Dr. Damian M. Grainger; Nicholas J. Turner; Jonathan Clayden
Chemistry - A European Journal 2014 Volume 20( Issue 41) pp:13084-13088
Publication Date(Web):
DOI:10.1002/chem.201404509
Abstract
Atropisomeric biaryls carrying ortho-hydroxymethyl and formyl groups were made enantioselectively by desymmetrisation of dialdehyde or diol substrates. The oxidation of the symmetrical diol substrates was achieved using a variant of galactose oxidase (GOase), and the reduction of the dialdehydes using a panel of ketoreductases. Either M or P enantiomers of the products could be formed, with absolute configurations assigned by time-dependent DFT calculations of circular dichroism spectra. The differing selectivities observed with different biaryl structures offer an insight into the detailed structure of the active site of the GOase enzyme.
Co-reporter:Dr. Liam Byrne;Dr. Jordi Solà;Dr. Thomas Boddaert;Dr. Tommaso Marcelli;Dr. Ralph W. Adams; Gareth A. Morris; Jonathan Clayden
Angewandte Chemie International Edition 2014 Volume 53( Issue 1) pp:151-155
Publication Date(Web):
DOI:10.1002/anie.201308264
Abstract
An N-terminal L-α-methylvaline dimer induces complete conformational control over the screw sense of an otherwise achiral helical peptide foldamer formed from the achiral quaternary amino acids Aib and Ac6c. The persistent right-handed screw-sense preference of the helix enables remote reactive sites to fall under the influence of the terminal chiral residues, and permits diastereoselective reactions such as alkene hydrogenation or iminium ion addition to take place with 1,16-, 1,31-, 1,46- and even 1,61-asymmetric induction. Stereochemical information may be communicated in this way over distances of up to 4 nm.
Co-reporter:Dr. Liam Byrne;Dr. Jordi Solà;Dr. Thomas Boddaert;Dr. Tommaso Marcelli;Dr. Ralph W. Adams; Gareth A. Morris; Jonathan Clayden
Angewandte Chemie 2014 Volume 126( Issue 1) pp:155-159
Publication Date(Web):
DOI:10.1002/ange.201308264
Abstract
An N-terminal L-α-methylvaline dimer induces complete conformational control over the screw sense of an otherwise achiral helical peptide foldamer formed from the achiral quaternary amino acids Aib and Ac6c. The persistent right-handed screw-sense preference of the helix enables remote reactive sites to fall under the influence of the terminal chiral residues, and permits diastereoselective reactions such as alkene hydrogenation or iminium ion addition to take place with 1,16-, 1,31-, 1,46- and even 1,61-asymmetric induction. Stereochemical information may be communicated in this way over distances of up to 4 nm.
Co-reporter:Rachel C. Atkinson, Daniel J. Leonard, Julien Maury, Daniele Castagnolo, Nicole Volz and Jonathan Clayden
Chemical Communications 2013 vol. 49(Issue 84) pp:9734-9736
Publication Date(Web):05 Sep 2013
DOI:10.1039/C3CC46193A
Dianionic enolates formed from N′-aryl urea derivatives of amino acids undergo intramolecular C-arylation by attack of the enolate anion on the N′-aryl ring, leading to a hydantoin derivative of a quaternary amino acid. In situ IR studies allow identification of four intermediates on the reaction pathway.
Co-reporter:Daniel J. Tetlow, Mark A. Vincent, Ian H. Hillier and Jonathan Clayden
Chemical Communications 2013 vol. 49(Issue 15) pp:1548-1550
Publication Date(Web):03 Jan 2013
DOI:10.1039/C2CC38704B
Deprotonation with strong bases of N-vinyl ureas carrying an N′-aryl substituent leads to migration of the N′-aryl group from N to C via an allyllithium; with weaker bases and electron-deficient aryl rings the direction of the migration reverses, and aryl substituents α to the urea N atom may migrate from C to N.
Co-reporter:Michael Tait, Morgan Donnard, Alberto Minassi, Julien Lefranc, Beatrice Bechi, Giorgio Carbone, Peter O’Brien, and Jonathan Clayden
Organic Letters 2013 Volume 15(Issue 1) pp:34-37
Publication Date(Web):December 19, 2012
DOI:10.1021/ol3029324
In the presence of (−)-sparteine or a (+)-sparteine surrogate, organolithiums add to N-alkenyl-N′-arylureas to give benzylic organolithiums in an enantioselective manner. Under the influence of DMPU, these organolithiums undergo rearrangement with migration of the N′-aryl ring from N to C, leading to the urea derivatives of enantiomerically enriched amines bearing tertiary substituents. Basic hydrolysis returns the functionalized amine, providing a new synthetic route to compounds with quaternary stereogenic centers bearing nitrogen.
Co-reporter:Jemma Senczyszyn, Heloise Brice, and Jonathan Clayden
Organic Letters 2013 Volume 15(Issue 8) pp:1922-1925
Publication Date(Web):April 3, 2013
DOI:10.1021/ol400571j
On treatment with acylating or sulfonylating agents, N-alkenyl pyridine carboxamides (N-pyridinecarbonyl enamines) undergo a dearomatizing cyclization initiated by pyridine acylation and followed by intramolecular trapping of the resulting pyridinium cation. The products are spirocyclic dihydropyridines which may be further elaborated to spirocyclic heterocycles with drug-like features.
Co-reporter:Daniele Castagnolo, Daniel J. Foley, Hatice Berber, Renzo Luisi, and Jonathan Clayden
Organic Letters 2013 Volume 15(Issue 9) pp:2116-2119
Publication Date(Web):April 15, 2013
DOI:10.1021/ol400570r
S-Alkenyl-N-arylthiocarbamates are formed from allylic alcohols by sigmatropic rearrangement and isomerization or C═C bond cleavage. They undergo carbolithiation with a range of organolithium reagents, generating benzyllithium intermediates in a stereospecific manner which may undergo N to C aryl migration to yield thiocarbamates with tertiary substituents. A simple base-promoted alcoholysis reveals a series of hindered tertiary thiols with branched carbon skeletons.
Co-reporter:Juliette Fremaux, Christel Dolain, Brice Kauffmann, Jonathan Clayden and Gilles Guichard
Chemical Communications 2013 vol. 49(Issue 67) pp:7415-7417
Publication Date(Web):21 Jun 2013
DOI:10.1039/C3CC40961A
The structures of various urea oligomers incorporating one or two central achiral 1,2-diamino-1,1-dimethylethane (DADME) units have been investigated in solution and in the crystalline state. These diamine monomers are analogous to the achiral helicogenic amino acid Aib (α-aminoisobutyric acid). Oligomers were found to fold into helical conformations with DADME units inducing local deviations from the canonical helix geometry of urea foldamers.
Co-reporter:Sarah J. Pike, Matteo De Poli, Wojciech Zawodny, James Raftery, Simon J. Webb and Jonathan Clayden
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 19) pp:3168-3176
Publication Date(Web):27 Mar 2013
DOI:10.1039/C3OB40463C
Ligating simple amino alcohol or amino ester monomers containing enantiotopic fluorine substituents to the C-terminus of a helical peptide places the fluorine atoms in diastereotopic environments, and gives two distinct and easily identifiable signals in the 19F NMR spectrum. In the case of a dynamically inverting helix built from achiral monomers, the chemical shift separation between the 19F signals provides a simple means of analysing the ratio of screw-sense conformers in the oligomer, in cases where an asymmetric bias leads to a screw-sense preference.
Co-reporter:Rebecca A. Harvey, Ol’ga Karlubíková, Sean Parris, Jonathan Clayden
Tetrahedron Letters 2013 Volume 54(Issue 31) pp:4064-4066
Publication Date(Web):31 July 2013
DOI:10.1016/j.tetlet.2013.05.098
Treatment of 3-allyloxyphenyl oxazolines with organolithium bases leads to allyllithiums which undergo dearomatising cyclisation. The resulting dearomatised anions may be quenched with electrophiles to form partially saturated benzopyran derivatives.Deprotonation of allyl phenyl ethers bearing oxazoline substituents on the phenyl ring leads to dearomatising cyclisation.
Co-reporter:Ugo Orcel;Dr. Matteo DePoli;Dr. Marta DeZotti; Jonathan Clayden
Chemistry - A European Journal 2013 Volume 19( Issue 48) pp:16357-16365
Publication Date(Web):
DOI:10.1002/chem.201302648
Abstract
The N-terminal nonapeptide domain of the fungal nonribosomal peptide antibiotics cephaibol A and cephaibol C (AcPheAib4LeuIvaGly- Aib) is reported to adopt a right-handed helical conformation in the crystalline state. However, this conformation is at odds with the left-handed helicity observed in solution in related synthetic oligomers capped with Ac-L-PheAib4 fragments. We report the synthesis of four diastereoisomers of the cephaibol N-terminal nonapeptide, and show by NMR and CD spectroscopy that the peptide containing the chiral amino acids Phe and Leu in the naturally occurring relative configuration exists in solution as an interconverting mixture of helical screw-sense conformers. In contrast, the nonapeptide containing the unnatural relative configuration at Phe and Leu adopts a single, stable helical screw-sense, which is left handed when the N-terminal Phe residue is L and right-handed when the N-terminal Phe residue is D.
Co-reporter:Matteo De Poli, Marta De Zotti, James Raftery, Juan A. Aguilar, Gareth A. Morris, and Jonathan Clayden
The Journal of Organic Chemistry 2013 Volume 78(Issue 6) pp:2248-2255
Publication Date(Web):January 14, 2013
DOI:10.1021/jo302705k
Oligomers of the achiral amino acid Aib adopt helical conformations in which the screw-sense may be controlled by a single N-terminal residue. Using crystallographic and NMR techniques, we show that the left- or right-handed sense of helical induction arises from the nature of the β-turn at the N terminus: the tertiary amino acid l-Val induces a left-handed type II β-turn in both the solid state and in solution, while the corresponding quaternary amino acid l-α-methylvaline induces a right-handed type III β-turn.
Co-reporter:Julien Lefranc ; Anne M. Fournier ; Gaëlle Mingat ; Simon Herbert ; Tommaso Marcelli
Journal of the American Chemical Society 2012 Volume 134(Issue 17) pp:7286-7289
Publication Date(Web):April 5, 2012
DOI:10.1021/ja301591m
Deprotonation of benzylic ureas, carbamates, and thiocarbamates bearing N′-alkenyl substituents generates carbanions which undergo intramolecular migration of the alkenyl group to the carbanionic center. Solvolysis of the urea products generates α-alkenylated amines. With an enantiomerically pure starting urea, migration proceeds stereospecifically, generating in enantiomerically enriched form products containing allylic quaternary stereogenic centers bearing N. Computational and in situ IR studies suggest that the reaction, formally a nucleophilic substitution at an sp2 carbon atom, proceeds by a concerted addition-elimination pathway.
Co-reporter:Anne M. Fournier and Jonathan Clayden
Organic Letters 2012 Volume 14(Issue 1) pp:142-145
Publication Date(Web):November 29, 2011
DOI:10.1021/ol2029355
Enol carbamates (O-vinylcarbamates) derived from aromatic or α,β-unsaturated compounds and bearing an N-aryl substituent undergo carbolithiation by nucleophilic attack at the (nominally nucleophilic) β position of the enol double bond. The resulting carbamate-stabilized allylic, propargylic, or benzylic organolithium rearranges with N→C migration of the N-aryl substituent, creating a quaternary carbon α to O. The products may be readily hydrolyzed to yield multiply branched tertiary alcohols in a one-pot tandem reaction, effectively a polarity-reversed nucleophilic β-alkylation–electrophilic α-arylation of an enol equivalent.
Co-reporter:Thomas Boddaert, Jordi Solà, Madeleine Helliwell and Jonathan Clayden
Chemical Communications 2012 vol. 48(Issue 28) pp:3397-3399
Publication Date(Web):31 Jan 2012
DOI:10.1039/C2CC00060A
1H NMR studies quantify the abilities of achiral amino acids to communicate a left-handed screw-sense preference from one helical Aib4 domain to another: certain quaternary amino acids (e.g. Ac6c) act as effective conductors of conformational preference while others (e.g. diphenylglycine) acts as insulators.
Co-reporter:Edmund W. D. Burke, Gareth A. Morris, Mark A. Vincent, Ian H. Hillier and Jonathan Clayden
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 4) pp:716-719
Publication Date(Web):2011/11/22
DOI:10.1039/C1OB06490H
The non-nucleoside reverse transcriptase inhibitor nevirapine displays in its room temperature 1H-NMR spectrum signals characteristic of a chiral compound. Following suggestions in the recent literature that nevirapine may display atropisomerism—and therefore be a chiral compound, due to slow interconversion between two enantiomeric conformers—we report the results of an NMR and computational study which reveal that while nevirapine does indeed possess two stable enantiomeric conformations, they interconvert with a barrier of about 76 kJ mol−1 at room temperature. Nevirapine has a half life for enantiomerisation at room temperature of the order of seconds, is not atropisomeric, and cannot exist as separable enantiomers.
Co-reporter:Damian M. Grainger;Alison Campbell Smith;Mark A. Vincent;Ian H. Hillier;Andrew E. H. Wheatley
European Journal of Organic Chemistry 2012 Volume 2012( Issue 4) pp:731-743
Publication Date(Web):
DOI:10.1002/ejoc.201101475
Abstract
In situ NMR and IR spectroscopy studies were carried out on the rearrangement of lithiated N-benzyl-N′-aryl ureas, which involves N-to-C aryl transfer with retention of configuration. The IR spectroscopy studies revealed that initial benzylic lithiation was followed by migration of the aryl ring to yield a lithiated urea product without a detectable dearomatised intermediate. Similar results were obtained by NMR spectroscopy, but when 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU) was added to the solvent mixture, a transient dearomatised intermediate was detectable during migration of a 1-naphthyl ring. DFT calculations highlight the importance of coordinated lithium cations, and their migration from one site to another, in the rearrangement. Rearrangement is initiated by migration of a solvated lithium cation from the anionic centre of the starting organolithium to a site close the adjacent phenyl ring, allowing retentive attack of the anionic centre on the more remote ring with movement of the solvated lithium cation to the remote ring stabilising the developing negative charge. A short-lived spirocyclic intermediate is predicted to undergo elimination by loss of the urea substituent, completing the migration. Coordination of the carbonyl group to a second solvated lithium cation appears to be essential for this step. Calculated shifts for this intermediate when a 1-naphthyl ring is migrating are consistent with the transient signals observed by NMR spectroscopy. Alternative pathways involving (1) invertive migration and (2) attack on the urea C=O group were also calculated and were found to require significantly higher energy transition states.
Co-reporter:Robert A. Brown;Dr. Tommaso Marcelli;Dr. Matteo DePoli;Dr. Jordi Solà; Jonathan Clayden
Angewandte Chemie 2012 Volume 124( Issue 6) pp:
Publication Date(Web):
DOI:10.1002/ange.201107583
Co-reporter:Robert A. Brown;Dr. Tommaso Marcelli;Dr. Matteo DePoli;Dr. Jordi Solà; Jonathan Clayden
Angewandte Chemie International Edition 2012 Volume 51( Issue 6) pp:
Publication Date(Web):
DOI:10.1002/anie.201107583
Co-reporter:Anne M. Fournier;Dr. Christopher J. Nichols;Dr. Mark A. Vincent; Ian H. Hillier; Jonathan Clayden
Chemistry - A European Journal 2012 Volume 18( Issue 51) pp:16478-16490
Publication Date(Web):
DOI:10.1002/chem.201201761
Abstract
Deprotonation of O-allyl, O-propargyl or O-benzyl carbamates in the presence of a lithium counterion leads to carbamate-stabilised organolithium compounds that may be quenched with electrophiles. We now report that when the allylic, propargylic or benzylic carbamate bears an N-aryl substituent, an aryl migration takes place, leading to stereochemical inversion and C-arylation of the carbamate α to oxygen. The aryl migration is an intramolecular SNAr reaction, despite the lack of anion-stabilising aryl substituents. Our in situ IR studies reveal a number of intermediates along the rearrangement pathway, including a “pre-lithiation complex,” the deprotonated carbamate, the rearranged anion, and the final arylated carbamate. No evidence was obtained for a dearomatised intermediate during the aryl migration. DFT calculations predict that during the reaction the solvated Li cation moves from the carbanion centre, thus freeing its lone pair for nucleophilic attack on the remote phenyl ring. This charge separation leads to several alternative conformations. The one having Li+ bound to the carbamate oxygen gives rise to the lowest-energy transition structure, and also leads to inversion of the configuration. In agreement with the IR studies, the DFT calculations fail to locate a dearomatised intermediate.
Co-reporter:Jordi Solà ; Gareth A. Morris
Journal of the American Chemical Society 2011 Volume 133(Issue 11) pp:3712-3715
Publication Date(Web):February 25, 2011
DOI:10.1021/ja1097034
While an unequal population of rapidly interconverting left- and right-handed conformers of a helical oligomer can be detected by circular dichroism, precise quantification of a conformer ratio has not previously been achieved. We demonstrate, using a set of labeled peptide analogues, that simple analysis of peak separation in their 13C NMR spectra at slow and fast exchange allows an accurate value for the ratio of helical conformers to be obtained. The method reports the ratio of conformers at the site of the label and can therefore be used to investigate local variations in helical conformational control.
Co-reporter:Julien Lefranc, Daniel J. Tetlow, Morgan Donnard, Alberto Minassi, Erik Gálvez, and Jonathan Clayden
Organic Letters 2011 Volume 13(Issue 2) pp:296-299
Publication Date(Web):December 17, 2010
DOI:10.1021/ol1027442
N-Vinyl ureas are emerging as a valuable class of compounds with both nucleophilic and electrophilic reactivity. They may be made by capturing the enamine tautomer of an imine with an isocyanate, a reaction which in general leads to the E isomer of the vinyl urea. Deprotonation of such a vinyl urea, or of an allyl urea, generates a dipole stabilized Z-allyl anion which may be protonated to return the Z-vinyl urea. Isomerization of an allyl urea with a Ru complex provides an alternative route to E-vinyl ureas.
Co-reporter:Gilles Lemière, Simon Sedehizadeh, Julie Toueg, Nadia Fleary-Roberts and Jonathan Clayden
Chemical Communications 2011 vol. 47(Issue 13) pp:3745-3747
Publication Date(Web):24 Feb 2011
DOI:10.1039/C1CC00048A
The alkynylpyrrolidine 4 was made via a dearomatising cyclisation of an aromatic amide, and was elaborated by stannylcupration and palladium-catalysed coupling to achieve the first total synthesis of three members of the isodomoic acid family; the same alkyne can be envisaged as a precursor to several more of this class of amnesic shellfish toxins.
Co-reporter:Paul MacLellan and Jonathan Clayden
Chemical Communications 2011 vol. 47(Issue 12) pp:3395-3397
Publication Date(Web):17 Feb 2011
DOI:10.1039/C0CC04912C
Lithiation of N-aryl S-α-alkylbenzyl thiocarbamates leads to rearrangement with migration of the N-aryl ring to the anionic centre α to S, a process which generally proceeds with ca. 98% retention of stereochemistry and returns chiral benzylic tertiary thiols in high enantiomeric ratios.
Co-reporter:James Clayton, Jonathan Clayden
Tetrahedron Letters 2011 Volume 52(Issue 19) pp:2436-2439
Publication Date(Web):11 May 2011
DOI:10.1016/j.tetlet.2011.02.091
Treatment of 4-, 3- or 2-aryl-4,5-diphenyloxazolines with isopropyllithium gives the products of dearomatising addition, fluorine-directed lithiation or nucleophilic aromatic substitution of fluoride depending on substitution pattern and conditions. In the case of the 4-fluoroaryl substrates, fluorinated 1,4-cyclohexadiene may be obtained in good yield.2-, 3- and 4-Fluoroaryloxazolines show varying reactivity towards organolithiums, undergoing either substitution, metallation or addition reactions.
Co-reporter:Jordi Solà;Madeleine Helliwell
Biopolymers 2011 Volume 95( Issue 1) pp:62-69
Publication Date(Web):
DOI:10.1002/bip.21535
Abstract
The structural influence of a single Gly residue inserted into an Aib16 homooligomer was studied in the solid state by X-ray crystallography. The peptides N3Aib8GlyAib8PheNH2 (1) and CbzPheAib8GlyAib8 (2) were found to adopt well-defined helical structures, which are broadly 310 helical. Indeed, 2 is the longest crystallographic 310 helix thus far reported. However, in the region of the central Gly residue, a loosening of the 310 structure is observed in both peptides, with 1 clearly showing local adoption of an α-helical structure in the region of residues 7–9. © 2010 Wiley Periodicals, Inc. Biopolymers 95: 62–69, 2011.
Co-reporter:Jordi Solà ; Madeleine Helliwell
Journal of the American Chemical Society 2010 Volume 132(Issue 13) pp:4548-4549
Publication Date(Web):March 16, 2010
DOI:10.1021/ja100662d
Conformational control over the screw sense of a helical 17-mer of achiral amino acids, Aib8GlyAib8, from a single chiral residue located at the N-terminus is better than that from a single amino acid located at the C-terminus. X-ray crystallography indicates that Aib8GlyAib8 forms the longest 310 helical structure observed crystallographically to date.
Co-reporter:Jonathan Clayden ; Morgan Donnard ; Julien Lefranc ; Alberto Minassi ;Daniel J. Tetlow
Journal of the American Chemical Society 2010 Volume 132(Issue 19) pp:6624-6625
Publication Date(Web):April 22, 2010
DOI:10.1021/ja1007992
Organolithiums add in an umpolung fashion to the β-carbon of N-carbamoyl enamines (N-vinyl ureas). The reaction proceeds with syn diastereospecificity and provides urea-stabilized, configurationally defined organolithiums. Facilitated by coordinating solvents (THF or DMPU), these undergo intramolecular attack on an N′-aryl group, resulting in retentive arylation of the organolithium and hence overall addition of an alkyl or aryl group to both carbon atoms of the urea-substituted alkene. Facile deprotection in hot butanol permits the rapid, multicomponent construction of heavily substituted amines.
Co-reporter:Anne M. Fournier, Robert A. Brown, William Farnaby, Hideki Miyatake-Ondozabal and Jonathan Clayden
Organic Letters 2010 Volume 12(Issue 10) pp:2222-2225
Publication Date(Web):April 20, 2010
DOI:10.1021/ol100627c
The first enantioselective synthesis of the antihistamine agent clemastine, as its (S,S)-stereoisomer, has been achieved by ether formation between a proline-derived chloroethylpyrrolidine and an enantiomerically enriched tertiary alcohol. The tertiary alcohol was formed from the carbamate derivative of α-methyl-p-chlorobenzyl alcohol by invertive aryl migration on lithiation. The (S,S)-stereochemistry of the product confirms the invertive nature of the rearrangement.
Co-reporter:Daniel J. Tetlow, Ulrich Hennecke, James Raftery, Michael J. Waring, David S. Clarke, and Jonathan Clayden
Organic Letters 2010 Volume 12(Issue 23) pp:5442-5445
Publication Date(Web):November 9, 2010
DOI:10.1021/ol102155h
On lithiation with lithium amides, N-allyl-N′-aryl ureas undergo rearrangement with transfer of the aryl ring from N to the allylic α carbon. From the α-arylated products, a further aryl transfer under the influence of a chiral lithium amide allows the enantioselective construction of 1,1-diarylallylamine derivatives. Stereoselectivity in these reactions results from the enantioselective formation of a planar chiral allyllithium under kinetic control.
Co-reporter:Jonathan Clayden, Stephen P. Fletcher, James Senior, Christopher P. Worrall
Tetrahedron: Asymmetry 2010 Volume 21(11–12) pp:1355-1360
Publication Date(Web):23 June 2010
DOI:10.1016/j.tetasy.2010.06.017
Phosphines and bisphosphines derived from hindered ortho-substituted diaryl ethers and diarylsulfones by lithiation are, with appropriate substitution patterns, resolvable atropisomeric ligands which form crystalline complexes with palladium dichloride.Appropriately substituted bisphosphinyldiaryl ethers are resolvable atropisomeric ligands which form stable wide bite angle complexes with Pd.
Co-reporter:Bo Yuan;Abigail Page;Christopher P. Worrall;Dr. Franck Escalettes;Dr. Simon C. Willies;Dr. Joseph J. W. McDouall; Nicholas J. Turner; Jonathan Clayden
Angewandte Chemie International Edition 2010 Volume 49( Issue 39) pp:7010-7013
Publication Date(Web):
DOI:10.1002/anie.201002580
Co-reporter:Dr. Jordi Solà;Dr. StephenP. Fletcher;Dr. Alejro Castellanos ; Jonathan Clayden
Angewandte Chemie International Edition 2010 Volume 49( Issue 38) pp:6836-6839
Publication Date(Web):
DOI:10.1002/anie.201001130
Co-reporter:Laurent Chabaud, Jonathan Clayden, Madeleine Helliwell, Abigail Page, James Raftery, Lluís Vallverdú
Tetrahedron 2010 66(34) pp: 6936-6957
Publication Date(Web):
DOI:10.1016/j.tet.2010.06.037
Co-reporter:Dr. Jordi Solà;Dr. StephenP. Fletcher;Dr. Alejro Castellanos ; Jonathan Clayden
Angewandte Chemie 2010 Volume 122( Issue 38) pp:6988-6991
Publication Date(Web):
DOI:10.1002/ange.201001130
Co-reporter:Jonathan Clayden
Chemical Society Reviews 2009 vol. 38(Issue 3) pp:817-829
Publication Date(Web):08 Dec 2008
DOI:10.1039/B801639A
By exploiting intramolecular interactions such as dipole repulsion, and by incorporating a terminal chiral controlling feature, the global conformation of a molecule may be governed. In such an environment, stereoselective reactions can occur at considerable distances from the source of stereochemical information, providing a simple method for information relay over scales of >1 nm (or about seven bond lengths). This tutorial review discusses the development of this idea, and describes examples which depend on relayed dipole repulsion and on the absolute control of helicity. Future prospects in the area employing control over extended helical foldamers are elaborated.
Co-reporter:Heloise Brice and Jonathan Clayden
Chemical Communications 2009 (Issue 15) pp:1964-1966
Publication Date(Web):12 Mar 2009
DOI:10.1039/B901558B
Isonicotinamides carrying N-furanylmethyl, N-pyrrolylalkyl or N-thiophenylmethyl substituents at nitrogen undergo cyclisation induced by an electrophile, giving spirocyclic compounds or doubly spirocyclic compounds in which both the nucleophilic and electrophilicheterocycles are dearomatised.
Co-reporter:Jonathan Clayden, Mark Pickworth and Lyn H. Jones
Chemical Communications 2009 (Issue 5) pp:547-549
Publication Date(Web):26 Nov 2008
DOI:10.1039/B817527F
The well-defined conformation of an N,N′-diarylurea allows a chiral sulfinyl substituent to influence diastereoselectivity in the formation of new stereogenic centres up to 14 bond lengths away.
Co-reporter:Jonathan Clayden, Stephen P. Fletcher, S. J. M. Rowbottom and Madeleine Helliwell
Organic Letters 2009 Volume 11(Issue 11) pp:2313-2316
Publication Date(Web):May 12, 2009
DOI:10.1021/ol9006635
The favored (RS*,M*) diastereoisomer of 2-aryl-pyridine 1 in the solution state results from intramolecular dipole−dipole interactions. In the crystalline state, intermolecular interactions dominate, and the conformation switches reversibly to (RS*,P*). Only racemic 1 exhibits this switching property: enantiomerically pure 1 exists as the (RS*,M*) diastereoisomer in both the solution and crystalline state.
Co-reporter:Jonathan Clayden, Stephen J. M. Rowbottom, Warren J. Ebenezer and Michael G. Hutchings
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 23) pp:4871-4880
Publication Date(Web):30 Sep 2009
DOI:10.1039/B911631A
Successive treatment of cyanuric chloride with two aromatic diamines, at least one of them sulfonated, yields water-soluble sulfonated azacalix[4]arenes which may be isolated by crystallisation. Functionalised azacalixarenes may be made by first displacing two chloro substituents from the cyclisation precursor. Attempted formation of an azacalix[6]arene led to a dimeric species for which two structures may be proposed, one of them an azacalixarene catenane.
Co-reporter:Jonathan Clayden ;Alejro Castellanos Dr.;Jordi Solà Dr. ;GarethA. Morris
Angewandte Chemie 2009 Volume 121( Issue 32) pp:6076-6079
Publication Date(Web):
DOI:10.1002/ange.200901892
Co-reporter:Jonathan Clayden ;James Senior ;Madeleine Helliwell Dr.
Angewandte Chemie 2009 Volume 121( Issue 34) pp:6388-6391
Publication Date(Web):
DOI:10.1002/ange.200901718
Co-reporter:Jonathan Clayden, Stephen J.M. Rowbottom, Michael G. Hutchings, Warren J. Ebenezer
Tetrahedron Letters 2009 50(27) pp: 3923-3925
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.04.068
Co-reporter:Jonathan Clayden ;Alejro Castellanos Dr.;Jordi Solà Dr. ;GarethA. Morris
Angewandte Chemie International Edition 2009 Volume 48( Issue 32) pp:5962-5965
Publication Date(Web):
DOI:10.1002/anie.200901892
Co-reporter:Jonathan Clayden ;James Senior ;Madeleine Helliwell Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 34) pp:6270-6273
Publication Date(Web):
DOI:10.1002/anie.200901718
Co-reporter:Jonathan Clayden, Lluís Vallverdú, James Clayton and Madeleine Helliwell
Chemical Communications 2008 (Issue 5) pp:561-563
Publication Date(Web):28 Nov 2007
DOI:10.1039/B716105K
Tertiary diamides of xanthene-1,8-dicarboxylic acid and biphenyl-2,2′-dicarboxylic acid exhibit a thermodynamic preference for anti stereochemistry which is inverted in the presence of Ti- or Sn-based Lewis acids, allowing interconversion between kinetically stable syn and anti diastereoisomeric atropisomers.
Co-reporter:Jonathan Clayden, Loïc Lemiègre, Mark Pickworth and Lyn Jones
Organic & Biomolecular Chemistry 2008 vol. 6(Issue 16) pp:2908-2913
Publication Date(Web):16 Jun 2008
DOI:10.1039/B802673D
Except in the most hindered of cases, N,N′-diaryl N,N′-dimethyl ureas adopt a conformation with the two aryl rings disposed cis to one another. Variable temperature NMR studies reveal the rate at which the Ar–N bonds rotate as well as the conformational preference of ortho disubstituted ureas in which more than one cis orientation is possible. In general, a conformation in which the aryl rings lie close in space but with their most bulky 2-substituents aligned anti is preferred, but with particularly bulky 2-substituents, conformations in which one of the aryl rings points away from the other may also be populated.
Co-reporter:Jonathan Clayden ;Sean Parris;Nuria Cabedo Dr.;AndrewH. Payne Dr.
Angewandte Chemie 2008 Volume 120( Issue 27) pp:5138-5140
Publication Date(Web):
DOI:10.1002/ange.200801078
Co-reporter:Jonathan Clayden Prof ;ChristopherP. Worrall;WesleyJ. Moran ;Madeleine Helliwell Dr.
Angewandte Chemie 2008 Volume 120( Issue 17) pp:3278-3281
Publication Date(Web):
DOI:10.1002/ange.200705660
Co-reporter:Jonathan Clayden Prof ;ChristopherP. Worrall;WesleyJ. Moran ;Madeleine Helliwell Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 17) pp:3234-3237
Publication Date(Web):
DOI:10.1002/anie.200705660
Co-reporter:Jonathan Clayden ;Sean Parris;Nuria Cabedo Dr.;AndrewH. Payne Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 27) pp:5060-5062
Publication Date(Web):
DOI:10.1002/anie.200801078
Co-reporter:Mark S. Betson, Ann Bracegirdle, Jonathan Clayden, Madeleine Helliwell, Andrew Lund, Mark Pickworth, Timothy J. Snape and Christopher P. Worrall
Chemical Communications 2007 (Issue 7) pp:754-756
Publication Date(Web):18 Dec 2006
DOI:10.1039/B614618J
The orientation of Ar–C, Ar–N and Ar–O bonds in biaryls, N,N′-diarylureas and diaryl ethers (whose conformers are distinguishable by NMR) may be controlled with a selectivity up to >95 : 5 by an adjacent stereogenic centre; the selectivity may be greater when a second stereogenic axis is inserted between the controlling centre and the slowly rotating bond.
Co-reporter:Jonathan Clayden and Wesley J. Moran
Organic & Biomolecular Chemistry 2007 vol. 5(Issue 7) pp:1028-1030
Publication Date(Web):27 Feb 2007
DOI:10.1039/B700353F
Rh(I)–catalysed [2 + 2 + 2] cycloaddition allows the synthesis of aryl ethers and diaryl methanes containing a high degree of steric hindrance from relatively simple diyne and alkyne precursors. The diarylmethanes made in this way show no evidence in their NMR spectra, however, of rotational restriction.
Co-reporter:Jonathan Clayden, Christopher C. Stimson and Martine Keenan
Chemical Communications 2006 (Issue 13) pp:1393-1394
Publication Date(Web):21 Feb 2006
DOI:10.1039/B600181E
Directed metallation and sulfinylation yields sulfoxides which undergo ipso nucleophilic aromatic substitution with tertiary and secondary alkyllithiums, giving aromatic rings bearing alkyl groups generally incompatible with directed metallation methods and with regioselectivity complementary with classical Friedel–Crafts substitution.
Co-reporter:Jonathan Clayden, Neil Westlund, Christopher S. Frampton and Madeleine Helliwell
Organic & Biomolecular Chemistry 2006 vol. 4(Issue 3) pp:455-461
Publication Date(Web):22 Dec 2005
DOI:10.1039/B514561A
Addition of laterally lithiated tertiary aromatic amides to benzaldimines controls the formation of a new stereogenic centre adjacent to the benzaldimine aromatic ring. Drawing on the fact that such amino-substituted stereogenic centres may themselves control the conformation of amides, with amido-substituted benzaldimines we found it becomes possible to relay stereochemistry from one amide to another via this intervening stereogenic centre. A group of dihydrostilbene-2,2′-dicarboxamide derivatives bearing one or two stereogenic axes are made by this method, which demonstrates the use of combined kinetic and thermodynamic control for the relay of stereochemical information.
Co-reporter:Jonathan Clayden, Yann J. Y. Foricher, Madeleine Helliwell, Paul Johnson, David Mitjans and Victoria Vinader
Organic & Biomolecular Chemistry 2006 vol. 4(Issue 3) pp:444-454
Publication Date(Web):22 Dec 2005
DOI:10.1039/B514558A
The orientation of a tertiary amide group adjacent to an aromatic ring may be governed by the stereochemistry of an adjacent chiral substituent. With a chiral substituent in both ortho positions, matched/mismatched pairs of isomers result. Evidence for matched stereochemistry is provided by the clean NMR spectra of single conformers, while mismatching gives poor or unexpected selectivities in the formation of chiral substituents, or mixtures of amide conformers. Attempts to use the match–mismatch effect to select for racemic pairs of enantiomeric substituents, and hence develop a “racemate-sequestering” reagent, are described, along with the use of “matching” to scavenge a single enantiomer of a diamine from material of incomplete enantiomeric purity.
Co-reporter:Mark S. Betson, Jonathan Clayden, Madeleine Helliwell, Paul Johnson, Lai Wah Lai, Jennifer H. Pink, Christopher C. Stimson, Neoclis Vassiliou, Neil Westlund, Samreen A. Yasin and Latifa H. Youssef
Organic & Biomolecular Chemistry 2006 vol. 4(Issue 3) pp:424-443
Publication Date(Web):22 Dec 2005
DOI:10.1039/B514557K
Tertiary aromatic amides bearing stereogenic centres ortho to the amide group may adopt two diastereoisomeric conformations which interconvert slowly on the NMR timescale at ambient temperature, and are therefore detectable by NMR. Certain classes of stereogenic centre—particularly sulfoxides, ephedrine-derived oxazolidines, and proline-derived imidazolidines—strongly bias the population of the two conformers. We propose a model, supported by molecular mechanics calculations, which rationalises the sense and magnitude of the conformational selectivity attained in terms of the steric and electronic properties of the controlling centre. The control over conformation may be exploited either by trapping the favoured conformer as an atropisomer, or by using it to relay information about the stereochemistry of the controlling centre.
Co-reporter:Jonathan Clayden and Neoclis Vassiliou
Organic & Biomolecular Chemistry 2006 vol. 4(Issue 14) pp:2667-2678
Publication Date(Web):15 Jun 2006
DOI:10.1039/B604548K
Stereochemistry is information, and stereoselective reactions are the means by which that information may be communicated within and between molecules. The control of remote stereogenic centres can be achieved by stereochemical relay, and the use of thermodynamic control over conformational preference is turning out to be a very powerful method for long-range transmission of stereochemical information.
Co-reporter:Jonathan Clayden, Lluís Vallverdú and Madeleine Helliwell
Organic & Biomolecular Chemistry 2006 vol. 4(Issue 11) pp:2106-2118
Publication Date(Web):25 Apr 2006
DOI:10.1039/B602912D
Benzanilides containing two or more potentially stereogenic amide axes exist in solution as mixtures of conformers which are detectable by NMR. For simple tertiary benzanilides carrying an ortho substituent on each ring, conformational control can be high (up to about 10 : 1) providing the substituents are large, indicating that the two axes are in conformational communication with one another. For more complex diamides, conformational communication breaks down, and mixtures of conformers are evident by NMR.
Co-reporter:Mark S. Betson Dr. ;Christopher P. Worrall;Simon Peace Dr.
Angewandte Chemie 2006 Volume 118(Issue 35) pp:
Publication Date(Web):28 JUL 2006
DOI:10.1002/ange.200601866
Fixierung möglich: Sogar bei Diarylether-Strukturen, die anders als die Struktur von Vancomycin nicht makrocyclisch sind, kann geeignete Substitution zu Atropisomerie führen. Stereochemische Stabilität um die Ar-OAr-Achse bei Raumtemperatur oder darüber ist möglich, wenn keiner der Ringe symmetrisch substituiert ist und zumindest ein Ring eine ortho-tert-Butyl-Gruppe oder etwas Ähnliches trägt.
Co-reporter:Mark S. Betson Dr. ;Christopher P. Worrall;Simon Peace Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 35) pp:
Publication Date(Web):28 JUL 2006
DOI:10.1002/anie.200601866
Bring on the substitute: Even outside of macrocyclic structures, such as vancomycin, appropriate substitution can give rise to atropisomerism in diaryl ethers. Stereochemical stability about the ArOAr axis at room temperature or above is possible when neither of the rings is symmetrically substituted and when at least one ring carries an ortho tert-butyl group or equivalent.
Co-reporter:Mark S. Betson, Jonathan Clayden, Madeleine Helliwell and David Mitjans
Organic & Biomolecular Chemistry 2005 vol. 3(Issue 21) pp:3898-3904
Publication Date(Web):28 Sep 2005
DOI:10.1039/B511452G
Atropisomeric aromatic amides bearing 2-sulfanyl groups are oxidised by m-CPBA to the corresponding sulfoxides apparently with very high diastereoselectivity. NMR studies and oxidations of chiral benzamides however indicate that the kinetic selectivity of the oxidation is in fact relatively poor, and that the final diastereoisomeric ratio (typically >99∶1) is under thermodynamic control, with relatively unhindered Ar–CO rotation readily converting the less stable to the more stable product diastereoisomer. Molecular mechanics indicates that the thermodynamic diastereoselectivity results principally from electrostatic repulsion between the CO and S–O dipoles.
Co-reporter:Jonathan Clayden,
Andrew Lund,
Lluís Vallverdú
and
Madeleine Helliwell
Nature 2004 431(7011) pp:966
Publication Date(Web):
DOI:10.1038/nature02933
Co-reporter:Matteo De Poli and Jonathan Clayden
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 5) pp:NaN843-843
Publication Date(Web):2013/11/25
DOI:10.1039/C3OB42167H
A single thionoglycine (glycine thioamide, –HNCH2C(S)–) residue inserted into a peptide foldamer provides both a pair of germinal protons for use as a 1H NMR stereochemical probe and a chromophore giving rise to a well defined Cotton effect in CD. Comparison of the response of these two features to a local helically chiral environment validates them as independent methods for quantifying the conformational screw-sense preference of a helical oligomer, in this case a peptide made of repeated Aib units. The sign of the Cotton effect provides a measure of the sign of the screw-sense preference, while both the chemical shift separation of the anisochronous signals of the glycine CH2 group and the magnitude of the Cotton effect give an estimate of the helicity excess of the oligomer. The thionoglycine unit is readily introduced synthetically by a thionation of a BocGlyAibOMe dipeptide.
Co-reporter:Edmund W. D. Burke, Gareth A. Morris, Mark A. Vincent, Ian H. Hillier and Jonathan Clayden
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 4) pp:NaN719-719
Publication Date(Web):2011/11/22
DOI:10.1039/C1OB06490H
The non-nucleoside reverse transcriptase inhibitor nevirapine displays in its room temperature 1H-NMR spectrum signals characteristic of a chiral compound. Following suggestions in the recent literature that nevirapine may display atropisomerism—and therefore be a chiral compound, due to slow interconversion between two enantiomeric conformers—we report the results of an NMR and computational study which reveal that while nevirapine does indeed possess two stable enantiomeric conformations, they interconvert with a barrier of about 76 kJ mol−1 at room temperature. Nevirapine has a half life for enantiomerisation at room temperature of the order of seconds, is not atropisomeric, and cannot exist as separable enantiomers.
Co-reporter:Jonathan Clayden, Stephen J. M. Rowbottom, Warren J. Ebenezer and Michael G. Hutchings
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 23) pp:NaN4880-4880
Publication Date(Web):2009/09/30
DOI:10.1039/B911631A
Successive treatment of cyanuric chloride with two aromatic diamines, at least one of them sulfonated, yields water-soluble sulfonated azacalix[4]arenes which may be isolated by crystallisation. Functionalised azacalixarenes may be made by first displacing two chloro substituents from the cyclisation precursor. Attempted formation of an azacalix[6]arene led to a dimeric species for which two structures may be proposed, one of them an azacalixarene catenane.
Co-reporter:Juliette Fremaux, Christel Dolain, Brice Kauffmann, Jonathan Clayden and Gilles Guichard
Chemical Communications 2013 - vol. 49(Issue 67) pp:NaN7417-7417
Publication Date(Web):2013/06/21
DOI:10.1039/C3CC40961A
The structures of various urea oligomers incorporating one or two central achiral 1,2-diamino-1,1-dimethylethane (DADME) units have been investigated in solution and in the crystalline state. These diamine monomers are analogous to the achiral helicogenic amino acid Aib (α-aminoisobutyric acid). Oligomers were found to fold into helical conformations with DADME units inducing local deviations from the canonical helix geometry of urea foldamers.
Co-reporter:Jonathan Clayden and Wesley J. Moran
Organic & Biomolecular Chemistry 2007 - vol. 5(Issue 7) pp:NaN1030-1030
Publication Date(Web):2007/02/27
DOI:10.1039/B700353F
Rh(I)–catalysed [2 + 2 + 2] cycloaddition allows the synthesis of aryl ethers and diaryl methanes containing a high degree of steric hindrance from relatively simple diyne and alkyne precursors. The diarylmethanes made in this way show no evidence in their NMR spectra, however, of rotational restriction.
Co-reporter:Jonathan Clayden, Loïc Lemiègre, Mark Pickworth and Lyn Jones
Organic & Biomolecular Chemistry 2008 - vol. 6(Issue 16) pp:NaN2913-2913
Publication Date(Web):2008/06/16
DOI:10.1039/B802673D
Except in the most hindered of cases, N,N′-diaryl N,N′-dimethyl ureas adopt a conformation with the two aryl rings disposed cis to one another. Variable temperature NMR studies reveal the rate at which the Ar–N bonds rotate as well as the conformational preference of ortho disubstituted ureas in which more than one cis orientation is possible. In general, a conformation in which the aryl rings lie close in space but with their most bulky 2-substituents aligned anti is preferred, but with particularly bulky 2-substituents, conformations in which one of the aryl rings points away from the other may also be populated.
Co-reporter:Heloise Brice and Jonathan Clayden
Chemical Communications 2009(Issue 15) pp:
Publication Date(Web):
DOI:10.1039/B901558B
Co-reporter:Bryden A. F. Le Bailly, Liam Byrne, Vincent Diemer, Mohammadali Foroozandeh, Gareth A. Morris and Jonathan Clayden
Chemical Science (2010-Present) 2015 - vol. 6(Issue 4) pp:NaN2322-2322
Publication Date(Web):2015/01/21
DOI:10.1039/C4SC03944K
Although foldamers, by definition, are extended molecular structures with a well-defined conformation, minor conformers must be populated at least to some extent in solution. We present a quantitative analysis of these minor conformers for a series of helical oligomers built from achiral but helicogenic α-amino acids. By measuring the chain length dependence or chain position dependence of NMR or CD quantities that measure screw-sense preference in a helical oligomer, we quantify values for the decay constant of a conformational signal as it passes through the molecular structure. This conformational signal is a perturbation of the racemic mixture of M and P helices that such oligomers typically adopt by the inclusion of an N or C terminal chiral inducer. We show that decay constants may be very low (<1% signal loss per residue) in non-polar solvents, and we evaluate the increase in decay constant that results in polar solvents, at higher temperatures, and with more conformationally flexible residues such as Gly. Decay constants are independent of whether the signal originates from the N or the C terminus. By interpreting the decay constant in terms of the probability with which conformations containing a screw-sense reversal are populated, we quantify the populations of these alternative minor conformers within the overall ensemble of secondary structures adopted by the foldamer. We deduce helical persistence lengths for Aib polymers that allow us to show that in a non-polar solvent a peptide helix, even in the absence of chiral residues, may continue with the same screw sense for approximately 200 residues.
Co-reporter:Romina Wechsel, Julien Maury, Juliette Fremaux, Scott P. France, Gilles Guichard and Jonathan Clayden
Chemical Communications 2014 - vol. 50(Issue 95) pp:NaN15009-15009
Publication Date(Web):2014/10/14
DOI:10.1039/C4CC06754A
The ability of urea-linked oligomers of achiral diamines (achiral analogues of the well-established chiral oligourea foldamers) to adopt helical conformations was explored spectroscopically. Up to four achiral units were ligated either to a well-formed helical trimer or to a single chiral diamine, and the extent to which they adopted a screw-sense preference was determined by NMR and CD. In the best performing cases, a trimeric chiral oligourea and even a single cis-cyclohexanediamine monomer induced folding into a helical conformation.
Co-reporter:Thomas Boddaert, Jordi Solà, Madeleine Helliwell and Jonathan Clayden
Chemical Communications 2012 - vol. 48(Issue 28) pp:NaN3399-3399
Publication Date(Web):2012/01/31
DOI:10.1039/C2CC00060A
1H NMR studies quantify the abilities of achiral amino acids to communicate a left-handed screw-sense preference from one helical Aib4 domain to another: certain quaternary amino acids (e.g. Ac6c) act as effective conductors of conformational preference while others (e.g. diphenylglycine) acts as insulators.
Co-reporter:Gilles Lemière, Simon Sedehizadeh, Julie Toueg, Nadia Fleary-Roberts and Jonathan Clayden
Chemical Communications 2011 - vol. 47(Issue 13) pp:NaN3747-3747
Publication Date(Web):2011/02/24
DOI:10.1039/C1CC00048A
The alkynylpyrrolidine 4 was made via a dearomatising cyclisation of an aromatic amide, and was elaborated by stannylcupration and palladium-catalysed coupling to achieve the first total synthesis of three members of the isodomoic acid family; the same alkyne can be envisaged as a precursor to several more of this class of amnesic shellfish toxins.
Co-reporter:Bryden A. F. Le Bailly and Jonathan Clayden
Chemical Communications 2014 - vol. 50(Issue 59) pp:NaN7952-7952
Publication Date(Web):2014/06/05
DOI:10.1039/C4CC03261F
The global screw-sense preference of an achiral helical oligomer may be controlled by a single chiral monomer located at one terminus. Remarkably, maximal control is induced in oligomers of the achiral quaternary amino acid Aib by a single C-terminal alaninamide residue, probably because the Ala side chain, though small, is compatible with a 310 helical conformation. The presence or absence of a C-terminal hydrogen bond donor determines the screw sense of the entire oligomer.
Co-reporter:Paul MacLellan and Jonathan Clayden
Chemical Communications 2011 - vol. 47(Issue 12) pp:NaN3397-3397
Publication Date(Web):2011/02/17
DOI:10.1039/C0CC04912C
Lithiation of N-aryl S-α-alkylbenzyl thiocarbamates leads to rearrangement with migration of the N-aryl ring to the anionic centre α to S, a process which generally proceeds with ca. 98% retention of stereochemistry and returns chiral benzylic tertiary thiols in high enantiomeric ratios.
Co-reporter:Rachel C. Atkinson, Daniel J. Leonard, Julien Maury, Daniele Castagnolo, Nicole Volz and Jonathan Clayden
Chemical Communications 2013 - vol. 49(Issue 84) pp:NaN9736-9736
Publication Date(Web):2013/09/05
DOI:10.1039/C3CC46193A
Dianionic enolates formed from N′-aryl urea derivatives of amino acids undergo intramolecular C-arylation by attack of the enolate anion on the N′-aryl ring, leading to a hydantoin derivative of a quaternary amino acid. In situ IR studies allow identification of four intermediates on the reaction pathway.
Co-reporter:Daniel J. Tetlow, Mark A. Vincent, Ian H. Hillier and Jonathan Clayden
Chemical Communications 2013 - vol. 49(Issue 15) pp:NaN1550-1550
Publication Date(Web):2013/01/03
DOI:10.1039/C2CC38704B
Deprotonation with strong bases of N-vinyl ureas carrying an N′-aryl substituent leads to migration of the N′-aryl group from N to C via an allyllithium; with weaker bases and electron-deficient aryl rings the direction of the migration reverses, and aryl substituents α to the urea N atom may migrate from C to N.
Co-reporter:Liam Byrne, Jordi Solà and Jonathan Clayden
Chemical Communications 2015 - vol. 51(Issue 54) pp:NaN10968-10968
Publication Date(Web):2015/06/02
DOI:10.1039/C5CC01790D
Helical peptides built principally from the achiral quaternary amino acid Aib but with an induced preferred screw-sense exhibit enantioselectivity in their chain-extension reactions when presented with a racemic tertiary amino acid. This is the first demonstration that secondary structure alone, in the absence of local chiral residues, can direct the enantioselectivity of peptide coupling.
Co-reporter:Gaëlle Mingat, Joseph J. W. McDouall and Jonathan Clayden
Chemical Communications 2014 - vol. 50(Issue 51) pp:NaN6757-6757
Publication Date(Web):2014/05/06
DOI:10.1039/C4CC02596B
Stereospecific [3,3]-sigmatropic rearrangement of O-substituted thiocarbamate derivatives of enantiopure allylic alcohols provides allylic thiocarbamates as single enantiomers. Intramolecular arylation by rearrangement of their allyllithium derivatives provides allylic tertiary thiols. Allylation and ring-closing metathesis gives 2,5-dihydrothiophenes containing sulfur-bearing quaternary centres.
Co-reporter:Jonathan Clayden
Chemical Society Reviews 2009 - vol. 38(Issue 3) pp:NaN829-829
Publication Date(Web):2008/12/08
DOI:10.1039/B801639A
By exploiting intramolecular interactions such as dipole repulsion, and by incorporating a terminal chiral controlling feature, the global conformation of a molecule may be governed. In such an environment, stereoselective reactions can occur at considerable distances from the source of stereochemical information, providing a simple method for information relay over scales of >1 nm (or about seven bond lengths). This tutorial review discusses the development of this idea, and describes examples which depend on relayed dipole repulsion and on the absolute control of helicity. Future prospects in the area employing control over extended helical foldamers are elaborated.
Co-reporter:Mark S. Betson, Ann Bracegirdle, Jonathan Clayden, Madeleine Helliwell, Andrew Lund, Mark Pickworth, Timothy J. Snape and Christopher P. Worrall
Chemical Communications 2007(Issue 7) pp:NaN756-756
Publication Date(Web):2006/12/18
DOI:10.1039/B614618J
The orientation of Ar–C, Ar–N and Ar–O bonds in biaryls, N,N′-diarylureas and diaryl ethers (whose conformers are distinguishable by NMR) may be controlled with a selectivity up to >95 : 5 by an adjacent stereogenic centre; the selectivity may be greater when a second stereogenic axis is inserted between the controlling centre and the slowly rotating bond.
Co-reporter:Jonathan Clayden, Lluís Vallverdú, James Clayton and Madeleine Helliwell
Chemical Communications 2008(Issue 5) pp:
Publication Date(Web):
DOI:10.1039/B716105K
Co-reporter:Jonathan Clayden, Mark Pickworth and Lyn H. Jones
Chemical Communications 2009(Issue 5) pp:NaN549-549
Publication Date(Web):2008/11/26
DOI:10.1039/B817527F
The well-defined conformation of an N,N′-diarylurea allows a chiral sulfinyl substituent to influence diastereoselectivity in the formation of new stereogenic centres up to 14 bond lengths away.
Co-reporter:Julien Maury, Bryden A. F. Le Bailly, James Raftery and Jonathan Clayden
Chemical Communications 2015 - vol. 51(Issue 59) pp:NaN11805-11805
Publication Date(Web):2015/06/05
DOI:10.1039/C5CC02995C
Linking together an oligourea and an oligoamide foldamer gives rise to a conformationally well-defined structure, despite the different hydrogen-bonding patterns in the two domains, provided the oligomers are ligated amide C terminus to urea N terminus. A powerful screw-sense preference induced at the N terminus of the resulting chimeric structure provides evidence for cooperative conformational interactions within the ‘block co-foldamer’.
Co-reporter:Daniele Castagnolo, Leonardo Degennaro, Renzo Luisi and Jonathan Clayden
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 8) pp:NaN2340-2340
Publication Date(Web):2014/12/16
DOI:10.1039/C4OB02329C
The addition of n-butyllithium to alkenylthiocarbamates in the presence of (−)-sparteine or the (+)-sparteine surrogate leads to asymmetric carbolithiation, and returns enantiomerically enriched thiocarbamate derivatives of secondary thiols. In THF, with the (+)-sparteine surrogate, in situ aryl migration leads to an enantiomerically enriched tertiary thiol derivative. Remarkably, the two pseudoenantiomeric chiral ligands do not always give enantiomeric products, probably as a result of a complex interplay of kinetic and thermodynamic control. In situ IR and NMR studies of a stable, hindered lithiated thiocarbamate demonstrated its chemical and configurational stability over a period of hours at 0 °C.
Co-reporter:Sarah J. Pike, James Raftery, Simon J. Webb and Jonathan Clayden
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 24) pp:NaN4131-4131
Publication Date(Web):2014/04/29
DOI:10.1039/C4OB00268G
The effect of Schellman motifs on the adoption of stable 310 helical conformations in a series of aminoisobutyric (Aib) oligomers has been studied in the solid state and solution. The destabilising effect of the Schellman motif (a local inversion of helical screw-sense due to a C-terminal ester residue) was quantified in the solid state using X-ray crystallography through analysis of the torsion angles and their deviation from those observed in an ideal 310 helix. Investigation of the intramolecular hydrogen-bonding interactions in the solid state led to the identification of a fully extended C5 conformation in one oligomer, which is a novel folding motif for Aib oligomers. The effect of ester groups with differing steric demands on intermolecular hydrogen-bonding contacts in the solid state was also ascertained. In solution, the adoption of a 310 conformation in Aib oligomers appeared to be more finely tuned, depending on a number of factors, including chain length and the steric demands of the C-terminal destabilising Schellman motif.
Co-reporter:Sarah J. Pike, Matteo De Poli, Wojciech Zawodny, James Raftery, Simon J. Webb and Jonathan Clayden
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 19) pp:NaN3176-3176
Publication Date(Web):2013/03/27
DOI:10.1039/C3OB40463C
Ligating simple amino alcohol or amino ester monomers containing enantiotopic fluorine substituents to the C-terminus of a helical peptide places the fluorine atoms in diastereotopic environments, and gives two distinct and easily identifiable signals in the 19F NMR spectrum. In the case of a dynamically inverting helix built from achiral monomers, the chemical shift separation between the 19F signals provides a simple means of analysing the ratio of screw-sense conformers in the oligomer, in cases where an asymmetric bias leads to a screw-sense preference.

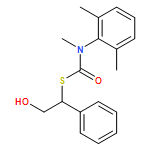
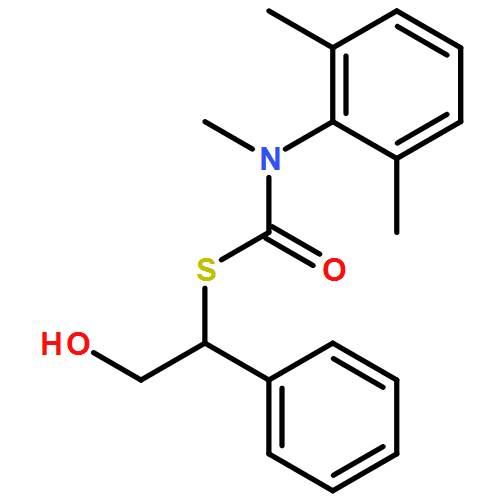
![1H-Imidazole-1-carbothioic acid, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3752800/1415351-98-2.png)
![1H-Imidazole-1-carbothioic acid, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3752800/1415351-98-2_b.png)
![Carbamothioic acid, N-methyl-N-1-naphthalenyl-, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3752800/1415351-96-0.png)
![Carbamothioic acid, N-methyl-N-1-naphthalenyl-, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3752800/1415351-96-0_b.png)
![Carbamothioic acid, N-(3-methoxyphenyl)-N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3752800/1415351-95-9.png)
![Carbamothioic acid, N-(3-methoxyphenyl)-N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3752800/1415351-95-9_b.png)
![Carbamothioic acid, N-(2-methoxyphenyl)-N-methyl-, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3752800/1415351-94-8.png)
![Carbamothioic acid, N-(2-methoxyphenyl)-N-methyl-, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3752800/1415351-94-8_b.png)
![Carbamothioic acid, N-methyl-N-(4-methylphenyl)-, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3752800/1415351-92-6.png)
![Carbamothioic acid, N-methyl-N-(4-methylphenyl)-, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3752800/1415351-92-6_b.png)


![Carbamothioic acid, N-(4-chlorophenyl)-N-methyl-, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3752800/1415351-91-5.png)
![Carbamothioic acid, N-(4-chlorophenyl)-N-methyl-, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3752800/1415351-91-5_b.png)
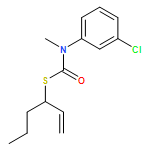
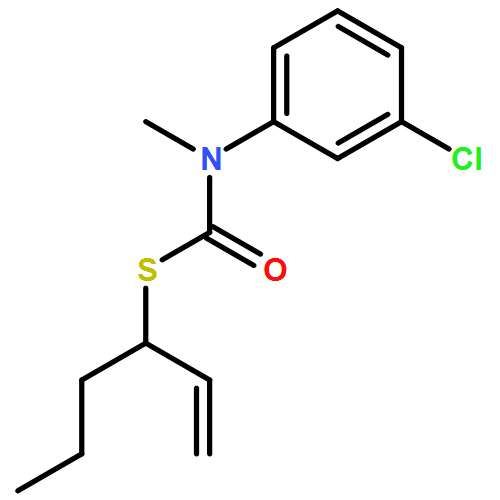
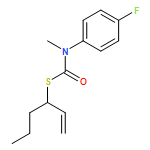
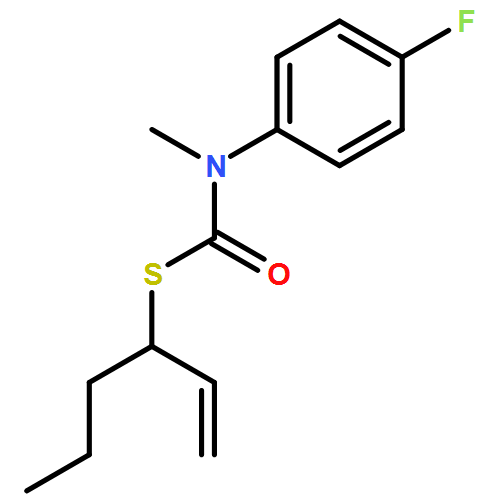
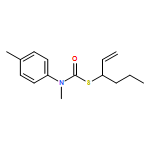
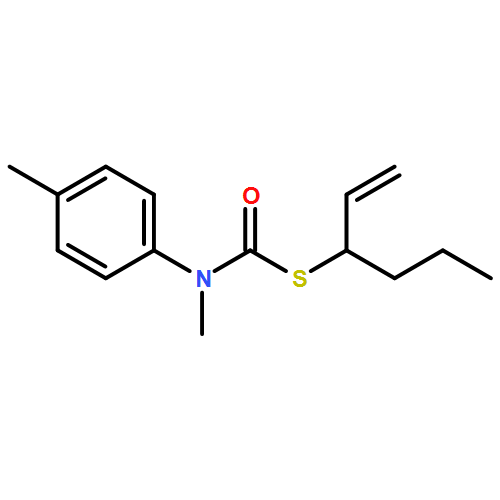
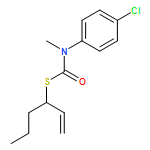
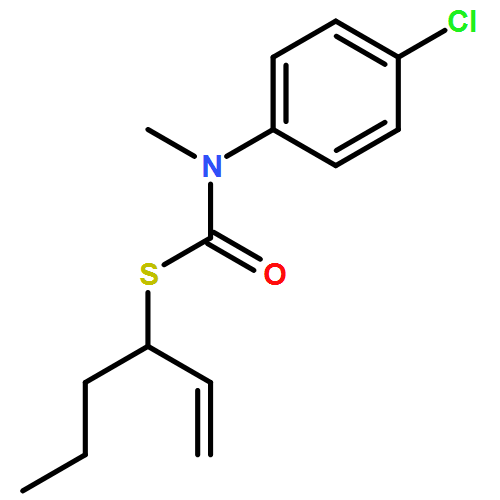
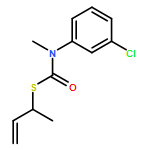
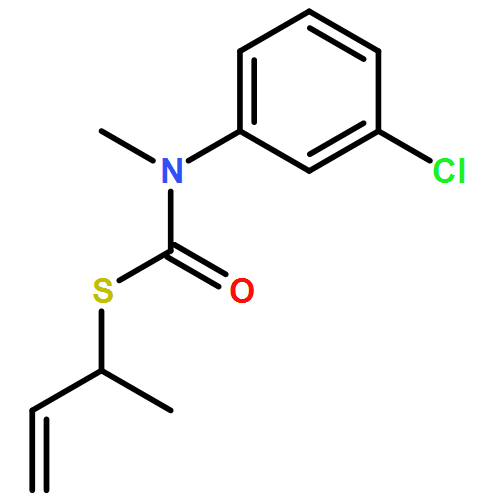
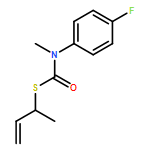
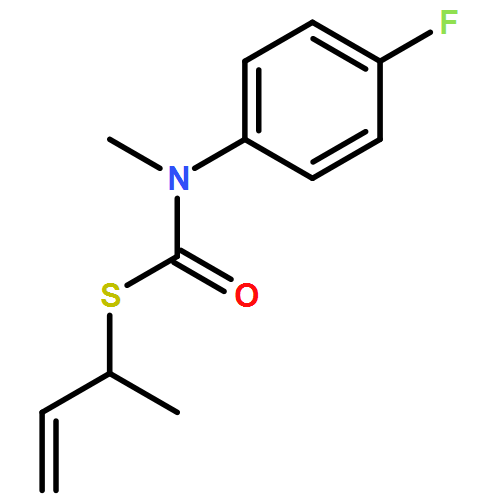
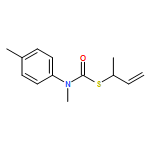
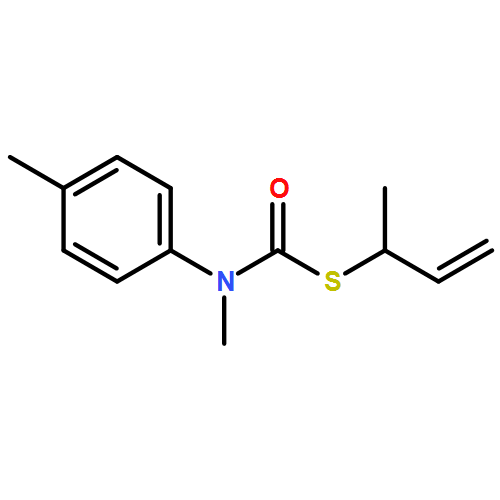
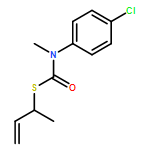
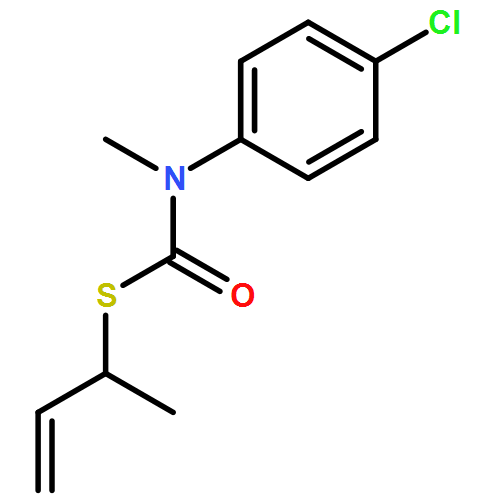

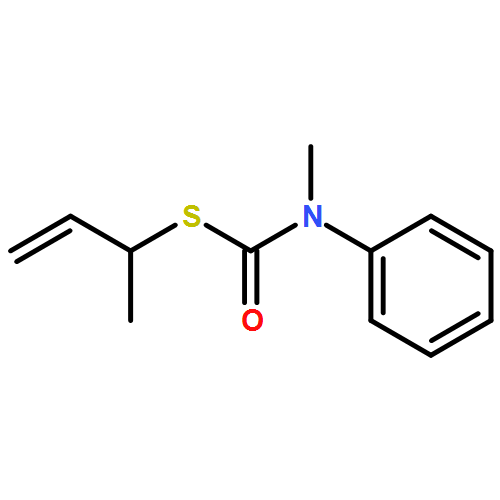
![1H-Imidazole-1-carbothioic acid, S-[(1S,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3752700/1415351-65-3.png)
![1H-Imidazole-1-carbothioic acid, S-[(1S,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3752700/1415351-65-3_b.png)
![1H-Imidazole-1-carbothioic acid, S-[(2E)-1-methyl-2-buten-1-yl] ester](/data/chemimg/3752700/1415351-64-2.png)
![1H-Imidazole-1-carbothioic acid, S-[(2E)-1-methyl-2-buten-1-yl] ester](/data/chemimg/3752700/1415351-64-2_b.png)

![Carbamothioic acid, N-methyl-, S-[(1R)-1-methyl-1-phenyl-2-propen-1-yl] ester](/data/chemimg/3752700/1371664-49-1.png)
![Carbamothioic acid, N-methyl-, S-[(1R)-1-methyl-1-phenyl-2-propen-1-yl] ester](/data/chemimg/3752700/1371664-49-1_b.png)
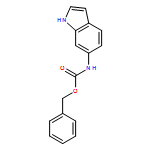
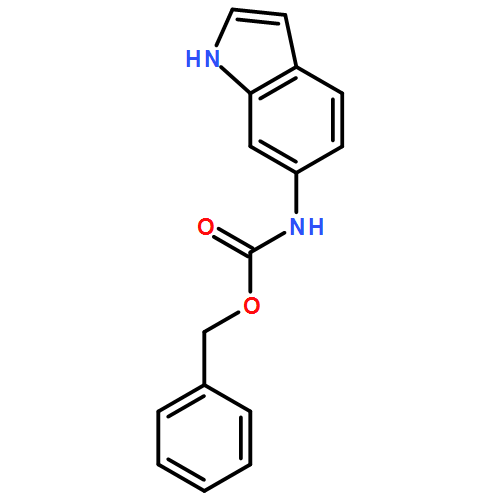
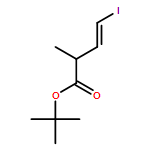
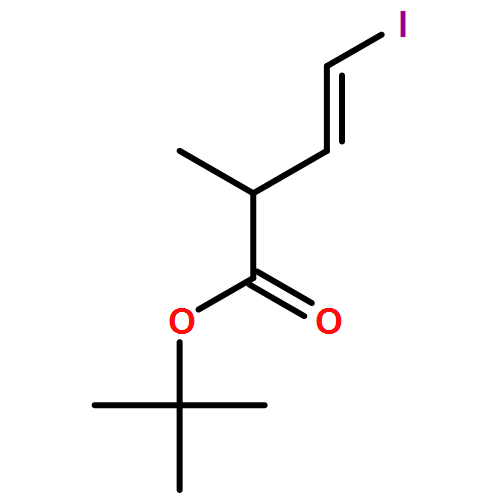
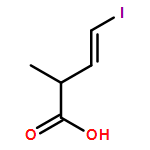
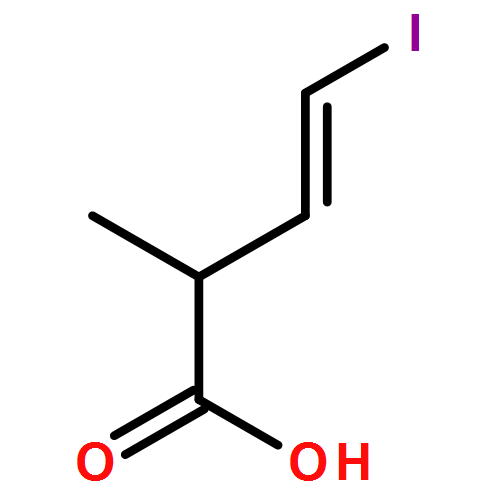
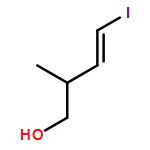
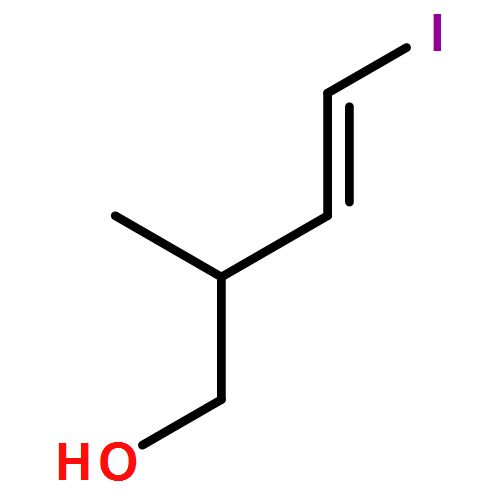
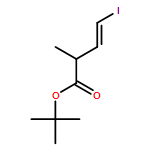
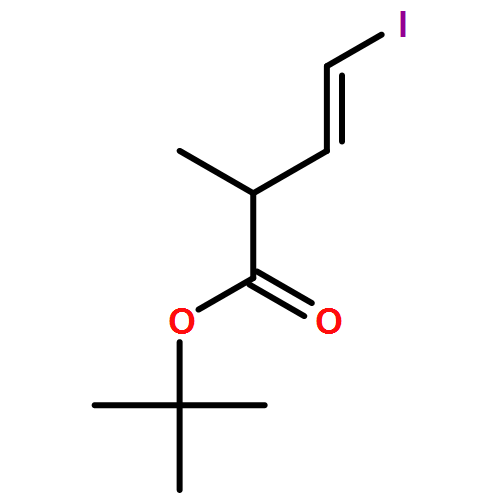
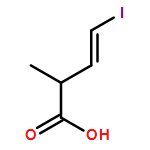
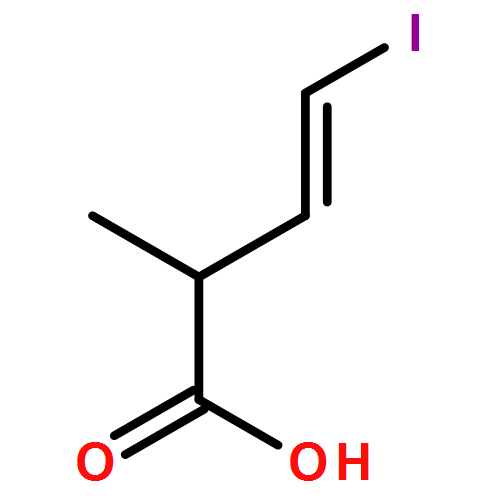
![Carbamic chloride, N-methyl-N-[4-(trifluoromethyl)phenyl]-](/data/chemimg/3752700/1235712-89-6.png)
![Carbamic chloride, N-methyl-N-[4-(trifluoromethyl)phenyl]-](/data/chemimg/3752700/1235712-89-6_b.png)
![L-Proline, 1-[(methylphenylamino)carbonyl]-](/data/chemimg/3752700/1217442-54-0.png)
![L-Proline, 1-[(methylphenylamino)carbonyl]-](/data/chemimg/3752700/1217442-54-0_b.png)
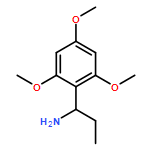
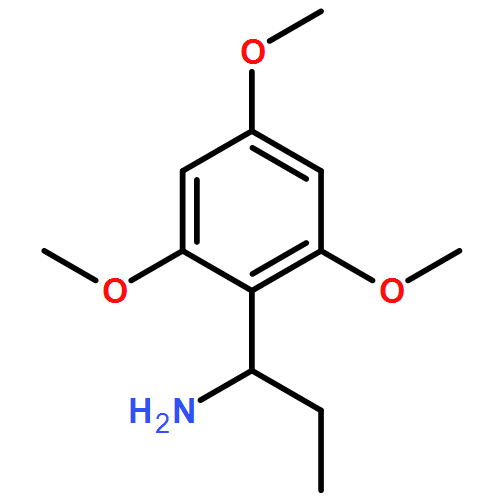
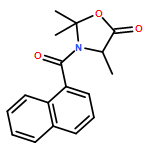
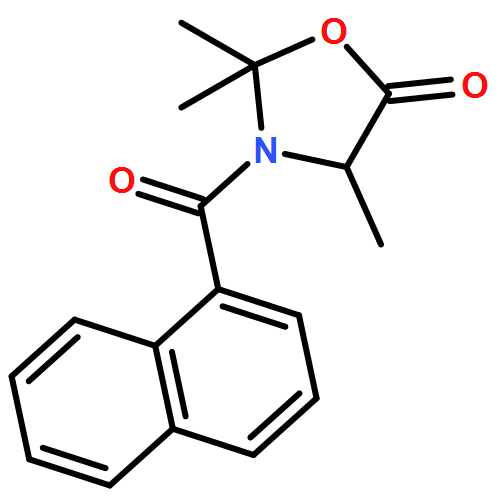
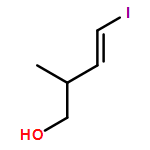








![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bS)-](/data/chemimg/103700/874948-63-7.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bS)-](/data/chemimg/103700/874948-63-7_b.png)
![Carbamic acid, [(1S)-2-[(1,1-dimethylethyl)amino]-1-methyl-2-oxoethyl]-, phenylmethyl ester (9CI)](/data/chemimg/3762200/871579-11-2.png)
![Carbamic acid, [(1S)-2-[(1,1-dimethylethyl)amino]-1-methyl-2-oxoethyl]-, phenylmethyl ester (9CI)](/data/chemimg/3762200/871579-11-2_b.png)
![Diphenanthro[4,3-d:3',4'-f][1,3,2]dioxaphosphepin,18-hydroxy-8,9-diphenyl-, 18-oxide, (8aS)-](/data/chemimg/115800/871130-17-5.png)
![Diphenanthro[4,3-d:3',4'-f][1,3,2]dioxaphosphepin,18-hydroxy-8,9-diphenyl-, 18-oxide, (8aS)-](/data/chemimg/115800/871130-17-5_b.png)



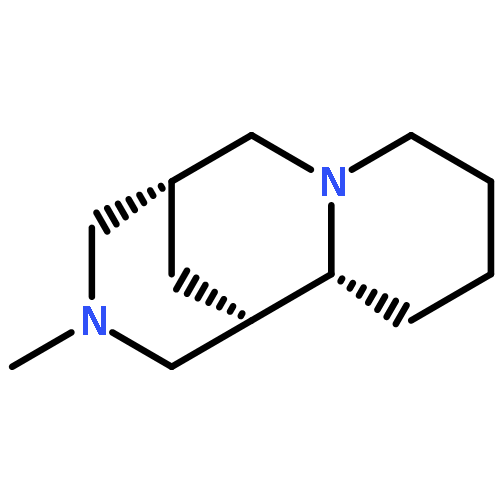
![CARBAMIC ACID, [(1S)-1-(AMINOMETHYL)-2-METHYLPROPYL]-, 1,1-DIMETHYLETHYL ESTER](http://img.cochemist.com/ccimg/400700/400652-49-5.png)
![CARBAMIC ACID, [(1S)-1-(AMINOMETHYL)-2-METHYLPROPYL]-, 1,1-DIMETHYLETHYL ESTER](http://img.cochemist.com/ccimg/400700/400652-49-5_b.png)


![Carbamic acid, [(1S)-1-methyl-2-[[[(1-methylethyl)amino]carbonyl]amino]ethyl]-, 1,1-dimethylethyl ester (9CI)](/data/chemimg/3755700/254101-05-8.png)
![Carbamic acid, [(1S)-1-methyl-2-[[[(1-methylethyl)amino]carbonyl]amino]ethyl]-, 1,1-dimethylethyl ester (9CI)](/data/chemimg/3755700/254101-05-8_b.png)
![Carbamic acid, N-[(2S)-2-[[(1,1-dimethylethoxy)carbonyl]amino]-3-methylbutyl]-, 2,5-dioxo-1-pyrrolidinyl ester](/data/chemimg/3755700/254100-97-5.png)
![Carbamic acid, N-[(2S)-2-[[(1,1-dimethylethoxy)carbonyl]amino]-3-methylbutyl]-, 2,5-dioxo-1-pyrrolidinyl ester](/data/chemimg/3755700/254100-97-5_b.png)
![Carbamic acid, N-[(2S)-2-[[(1,1-dimethylethoxy)carbonyl]amino]propyl]-, 2,5-dioxo-1-pyrrolidinyl ester](/data/chemimg/3755700/254100-96-4.png)
![Carbamic acid, N-[(2S)-2-[[(1,1-dimethylethoxy)carbonyl]amino]propyl]-, 2,5-dioxo-1-pyrrolidinyl ester](/data/chemimg/3755700/254100-96-4_b.png)
![Carbamic acid,[2-[[[(2,5-dioxo-1-pyrrolidinyl)oxy]carbonyl]amino]ethyl]-, 1,1-dimethylethylester (9CI)](http://img.cochemist.com/ccimg/254200/254100-95-3.png)
![Carbamic acid,[2-[[[(2,5-dioxo-1-pyrrolidinyl)oxy]carbonyl]amino]ethyl]-, 1,1-dimethylethylester (9CI)](http://img.cochemist.com/ccimg/254200/254100-95-3_b.png)
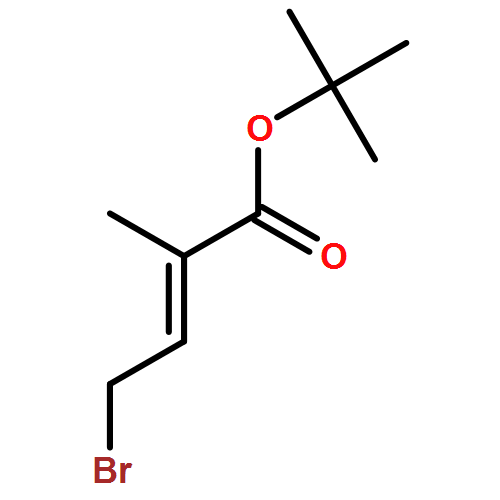


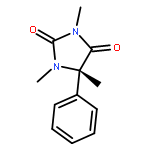
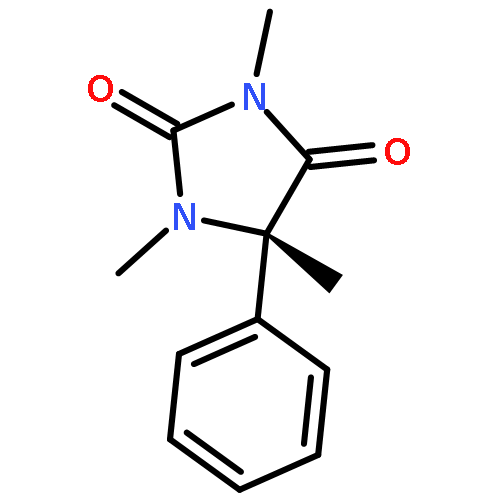
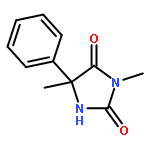
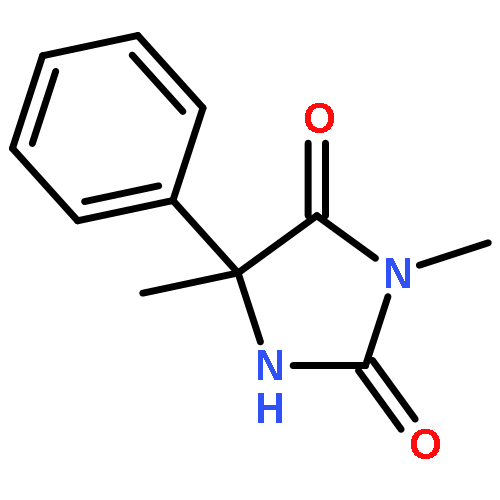
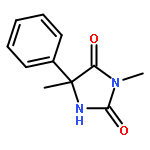
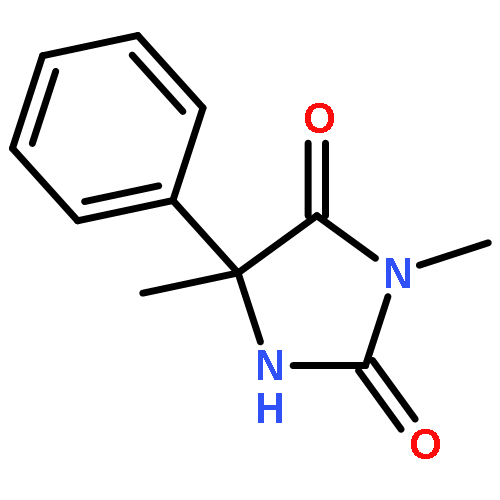
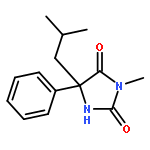
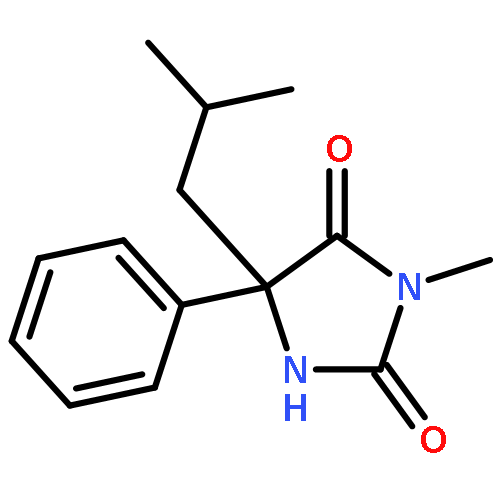
![Glycine, N-3-butenyl-N-[(1R)-1-phenylethyl]-, methyl ester](http://img.cochemist.com/ccimg/186700/186653-18-9.png)
![Glycine, N-3-butenyl-N-[(1R)-1-phenylethyl]-, methyl ester](http://img.cochemist.com/ccimg/186700/186653-18-9_b.png)



![L-Leucine, N-[(methylphenylamino)carbonyl]-](http://img.cochemist.com/ccimg/168500/168470-92-6.png)
![L-Leucine, N-[(methylphenylamino)carbonyl]-](http://img.cochemist.com/ccimg/168500/168470-92-6_b.png)
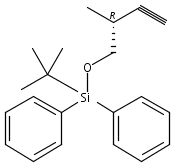
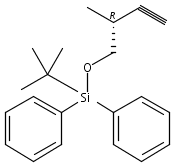


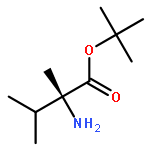
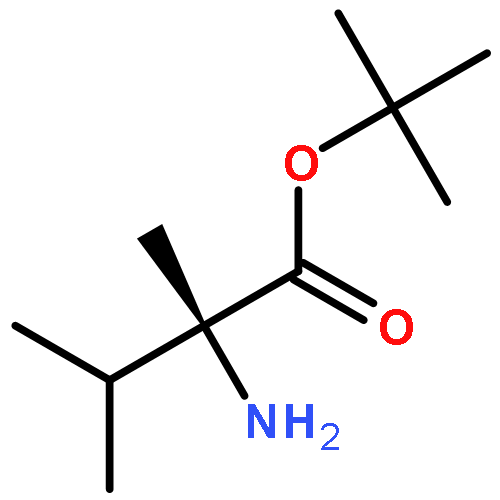




![2-Propen-1-amine, N-[(4-chlorophenyl)methylene]-](http://img.cochemist.com/ccimg/131500/131480-14-3.png)
![2-Propen-1-amine, N-[(4-chlorophenyl)methylene]-](http://img.cochemist.com/ccimg/131500/131480-14-3_b.png)
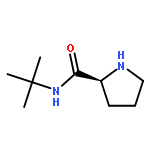
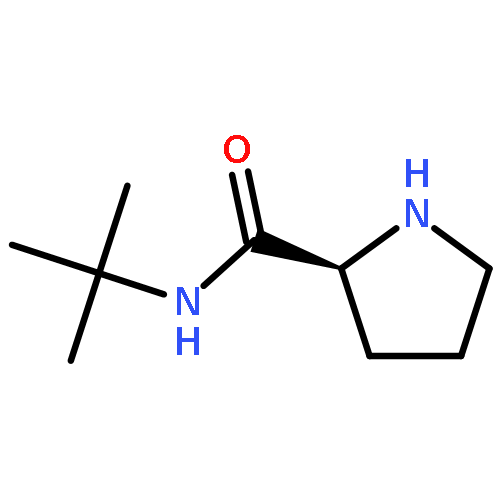

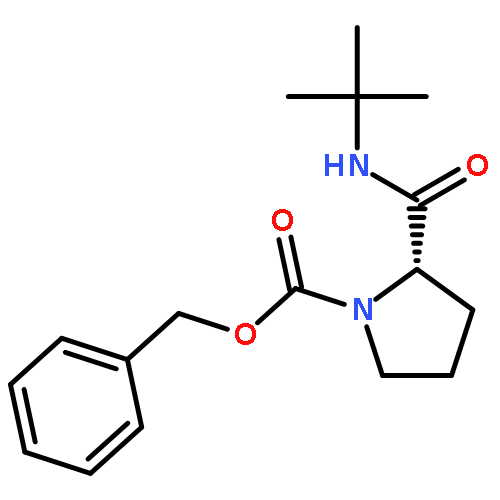
![1-Propanol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-, (R)-](http://img.cochemist.com/ccimg/112100/112057-64-4.png)
![1-Propanol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-, (R)-](http://img.cochemist.com/ccimg/112100/112057-64-4_b.png)

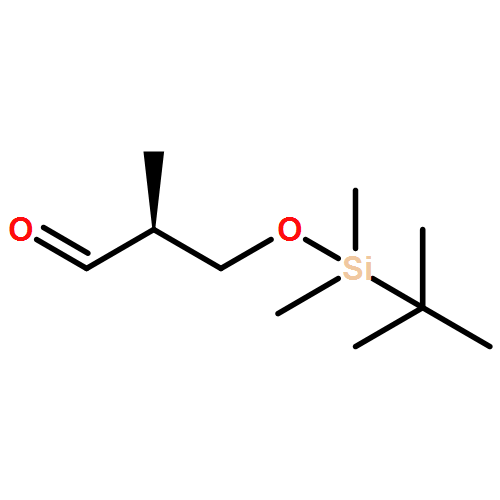
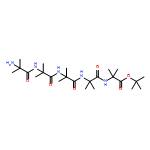
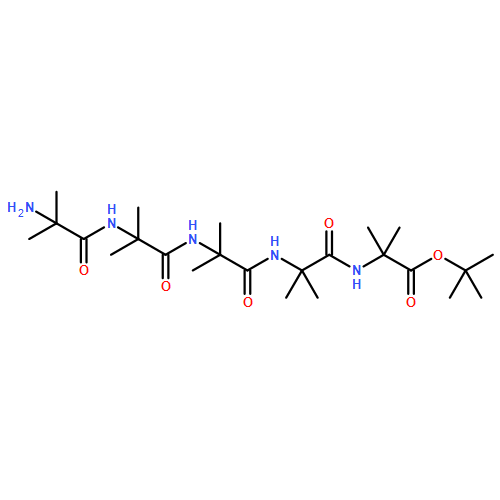

![1-Propanol, 3-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-2-methyl-, (2R)-](/data/chemimg/3752600/95514-04-8.png)
![1-Propanol, 3-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-2-methyl-, (2R)-](/data/chemimg/3752600/95514-04-8_b.png)
![Propanal, 3-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-2-methyl-, (S)-](http://img.cochemist.com/ccimg/92900/92817-88-4.png)
![Propanal, 3-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-2-methyl-, (S)-](http://img.cochemist.com/ccimg/92900/92817-88-4_b.png)


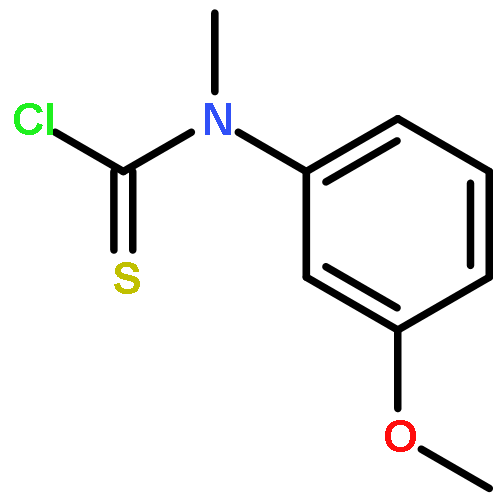
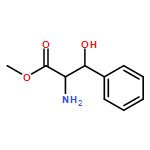
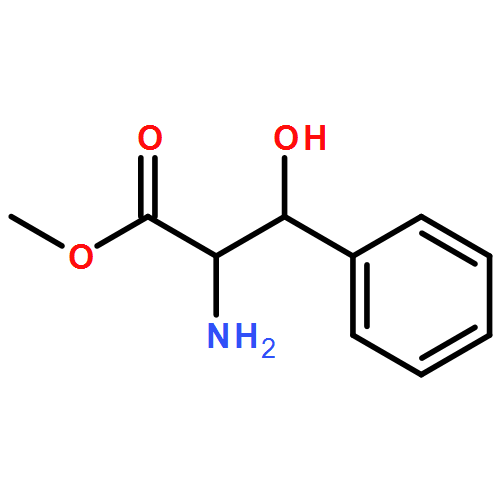




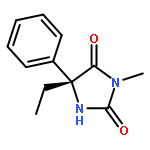
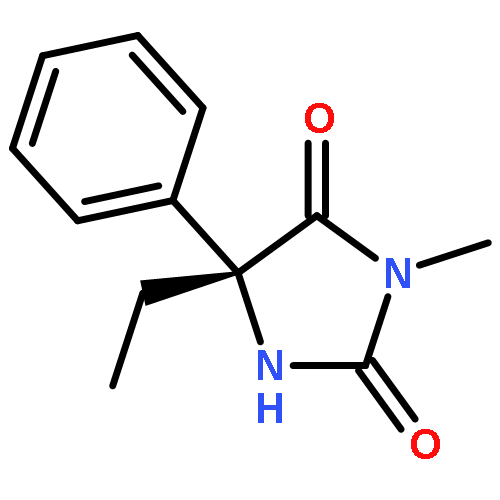
![Benzene, 1,1'-[(1S,2S)-1,2-dimethoxy-1,2-ethanediyl]bis-](http://img.cochemist.com/ccimg/62900/62860-42-8.png)
![Benzene, 1,1'-[(1S,2S)-1,2-dimethoxy-1,2-ethanediyl]bis-](http://img.cochemist.com/ccimg/62900/62860-42-8_b.png)



![BENZYL N-[1-(TERT-BUTYLAMINO)-3-METHYL-1-OXOBUTAN-2-YL]CARBAMATE](/data/chemimg/608700/61274-17-7.png)
![BENZYL N-[1-(TERT-BUTYLAMINO)-3-METHYL-1-OXOBUTAN-2-YL]CARBAMATE](/data/chemimg/608700/61274-17-7_b.png)


![Pyrrolidine, 1-[(2S)-2-amino-1-oxopropyl]-](http://img.cochemist.com/ccimg/56400/56384-04-4.png)
![Pyrrolidine, 1-[(2S)-2-amino-1-oxopropyl]-](http://img.cochemist.com/ccimg/56400/56384-04-4_b.png)



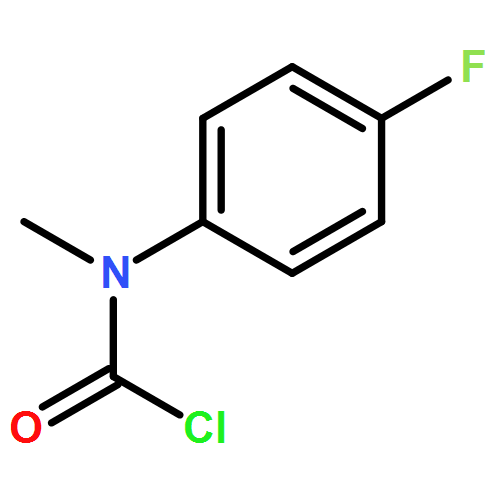




![2-[(4-METHYL-2-NITROPHENYL)DIAZENYL]-N-(2-METHYLPHENYL)-3-OXOBUTA<WBR />NAMIDE](http://img.cochemist.com/ccimg/36300/36291-48-2.png)
![2-[(4-METHYL-2-NITROPHENYL)DIAZENYL]-N-(2-METHYLPHENYL)-3-OXOBUTA<WBR />NAMIDE](http://img.cochemist.com/ccimg/36300/36291-48-2_b.png)

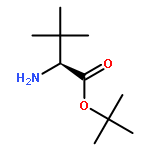
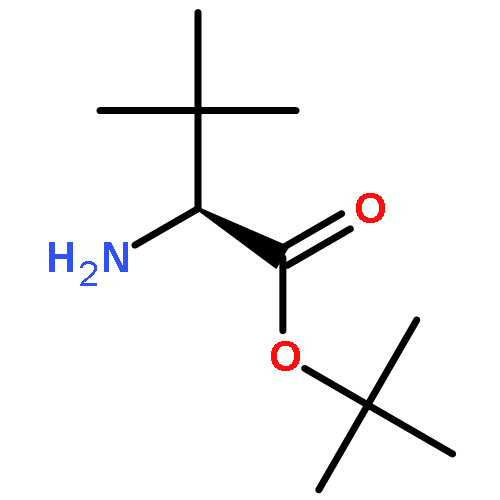
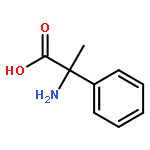
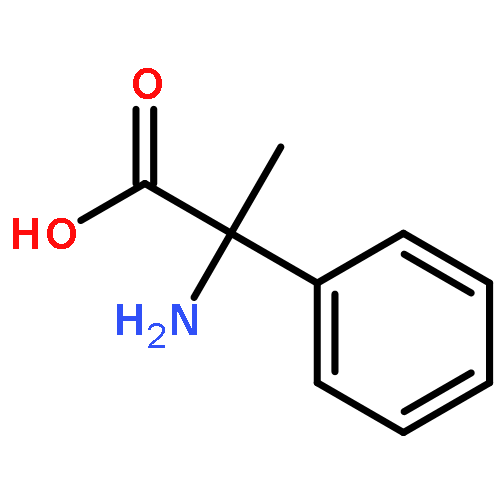


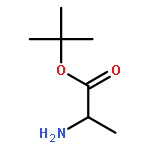
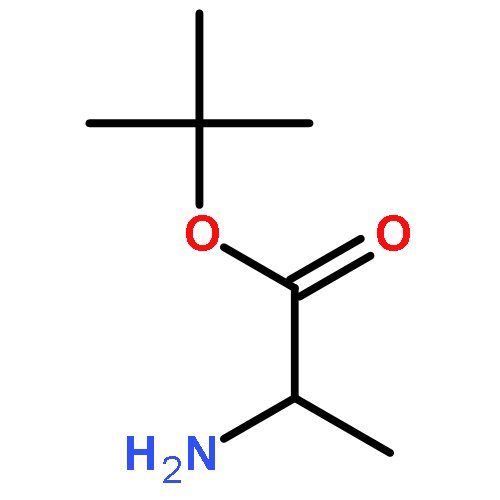


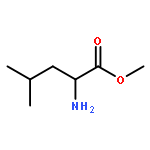
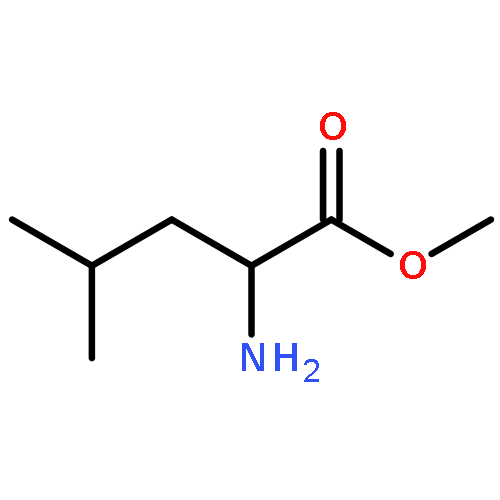
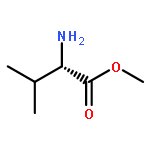
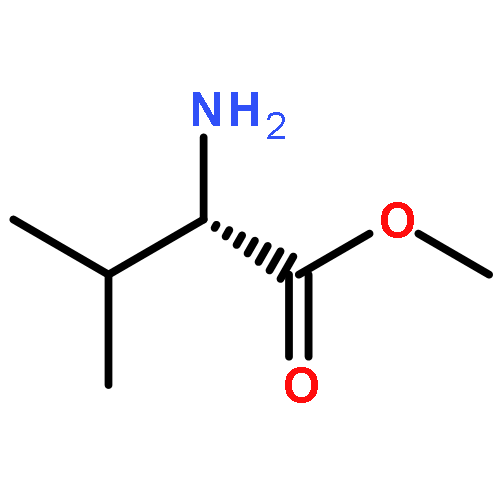


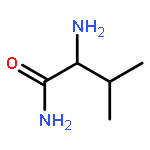
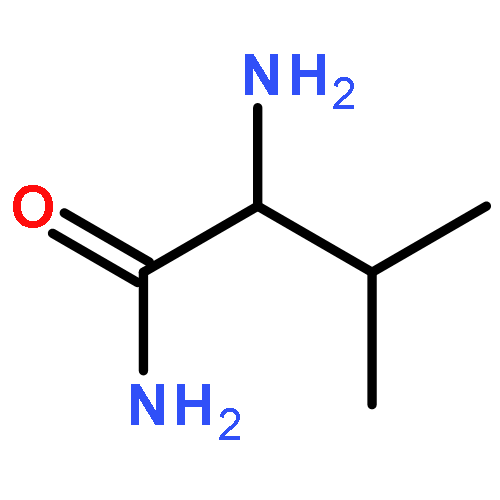

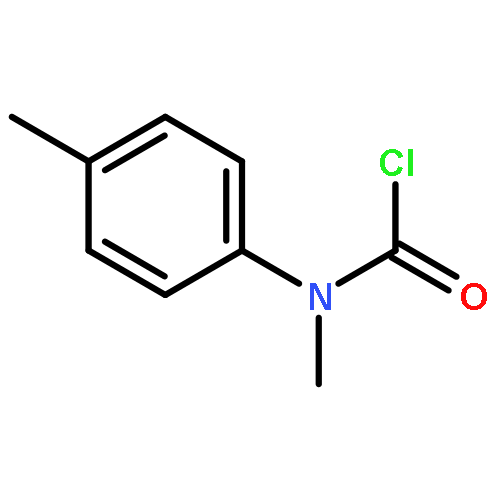
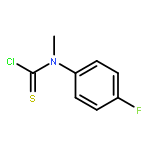

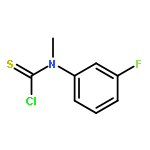




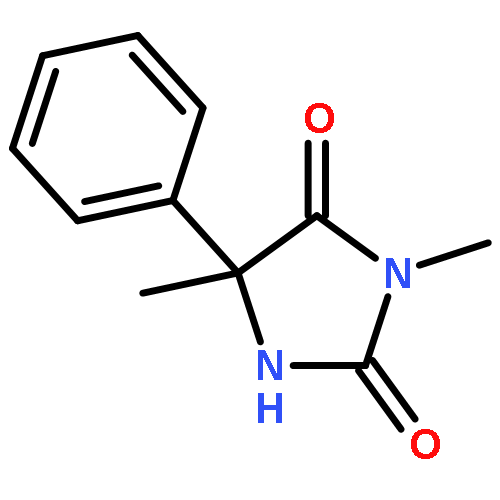
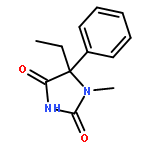
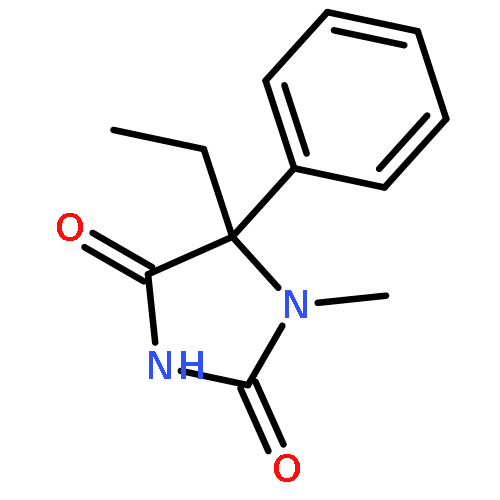
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4.png)
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4_b.png)




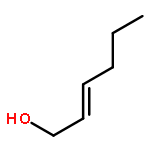
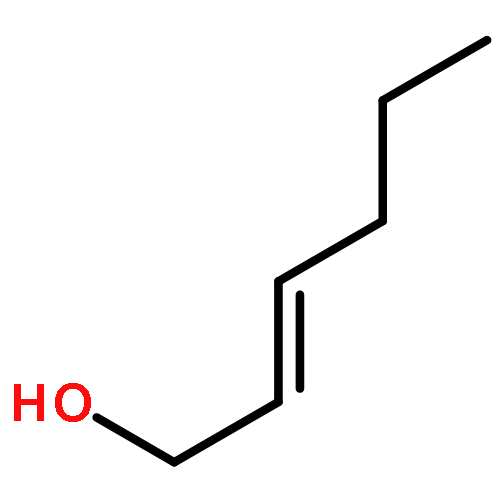



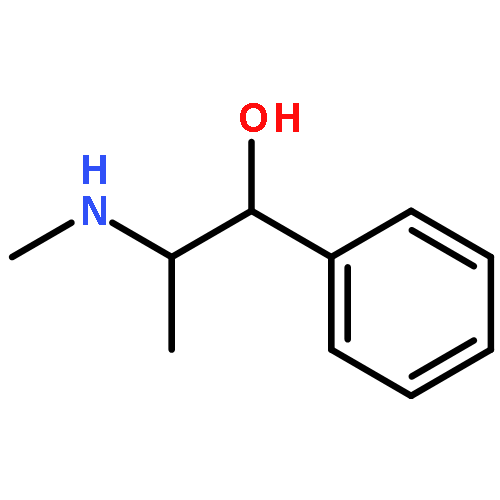


![Benzene, 1-fluoro-3-[1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-](/data/chemimg/3752800/1558812-46-6.png)
![Benzene, 1-fluoro-3-[1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-](/data/chemimg/3752800/1558812-46-6_b.png)
![Benzene, 1-fluoro-3-[(1R)-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-](/data/chemimg/3752800/1558812-45-5.png)
![Benzene, 1-fluoro-3-[(1R)-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-](/data/chemimg/3752800/1558812-45-5_b.png)
![Propanamide, 2-azido-N-[1-(4,5-dihydro-4,4-dimethyl-5-oxo-2-oxazolyl)-1-methylethyl]-2-methyl-](/data/chemimg/3752600/1462343-89-0.png)
![Propanamide, 2-azido-N-[1-(4,5-dihydro-4,4-dimethyl-5-oxo-2-oxazolyl)-1-methylethyl]-2-methyl-](/data/chemimg/3752600/1462343-89-0_b.png)
![Propanamide, 2-azido-N-[2-[[1-(4,5-dihydro-4,4-dimethyl-5-oxo-2-oxazolyl)-1-methylethyl]amino]-1,1-dimethyl-2-oxoethyl]-2-methyl-](/data/chemimg/3752600/1187839-36-6.png)
![Propanamide, 2-azido-N-[2-[[1-(4,5-dihydro-4,4-dimethyl-5-oxo-2-oxazolyl)-1-methylethyl]amino]-1,1-dimethyl-2-oxoethyl]-2-methyl-](/data/chemimg/3752600/1187839-36-6_b.png)










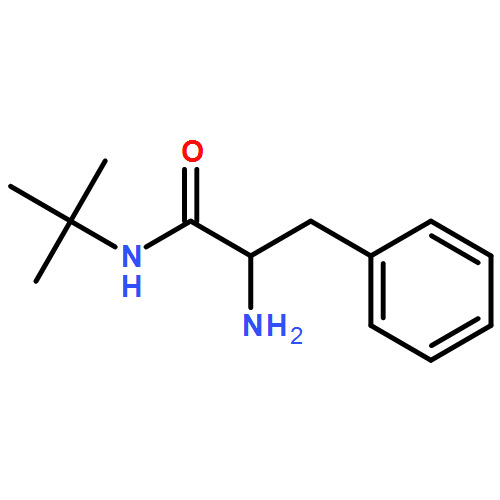
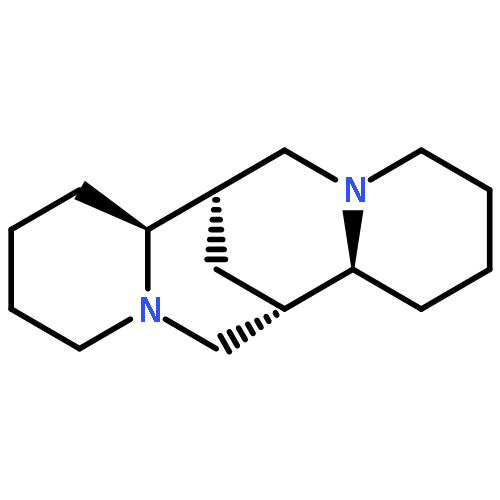


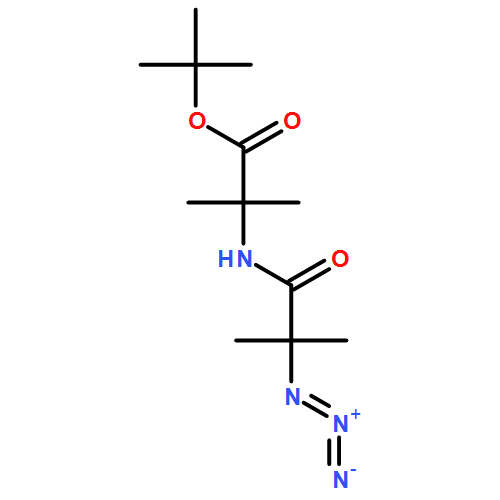
![Carbamic acid, N-[(1S)-1-(fluorocarbonyl)-1,2-dimethylpropyl]-, phenylmethyl ester](/data/chemimg/3752400/175224-23-4.png)
![Carbamic acid, N-[(1S)-1-(fluorocarbonyl)-1,2-dimethylpropyl]-, phenylmethyl ester](/data/chemimg/3752400/175224-23-4_b.png)
![L-Isovaline, 3-methyl-N-[(phenylmethoxy)carbonyl]-](/data/chemimg/3752600/137584-40-8.png)
![L-Isovaline, 3-methyl-N-[(phenylmethoxy)carbonyl]-](/data/chemimg/3752600/137584-40-8_b.png)
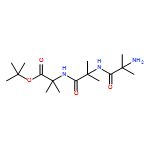

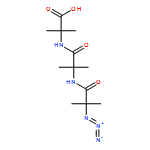
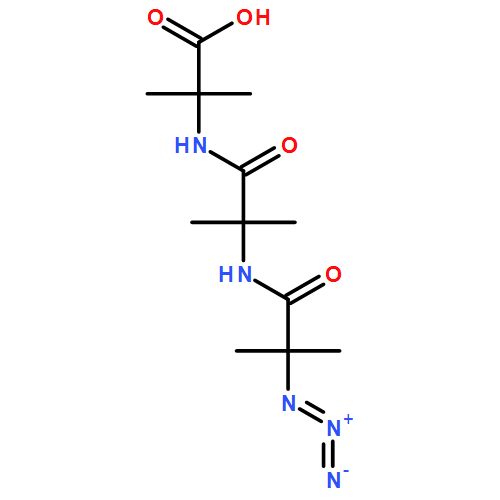


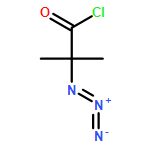
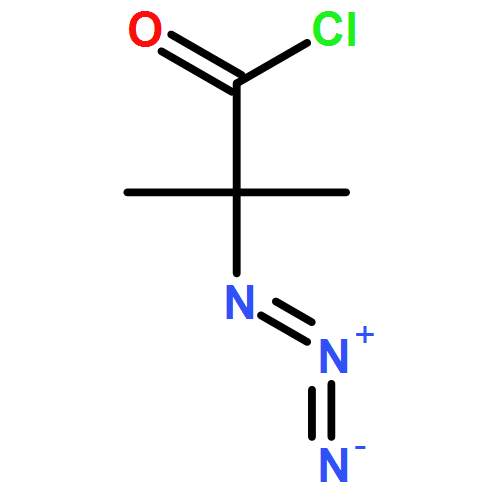
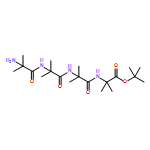

![Carbamothioic acid, N-methyl-N-phenyl-, S-[(1R)-1-phenylethyl] ester](/data/chemimg/3756100/1670221-07-4.png)
![Carbamothioic acid, N-methyl-N-phenyl-, S-[(1R)-1-phenylethyl] ester](/data/chemimg/3756100/1670221-07-4_b.png)
![Pyrrolidine, 1-[[dimethyl[(1E,3E)-5-(methylthio)-1,3-pentadien-1-yl]silyl]methyl]-](/data/chemimg/3754200/1644149-51-8.png)
![Pyrrolidine, 1-[[dimethyl[(1E,3E)-5-(methylthio)-1,3-pentadien-1-yl]silyl]methyl]-](/data/chemimg/3754200/1644149-51-8_b.png)
![Pyrrolidine, 1-[[dimethyl[(1E)-3-(methylthio)-1,4-pentadien-1-yl]silyl]methyl]-](/data/chemimg/3754200/1644149-50-7.png)
![Pyrrolidine, 1-[[dimethyl[(1E)-3-(methylthio)-1,4-pentadien-1-yl]silyl]methyl]-](/data/chemimg/3754200/1644149-50-7_b.png)
![Pyrrolidine, 1-[[dimethyl[(2E)-1-(methylthio)-2,4-pentadien-1-yl]silyl]methyl]-](/data/chemimg/3754200/1644149-49-4.png)
![Pyrrolidine, 1-[[dimethyl[(2E)-1-(methylthio)-2,4-pentadien-1-yl]silyl]methyl]-](/data/chemimg/3754200/1644149-49-4_b.png)
![Pyrrolidine, 1-[[[(1E)-3-ethenyl-1,5-hexadien-1-yl]dimethylsilyl]methyl]-](/data/chemimg/3754200/1644149-48-3.png)
![Pyrrolidine, 1-[[[(1E)-3-ethenyl-1,5-hexadien-1-yl]dimethylsilyl]methyl]-](/data/chemimg/3754200/1644149-48-3_b.png)
![Pyrrolidine, 1-[[(3-ethyl-2,4-pentadien-1-yl)dimethylsilyl]methyl]-](/data/chemimg/3754200/1644149-47-2.png)
![Pyrrolidine, 1-[[(3-ethyl-2,4-pentadien-1-yl)dimethylsilyl]methyl]-](/data/chemimg/3754200/1644149-47-2_b.png)
![Pyrrolidine, 1-[[[(1E)-3-ethyl-1,3-pentadien-1-yl]dimethylsilyl]methyl]-](/data/chemimg/3754200/1644149-46-1.png)
![Pyrrolidine, 1-[[[(1E)-3-ethyl-1,3-pentadien-1-yl]dimethylsilyl]methyl]-](/data/chemimg/3754200/1644149-46-1_b.png)
![Pyrrolidine, 1-[[[(1E)-3-ethyl-1,4-pentadien-1-yl]dimethylsilyl]methyl]-](/data/chemimg/3754200/1644149-45-0.png)
![Pyrrolidine, 1-[[[(1E)-3-ethyl-1,4-pentadien-1-yl]dimethylsilyl]methyl]-](/data/chemimg/3754200/1644149-45-0_b.png)
![Pyrrolidine, 1-[[(1E,3E)-1,3-hexadien-1-yldimethylsilyl]methyl]-](/data/chemimg/3754200/1644149-44-9.png)
![Pyrrolidine, 1-[[(1E,3E)-1,3-hexadien-1-yldimethylsilyl]methyl]-](/data/chemimg/3754200/1644149-44-9_b.png)
![Pyrrolidine, 1-[[dimethyl[(1E)-3-methyl-1,4-pentadien-1-yl]silyl]methyl]-](/data/chemimg/3754200/1644149-43-8.png)
![Pyrrolidine, 1-[[dimethyl[(1E)-3-methyl-1,4-pentadien-1-yl]silyl]methyl]-](/data/chemimg/3754200/1644149-43-8_b.png)
![Pyrrolidine, 1-[[dimethyl[(2E)-1-methyl-2,4-pentadien-1-yl]silyl]methyl]-](/data/chemimg/3754200/1644149-42-7.png)
![Pyrrolidine, 1-[[dimethyl[(2E)-1-methyl-2,4-pentadien-1-yl]silyl]methyl]-](/data/chemimg/3754200/1644149-42-7_b.png)
![Pyrrolidine, 1-[[dimethyl[(1E,3E)-5-(trimethylsilyl)-1,3-pentadien-1-yl]silyl]methyl]-](/data/chemimg/3754200/1644149-41-6.png)
![Pyrrolidine, 1-[[dimethyl[(1E,3E)-5-(trimethylsilyl)-1,3-pentadien-1-yl]silyl]methyl]-](/data/chemimg/3754200/1644149-41-6_b.png)
![Ethanamine, N-[[dimethyl(2E)-2,4-pentadien-1-ylsilyl]methyl]-2-methoxy-N-(2-methoxyethyl)-](/data/chemimg/3754200/1644149-39-2.png)
![Ethanamine, N-[[dimethyl(2E)-2,4-pentadien-1-ylsilyl]methyl]-2-methoxy-N-(2-methoxyethyl)-](/data/chemimg/3754200/1644149-39-2_b.png)
![Pyrrolidine, 1-[[dimethyl(2E)-2,4-pentadien-1-ylsilyl]methyl]-](/data/chemimg/3754200/1644149-37-0.png)
![Pyrrolidine, 1-[[dimethyl(2E)-2,4-pentadien-1-ylsilyl]methyl]-](/data/chemimg/3754200/1644149-37-0_b.png)
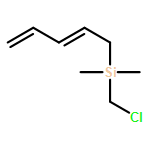
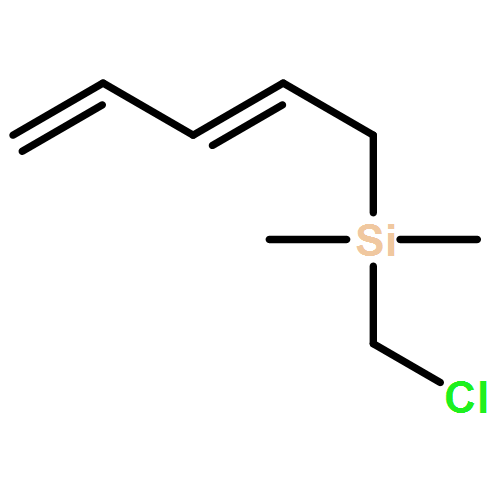

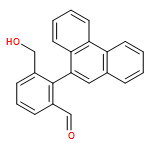
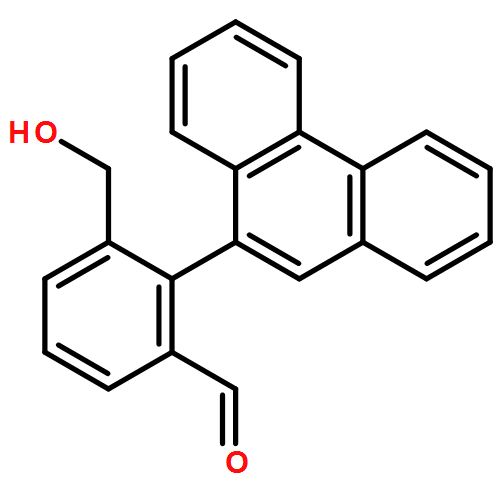
![Phenanthrene, 9-[2,6-bis(dibromomethyl)phenyl]-](/data/chemimg/3756100/1631038-39-5.png)
![Phenanthrene, 9-[2,6-bis(dibromomethyl)phenyl]-](/data/chemimg/3756100/1631038-39-5_b.png)

![Naphthalene, 1-[2,6-bis(dibromomethyl)phenyl]-](/data/chemimg/3756000/1631038-13-5.png)
![Naphthalene, 1-[2,6-bis(dibromomethyl)phenyl]-](/data/chemimg/3756000/1631038-13-5_b.png)
![1H-Imidazole-1-carbothioic acid, S-[(1R,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-31-4.png)
![1H-Imidazole-1-carbothioic acid, S-[(1R,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-31-4_b.png)
![1H-Indole-1-carboxylic acid, 6-[(1S,2E)-3-cyclohexyl-1-methyl-1-[[(methylamino)carbonyl]thio]-2-propen-1-yl]-, 1,1-dimethylethyl ester](/data/chemimg/3754600/1613417-30-3.png)
![1H-Indole-1-carboxylic acid, 6-[(1S,2E)-3-cyclohexyl-1-methyl-1-[[(methylamino)carbonyl]thio]-2-propen-1-yl]-, 1,1-dimethylethyl ester](/data/chemimg/3754600/1613417-30-3_b.png)
![1H-Indole-1-carboxylic acid, 6-[(2E)-3-cyclohexyl-1-methyl-1-[[(methylamino)carbonyl]thio]-2-propen-1-yl]-, 1,1-dimethylethyl ester](/data/chemimg/3754600/1613417-29-0.png)
![1H-Indole-1-carboxylic acid, 6-[(2E)-3-cyclohexyl-1-methyl-1-[[(methylamino)carbonyl]thio]-2-propen-1-yl]-, 1,1-dimethylethyl ester](/data/chemimg/3754600/1613417-29-0_b.png)
![Carbamothioic acid, N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-methyl-1-(2-pyridinyl)-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-28-9.png)
![Carbamothioic acid, N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-methyl-1-(2-pyridinyl)-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-28-9_b.png)
![Carbamothioic acid, N-methyl-, S-[(2E)-3-cyclohexyl-1-methyl-1-(2-pyridinyl)-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-27-8.png)
![Carbamothioic acid, N-methyl-, S-[(2E)-3-cyclohexyl-1-methyl-1-(2-pyridinyl)-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-27-8_b.png)
![Carbamothioic acid, N-methyl-, S-[(1R,2E)-3-cyclohexyl-1-methyl-1-(1-naphthalenyl)-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-26-7.png)
![Carbamothioic acid, N-methyl-, S-[(1R,2E)-3-cyclohexyl-1-methyl-1-(1-naphthalenyl)-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-26-7_b.png)
![Carbamothioic acid, N-methyl-, S-[(2E)-3-cyclohexyl-1-methyl-1-(1-naphthalenyl)-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-25-6.png)
![Carbamothioic acid, N-methyl-, S-[(2E)-3-cyclohexyl-1-methyl-1-(1-naphthalenyl)-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-25-6_b.png)
![Carbamothioic acid, N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-(2-methoxyphenyl)-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-24-5.png)
![Carbamothioic acid, N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-(2-methoxyphenyl)-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-24-5_b.png)
![Carbamothioic acid, N-methyl-, S-[(2E)-3-cyclohexyl-1-(2-methoxyphenyl)-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-23-4.png)
![Carbamothioic acid, N-methyl-, S-[(2E)-3-cyclohexyl-1-(2-methoxyphenyl)-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-23-4_b.png)
![Carbamothioic acid, N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-(3-methoxyphenyl)-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-22-3.png)
![Carbamothioic acid, N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-(3-methoxyphenyl)-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-22-3_b.png)
![Carbamothioic acid, N-methyl-, S-[(2E)-3-cyclohexyl-1-(3-methoxyphenyl)-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-21-2.png)
![Carbamothioic acid, N-methyl-, S-[(2E)-3-cyclohexyl-1-(3-methoxyphenyl)-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-21-2_b.png)
![Carbamothioic acid, N-methyl-, S-[(1S,2E)-1-(4-chlorophenyl)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-20-1.png)
![Carbamothioic acid, N-methyl-, S-[(1S,2E)-1-(4-chlorophenyl)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-20-1_b.png)
![Carbamothioic acid, N-methyl-, S-[(2E)-1-(4-chlorophenyl)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-19-8.png)
![Carbamothioic acid, N-methyl-, S-[(2E)-1-(4-chlorophenyl)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-19-8_b.png)
![Carbamothioic acid, N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-(4-fluorophenyl)-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-18-7.png)
![Carbamothioic acid, N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-(4-fluorophenyl)-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-18-7_b.png)
![Carbamothioic acid, N-methyl-, S-[(2E)-3-cyclohexyl-1-(4-fluorophenyl)-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-17-6.png)
![Carbamothioic acid, N-methyl-, S-[(2E)-3-cyclohexyl-1-(4-fluorophenyl)-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-17-6_b.png)
![Carbamothioic acid, N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-methyl-1-(4-methylphenyl)-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-16-5.png)
![Carbamothioic acid, N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-methyl-1-(4-methylphenyl)-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-16-5_b.png)
![Carbamothioic acid, N-methyl-, S-[(1R,2E)-3-cyclohexyl-1-methyl-1-(4-methylphenyl)-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-15-4.png)
![Carbamothioic acid, N-methyl-, S-[(1R,2E)-3-cyclohexyl-1-methyl-1-(4-methylphenyl)-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-15-4_b.png)
![Carbamothioic acid, N-methyl-, S-[(2E)-3-cyclohexyl-1-methyl-1-(4-methylphenyl)-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-14-3.png)
![Carbamothioic acid, N-methyl-, S-[(2E)-3-cyclohexyl-1-methyl-1-(4-methylphenyl)-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-14-3_b.png)
![Carbamothioic acid, N-methyl-, S-[(2E)-3-cyclohexyl-1-(4-methoxyphenyl)-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-13-2.png)
![Carbamothioic acid, N-methyl-, S-[(2E)-3-cyclohexyl-1-(4-methoxyphenyl)-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-13-2_b.png)
![Carbamothioic acid, N-1H-indol-6-yl-N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-12-1.png)
![Carbamothioic acid, N-1H-indol-6-yl-N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-12-1_b.png)
![Carbamothioic acid, N-1H-indol-6-yl-N-methyl-, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-11-0.png)
![Carbamothioic acid, N-1H-indol-6-yl-N-methyl-, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-11-0_b.png)
![1H-Indole-1-carboxylic acid, 6-[[[[(1S,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl]thio]carbonyl]methylamino]-, 1,1-dimethylethyl ester](/data/chemimg/3754600/1613417-10-9.png)
![1H-Indole-1-carboxylic acid, 6-[[[[(1S,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl]thio]carbonyl]methylamino]-, 1,1-dimethylethyl ester](/data/chemimg/3754600/1613417-10-9_b.png)
![1H-Indole-1-carboxylic acid, 6-[[[[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl]thio]carbonyl]methylamino]-, 1,1-dimethylethyl ester](/data/chemimg/3754600/1613417-09-6.png)
![1H-Indole-1-carboxylic acid, 6-[[[[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl]thio]carbonyl]methylamino]-, 1,1-dimethylethyl ester](/data/chemimg/3754600/1613417-09-6_b.png)
![Carbamothioic acid, N-methyl-N-2-pyridinyl-, S-[(1S,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-08-5.png)
![Carbamothioic acid, N-methyl-N-2-pyridinyl-, S-[(1S,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-08-5_b.png)
![Carbamothioic acid, N-methyl-N-2-pyridinyl-, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-07-4.png)
![Carbamothioic acid, N-methyl-N-2-pyridinyl-, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-07-4_b.png)
![Carbamothioic acid, N-methyl-N-1-naphthalenyl-, S-[(1R,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-06-3.png)
![Carbamothioic acid, N-methyl-N-1-naphthalenyl-, S-[(1R,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-06-3_b.png)
![Carbamothioic acid, N-(2-methoxyphenyl)-N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-05-2.png)
![Carbamothioic acid, N-(2-methoxyphenyl)-N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-05-2_b.png)
![Carbamothioic acid, N-(3-methoxyphenyl)-N-methyl-, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-04-1.png)
![Carbamothioic acid, N-(3-methoxyphenyl)-N-methyl-, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-04-1_b.png)
![Carbamothioic acid, N-(4-chlorophenyl)-N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-03-0.png)
![Carbamothioic acid, N-(4-chlorophenyl)-N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-03-0_b.png)
![Carbamothioic acid, N-(4-fluorophenyl)-N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-02-9.png)
![Carbamothioic acid, N-(4-fluorophenyl)-N-methyl-, S-[(1S,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-02-9_b.png)
![Carbamothioic acid, N-(4-fluorophenyl)-N-methyl-, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-01-8.png)
![Carbamothioic acid, N-(4-fluorophenyl)-N-methyl-, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-01-8_b.png)
![Carbamothioic acid, N-methyl-N-(4-methylphenyl)-, S-[(1S,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-00-7.png)
![Carbamothioic acid, N-methyl-N-(4-methylphenyl)-, S-[(1S,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613417-00-7_b.png)
![Carbamothioic acid, N-methyl-N-(4-methylphenyl)-, S-[(1R,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613416-99-1.png)
![Carbamothioic acid, N-methyl-N-(4-methylphenyl)-, S-[(1R,2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613416-99-1_b.png)
![Carbamothioic acid, N-(4-methoxyphenyl)-N-methyl-, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613416-98-0.png)
![Carbamothioic acid, N-(4-methoxyphenyl)-N-methyl-, S-[(2E)-3-cyclohexyl-1-methyl-2-propen-1-yl] ester](/data/chemimg/3754600/1613416-98-0_b.png)
![1H-Indole-1-carboxylic acid, 6-[(1R,2E)-3-cyclohexyl-1-mercapto-1-methyl-2-propen-1-yl]-, 1,1-dimethylethyl ester](/data/chemimg/3754600/1613416-97-9.png)
![1H-Indole-1-carboxylic acid, 6-[(1R,2E)-3-cyclohexyl-1-mercapto-1-methyl-2-propen-1-yl]-, 1,1-dimethylethyl ester](/data/chemimg/3754600/1613416-97-9_b.png)
![1H-Indole-1-carboxylic acid, 6-[(2E)-3-cyclohexyl-1-mercapto-1-methyl-2-propen-1-yl]-, 1,1-dimethylethyl ester](/data/chemimg/3754600/1613416-96-8.png)
![1H-Indole-1-carboxylic acid, 6-[(2E)-3-cyclohexyl-1-mercapto-1-methyl-2-propen-1-yl]-, 1,1-dimethylethyl ester](/data/chemimg/3754600/1613416-96-8_b.png)
![1-Naphthalenemethanethiol, α-[(1E)-2-cyclohexylethenyl]-α-methyl-, (αR)-](/data/chemimg/3754600/1613416-95-7.png)
![1-Naphthalenemethanethiol, α-[(1E)-2-cyclohexylethenyl]-α-methyl-, (αR)-](/data/chemimg/3754600/1613416-95-7_b.png)
![1-Naphthalenemethanethiol, α-[(1E)-2-cyclohexylethenyl]-α-methyl-](/data/chemimg/3754600/1613416-94-6.png)
![1-Naphthalenemethanethiol, α-[(1E)-2-cyclohexylethenyl]-α-methyl-](/data/chemimg/3754600/1613416-94-6_b.png)
![Benzenemethanethiol, α-[(1E)-2-cyclohexylethenyl]-2-methoxy-α-methyl-, (αS)-](/data/chemimg/3754600/1613416-93-5.png)
![Benzenemethanethiol, α-[(1E)-2-cyclohexylethenyl]-2-methoxy-α-methyl-, (αS)-](/data/chemimg/3754600/1613416-93-5_b.png)
![Benzenemethanethiol, α-[(1E)-2-cyclohexylethenyl]-2-methoxy-α-methyl-](/data/chemimg/3754600/1613416-92-4.png)
![Benzenemethanethiol, α-[(1E)-2-cyclohexylethenyl]-2-methoxy-α-methyl-](/data/chemimg/3754600/1613416-92-4_b.png)
![Benzenemethanethiol, α-[(1E)-2-cyclohexylethenyl]-3-methoxy-α-methyl-, (αS)-](/data/chemimg/3754600/1613416-91-3.png)
![Benzenemethanethiol, α-[(1E)-2-cyclohexylethenyl]-3-methoxy-α-methyl-, (αS)-](/data/chemimg/3754600/1613416-91-3_b.png)
![Benzenemethanethiol, α-[(1E)-2-cyclohexylethenyl]-3-methoxy-α-methyl-](/data/chemimg/3754600/1613416-90-2.png)
![Benzenemethanethiol, α-[(1E)-2-cyclohexylethenyl]-3-methoxy-α-methyl-](/data/chemimg/3754600/1613416-90-2_b.png)
![Benzenemethanethiol, 4-chloro-α-[(1E)-2-cyclohexylethenyl]-α-methyl-](/data/chemimg/3754600/1613416-89-9.png)
![Benzenemethanethiol, 4-chloro-α-[(1E)-2-cyclohexylethenyl]-α-methyl-](/data/chemimg/3754600/1613416-89-9_b.png)
![Benzenemethanethiol, α-[(1E)-2-cyclohexylethenyl]-4-fluoro-α-methyl-, (αS)-](/data/chemimg/3754600/1613416-88-8.png)
![Benzenemethanethiol, α-[(1E)-2-cyclohexylethenyl]-4-fluoro-α-methyl-, (αS)-](/data/chemimg/3754600/1613416-88-8_b.png)
![Benzenemethanethiol, α-[(1E)-2-cyclohexylethenyl]-α,4-dimethyl-, (αR)-](/data/chemimg/3754600/1613416-87-7.png)
![Benzenemethanethiol, α-[(1E)-2-cyclohexylethenyl]-α,4-dimethyl-, (αR)-](/data/chemimg/3754600/1613416-87-7_b.png)
![Benzenemethanethiol, α-[(1E)-2-cyclohexylethenyl]-α,4-dimethyl-](/data/chemimg/3754600/1613416-86-6.png)
![Benzenemethanethiol, α-[(1E)-2-cyclohexylethenyl]-α,4-dimethyl-](/data/chemimg/3754600/1613416-86-6_b.png)
![Benzene, 1-chloro-3-[(1R)-1-ethenyl-1-(2-propen-1-ylthio)butyl]-](/data/chemimg/3754600/1613416-85-5.png)
![Benzene, 1-chloro-3-[(1R)-1-ethenyl-1-(2-propen-1-ylthio)butyl]-](/data/chemimg/3754600/1613416-85-5_b.png)
![Benzene, 1-chloro-3-[1-ethenyl-1-(2-propen-1-ylthio)butyl]-](/data/chemimg/3754600/1613416-84-4.png)
![Benzene, 1-chloro-3-[1-ethenyl-1-(2-propen-1-ylthio)butyl]-](/data/chemimg/3754600/1613416-84-4_b.png)
![1H-Indole-1-carboxylic acid, 6-[(2E)-3-cyclohexyl-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-, 1,1-dimethylethyl ester](/data/chemimg/3754600/1613416-83-3.png)
![1H-Indole-1-carboxylic acid, 6-[(2E)-3-cyclohexyl-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-, 1,1-dimethylethyl ester](/data/chemimg/3754600/1613416-83-3_b.png)
![Naphthalene, 1-[(1E)-3-cyclohexyl-1-methyl-3-(2-propen-1-ylthio)-1-propen-1-yl]-](/data/chemimg/3754600/1613416-82-2.png)
![Naphthalene, 1-[(1E)-3-cyclohexyl-1-methyl-3-(2-propen-1-ylthio)-1-propen-1-yl]-](/data/chemimg/3754600/1613416-82-2_b.png)
![Naphthalene, 1-[(1R,2E)-3-cyclohexyl-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-](/data/chemimg/3754600/1613416-81-1.png)
![Naphthalene, 1-[(1R,2E)-3-cyclohexyl-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-](/data/chemimg/3754600/1613416-81-1_b.png)
![Naphthalene, 1-[(2E)-3-cyclohexyl-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-](/data/chemimg/3754600/1613416-80-0.png)
![Naphthalene, 1-[(2E)-3-cyclohexyl-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-](/data/chemimg/3754600/1613416-80-0_b.png)
![Benzene, 1-[(1S,2E)-3-cyclohexyl-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-2-methoxy-](/data/chemimg/3754600/1613416-79-7.png)
![Benzene, 1-[(1S,2E)-3-cyclohexyl-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-2-methoxy-](/data/chemimg/3754600/1613416-79-7_b.png)
![Benzene, 1-[(2E)-3-cyclohexyl-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-2-methoxy-](/data/chemimg/3754600/1613416-78-6.png)
![Benzene, 1-[(2E)-3-cyclohexyl-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-2-methoxy-](/data/chemimg/3754600/1613416-78-6_b.png)
![Benzene, 1-[(1R,2E)-3-cyclohexyl-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-3-methoxy-](/data/chemimg/3754600/1613416-77-5.png)
![Benzene, 1-[(1R,2E)-3-cyclohexyl-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-3-methoxy-](/data/chemimg/3754600/1613416-77-5_b.png)
![Benzene, 1-[(2E)-3-cyclohexyl-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-3-methoxy-](/data/chemimg/3754600/1613416-76-4.png)
![Benzene, 1-[(2E)-3-cyclohexyl-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-3-methoxy-](/data/chemimg/3754600/1613416-76-4_b.png)
![Benzene, 1-[(1R,2E)-3-cyclohexyl-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-4-methyl-](/data/chemimg/3754600/1613416-75-3.png)
![Benzene, 1-[(1R,2E)-3-cyclohexyl-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-4-methyl-](/data/chemimg/3754600/1613416-75-3_b.png)
![Benzene, 1-[(2E)-3-cyclohexyl-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-4-methyl-](/data/chemimg/3754600/1613416-74-2.png)
![Benzene, 1-[(2E)-3-cyclohexyl-1-methyl-1-(2-propen-1-ylthio)-2-propen-1-yl]-4-methyl-](/data/chemimg/3754600/1613416-74-2_b.png)


![1H-Indole-1-carboxylic acid, 6-[(2R)-2,5-dihydro-2-methyl-2-thienyl]-, 1,1-dimethylethyl ester](/data/chemimg/3754600/1613416-69-5.png)
![1H-Indole-1-carboxylic acid, 6-[(2R)-2,5-dihydro-2-methyl-2-thienyl]-, 1,1-dimethylethyl ester](/data/chemimg/3754600/1613416-69-5_b.png)
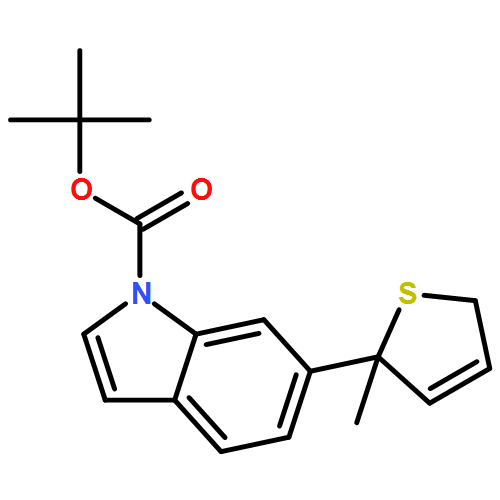
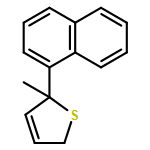
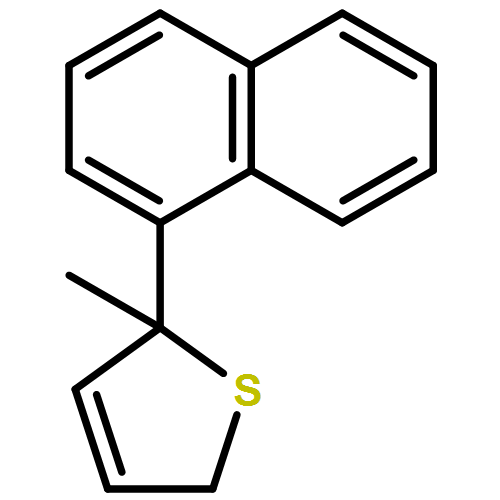
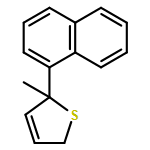
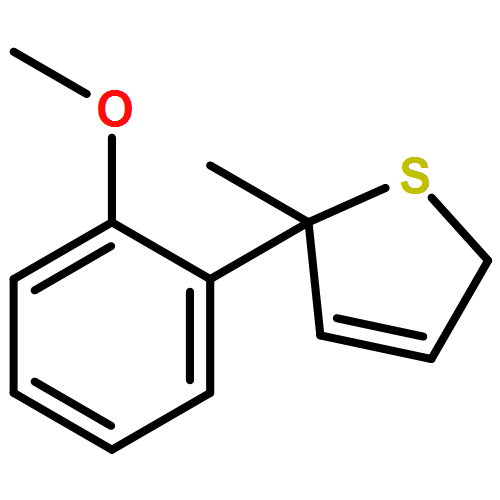
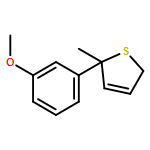
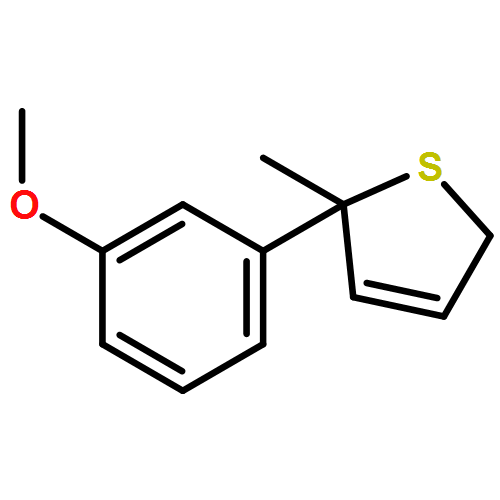
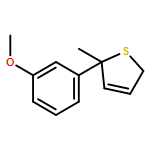
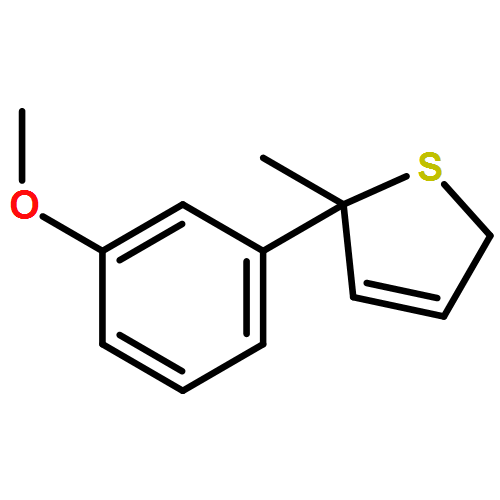
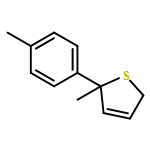
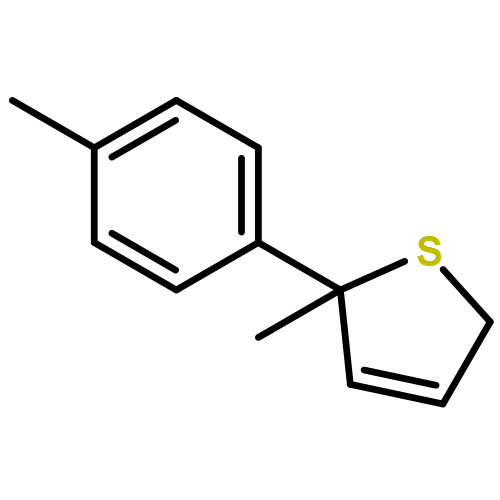
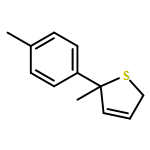
![L-Lysine, N6-[(1,1-dimethylethoxy)carbonyl]-N2-[(methylphenylamino)carbonyl]-, methyl ester](/data/chemimg/3752800/1462366-90-0.png)
![L-Lysine, N6-[(1,1-dimethylethoxy)carbonyl]-N2-[(methylphenylamino)carbonyl]-, methyl ester](/data/chemimg/3752800/1462366-90-0_b.png)
![L-Methionine, N-[(methylphenylamino)carbonyl]-, methyl ester](/data/chemimg/3752800/1462366-75-1.png)
![L-Methionine, N-[(methylphenylamino)carbonyl]-, methyl ester](/data/chemimg/3752800/1462366-75-1_b.png)
![L-Leucine, N-[(methylphenylamino)carbonyl]-, methyl ester](/data/chemimg/3752800/1462366-45-5.png)
![L-Leucine, N-[(methylphenylamino)carbonyl]-, methyl ester](/data/chemimg/3752800/1462366-45-5_b.png)
![L-Alanine, N-methyl-N-[(methylphenylamino)carbonyl]-](/data/chemimg/3752800/1462365-40-7.png)
![L-Alanine, N-methyl-N-[(methylphenylamino)carbonyl]-](/data/chemimg/3752800/1462365-40-7_b.png)