Co-reporter:Lili Ye, Long Zhao, Lidong Zhang, and Fei Qi
The Journal of Physical Chemistry A 2012 Volume 116(Issue 1) pp:55-63
Publication Date(Web):December 12, 2011
DOI:10.1021/jp207978n
The unimolecular decomposition processes of ethylene glycol have been investigated with the QCISD(T) method with geometries optimized at the B3LYP/6-311++G(d,p) level. Among the decomposition channels identified, the H2O-elimination channels have the lowest barriers, and the C–C bond dissociation is the lowest-energy dissociation channel among the barrierless reactions (the direct bond cleavage reactions). The temperature and pressure dependent rate constant calculations show that the H2O-elimination reactions are predominant at low temperature, whereas at high temperature, the direct C–C bond dissociation reaction is dominant. At 1 atm, in the temperature range 500–2000 K, the calculated rate constant is expressed to be 7.63 × 1047T–10.38 exp(−42262/T) for the channel CH2OHCH2OH → CH2CHOH + H2O, and 2.48 × 1051T–11.58 exp(−43593/T) for the channel CH2OHCH2OH → CH3CHO + H2O, whereas for the direct bond dissociation reaction CH2OHCH2OH → CH2OH + CH2OH the rate constant expression is 1.04 × 1071T–16.16 exp(−52414/T).
Co-reporter:Lili Ye, Feng Zhang, Lidong Zhang, and Fei Qi
The Journal of Physical Chemistry A 2012 Volume 116(Issue 18) pp:4457-4465
Publication Date(Web):April 20, 2012
DOI:10.1021/jp301424k
Polyols, a typical type of alcohol containing multiple hydroxyl groups, are being regarded as a new generation of a green energy platform. In this paper, the decomposition mechanisms for three polyol molecules, i.e., 1,2-propanediol, 1,3-propanediol, and glycerol, have been investigated by quantum chemistry calculations. The potential energy surfaces of propanediols and glycerol have been built by the QCISD(T) and CBS-QB3 methods, respectively. For the three molecules studied, the H2O-elimination and C–C bond dissociation reactions show great importance among all of the unimolecular decomposition channels. Rate constant calculations further demonstrate that the H2O-elimination reactions are predominant at low temperature and pressure, whereas the direct C–C bond dissociation reactions prevail at high temperature and pressure. The temperature and pressure dependence of calculated rate constants was demonstrated by the fitted Arrhenius equations. This work aims to better understand the thermal decomposition process of polyols and provide useful thermochemical and kinetic data for kinetic modeling of polyols-derived fuel combustion.
Co-reporter:Long Zhao, Lili Ye, Feng Zhang, and Lidong Zhang
The Journal of Physical Chemistry A 2012 Volume 116(Issue 37) pp:9238-9244
Publication Date(Web):August 21, 2012
DOI:10.1021/jp305885s
Pentanol is one of the promising “next generation” alcohol fuels with high energy density and low hygroscopicity. In the present work, dominant reaction channels of thermal decomposition of three isomers of pentanol: 1-pentanol, 2-methyl-1-butanol, and 3-methyl-1-butanol were investigated by CBS-QB3 calculations. Subsequently, the temperature- and pressure-dependent rate constants for these channels were computed by RRKM/master equation simulations. The difference between the thermal decomposition behavior of pentanol and butanol were discussed, while butanol as another potential alternative alcohol fuel has been extensively studied both experimentally and theoretically. Rate constants of barrierless bond dissociation reactions of pentanol isomers were treated by the variational transition state theory. The comparison between various channels revealed that the entropies of variational transition states significantly impact the rate constants of pentanol decomposition reactions. This work provides sound quality kinetic data for major decomposition channels of three pentanol isomers in the temperature range of 800–2000 K with pressure varying from 7.6 to 7.6 × 104 Torr, which might be valuable for developing detailed kinetic models for pentanol combustion.
Co-reporter:Dongna Chen, Hanfeng Jin, Zhandong Wang, Lidong Zhang, and Fei Qi
The Journal of Physical Chemistry A 2011 Volume 115(Issue 5) pp:602-611
Publication Date(Web):January 5, 2011
DOI:10.1021/jp1099305
Alkyl hydroperoxides are found to be important intermediates in the combustion and oxidation processes of hydrocarbons. However, studies of ethyl hydroperoxide (CH3CH2OOH) are limited. In this work, kinetics and mechanisms for unimolecular decomposition of CH3CH2OOH have been investigated. The potential energy surface of decomposition reactions have first been predicted at the CCSD(T)/6-311+G(3df,2p)//B3LYP/6-311G(d,p) level. The results show that the formation of CH3CH2O + OH via O−O direct bond dissociation is dominant, the branching ratio of which is over 99% in the whole temperature range from 300 to 1000 K, and its rate constant can be expressed as k1 = 9.26 × 1052T−11.91exp(−26879/T) s−1 at 1 atm. The rate constants of the reaction CH3CH2OOH → CH3CH2O + OH at different temperatures and pressures have been calculated, which can help us to comprehend the reactions of CH3CH2OOH at experimental conditions.
Co-reporter:Tao Yuan, Lidong Zhang, Zhongyue Zhou, Mingfeng Xie, Lili Ye, and Fei Qi
The Journal of Physical Chemistry A 2011 Volume 115(Issue 9) pp:1593-1601
Publication Date(Web):February 15, 2011
DOI:10.1021/jp109640z
An experimental study of n-heptane pyrolysis (2.0% n-heptane in argon) has been performed at low pressure (400 Pa) within the temperature range from 780 to 1780 K. The pyrolysis products were detected by using synchrotron vacuum ultraviolet photoionization mass spectrometry (SVUV−PIMS). Photoionization mass spectra and photoionization efficiency spectra were measured to identify pyrolysis products, especially radicals and isomers. Mole fraction profiles of pyrolysis products versus temperature were also measured, indicating that H2, CH4, C2H2, and C2−C6 alkenes are major pyrolysis products of n-heptane. Meanwhile, the thermal decomposition pathways of n-heptane have been investigated using theoretical calculation. The calculation results are in good agreement with the experimental measurement. On the basis of the experimental observation and theoretical calculation, the pyrolysis channels of unimolecular dissociation are proposed to understand the pyrolysis process of n-heptane.
Co-reporter:Lili Xing, Feng Zhang, Lidong Zhang
Proceedings of the Combustion Institute (2017) Volume 36(Issue 1) pp:179-186
Publication Date(Web):1 January 2017
DOI:10.1016/j.proci.2016.08.050
The oxidation mechanism of cyclohexylmethyl radical (cy-C6H11CH2), a prototypical alkyl-substituted cycloalkyl radical, has been investigated by high level quantum chemical calculations, and the chemical kinetics was studied by the variational transition state theory and the Rice–Ramsperger–Kassel–Marcus/Master-Equation theory. The relationship between molecular structure and reactivity was explored for the cyclohexylmethyl peroxy radical, which was also compared with chain-like alkyl peroxy radicals. It is shown that the 1,5 H-shift reaction is more competitive than the 1,6 H-shift for the cy-C6H11CH2OO radical, and at high temperatures the concerted elimination reaction channel forming HO2 and methylenecyclohexane bimolecular products becomes more important. Comparing with cyclohexane, the presence of a methyl group in cyclic alkanes prompts the 1,5-H shift of the corresponding peroxy radical and accelerates the overall low temperature chain branching reaction rate. The current study extends kinetic data involved in cy-C6H11CH2 oxidation including cy-C6H11CH2 and O2 recombination, subsequent isomerization and dissociation of the cy-C6H11CH2OO radical over wide pressure and temperature range.
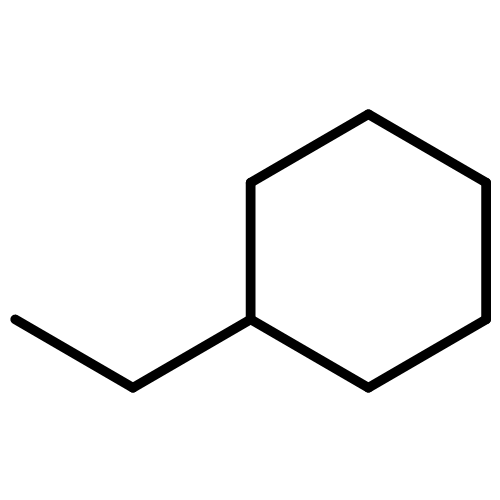
Co-reporter:Zhandong Wang, Huiting Bian, Yu Wang, Lidong Zhang, ... Fei Qi
Proceedings of the Combustion Institute (2015) Volume 35(Issue 1) pp:367-375
Publication Date(Web):1 January 2015
DOI:10.1016/j.proci.2014.05.119
To get a better understanding of the combustion chemistry of cycloalkanes with long side chain, the pyrolysis of ethylcyclohexane (ECH) was studied in a flow reactor at atmospheric pressure. The pyrolysis species were analyzed by two methods, synchrotron vacuum ultraviolet photoionization mass spectrometry and gas chromatography. Dozens of species were identified and quantified, including lots of isomers. The emphasis of this study is to investigate the primary decomposition of ECH, including its initial decomposition, isomerization, and further reactions of the cyclic C8H15 radicals formed from the H-abstraction of ECH. The observation of C8H16 alkene indicates the existence of ring-opening isomerization reaction of ECH. The ring-opening isomerization reaction of cyclic C8H15 radicals produces alkenyl radicals, whose further decomposition constitutes the various chain and branched intermediates in ECH pyrolysis. The formation of isoprene and vinylcyclopentane is discussed, which highlights isomerization reactions of radical addition on the double bond of alkenyl radicals, such as oct-6-en-1-yl and oct-5-en-1-yl radicals. The theoretical calculation on the reaction pathways of oct-5-en-1-yl radical also shows that its internal H-migration pathway via eight-membered ring might be competitive to the one via five-membered ring. On the other hand, the decomposition of cyclic C8H15 radicals causes the formation of cyclic intermediates, i.e. C8H14 alkenes, methylenecyclohexane and cyclohexene, which are potential aromatic precursors.