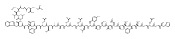Co-reporter:Zhengyu Wang, Xiaofan Shi, Huan Zhang, Liang Yu, Yanhua Cheng, Hefeng Zhang, Huibin Zhang, Jinpei Zhou, Jing Chen, Xu Shen, Wenhu Duan
European Journal of Medicinal Chemistry 2017 Volume 139(Volume 139) pp:
Publication Date(Web):20 October 2017
DOI:10.1016/j.ejmech.2017.07.051
•A series of cycloalkyl-fused N-thiazol-2-yl-benzamides was investigated as tissue non-specific partial GK activators.•Compound 72 showed a good balance between in vitro potency and enzyme kinetic parameters.•Chronic treatment of compound 72 demonstrated potent activity in the OGTT in diabetic db/db mice.•Chronic oral administration of 72 showed no obvious effects on CHO, HDL-C and LDL-C when compared with the vehicle group.•Acute treatment of compound 72 did not induce hypoglycemia in C57BL/6J mice even at 200 mg/kg via oral administration.Glucokinase (GK) activators are being developed for the treatment of type 2 diabetes mellitus (T2DM). However, existing GK activators have risks of hypoglycemia caused by over-activation of GK in islet cells and dyslipidemia caused by over-activation of intrahepatic GK. In the effort to mitigate risks of hypoglycemia and dyslipidemia while maintaining the promising efficacy of GK activator, we investigated a series of cycloalkyl-fused N-thiazol-2-yl-benzamides as tissue non-specific partial GK activators, which led to the identification of compound 72 that showed a good balance between in vitro potency and enzyme kinetic parameters, and protected β-cells from streptozotocin-induced apoptosis. Chronic treatment of compound 72 demonstrated its potent activity in regulation of glucose homeostasis and low risk of dyslipidemia with diabetic db/db mice in oral glucose tolerance test (OGTT). Moreover, acute treatment of compound 72 did not induce hypoglycemia in C57BL/6J mice even at 200 mg/kg via oral administration.Download high-res image (352KB)Download full-size image
Co-reporter:Yifei Yang, Lingyun Yang, Yaodan Han, Zhenwei Wu, Pan Chen, Huibin Zhang, Jinpei Zhou
Bioorganic Chemistry 2017 Volume 72(Volume 72) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.bioorg.2017.03.009
•Novel glycyrrhetic derivatives were designed and synthesized.•Levels of serum marker enzymes was evaluated.•Histopathological studies was performed.Glycyrrhetic acid (GA), the main hydrolysate of glycyrrhizic acid extracted from the roots of the Chinese herb Glycyrrhiza glabra, was reported to be accumulated in hepatocytes due to the extensive distribution of GA receptors in liver. A series of hepatocyte-specific derivatives on the basis of anetholtrithione and glycyrrhizic were designed and synthesized. The potential beneficial effect was evaluated in carbon tetrachloride (CCl4)-induced liver injury model. In addition, the hepatoprotective activity of these derivatives was assessed by measuring levels of serum marker enzymes, including serum glutamate oxaloacetate transaminase (GOT), serum glutamate pyruvate transaminase (GPT), alkaline phosphatase (AKP), lactate dehydrogenase (LDH) and the ratio of GSH to GSSG. Gratifyingly, compounds 5a–c (100 mg/kg, p.o.) markedly prevented CCl4-induced elevation of levels of serum GPT, GOT. A comparative histopathological study of liver exhibited almost a normal liver lobular architecture and cell structure of the livers, as compared to CCl4-treated group. These findings were confirmed with the histopathological observations, where hepatocyte-specific glycyrrhetic acid derivatives 5a–c were capable of reversing the toxic effects of CCl4 on hepatocytes.Download high-res image (49KB)Download full-size image
Co-reporter:Leilei Zhao, Yifei Yang, Yahui Guo, Lingyun Yang, Jian Zhang, Jinpei Zhou, Huibin Zhang
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 8(Issue 8) pp:
Publication Date(Web):15 April 2017
DOI:10.1016/j.bmc.2017.03.008
BRD4 is an attractive target for antitumor due to its important role in regulation of gene transcription. In this paper, we synthesized a series of 7-methylimidazo[1,5-a]pyrazin-8(7H)-one derivatives as potent BRD4 inhibitors and evaluated their BRD4 inhibitory activities in vitro and anti-proliferation effects on tumor cells. Gratifyingly, compound 10j exhibited robust potency of BRD4(1) and BRD4(2) inhibition with IC50 values of 130 and 76 nM respectively. Docking studies were performed to explain the structure-activity relationship. Furthermore, compound 10j potently inhibited cell proliferation in BRD4-sensitive cell lines HL-60 and MV4-11 with IC50 value of 0.57 and 0.18 μM respectively. Activity on BRD4-independent K562 cell was weaker than on BRD4-sensitive lines. Overall, these results suggest that compound 10j is a potential BRD4 inhibitor deserving further investigation for cancer treatment.Download high-res image (101KB)Download full-size image
Co-reporter:Yifei Yang, Yuan Zhang, LingYun Yang, Leilei Zhao, Lianghui Si, Huibin Zhang, Qingsong Liu, Jinpei Zhou
Bioorganic Chemistry 2017 Volume 70(Volume 70) pp:
Publication Date(Web):1 February 2017
DOI:10.1016/j.bioorg.2016.12.002
•Novel imidazopyridine c-Met inhibitors were designed and synthesized.•Molecular docking was performed.•Cell proliferation inhibition was evaluated in the TRP-Met-BaF3 cell line.•Binding activity in c-Met kinase was determined by ELISA assay.Receptor tyrosine kinase c-Met acts as an alternative angiogenic pathway in the process and contents of cancers. A series of imidazopyridine derivatives were designed and synthesized according to the established docking studies as possible c-Met inhibitors. Most of these imidazopyridine derivatives displayed nanomolar potency against c-Met in both biochemical enzymatic screens and cellular pharmacology studies. Especially, compound 7 g exhibited the most inhibitory activity against c-Met with IC50 of 53.4 nM and 253 nM in enzymatic and cellular level, respectively. Following that, the compound 7 g was docked into the protein of c-Met and the structure-activity relationship was analyzed in detail. These findings indicated that the novel imidazopyridine derivative compound 7 g was a potential c-Met inhibitor deserving further investigation for cancer treatment.Download high-res image (159KB)Download full-size image
Co-reporter:Wei Yan;Zhaoru Huang;Zhengyu Wang;Sufen Cao;Linjiang Tong;Tao Zhang;Chen Wang;Lin Zhou;Jian Ding;Cheng Luo;Jinpei Zhou;Hua Xie;Wenhu Duan
Chemical Biology & Drug Design 2016 Volume 87( Issue 5) pp:694-703
Publication Date(Web):
DOI:10.1111/cbdd.12703
In this study, we described the design, synthesis, and biological evaluation of 1,3-diaryl-pyridones as vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitors. The 1,3-diaryl-pyridones were synthesized via Chan-Lam and Suzuki coupling reactions. Two representative compounds, 17 and 35h, displayed excellent enzymatic inhibitory activities, with IC50 values of 3.5 and 3.0 nm, respectively. Furthermore, compounds 17 and 35h blocked the tube formation and suppressed the VEGF-induced phosphorylation of VEGFR-2 and downstream extracellular signal-regulated kinases (Erk) in human umbilical vein endothelial cells (HUVECs) at 10 nm concentration. The docking simulation showed that compound 17 bound well into the active site of VEGFR-2 via two hydrogen bonds and hydrophobic interactions.
Co-reporter:Yifei Yang, Leilei Zhao, Bin Xu, LingYun Yang, Jian Zhang, Huibin Zhang, Jinpei Zhou
Bioorganic Chemistry 2016 Volume 68() pp:236-244
Publication Date(Web):October 2016
DOI:10.1016/j.bioorg.2016.08.009
•Novel dihydroquinoxalinone BRD4 inhibitors were designed and synthesized.•Molecular docking was performed.•Binding activity in BRD4 was determined by AlphaScreen assay.•Cell viability assay was evaluated in the MV-4-11 cell line.BRD4 plays a key role in transcriptional regulation. Recent biological and pharmacological studies have demonstrated that bromodomain-containing protein 4 (BRD4) is a viable drug target for cancer treatment. In this study, we synthesized a series of dihydroquinoxalinone derivatives and evaluated their BRD4 inhibitory activities, obtaining compound 5i with IC50 value of 73 nM of binding activity in BRD4(1) and 258 nM of cellular activity in MV-4-11 cancer cell lines. Docking studies were performed to explain the structure-activity relationship. Based on its potent biochemical and anti-proliferative activity, the novel BRD4 inhibitor compound 5i, is a promising lead compound for further investigation.
Co-reporter:Xiaoming Zha;Liming Wu;Siyuan Xu;Fangxia Zou;Jiayue Xi
Medicinal Chemistry Research 2016 Volume 25( Issue 12) pp:2822-2831
Publication Date(Web):2016 December
DOI:10.1007/s00044-016-1706-8
Lysine specific demethylase 1 plays a crucial role in regulating histone methylation at residues K4 and K9 on histone H3 and over-expresses in a variety of cancers. Here we designed, synthesized and evaluated a series of N-(3-substituted-phenyl)benzenesulfonamides as reversible lysine specific demethylase 1 inhibitors. All the compounds exhibited lysine specific demethylase 1 inhibition with the half maximal inhibitory concentration (IC50) values between 7.5 and 68 μM. Three most active compounds 2a, 2c and 2i displayed only modest effect on flavin adenine dinucleotide-dependent enzymes mono-amine oxidases A and B, and their reversibilities of lysine specific demethylase 1 inhibition were confirmed. Molecular docking was also carried out to predict the binding mode of 2a into the active site of lysine specific demethylase 1. Taken together, N-(3-substituted-phenyl)benzenesulfonamides including 2a represent a new class of selective and reversible lysine specific demethylase 1 inhibitors with pharmaceutical research.
Co-reporter:Yifei Yang;Fangxia Zou;Leilei Zhao;Yulan Cheng
Medicinal Chemistry Research 2016 Volume 25( Issue 4) pp:585-595
Publication Date(Web):2016 April
DOI:10.1007/s00044-016-1513-2
Bromodomain-containing protein is involved in many essential cellular processes, such as chromosomes for cell cycle progression, cellular viability and embryonic stem cell regulation, which plays a significant role in cancers and lysine acetylation. However, there is no information available regarding the discovery for structurally novel existing BRD4(1) inhibitors up to date. Therefore, we collected reported compounds from GSK library to generate ligand-based pharmacophore and used 11 BRD4(1)-inhibitor co-crystal structures to establish our structure-based pharmacophore for multiple virtual screening of potent BRD4(1) inhibitors. These results may provide important information for further design and optimization of novel BRD4(1) inhibitors in cancer treatment. The results of this study will not only provide a better understanding of BRD4(1) inhibitors interaction, but will also assist the development of new potent hits for BRD4(1).
Co-reporter:Wukun Liu, Jie Hua, Jinpei Zhou, Huibin Zhang, Haiyang Zhu, Yanhua Cheng, Ronald Gust
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 15) pp:5008-5012
Publication Date(Web):1 August 2012
DOI:10.1016/j.bmcl.2012.06.014
Twenty scopoletin derivatives were developed by a systematic combinatorial chemical approach and their chemical structures were confirmed by MS, IR, 1H NMR spectra and elemental analysis. Primary screening against mammary (MCF-7 and MDA-MB 231) and colon (HT-29) carcinoma cells indicated that five compounds (8d, 8g, 8j, 11b and 11g) displayed high antitumor potencies with IC50 values below 20 μM whereas scopoletin showed IC50 values above 100 μM. Moreover, the most promising compound 11g was more active than 5-fluorouracil. These results clearly indicated that the modification of the scopoletin structure could greatly increase its antitumor activity in vitro.Two series of scopoletin derivatives were synthesized and investigated for cytotoxicity against mammary (MCF-7, MDA-MB 231) and colon (HT-29) carcinoma cell lines.
Co-reporter:Wukun Liu, Jinpei Zhou, Tong Zhang, Huibin Zhang, Haiyang Zhu, Yanhua Cheng, Ronald Gust
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 19) pp:6076-6080
Publication Date(Web):1 October 2012
DOI:10.1016/j.bmcl.2012.08.041
Cyanoguanidine derivatives of loratadine (3a–i) were synthesized and screened for antitumor and anti-inflammatory activity. The most promising compound 3c (R = n-C8H17) possessed at least twofold higher in vitro cytotoxicity than 5-fluorouracil against mammary (MCF-7 and MDA-MB 231) as well as colon (HT-29) carcinoma cells. The mode of action, however, is so far unclear. The participation of the COX-1/2 enzymes on the cytotoxicity, however, is very unlikely. Nevertheless all compounds showed stronger in vivo anti-inflammatory activity than ibuprofen in the xylene-induced ear swelling assay in mice.
Co-reporter:Wukun Liu, Jinpei Zhou, Tong Zhang, Haiyang Zhu, Hai Qian, Huibin Zhang, Wenlong Huang, Ronald Gust
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 8) pp:2701-2704
Publication Date(Web):15 April 2012
DOI:10.1016/j.bmcl.2012.03.002
Thiourea derivatives (6a–e) were developed and screened for antitumor and anti-inflammatory activity. Most of the compounds exhibited growth inhibitory effects comparable to 5-fluorouracil in vitro against mammary (MCF-7 and MDA-MB 231) as well as colon (HT-29) carcinoma cells. They also showed stronger anti-inflammatory activity than ibuprofen in vivo in the xylene-induced ear swelling assay in mice.
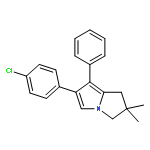
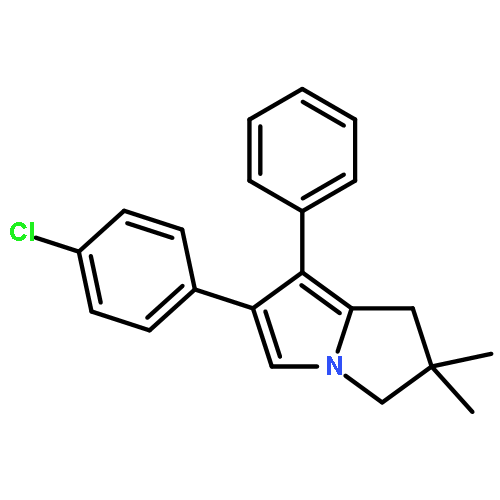
Co-reporter:Wukun Liu, Jinpei Zhou, Kerstin Bensdorf, Huibin Zhang, Haoran Liu, Yubin Wang, Hai Qian, Yanchun Zhang, Anja Wellner, Gerhard Rubner, Wenlong Huang, Cancheng Guo, Ronald Gust
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 3) pp:907-913
Publication Date(Web):March 2011
DOI:10.1016/j.ejmech.2011.01.002
A series of C5-substituted licofelone ([2,2-dimethyl-6-(4-chlorophenyl)-7-phenyl-2,3-dihydro-1H-pyrrolizin-5-yl]acetic acid) derivatives were developed by a parallel synthesis approach and investigated for cytotoxicity against MCF-7 and MDA-MB-231 cells as well as for anti-inflammatory potency in vitro and in vivo. Dependent on the C5-substituent, the compounds showed high selectivity for MCF-7 cells. Especially 2-oxoethyl benzoate derivatives were inactive at the MDA-MB-231 cell line and as active as 5-FU at MCF-7 cells. C5-acetyl (8a), –2-oxoethyl formiate (8e), –2-oxoethyl acetate (8f) and –2-oxoethyl propionate (8g) derivatives showed growth inhibition at both cell lines, comparable with cisplatin. Modifications significantly reduced the inhibitory potency at COX-1 and COX-2 in vitro and in the xylene-induced ear swelling assay in mice. Only compound 8a was equipotent to licofelone, ibuprofen and celecoxibe in vivo.Research highlights► Licofelone ([2,2-dimethyl-6-(4-chlorophenyl)-7-phenyl-2,3-dihydro-1H-pyrrolizin-5-yl]acetic acid) derivation at C5. ► Increased selectivity for MCF-7 cells especially determined for 2-oxoethyl benzoate derivatives. ► Modifications significantly reduced anti-inflammatory effects in vitro and in vivo.
Co-reporter:Wukun Liu;Jinpei Zhou;Fan Qi;Kerstin Bensdorf;Zhiyu Li;Huibin Zhang;Hai Qian;Wenlong Huang;Xueting Cai;Peng Cao;Anja Wellner;Ronald Gust
Archiv der Pharmazie 2011 Volume 344( Issue 7) pp:451-458
Publication Date(Web):
DOI:10.1002/ardp.201000281
Abstract
In an attempt to develop potent and selective anti-tumor drugs, a series of novel 2-amino-thiazole-5-carboxylic acid phenylamide derivatives were designed based on the structure of dasatinib. All compounds were synthesized by a systematic combinatorial chemical approach. Biological evaluation revealed that N-(2-chloro-6-methylphenyl)-2-(2-(4-methylpiperazin-1-yl)acetamido)thiazole-5-carboxamide (6d) exhibited high antiproliferative potency on human K563 leukemia cells comparable to dasatinib. Against mammary and colon carcinoma cells 6d was either inactive (MDA-MB 231) or distinctly less active (MCF-7 and HT-29: IC50 = 20.2 and 21.6 µM, respectively). Dasatinib showed at each cell line IC50 < 1 µM. The results of this structure activity relationship study clearly documented that the pyrimidin-4-ylamino core of dasatinib is responsible for the anti-tumor activity against non-leukemia cell lines.
Co-reporter:Hai Qian, Wei Chen, Jin Pei Zhou, Wen Long Huang, Hui Bin Zhang, Jing Jin
Chinese Chemical Letters 2010 Volume 21(Issue 4) pp:388-390
Publication Date(Web):April 2010
DOI:10.1016/j.cclet.2009.12.016
A microwave-assisted solid phase synthesis for endothelin 1 is presented. Reduced endothelin 1 was synthesized efficiently on Wang resin under microwave irradiation using Fmoc/tBu orthogonal protection strategy. The whole peptide was cleaved from the resin and two disulphide bridges were formed under air oxidation at room temperature. The purity and efficiency of synthesizing the peptide is much higher than other methods used before.
Co-reporter:Hui-bin Zhang, Ya-an Zhang, Guan-zhong Wu, Jin-pei Zhou, Wen-long Huang, Xiao-wen Hu
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 6) pp:1740-1744
Publication Date(Web):15 March 2009
DOI:10.1016/j.bmcl.2009.01.082
A novel class of sulfonylurea and thiourea derivatives substituted with benzenesulfonamide groups were designed and synthesized. The target compounds were assayed for the effects on the insulin release of isolated rat pancreatic islets and the glucose transport in adipocytes of rats. Some of them exhibited high potency. Compound 10 also had potent antiplatelet activity and showed an excellent property to protect collagen–epinephrine-induced mice mortality as well as plasma glucose-lowering activity in vivo. The preliminary pharmacological profile of compound 10 showed that it might be useful in the treatment of diabetics with cardiovascular and nephropathy complications.Compound 10 (R1 = Br, R2 = H, R3 = CH3) with excellent hypoglycemic and antithrombotic properties, might be useful in the treatment of diabetics with cardiovascular and nephropathy complications.

![tert-butyl 4-(1-(2-ethoxyethyl)-1H-benzo[d]imidazol-2-yl)piperidine-1-carboxylate](http://img.cochemist.com/ccimg/1181300/1181267-36-6.png)
![tert-butyl 4-(1-(2-ethoxyethyl)-1H-benzo[d]imidazol-2-yl)piperidine-1-carboxylate](http://img.cochemist.com/ccimg/1181300/1181267-36-6_b.png)




![Quinoline, 7-methoxy-4-[(6-phenyl-1,2,4-triazolo[4,3-b]pyridazin-3-yl)methoxy]-](/data/chemimg/314900/1002304-34-8.png)
![Quinoline, 7-methoxy-4-[(6-phenyl-1,2,4-triazolo[4,3-b]pyridazin-3-yl)methoxy]-](/data/chemimg/314900/1002304-34-8_b.png)
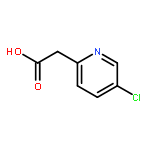
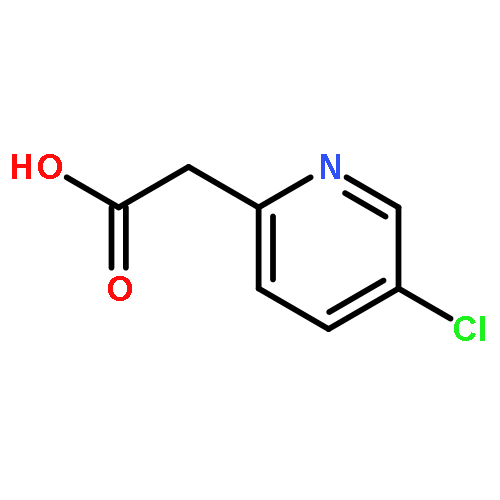


![6-(Methoxycarbonyl)-2-(4-methylphenyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/917300/917252-78-9.png)
![6-(Methoxycarbonyl)-2-(4-methylphenyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/917300/917252-78-9_b.png)
![Ethanol, 2-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]-](/data/chemimg/1005600/901792-84-5.png)
![Ethanol, 2-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]-](/data/chemimg/1005600/901792-84-5_b.png)
![2-(2-Methylphenyl)imidazo[1,2-a]pyridine-3-carbaldehyde](http://img.cochemist.com/ccimg/898400/898389-26-9.png)
![2-(2-Methylphenyl)imidazo[1,2-a]pyridine-3-carbaldehyde](http://img.cochemist.com/ccimg/898400/898389-26-9_b.png)
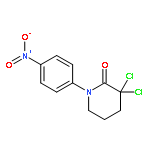
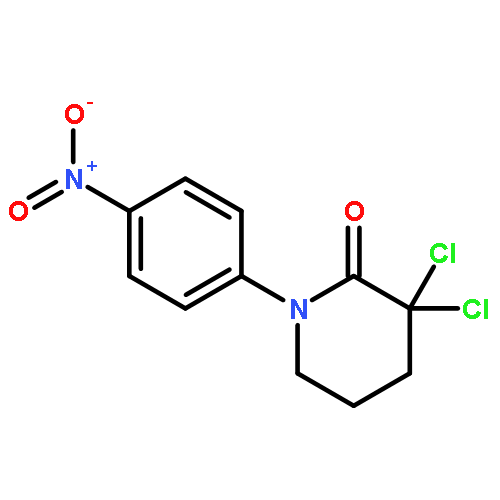
![Pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione,7-amino-6-(aminomethyl)-5-(2-bromophenyl)-1,3-dimethyl-](http://img.cochemist.com/ccimg/874000/873934-77-1.png)
![Pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione,7-amino-6-(aminomethyl)-5-(2-bromophenyl)-1,3-dimethyl-](http://img.cochemist.com/ccimg/874000/873934-77-1_b.png)
![2(1H)-Pyrimidinone, 1-[1-[(2S)-3-[(6-chloro-2-naphthalenyl)sulfonyl]-2-hydroxy-1-oxopropyl]-4-piperidinyl]tetrahydro](http://img.cochemist.com/ccimg/870300/870262-90-1.png)
![2(1H)-Pyrimidinone, 1-[1-[(2S)-3-[(6-chloro-2-naphthalenyl)sulfonyl]-2-hydroxy-1-oxopropyl]-4-piperidinyl]tetrahydro](http://img.cochemist.com/ccimg/870300/870262-90-1_b.png)
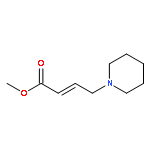
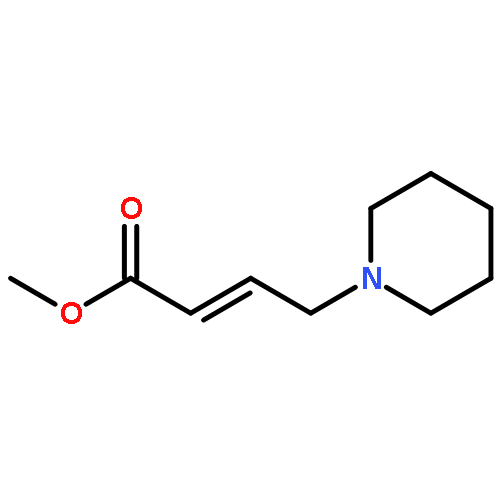
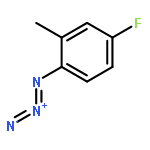
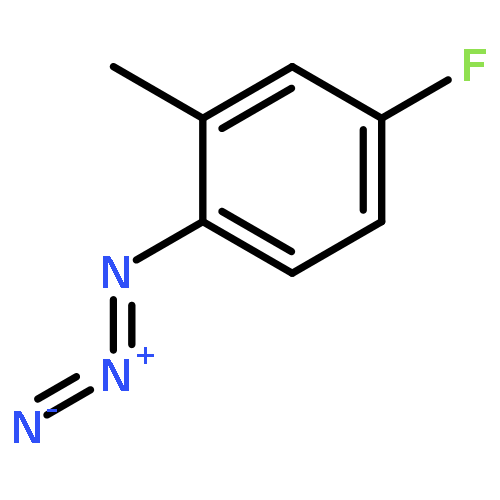
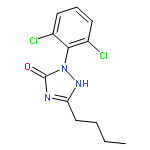
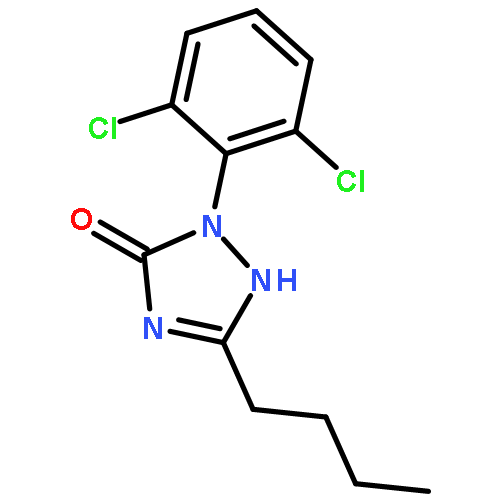
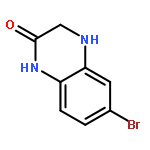
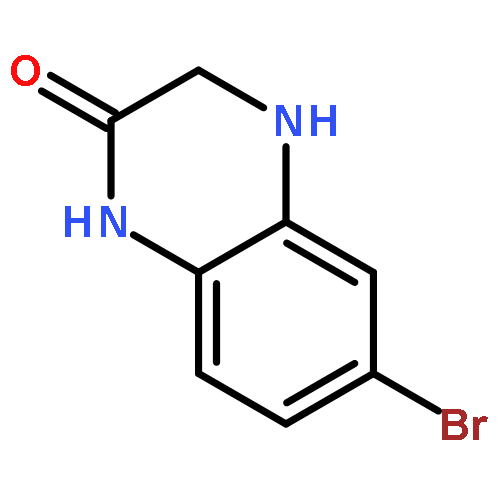
![(2-(4-BROMOPHENYL)IMIDAZO[1,2-A]PYRIDIN-3-YL)METHANAMINE](http://img.cochemist.com/ccimg/824500/824413-92-5.png)
![(2-(4-BROMOPHENYL)IMIDAZO[1,2-A]PYRIDIN-3-YL)METHANAMINE](http://img.cochemist.com/ccimg/824500/824413-92-5_b.png)
![7-Methyl-2-(4-methylphenyl)imidazo[1,2-a]pyridine-3-carbaldehyde](http://img.cochemist.com/ccimg/727700/727652-07-5.png)
![7-Methyl-2-(4-methylphenyl)imidazo[1,2-a]pyridine-3-carbaldehyde](http://img.cochemist.com/ccimg/727700/727652-07-5_b.png)




![8-Methyl-2-(4-methylphenyl)imidazo[1,2-a]pyridine-3-carbaldehyde](http://img.cochemist.com/ccimg/524800/524724-74-1.png)
![8-Methyl-2-(4-methylphenyl)imidazo[1,2-a]pyridine-3-carbaldehyde](http://img.cochemist.com/ccimg/524800/524724-74-1_b.png)
![Thiazolidine, 3-[(3R)-3-amino-1-oxo-4-phenylbutyl]-](http://img.cochemist.com/ccimg/479600/479585-68-7.png)
![Thiazolidine, 3-[(3R)-3-amino-1-oxo-4-phenylbutyl]-](http://img.cochemist.com/ccimg/479600/479585-68-7_b.png)
![2-(3-methylphenyl)-Imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/476000/475992-43-9.png)
![2-(3-methylphenyl)-Imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/476000/475992-43-9_b.png)
![2-Butanol, 4-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]-](/data/chemimg/3196400/452095-52-2.png)
![2-Butanol, 4-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]-](/data/chemimg/3196400/452095-52-2_b.png)
![6-Methyl-2-p-tolyl-imidazo[1,2-a]pyridine-3-carboxaldehyde](http://img.cochemist.com/ccimg/400800/400777-11-9.png)
![6-Methyl-2-p-tolyl-imidazo[1,2-a]pyridine-3-carboxaldehyde](http://img.cochemist.com/ccimg/400800/400777-11-9_b.png)
![1-Propanol, 3-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]-](/data/chemimg/3151700/361541-87-9.png)
![1-Propanol, 3-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]-](/data/chemimg/3151700/361541-87-9_b.png)
![4-chloro-N-[2-(4-sulfamoylphenyl)ethyl]benzenesulfonamide](http://img.cochemist.com/ccimg/349700/349620-56-0.png)
![4-chloro-N-[2-(4-sulfamoylphenyl)ethyl]benzenesulfonamide](http://img.cochemist.com/ccimg/349700/349620-56-0_b.png)
![Benzenesulfonamide, N-[2-[4-(aminosulfonyl)phenyl]ethyl]-](http://img.cochemist.com/ccimg/349700/349620-34-4.png)
![Benzenesulfonamide, N-[2-[4-(aminosulfonyl)phenyl]ethyl]-](http://img.cochemist.com/ccimg/349700/349620-34-4_b.png)


![4-Imidazolidinone, 5-[2-(methylthio)ethyl]-3-(phenylmethyl)-2-thioxo-](http://img.cochemist.com/ccimg/256400/256377-35-2.png)
![4-Imidazolidinone, 5-[2-(methylthio)ethyl]-3-(phenylmethyl)-2-thioxo-](http://img.cochemist.com/ccimg/256400/256377-35-2_b.png)
![Thieno[3,2-b]pyridine-2-sulfonamide,N-[(3S)-1-[(1-amino-7-isoquinolinyl)methyl]-2-oxo-3-pyrrolidinyl]-](http://img.cochemist.com/ccimg/250700/250645-18-2.png)
![Thieno[3,2-b]pyridine-2-sulfonamide,N-[(3S)-1-[(1-amino-7-isoquinolinyl)methyl]-2-oxo-3-pyrrolidinyl]-](http://img.cochemist.com/ccimg/250700/250645-18-2_b.png)


![1-[3-(Aminomethyl)phenyl]-N-[3-fluoro-2'-(methylsulfonyl)biphenyl-4-yl]-3-(trifluoromethyl)-1H-pyrazole-5-carboxamide](http://img.cochemist.com/ccimg/210000/209957-47-1.png)
![1-[3-(Aminomethyl)phenyl]-N-[3-fluoro-2'-(methylsulfonyl)biphenyl-4-yl]-3-(trifluoromethyl)-1H-pyrazole-5-carboxamide](http://img.cochemist.com/ccimg/210000/209957-47-1_b.png)



















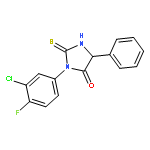
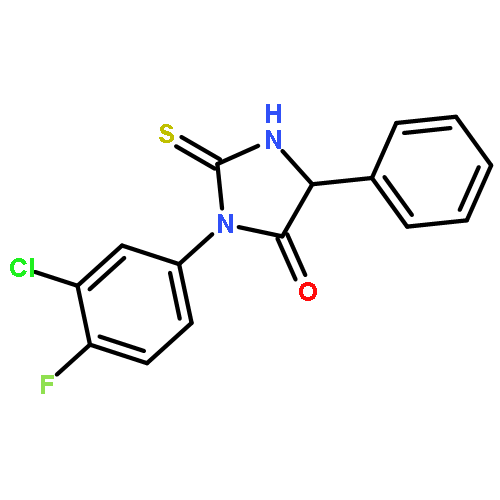
![4-Imidazolidinone, 5-phenyl-2-thioxo-3-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/143300/143247-33-0.png)
![4-Imidazolidinone, 5-phenyl-2-thioxo-3-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/143300/143247-33-0_b.png)
![4-Imidazolidinone, 5-methyl-2-thioxo-3-[3-(trifluoromethyl)phenyl]-, (±)-](http://img.cochemist.com/ccimg/143300/143247-29-4.png)
![4-Imidazolidinone, 5-methyl-2-thioxo-3-[3-(trifluoromethyl)phenyl]-, (±)-](http://img.cochemist.com/ccimg/143300/143247-29-4_b.png)






![Benzoic acid, 4-[(methoxyamino)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/137100/137042-75-2.png)
![Benzoic acid, 4-[(methoxyamino)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/137100/137042-75-2_b.png)
![5H-Benzo[5,6]cyclohepta[1,2-b]pyridin-11-ol, 8-chloro-6,11-dihydro-](http://img.cochemist.com/ccimg/133400/133330-72-0.png)
![5H-Benzo[5,6]cyclohepta[1,2-b]pyridin-11-ol, 8-chloro-6,11-dihydro-](http://img.cochemist.com/ccimg/133400/133330-72-0_b.png)


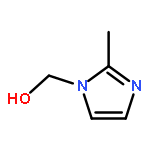
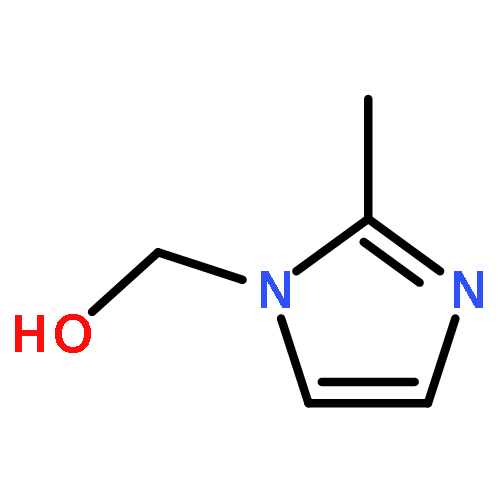
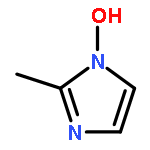
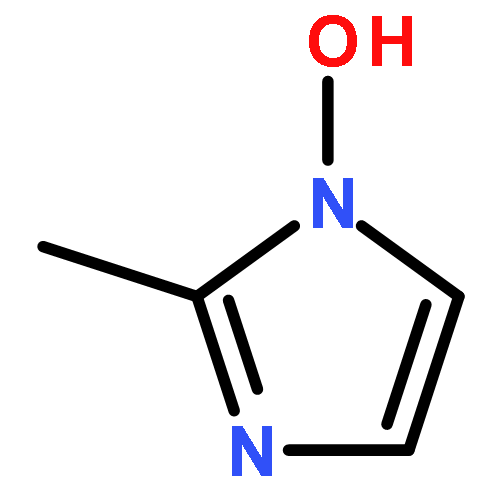


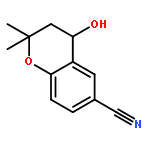
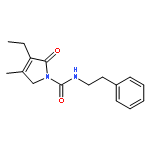
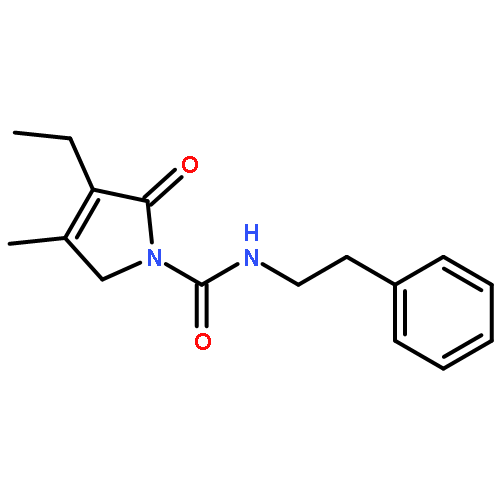
![Benzenesulfonylchloride,4-[2-[[(3-ethyl-2,5-dihydro-4-methyl-2-oxo-1H-pyrrol-1-yl)carbonyl]amino]ethyl]-](http://img.cochemist.com/ccimg/119100/119043-16-2.png)
![Benzenesulfonylchloride,4-[2-[[(3-ethyl-2,5-dihydro-4-methyl-2-oxo-1H-pyrrol-1-yl)carbonyl]amino]ethyl]-](http://img.cochemist.com/ccimg/119100/119043-16-2_b.png)
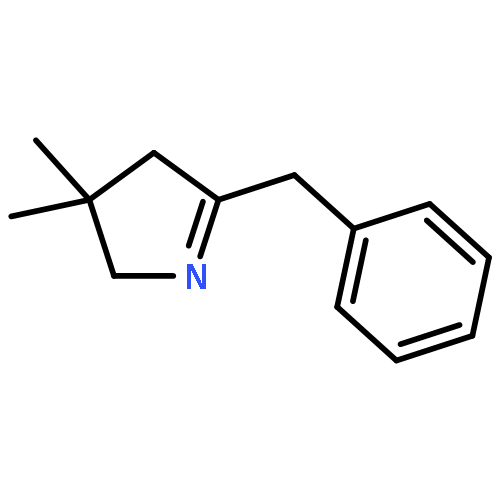
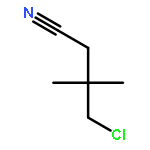
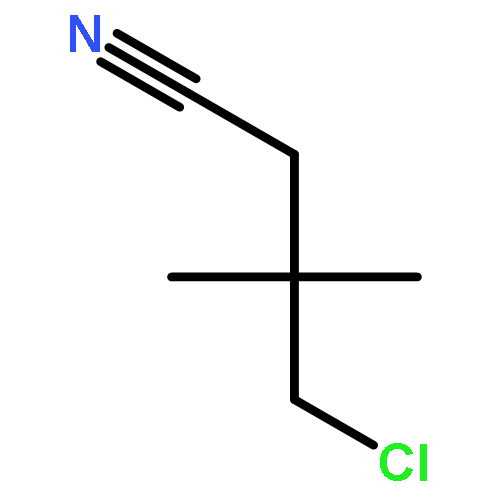
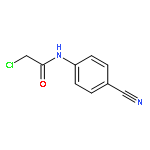
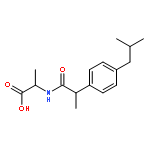
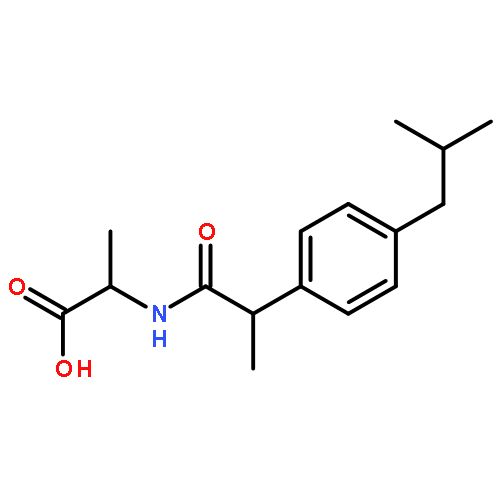
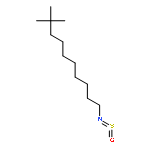
![2-[2-[4-(2-methylpropyl)phenyl]propanoylamino]acetic Acid](http://img.cochemist.com/ccimg/110500/110467-58-8.png)
![2-[2-[4-(2-methylpropyl)phenyl]propanoylamino]acetic Acid](http://img.cochemist.com/ccimg/110500/110467-58-8_b.png)


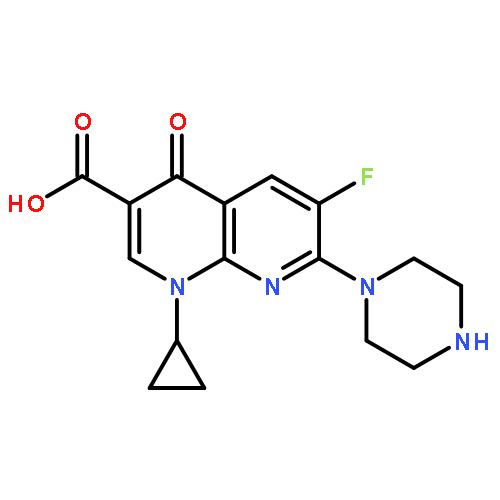
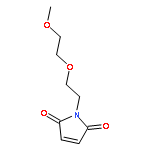
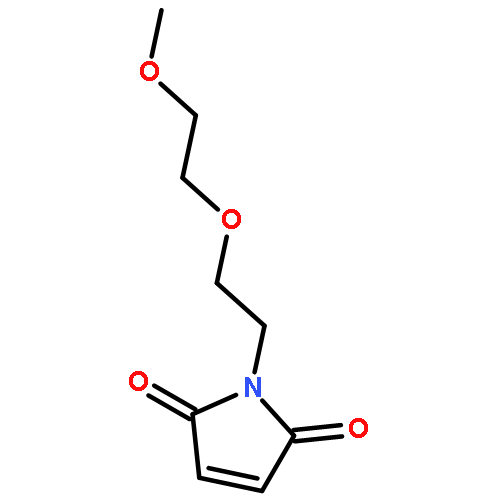
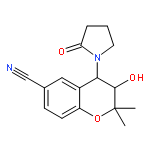
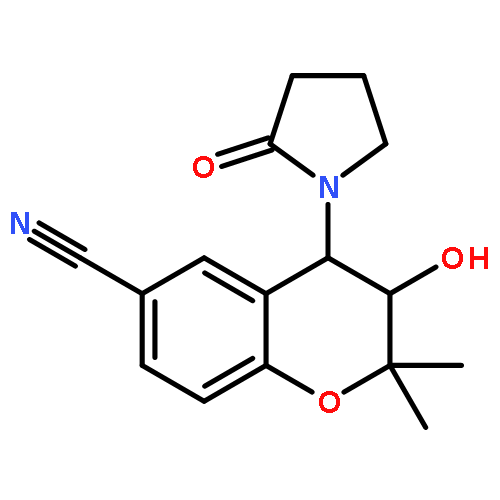
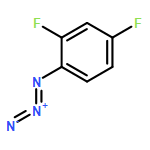

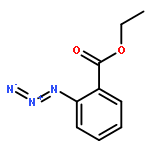
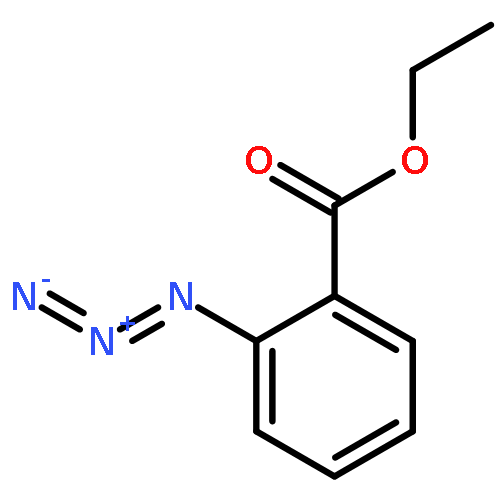
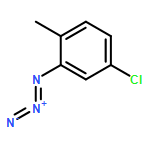
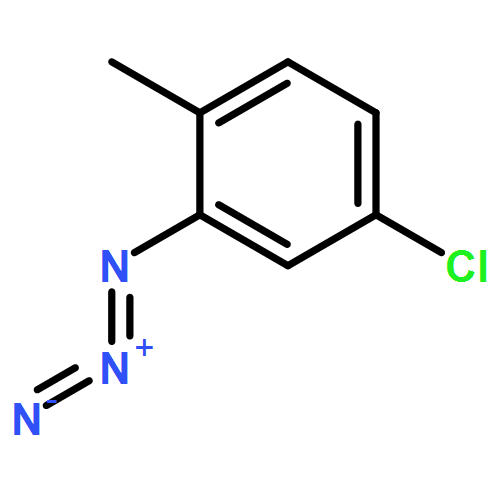
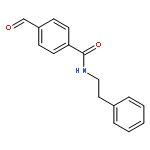
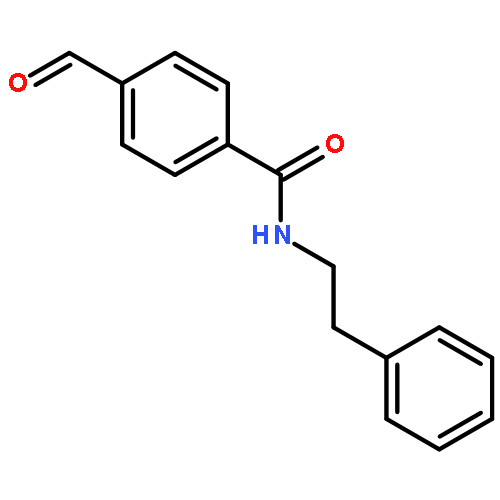
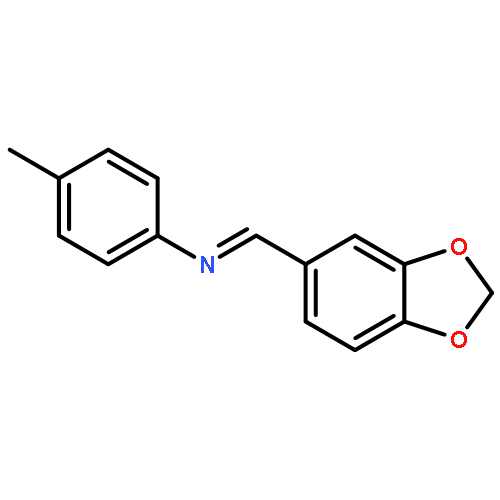
![BENZENAMINE, 4-METHOXY-N-[(4-METHOXYPHENYL)METHYLENE]-, (Z)-](http://img.cochemist.com/ccimg/83400/83306-67-6.png)
![BENZENAMINE, 4-METHOXY-N-[(4-METHOXYPHENYL)METHYLENE]-, (Z)-](http://img.cochemist.com/ccimg/83400/83306-67-6_b.png)



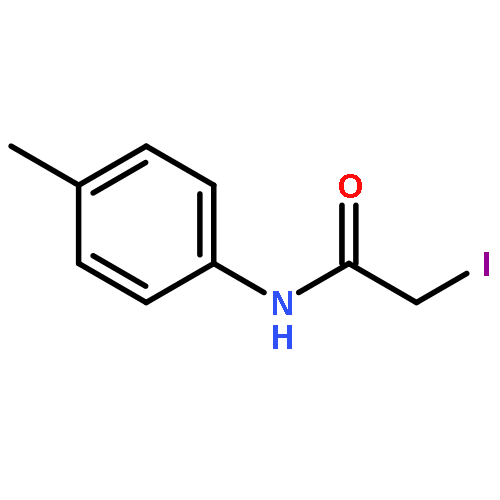
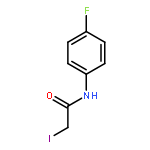
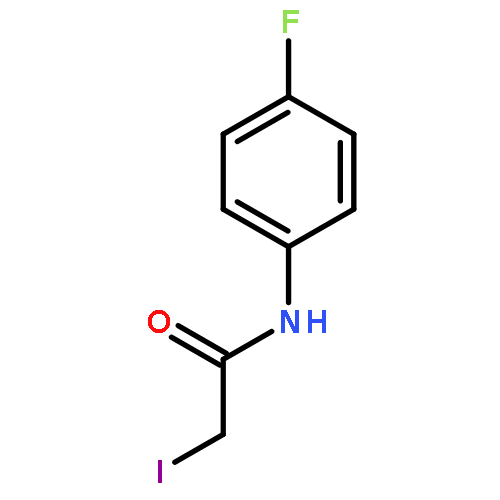

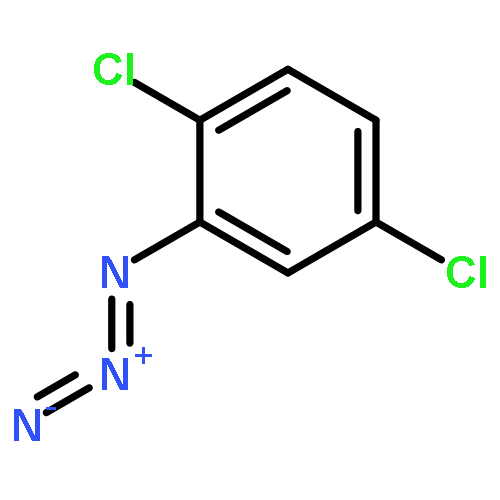


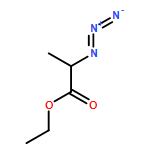
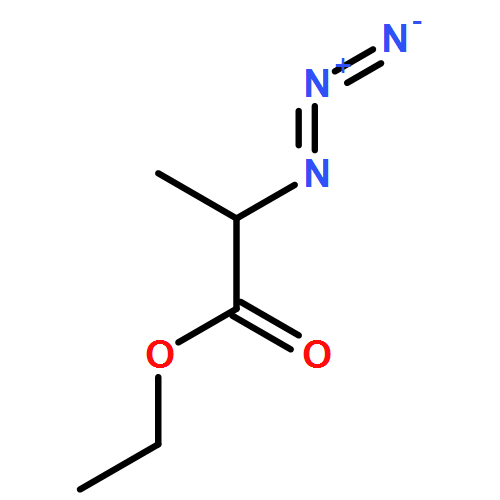
![IMIDAZO[1,2-A]PYRIDINE, 7-METHYL-2-(4-METHYLPHENYL)-](http://img.cochemist.com/ccimg/66000/65964-61-6.png)
![IMIDAZO[1,2-A]PYRIDINE, 7-METHYL-2-(4-METHYLPHENYL)-](http://img.cochemist.com/ccimg/66000/65964-61-6_b.png)
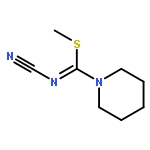
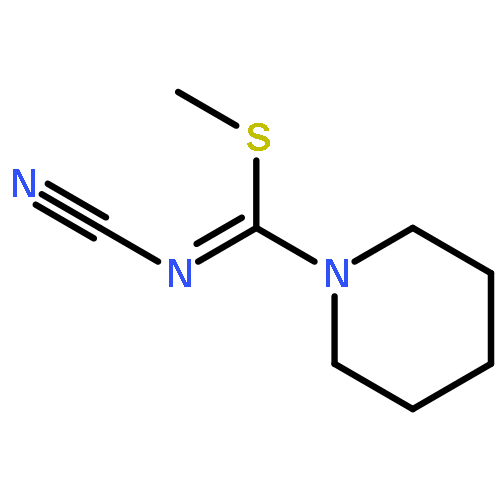
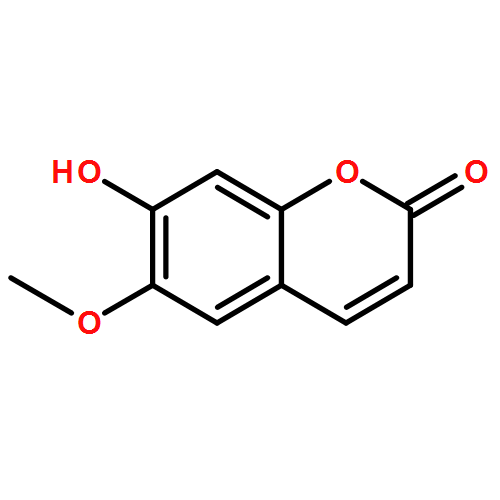
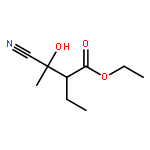
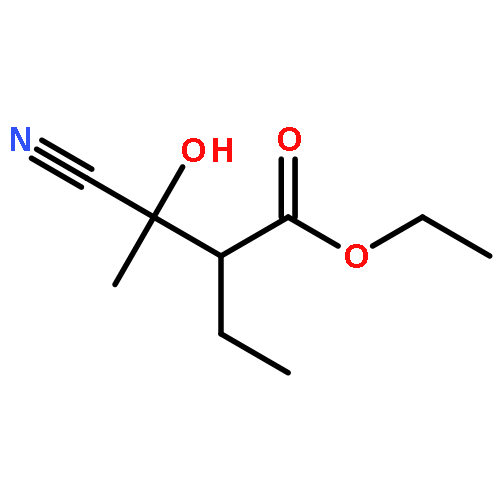
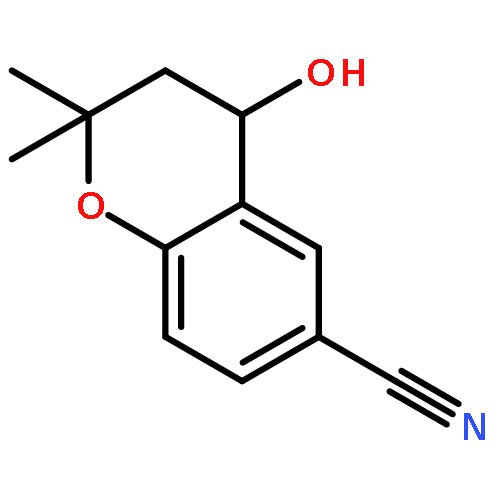
![2H-Oxireno[c][1]benzopyran-6-carbonitrile, 1a,7b-dihydro-2,2-dimethyl-](http://img.cochemist.com/ccimg/65100/65018-90-8.png)
![2H-Oxireno[c][1]benzopyran-6-carbonitrile, 1a,7b-dihydro-2,2-dimethyl-](http://img.cochemist.com/ccimg/65100/65018-90-8_b.png)





![2-(2,4-Dichlorophenyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/63200/63111-80-8.png)
![2-(2,4-Dichlorophenyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/63200/63111-80-8_b.png)
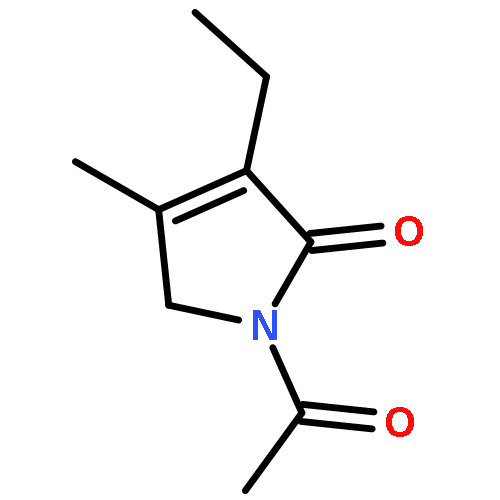
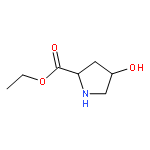
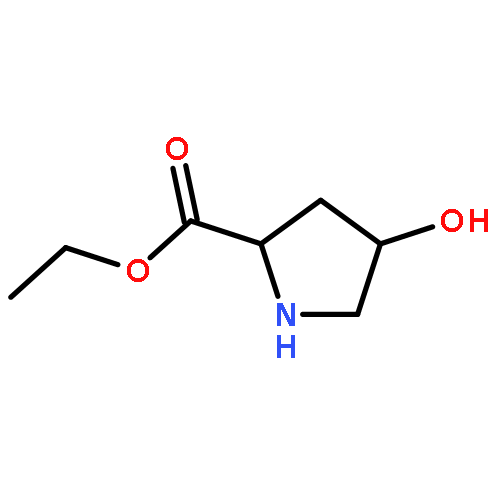




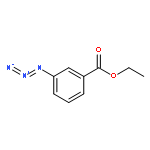
![Pyrrolidine, 1-[(2S)-2-amino-3-methyl-1-oxobutyl]-](http://img.cochemist.com/ccimg/54200/54164-07-7.png)
![Pyrrolidine, 1-[(2S)-2-amino-3-methyl-1-oxobutyl]-](http://img.cochemist.com/ccimg/54200/54164-07-7_b.png)

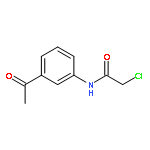
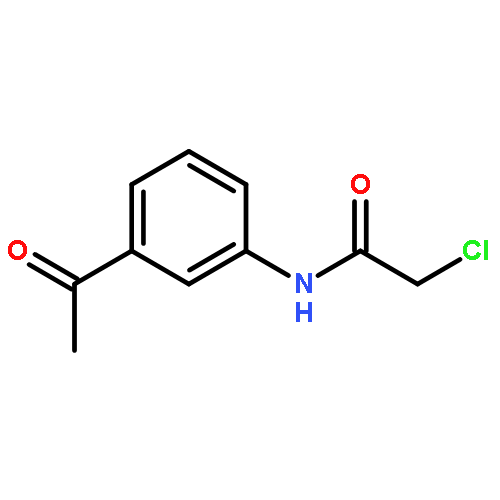

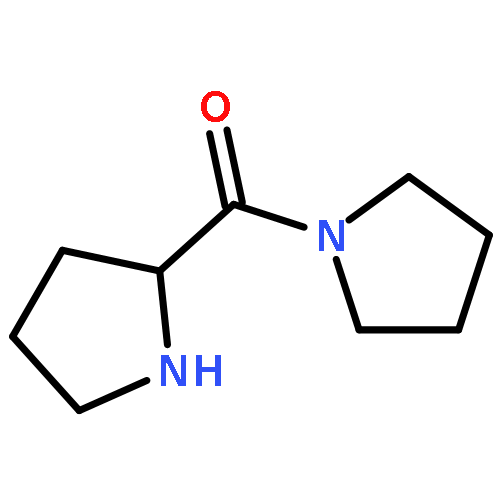
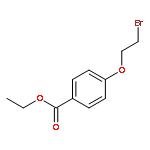
![2-ETHYL-8-METHOXY-2,3,4,5-TETRAHYDRO-1H-PYRIDO[4,3-B]INDOLE](http://img.cochemist.com/ccimg/39200/39123-65-4.png)
![2-ETHYL-8-METHOXY-2,3,4,5-TETRAHYDRO-1H-PYRIDO[4,3-B]INDOLE](http://img.cochemist.com/ccimg/39200/39123-65-4_b.png)
![8-CHLORO-6,11-DIHYDRO-11-(1-METHYL-4-PIPERIDINYL)-5H-BENZO[5,6]CYCLOHEPTA[1,2-B]PYRIDIN-11-OL](http://img.cochemist.com/ccimg/38100/38089-93-9.png)
![8-CHLORO-6,11-DIHYDRO-11-(1-METHYL-4-PIPERIDINYL)-5H-BENZO[5,6]CYCLOHEPTA[1,2-B]PYRIDIN-11-OL](http://img.cochemist.com/ccimg/38100/38089-93-9_b.png)












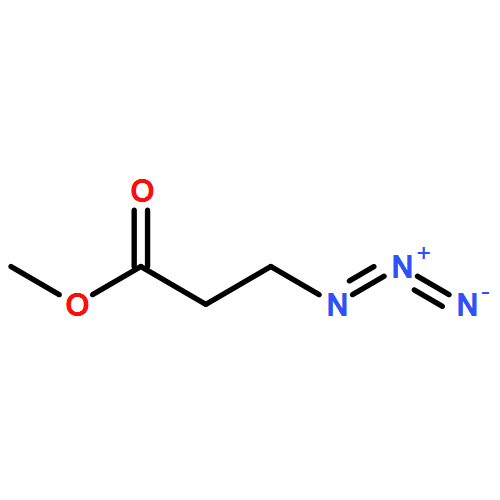



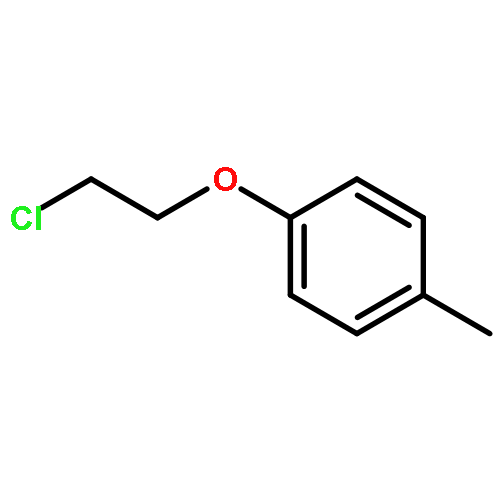
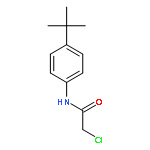
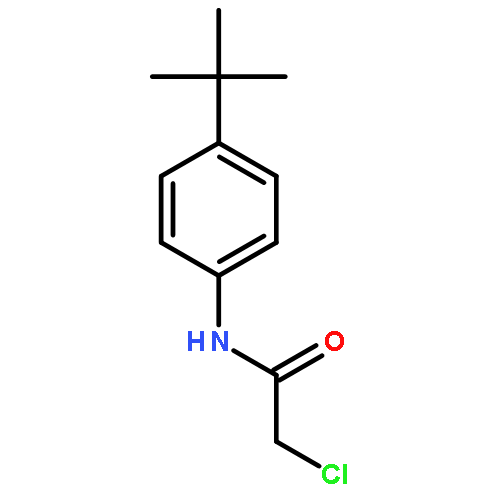

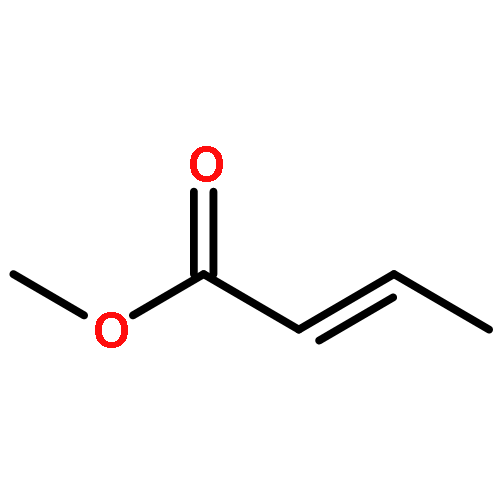
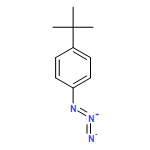

![Benzenamine,4-chloro-N-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/15500/15485-22-0.png)
![Benzenamine,4-chloro-N-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/15500/15485-22-0_b.png)
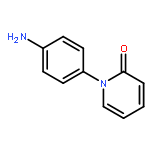
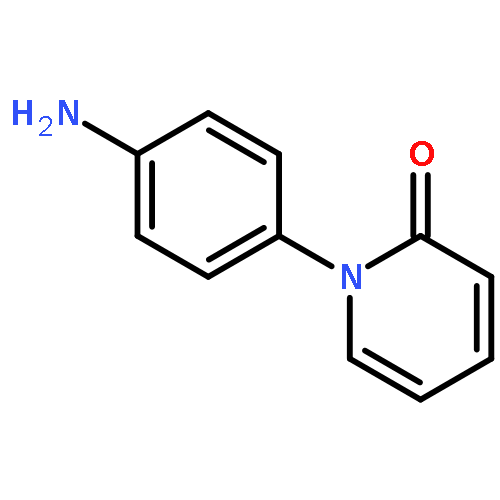
![Benzonitrile,4-[[(4-methoxyphenyl)methylene]amino]-](http://img.cochemist.com/ccimg/13100/13036-19-6.png)
![Benzonitrile,4-[[(4-methoxyphenyl)methylene]amino]-](http://img.cochemist.com/ccimg/13100/13036-19-6_b.png)




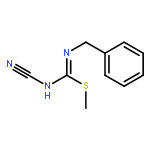
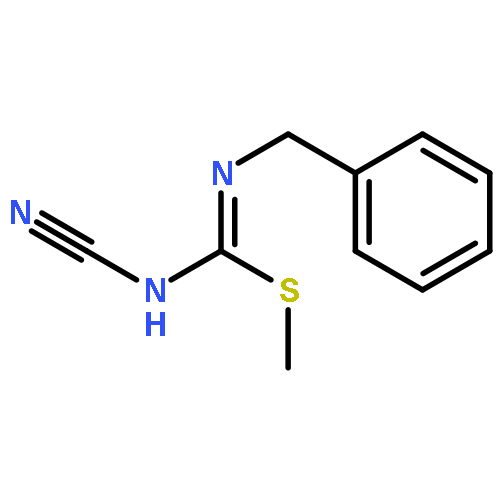
![7,8-dimethyl-2H-[1,3,5]triazino[2,1-b][1,3]benzothiazole-2,4(3H)-dione](http://img.cochemist.com/ccimg/5900/5848-27-1.png)
![7,8-dimethyl-2H-[1,3,5]triazino[2,1-b][1,3]benzothiazole-2,4(3H)-dione](http://img.cochemist.com/ccimg/5900/5848-27-1_b.png)
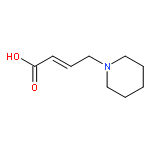
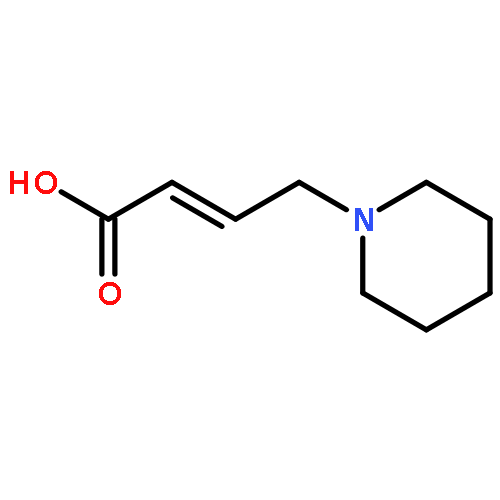


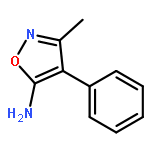
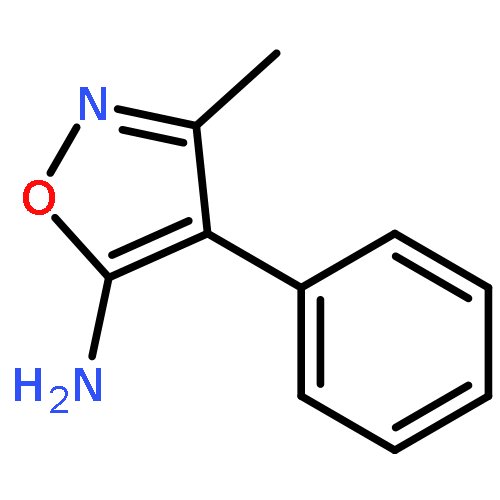
![2-Phenylimidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/4200/4105-21-9.png)
![2-Phenylimidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/4200/4105-21-9_b.png)
![5H-Benzo[5,6]cyclohepta[1,2-b]pyridine, 6,11-dihydro-](http://img.cochemist.com/ccimg/4000/3964-78-1.png)
![5H-Benzo[5,6]cyclohepta[1,2-b]pyridine, 6,11-dihydro-](http://img.cochemist.com/ccimg/4000/3964-78-1_b.png)
![2-Phenylimidazo[1,2-a]pyridine-3-carbaldehyde](http://img.cochemist.com/ccimg/3700/3672-39-7.png)
![2-Phenylimidazo[1,2-a]pyridine-3-carbaldehyde](http://img.cochemist.com/ccimg/3700/3672-39-7_b.png)
![4H-PYRAZOLO[1,5-C][1,3]THIAZOLE-2-CARBALDEHYDE](http://img.cochemist.com/ccimg/3300/3296-03-5.png)
![4H-PYRAZOLO[1,5-C][1,3]THIAZOLE-2-CARBALDEHYDE](http://img.cochemist.com/ccimg/3300/3296-03-5_b.png)
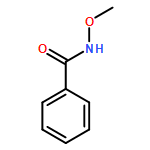
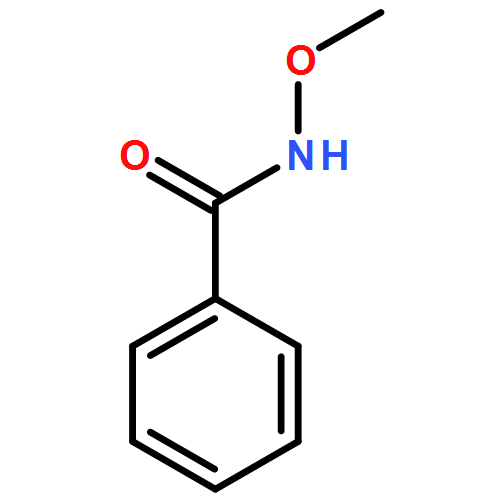


![Benzenamine, N-[(4-methoxyphenyl)methylene]-4-methyl-, (E)-](http://img.cochemist.com/ccimg/2000/1968-81-6.png)
![Benzenamine, N-[(4-methoxyphenyl)methylene]-4-methyl-, (E)-](http://img.cochemist.com/ccimg/2000/1968-81-6_b.png)
![Benzenamine, 4-methoxy-N-[(4-methoxyphenyl)methylene]-](/data/chemimg/922100/1749-08-2.png)
![Benzenamine, 4-methoxy-N-[(4-methoxyphenyl)methylene]-](/data/chemimg/922100/1749-08-2_b.png)
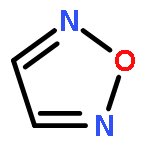
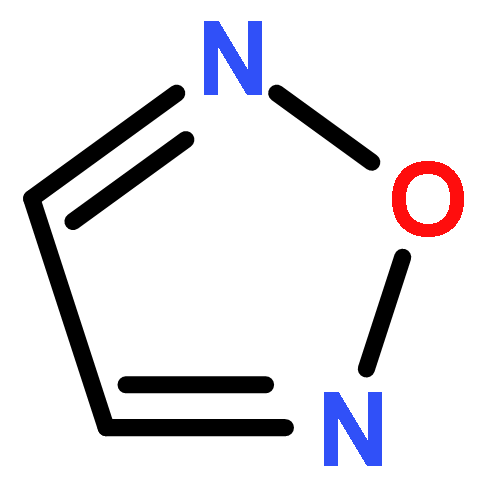


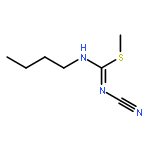
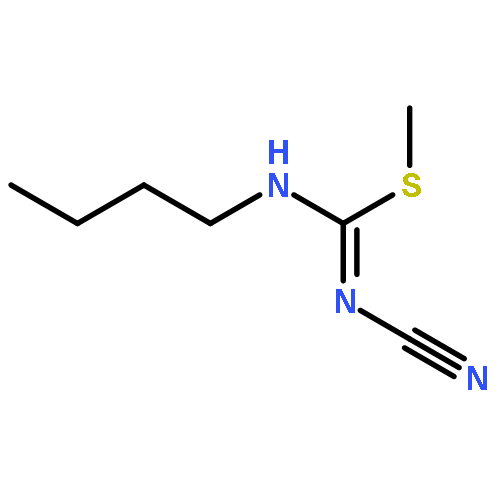
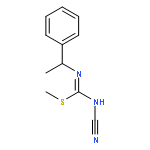
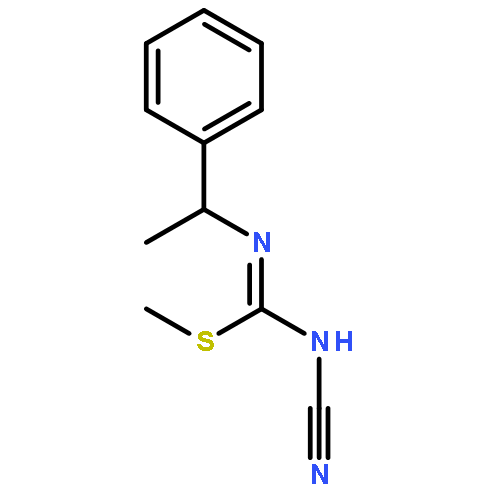
![2-[(3-CYANO-5,7-DIMETHYL-2-QUINOLINYL)AMINO]ETHYL 3-METHOXYBENZOA<WBR />TE](http://img.cochemist.com/ccimg/100/74-40-8.png)
![2-[(3-CYANO-5,7-DIMETHYL-2-QUINOLINYL)AMINO]ETHYL 3-METHOXYBENZOA<WBR />TE](http://img.cochemist.com/ccimg/100/74-40-8_b.png)
![(S)-tert-Butyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate](http://img.cochemist.com/ccimg/1268600/1268524-70-4.png)
![(S)-tert-Butyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate](http://img.cochemist.com/ccimg/1268600/1268524-70-4_b.png)
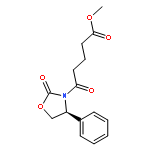
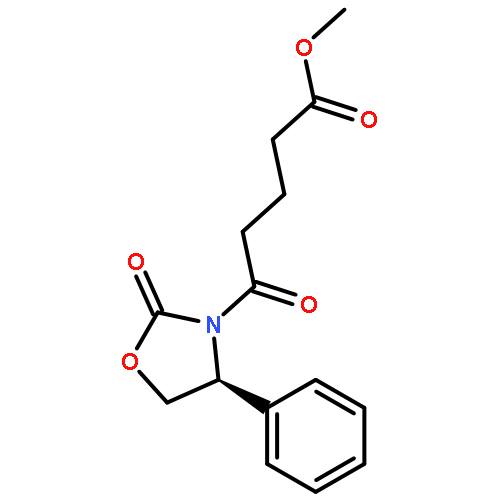
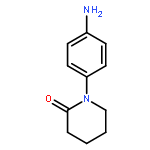
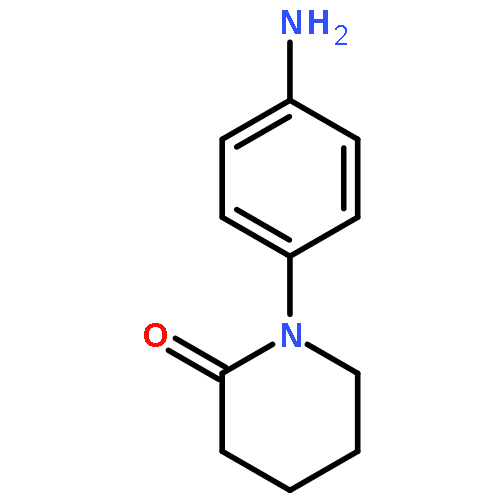
![4-bromo-N-[2-(4-sulfamoylphenyl)ethyl]benzenesulfonamide](http://img.cochemist.com/ccimg/349700/349614-35-3.png)
![4-bromo-N-[2-(4-sulfamoylphenyl)ethyl]benzenesulfonamide](http://img.cochemist.com/ccimg/349700/349614-35-3_b.png)