Co-reporter:Heng Song, Ke Ye, Peiyu Geng, Xiao Han, Rongzhen Liao, Chen-Ho Tung, and Wenguang Wang
ACS Catalysis November 3, 2017 Volume 7(Issue 11) pp:7709-7709
Publication Date(Web):October 3, 2017
DOI:10.1021/acscatal.7b02527
This paper describes a cooperative iron–thiolate catalyst Cp*Fe(1,2-Ph2PC6H4S)(NCMe) (Cp*– = C5Me5–, [1(NCMe)]) for regioselective hydroboration of aryl epoxide by pinacolborane (HBpin). The critical catalytic step involves the direct addition of epoxide to the catalyst rather than activation of the B–H bond of HBpin. Through iron–thiolate cooperation, [1(NCMe)] opens the aryl epoxide rings affording ferrous–alkoxide compounds. Notably, the ferrous–alkoxide intermediate (4) was structurally characterized after its isolation from the reaction of [1(NCMe)] with trans-2,3-diphenyloxirane. The more Lewis acidic hydroboranes such as H3B·THF and 9-BBN (BBN = borabicyclononane) can also be captured by [1(NCMe)]. The resulting iron–borane adducts [1H(BH2)] and [1H(BBN)] feature an agnostic Fe···B–H interaction. DFT calculations indicate that the addition of HBpin across the iron–thiolate sites is endergonic by 12.9 kcal/mol, whereas it is exergonic by 20.2 kcal/mol with BH3 and 4.6 kcal/mol with 9-BBN. Combining the experimental data with theoretical studies, a mechanism of the substrate activation by [1(NCMe)], followed by HBpin addition, is proposed for the catalysis.Keywords: epoxide hydroboration; ferrous−alkoxide; ferrous−borane adduct; iron catalysis; metal−ligand cooperation;
Co-reporter:Xin Yu, Chen-Ho Tung, and Wenguang Wang, Mioy T. Huynh, Danielle L. Gray, Sharon Hammes-Schiffer, and Thomas B. Rauchfuss
Organometallics June 12, 2017 Volume 36(Issue 11) pp:2245-2245
Publication Date(Web):May 18, 2017
DOI:10.1021/acs.organomet.7b00297
This study describes the structural, spectroscopic, and electrochemical properties of electronically unsymmetrical diiron hydrides. The terminal hydride Cp*Fe(pdt)Fe(dppe)(CO)H ([1(t-H)]0, Cp*– = Me5C5–, pdt2– = CH2(CH2S–)2, dppe = Ph2PC2H4PPh2) was prepared by hydride reduction of [Cp*Fe(pdt)Fe(dppe)(CO)(NCMe)]+. As established by X-ray crystallography, [1(t-H)]0 features a terminal hydride ligand. Unlike previous examples of terminal diiron hydrides, [1(t-H)]0 does not isomerize to the bridging hydride [1(μ-H)]0. Oxidation of [1(t-H)]0 gives [1(t-H)]+, which was also characterized crystallographically as its BF4– salt. Density functional theory (DFT) calculations indicate that [1(t-H)]+ is best described as containing an Cp*FeIII center. In solution, [1(t-H)]+ isomerizes to [1(μ-H)]+, as anticipated by DFT. Reduction of [1(μ-H)]+ by Cp2Co afforded the diferrous bridging hydride [1(μ-H)]0. Electrochemical measurements and DFT calculations indicate that the couples [1(t-H)]+/0 and [1(μ-H)]+/0 differ by 210 mV. Qualitative measurements indicate that [1(t-H)]0 and [1(μ-H)]0 are close in free energy. Protonation of [1(t-H)]0 in MeCN solution affords H2 even with weak acids via hydride transfer. In contrast, protonation of [1(μ-H)]0 yields 0.5 equiv of H2 by a proposed protonation-induced electron transfer process. Isotopic labeling indicates that μ-H/D ligands are inert.
Co-reporter:Ailing Zhang, Sakthi Raje, Jianguo Liu, Xiaoyan Li, Raja Angamuthu, Chen-Ho Tung, and Wenguang Wang
Organometallics August 28, 2017 Volume 36(Issue 16) pp:3135-3135
Publication Date(Web):August 10, 2017
DOI:10.1021/acs.organomet.7b00472
The insertion of CO into the Ni–C bond of synthetic Ni(II)–CH3 cationic complex ([1-CH3]+) affords a nickel–acetyl complex ([1-COCH3]+). Reduction of resultant [1-COCH3]+ by borohydrides produces CH3CHO, CH3CH2OH, and an Ni(0) compound ([1]0), which can react with CH3I to regenerate [1-CH3]+. By conducting deuterium labeling experiments, we have demonstrated that CH3CHO is the primary product from CH3CH2OH in such CO transformation reactions. In the reduction of [1-COCH3]+, the formation of CH3CHO competes with the loss of CH4, which leads to a Ni(0)–CO compound ([1-CO]0) as a minor product. Our results establish fundamental steps in the exploration of nickel-mediated CO transformation to valuable chemicals.
Co-reporter:Xiaoxiao Chu;Xin Xu;Hao Su;Sakthi Raje;Raja Angamuthu;Chen-Ho Tung
Inorganic Chemistry Frontiers 2017 vol. 4(Issue 4) pp:706-711
Publication Date(Web):2017/04/11
DOI:10.1039/C6QI00536E
Heterometallic complexes exhibit the advantages of exceptional crystal structures and electronic performance over monometallic ones in mimicking the active site of metalloenzymes such as [NiFe] hydrogenase and acetyl-coenzyme A synthase/carbon monoxide dehydrogenase (ACS/CODH). Herein, we discovered a new mild route to synthesize [NiCu] binuclear complexes [(dppe)Ni(μ-pdt)Cu(dppe)](BF4) (dppe = 1,2-C2H4(PPh2)2, 1), [(dppe)Ni(μ-pdt)Cu(dppbz)]BF4 (dppbz = 1,2-C6H4(PPh2)2; 2), and [(dppe)Ni(μ-pdt)Cu(dcpe)]BF4 (dcpe = 1,2-C2H4(PCy2)2; 3) through Ni(pdt)(dppe) (pdt = (SC3H6S)2−) and [Cu(P–P)(NCMe)2]BF4 (P–P = diphosphine chelating ligand). The structures of these models in the [(dppe)Ni(pdt)Cu(P–P)]+ type are composed of Cu(I) in tetrahedral and Ni(II) in planar conformations. DFT calculations suggest that the HOMO corresponds to the delocalized π-orbital of the P–Cu–S system, while the LUMO is primarily composed of Ni, P, and S atoms with antibonding character.
Co-reporter:Xiaoxiao Chu;Xin Yu;Sakthi Raje;Raja Angamuthu;Jianping Ma;Chen-Ho Tung
Dalton Transactions 2017 vol. 46(Issue 40) pp:13681-13685
Publication Date(Web):2017/10/17
DOI:10.1039/C7DT02892J
A [NiFe] complex [(dppe)Ni(pdt)FeCp*(CO)]+ was isolated and characterized as two isomers [1(CO)]+ and [1′(CO)]+. Heating the solution of [1(CO)]+ allowed it to convert into [1′(CO)]+. The one-electron oxidation of [1′(CO)]+ to [1′(CO)]2+ induced fluxional CO movement providing [1(CO)]2+. Recovery of [1(CO)]+ was realized by the one-electron reduction of [1(CO)]2+.
Co-reporter:Jia-Heng Xu, Ling-Yu Guo, Hai-Feng Su, Xiang Gao, Xiao-Fan Wu, Wen-Guang Wang, Chen-Ho Tung, and Di Sun
Inorganic Chemistry 2017 Volume 56(Issue 3) pp:
Publication Date(Web):January 24, 2017
DOI:10.1021/acs.inorgchem.6b02698
Inspired by the transition-metal-oxo cubical Mn4CaO5 in photosystem II, we herein report a disc-like heptanuclear mixed-valent cobalt cluster, [CoII5CoIII2(mdea)4(N3)2(CH3CN)6(OH)2(H2O)2·4ClO4] (1, H2mdea = N-methyldiethanolamine), for photocatalytic oxygen evolution. The topology of the Co7 core resembles a small piece of cobaltate protected by terminal H2O, N3–, CH3CN, and multidentate N-methyldiethanolamine at the periphery. Under the optimal photocatalytic conditions, 1 exhibits water oxidation activity with a turnover number (TON) of 210 and a turnover frequency (TOFinitial) of 0.23 s–1. Importantly, electrospray mass spectrometry (ESI-MS) was used to not only identify the possible main active species in the water oxidation reaction but also monitor the evolutions of oxidation states of cobalt during the photocatalytic reactions. These results shed light on the design concept of new water oxidation catalysts and mechanism-related issues such as the key active intermediate and oxidation state evolution in the oxygen evolution process. The magnetic properties of 1 were also discussed in detail.
Co-reporter:Jiaheng Xu;Zhi Wang;Wenguang Yu;Dr. Di Sun;Dr. Qing Zhang;Dr. Chen-Ho Tung;Dr. Wenguang Wang
ChemSusChem 2016 Volume 9( Issue 10) pp:1146-1152
Publication Date(Web):
DOI:10.1002/cssc.201600101
Abstract
Two Kagóme cobalt(II)-organic layers of [Co3(μ3-OH)2(bdc)2]n (1) and [Co3(μ3-OH)2(chdc)2]n (2) (bdc=o-benzenedicarboxylate and chdc=1,2-cyclohexanedicarboxylate) that bear bridging OH− ligands were explored as water oxidation catalysts (WOCs) for photocatalytic O2 production. The activities of 1 and 2 towards H2O oxidation were assessed by monitoring the in situ O2 concentration versus time in the reaction medium by utilizing a Clark-type oxygen electrode under photochemical conditions. The oxygen evolution rate (R ) was 24.3 μmol s−1 g−1 for 1 and 48.8 μmol s−1 g−1 for 2 at pH 8.0. Photocatalytic reaction studies show that 1 and 2 exhibit enhanced activities toward the oxidation of water compared to commercial nanosized Co3O4. In scaled-up photoreactions, the pH value of the reaction medium decreased from 8.0 to around 7.0 after 20 min and the O2 production ceased. Based on the amounts of the sacrificial oxidant (K2S2O8) used, the yield of O2 produced is 49.6 % for 2 and 29.8 % for 1. However, the catalyst can be recycled without a significant loss of catalytic activity. Spectroscopic studies suggest that the structure and composition of recycled 1 and 2 are maintained. In isotope-labeling H218O (97 % enriched) experiments, the distribution of 16O16O/16O18O/18O18O detected was 0:7.55:92.45, which is comparable to the theoretical values of 0.09:5.82:94.09. This work not only provides new catalysts that resemble ligand-protected cobalt oxide materials but also establishes the significance of the existence of OH− (or H2O) binding sites at the metal center in WOCs.
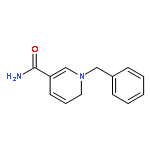
Co-reporter:Fanjun Zhang, Jiong Jia, Shuli Dong, Wenguang Wang, and Chen-Ho Tung
Organometallics 2016 Volume 35(Issue 8) pp:1151-1159
Publication Date(Web):April 5, 2016
DOI:10.1021/acs.organomet.6b00179
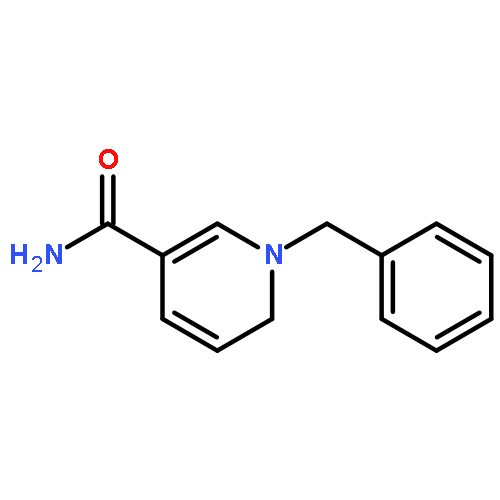
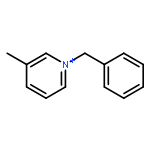
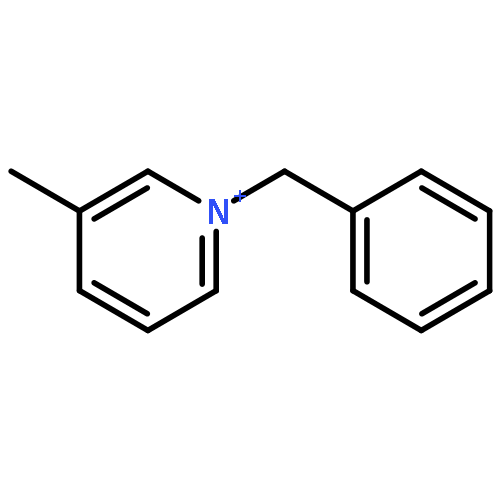
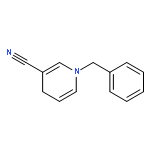
Iron(II) hydride complexes of the “piano-stool” type, Cp*(P-P)FeH (P-P = dppe (1H), dppbz (2H), dppm (3H), dcpe (4H)) were examined as hydride donors in the reduction of N-benzylpyridinium and acridinium salts. Two pathways of hydride transfer, “single-step H–” transfer to pyridinium and a “two-step (e–/H•)” transfer for acridinium reduction, were observed. When 1-benzylnicotinamide cation (BNA+) was used as an H– acceptor, kinetic studies suggested that BNA+ was reduced at the C6 position, affording 1,6-BNAH, which can be converted to the more thermally stable 1,4-product. The rate constant k of H– transfer was very sensitive to the bite angle of P–Fe–P in Cp*(P-P)FeH and ranged from 3.23 × 10–3 M–1 s–1 for dppe to 1.74 × 10–1 M–1 s–1 for dppm. The results obtained from reduction of a range of N-benzylpyridinium derivatives suggest that H– transfer is more likely to be charge controlled. In the reduction of 10-methylacridinium ion (Acr+), the reaction was initiated by an e– transfer (ET) process and then followed by rapid disproportionation reactions to produce Acr2 dimer and release of H2. To achieve H• transfer after ET from [Cp*(P-P)FeH]+ to acridine radicals, the bulkier acridinium salt 9-phenyl-10-methylacridinium (PhAcr+) was selected as an acceptor. More evidence for this “two-step (e–/H•)” process was derived from the characterization of PhAcr• and [4H]+ radicals by EPR spectra and by the crystallographic structure confirmation of [4H]+. Our mechanistic understanding of fundamental H– transfer from iron(II) hydrides to NAD+ analogues provides insight into establishing attractive bio-organometallic transformation cycles driven by iron catalysis.
Co-reporter:Xiaoxiao Chu, Xin Xu, Hao Su, Sakthi Raje, Raja Angamuthu, Chen-Ho Tung and Wenguang Wang
Inorganic Chemistry Frontiers 2017 - vol. 4(Issue 4) pp:NaN711-711
Publication Date(Web):2017/02/07
DOI:10.1039/C6QI00536E
Heterometallic complexes exhibit the advantages of exceptional crystal structures and electronic performance over monometallic ones in mimicking the active site of metalloenzymes such as [NiFe] hydrogenase and acetyl-coenzyme A synthase/carbon monoxide dehydrogenase (ACS/CODH). Herein, we discovered a new mild route to synthesize [NiCu] binuclear complexes [(dppe)Ni(μ-pdt)Cu(dppe)](BF4) (dppe = 1,2-C2H4(PPh2)2, 1), [(dppe)Ni(μ-pdt)Cu(dppbz)]BF4 (dppbz = 1,2-C6H4(PPh2)2; 2), and [(dppe)Ni(μ-pdt)Cu(dcpe)]BF4 (dcpe = 1,2-C2H4(PCy2)2; 3) through Ni(pdt)(dppe) (pdt = (SC3H6S)2−) and [Cu(P–P)(NCMe)2]BF4 (P–P = diphosphine chelating ligand). The structures of these models in the [(dppe)Ni(pdt)Cu(P–P)]+ type are composed of Cu(I) in tetrahedral and Ni(II) in planar conformations. DFT calculations suggest that the HOMO corresponds to the delocalized π-orbital of the P–Cu–S system, while the LUMO is primarily composed of Ni, P, and S atoms with antibonding character.
.jpg)






![Phosphine, [2-(chloromethyl)phenyl]diphenyl-](/data/chemimg/1143800/167706-40-3.png)
![Phosphine, [2-(chloromethyl)phenyl]diphenyl-](/data/chemimg/1143800/167706-40-3_b.png)