Co-reporter:Rosa E. Bulo, Carine Michel, Paul Fleurat-Lessard, and Philippe Sautet
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 12) pp:5567-5577
Publication Date(Web):October 17, 2013
DOI:10.1021/ct4005596
This paper critically evaluates the state of the art in combined quantum mechanical/molecular mechanical (QM/MM) approaches to the computational description of chemistry in water and supplies guidelines for the setup of customized multiscale simulations of aqueous processes. We differentiate between structural and dynamic performance, since some tasks, e.g., the reproduction of NMR or UV–vis spectra, require only structural accuracy, while others, i.e., reaction mechanisms, require accurate dynamic data as well. As a model system for aqueous solutions in general, the approaches were tested on a QM water cluster in an environment of MM water molecules. The key difficulty is the description of the possible diffusion of QM molecules into the MM region and vice versa. The flexible inner region ensemble separator (FIRES) approach constrains QM solvent molecules within an active (QM) region. Sorted adaptive partitioning (SAP), difference-based adaptive solvation (DAS), and buffered-force (BF) are all adaptive approaches that use a buffer zone in which solvent molecules gradually adapt from QM to MM (or vice versa). The costs of SAP and DAS are relatively high, while BF is fast but sacrifices conservation of both energy and momentum. Simulations in the limit of an infinitely small buffer zone, where DAS and SAP become equivalent, are discussed as well and referred to as ABRUPT. The best structural accuracy is obtained with DAS, BF, and ABRUPT, all three of similar quality. FIRES performs very well for dynamic properties localized deep within the QM region. By means of elimination DAS emerges as the best overall compromise between structural and dynamic performance. Eliminating the buffer zone (ABRUPT) improves efficiency and still leads to surprisingly good results. While none of the many new flavors are perfect, all together this new field already allows accurate description of a wide range of structural and dynamic properties of aqueous solutions.

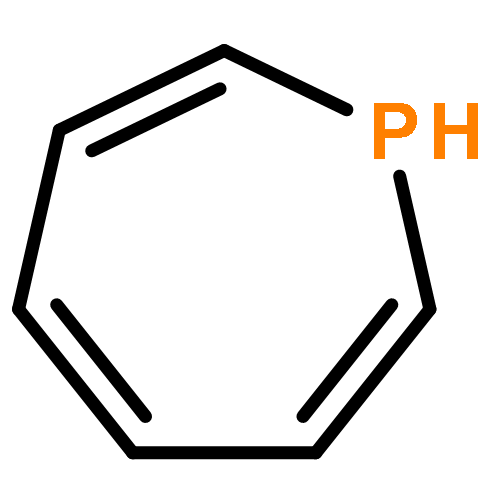
![7-Phosphabicyclo[4.1.0]hepta-1,3,5-triene](http://img.cochemist.com/ccimg/852300/852242-61-6.png)
![7-Phosphabicyclo[4.1.0]hepta-1,3,5-triene](http://img.cochemist.com/ccimg/852300/852242-61-6_b.png)
![BICYCLO[6.1.0]NONA-2,4,6-TRIENE](http://img.cochemist.com/ccimg/700/696-76-4.png)
![BICYCLO[6.1.0]NONA-2,4,6-TRIENE](http://img.cochemist.com/ccimg/700/696-76-4_b.png)
![7-THIABICYCLO[4.1.0]HEPTA-1,3,5-TRIENE](http://img.cochemist.com/ccimg/65400/65330-66-7.png)
![7-THIABICYCLO[4.1.0]HEPTA-1,3,5-TRIENE](http://img.cochemist.com/ccimg/65400/65330-66-7_b.png)


![7-Phosphabicyclo[2.2.1]hepta-2,5-diene](http://img.cochemist.com/ccimg/152100/152072-08-7.png)
![7-Phosphabicyclo[2.2.1]hepta-2,5-diene](http://img.cochemist.com/ccimg/152100/152072-08-7_b.png)




![Bicyclo[4.4.1]undeca-1,3,5,7,9-pentaene](http://img.cochemist.com/ccimg/2500/2443-46-1.png)
![Bicyclo[4.4.1]undeca-1,3,5,7,9-pentaene](http://img.cochemist.com/ccimg/2500/2443-46-1_b.png)
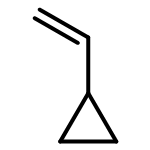





![bicyclo[4.2.1]nona-2,4,7-triene](http://img.cochemist.com/ccimg/5300/5240-87-9.png)
![bicyclo[4.2.1]nona-2,4,7-triene](http://img.cochemist.com/ccimg/5300/5240-87-9_b.png)