Co-reporter:Yuka Takahashi, Hirotaka Ikeda, Yuki Kanase, Kosho Makino, Hidetsugu Tabata, Tetsuta Oshitari, Satoshi Inagaki, Yuko Otani, Hideaki Natsugari, Hideyo Takahashi, and Tomohiko Ohwada
The Journal of Organic Chemistry November 3, 2017 Volume 82(Issue 21) pp:11370-11370
Publication Date(Web):October 2, 2017
DOI:10.1021/acs.joc.7b01759
The conformational properties of N-acyl azoles (imidazole, pyrazole, and triazole) were examined. The N-2′,4′,6′-trichlorobenzoyl azoles were stable in methanol at room temperature, and no hydrolyzed products were observed over 7 days in the presence of 5% trifluoroacetic acid or 5% triethylamine in CDCl3. The high stability may be explained by the double-bond amide character caused by the steric hindrance due to the ortho-substituents in the benzoyl group. While specific E-amide preferences were observed in N-acyl pyrazoles/triazoles, the amides of the imidazoles gave a mixture of E and Z. One of the conceivable ideas to rationalize this conformational preference may be repulsive interaction between two sets of lone-pair electrons on the pyrazole 2-nitrogen (nN) and the carbonyl oxygen atoms (nO) in the Z-conformation of N-acyl pyrazoles/triazoles. However, analysis of orbital interactions suggested that in the case of the E-conformation of N-acyl pyrazoles, such electron repulsion is small because of distance. The interbond energy calculations suggested that the Z-conformer is involved in strong vicinal σ–σ repulsion along the amide linkage between the σN1N2 and σC1C2 orbitals in the anti-periplanar arrangement and between the σN1C5 and σC1C2 orbitals in the syn-periplanar arrangement, which lead to the overwhelming E-preference in N-acyl pyrazoles/triazoles. In the case of N-acyl imidazoles, similar vicinal σ–σ repulsions were counterbalanced, leading to a weak preference for the E-conformer over the Z-conformer. The chemically stable and E-preferring N-acyl azoles may be utilized as scaffolds in future drug design.
Co-reporter:Yingtang Ning, Yuko Otani, and Tomohiko Ohwada
The Journal of Organic Chemistry June 16, 2017 Volume 82(Issue 12) pp:6313-6313
Publication Date(Web):May 23, 2017
DOI:10.1021/acs.joc.7b00904
An experimental study of base-induced transformation reaction of 2-acyl-3-alkyl-2H-azirines to oxazoles indicated that a deprotonation-initiated mechanism is involved, in addition to nucleophilic addition to the imine functionality. Calculations suggested the participation of a ketenimine (ethenimine) intermediate generated by azirine ring opening of the carbanion intermediate formed by α-deprotonation of 2H-azirine. The ketenimine intermediate possessing methyl substituents at C(3) appears to be more stable than the tautomeric nitrile ylide which was proposed to be involved in photoinduced and pyrolysis reactions of 2-acyl-3-alkyl-2H-azirines to afford oxazoles. Thus, intermediacy of ketenimine is consistent with both experimental and computational results, at least under strongly basic reaction conditions.
Co-reporter:Makafui Gasonoo, Akinari Sumita, Kenneth N. Boblak, Kristen Giuffre, Tomohiko Ohwada, and Douglas A Klumpp
The Journal of Organic Chemistry June 16, 2017 Volume 82(Issue 12) pp:6044-6044
Publication Date(Web):May 30, 2017
DOI:10.1021/acs.joc.7b00311
A superacid-promoted method for the synthesis of 9,9-diarylfluorenes is described. The chemistry involves cyclizations and arylations with biphenyl-substituted heterocyclic ketones and a mechanism is proposed involving superelectrophilic intermediates. The key reactive intermediates–dicationic and trication fluorenyl cations have been observed by low-temperature NMR and the mechanism has been further studied using DFT calculations.
Co-reporter:Akinari Sumita;Makafui Gasonoo;Kenneth J. Boblak; Tomohiko Ohwada; Douglas A Klumpp
Chemistry - A European Journal 2017 Volume 23(Issue 11) pp:2566-2570
Publication Date(Web):2017/02/21
DOI:10.1002/chem.201606036
AbstractA series of 9-fluorenyl cations has been studied and it is shown that increasing charge on a heterocyclic substituent group enhances the anti-aromatic character of the carbocation system. Similarly, a series of dibenzosuberenyl cations has been studied and increasing charge on a substituent group is shown to enhance aromatic character in the carbocation system. These studies include the direct observations of dicationic and tricationic species using stable-ion conditions and low temperature NMR. The structures of these ions were further characterized using DFT calculations, confirming that highly charged organic ions may exhibit unusual distributions of π-electrons and delocalization of electrons in 4n or 4n+2 π-systems.
Co-reporter:Yingtang Ning;Tomoya Fukuda;Hirotaka Ikeda;Yuko Otani;Masatoshi Kawahata;Kentaro Yamaguchi
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 6) pp:1381-1392
Publication Date(Web):2017/02/07
DOI:10.1039/C6OB02719A
Neighboring group participation is defined as the action of a substituent to stabilize a transition state or an intermediate by forming a bond or a partial bond with the reaction center. In addition to the primary interaction with the nearest neighboring group, secondary interactions involving another neighboring group(s) could also occur in principle. Here, we revisit this issue by examining the influence of secondary interactions on the stability and reactivity of the putative iminylium cation intermediates, formed by N–O bond cleavage of 1-tetralone oxime systems. A direct observation of a peri-bromo-iminylium intermediate in solution supported the involvement of iminylium cations and the stabilizing effect of secondary interactions arising from a distal tandem substituent. Both experimental and computational findings support the idea that secondary interactions of a tandem-neighboring group on the primary peri-heteroatom (Br, Cl, and O(Me))-iminylium bonding interaction, i.e., a weak halogen bonding interaction (ester (nitro) oxygen–halogen bonding) and an unprecedented hydrogen bonding interaction between a nitro oxygen atom and a CH3O hydrogen atom, are crucial determinants of the reaction pathway, leading to either overwhelmingly selective syn-migration of the oxime functionality or covalent bond formation under acid-catalyzed Beckmann rearrangement conditions.
Co-reporter:Akinari Sumita;Yuko Otani
Chemical Communications 2017 vol. 53(Issue 9) pp:1482-1485
Publication Date(Web):2017/01/26
DOI:10.1039/C6CC09618B
We describe the chemoselective conversion of carboxylic acids to functional aromatic ketones promoted by a tailored organophosphate ester in the presence of a Brønsted acid. The protonated phosphate ester reacts with the carboxylic acid to form acyl phosphate, which reacts with benzenes to give aromatic ketones, probably through the acylium ion or its equivalent. The reaction time is short even at room temperature, and the reaction is compatible with various other functional groups, including amines, olefins, esters, amides and nitriles.
Co-reporter:Siyuan Wang, Tohru Taniguchi, Kenji Monde, Masatoshi Kawahata, Kentaro Yamaguchi, Yuko Otani and Tomohiko Ohwada
Chemical Communications 2016 vol. 52(Issue 21) pp:4018-4021
Publication Date(Web):18 Feb 2016
DOI:10.1039/C6CC00284F
Nitrogen-pyramidalization of amide increases electron density on nitrogen and decreases that on carbonyl oxygen. We identified hydrogen-bonding to carbonyl of nitrogen-pyramidalized bicyclic β-proline derivatives by crystallography, and by NMR and vibrational circular dichroism (VCD) spectroscopy in solution. Such hydrogen-bonding can switch the preferred nitrogen-pyramidalization direction, as detected by VCD spectroscopy.
Co-reporter:Sejin Jung; Asuka Inoue; Sho Nakamura; Takayuki Kishi; Akiharu Uwamizu; Misa Sayama; Masaya Ikubo; Yuko Otani; Kuniyuki Kano; Kumiko Makide; Junken Aoki
Journal of Medicinal Chemistry 2016 Volume 59(Issue 8) pp:3750-3776
Publication Date(Web):April 14, 2016
DOI:10.1021/acs.jmedchem.5b01925
Lysophosphatidylserine (LysoPS) is an endogenous lipid mediator that specifically activates membrane proteins of the P2Y and its related families of G protein-coupled receptors (GPCR), GPR34 (LPS1), P2Y10 (LPS2), and GPR174 (LPS3). Here, in order to increase potency and receptor selectivity, we designed and synthesized LysoPS analogues containing the conformational constraints of the glycerol moiety. These reduced structural flexibility by fixation of the glycerol framework of LysoPS using a 2-hydroxymethyl-3-hydroxytetrahydropyran skeleton, and related structures identified compounds which exhibited high potency and selectivity for activation of GPR34 or P2Y10. Morphing of the structural shape of the 2-hydroxymethyl-3-hydroxytetrahydropyran skeleton into a planar benzene ring enhanced the P2Y10 activation potentcy rather than the GPR34 activation.
Co-reporter:Yong-Mei Cui, Xin-Lan Liu, Wen-Ming Zhang, Hai-Xia Lin, Tomohiko Ohwada, Katsutoshi Ido, Kohei Sawada
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 2) pp:283-287
Publication Date(Web):15 January 2016
DOI:10.1016/j.bmcl.2015.12.038
A series of N-acylaminoalkyloxime derivatives of dehydroabietic acid were synthesized and evaluated for BK channel-opening activities in an assay system of CHO-K1 cells expressing hBKα channels. The structure-activity relationship study revealed that a non-covalent interaction between the S atom of the 2-thiophene and the carbonyl O atom may contribute to conformation restriction for interaction with the ion channel. This research could guide the design and synthesis of novel abietane-based BK channel opener.A series of N-acylaminoalkyloxime derivatives of dehydroabietic acid were synthesized and evaluated for BK channel-opening activities in an assay system of CHO-K1 cells expressing hBKα channels.
Co-reporter:Masaya Ikubo; Asuka Inoue; Sho Nakamura; Sejin Jung; Misa Sayama; Yuko Otani; Akiharu Uwamizu; Keisuke Suzuki; Takayuki Kishi; Akira Shuto; Jun Ishiguro; Michiyo Okudaira; Kuniyuki Kano; Kumiko Makide; Junken Aoki
Journal of Medicinal Chemistry 2015 Volume 58(Issue 10) pp:4204-4219
Publication Date(Web):May 13, 2015
DOI:10.1021/jm5020082
Lysophosphatidylserine (LysoPS) is an endogenous lipid mediator generated by hydrolysis of membrane phospholipid phosphatidylserine. Recent ligand screening of orphan G-protein-coupled receptors (GPCRs) identified two LysoPS-specific human GPCRs, namely, P2Y10 (LPS2) and GPR174 (LPS3), which, together with previously reported GPR34 (LPS1), comprise a LysoPS receptor family. Herein, we examined the structure–activity relationships of a series of synthetic LysoPS analogues toward these recently deorphanized LysoPS receptors, based on the idea that LysoPS can be regarded as consisting of distinct modules (fatty acid, glycerol, and l-serine) connected by phosphodiester and ester linkages. Starting from the endogenous ligand (1-oleoyl-LysoPS, 1), we optimized the structure of each module and the ester linkage. Accordingly, we identified some structural requirements of each module for potency and for receptor subtype selectivity. Further assembly of individually structure-optimized modules yielded a series of potent and LysoPS receptor subtype-selective agonists, particularly for P2Y10 and GPR174.
Co-reporter:Hiroaki Kurouchi;Akinari Sumita;Dr. Yuko Otani ;Dr. Tomohiko Ohwada
Chemistry - A European Journal 2014 Volume 20( Issue 28) pp:8682-8690
Publication Date(Web):
DOI:10.1002/chem.201402447
Abstract
We found that phenethylcarbamates that bear ortho-salicylate as an ether group (carbamoyl salicylates) dramatically accelerate OC bond dissociation in strong acid to facilitate generation of isocyanate cation (N-protonated isocyanates), which undergo subsequent intramolecular aromatic electrophilic cyclization to give dihydroisoquinolones. To generate isocyanate cations from carbamates in acidic media as electrophiles for aromatic substitution, protonation at the ether oxygen, the least basic heteroatom, is essential to promote CO bond cleavage. However, the carbonyl oxygen of carbamates, the most basic site, is protonated exclusively in strong acids. We found that the protonation site can be shifted to an alternative basic atom by linking methyl salicylate to the ether oxygen of carbamate. The methyl ester oxygen ortho to the phenolic (ether) oxygen of salicylate is as basic as the carbamate carbonyl oxygen, and we found that monoprotonation at the methyl ester oxygen in strong acid resulted in the formation of an intramolecular cationic hydrogen bond (>CO+H⋅⋅⋅O<) with the phenolic ether oxygen. This facilitates OC bond dissociation of phenethylcarbamates, thereby promoting isocyanate cation formation. In contrast, superacid-mediated diprotonation at the methyl ester oxygen of the salicylate and the carbonyl oxygen of the carbamate afforded a rather stable dication, which did not readily undergo CO bond dissociation. This is an unprecedented and unknown case in which the monocation has greater reactivity than the dication.
Co-reporter:Siyuan Wang, Yuko Otani, Xin Liu, Masatoshi Kawahata, Kentaro Yamaguchi, and Tomohiko Ohwada
The Journal of Organic Chemistry 2014 Volume 79(Issue 11) pp:5287-5300
Publication Date(Web):May 5, 2014
DOI:10.1021/jo500916j
Because homooligomers of 7-azabicyclo[2.2.1]heptane-2-endo-carboxylic acid, a bridged β-proline analogue with a substituent installed at the remote C4-bridgehead position, completely biased the amide cis–trans equilibrium to the cis-amide structure, we expected that introduction of a substituent at the C1-bridgehead position adjacent to the carboxylic acid moiety, rather than the remote C4-bridgehead position, would tip the cis–trans amide equilibrium toward trans-amide structure without the aid of hydrogen bonding. Thus, in this work we established an efficient synthetic route to an optically active bicyclic analogue of 1,1-disubstituted β-proline, bearing a substituent at the C1-bridgehead position. Crystallographic, spectroscopic, and computational studies showed that indeed oligomers of this analogue take a consistent helical structure involving all-trans-amide linkages, independently of the number of residues, from the dimer up to the octamer. Oligomers composed of (R)-β-amino acid units form an extended left-handed helix with about 2.7 residues per turn and an approximately 4.0 Å rise per residue, characterized by complete lack of main-chain hydrogen bonding. This unique helical structure shows some similarity in shape to the trans-amide-based polyproline II (PPII) helix. The present helix was stable in various kinds of solvents such as alcohols. The present work provided a fundamental structural basis for future applications.
Co-reporter:Akinari Sumita;Hiroaki Kurouchi;Dr. Yuko Otani ;Dr. Tomohiko Ohwada
Chemistry – An Asian Journal 2014 Volume 9( Issue 10) pp:2995-3004
Publication Date(Web):
DOI:10.1002/asia.201402625
Abstract
Carbamates have been used as precursors of isocyanates, but heating in the presence of strong acids is required because cleavage of the CO bond in carbamates is energy-demanding even in acid media. Direct amidation of aromatic compounds by isocyanate cations generated at room temperature from carbamoyl salicylates in trifluoromethanesulfonic acid (TfOH) was examined. Carbamates with ortho-salicylate as an ether group (carbamoyl salicylates) showed dramatically accelerated OC bond dissociation in TfOH, which resulted in facile generation of the isocyanate cation. These chemoselective intermolecular aromatic amidation reactions proceeded even at room temperature and showed good compatibility with other electrophilic functionalities and high discrimination between N-monosubstituted carbamate and N,N-disubstituted carbamate. The reaction rates of secondary and tertiary amide formation were markedly different, and this difference was utilized to achieve successive (tandem) amidation reactions of molecules with an N-monosubstituted carbamate and an N,N-disubstituted carbamate with two kinds of aromatic compounds.
Co-reporter:Tomohiko Ohwada;Yoji Kabasawa;Norihiko Tani;Yuko Otani;Yuko Sakamaki;Masatoshi Kawahata;Kentaro Yamaguchi
PNAS 2013 Volume 110 (Issue 11 ) pp:4206-4211
Publication Date(Web):2013-03-12
DOI:10.1073/pnas.1300381110
Neighboring group participation is one of the fundamental interactions in organic reactions and can influence the reaction
rate, stereoselectivity, and reaction pathway through transient carbon-carbon or carbon-heteroatom bond formation. The latter
category includes cyclic three- and five-membered bromonium ions, wherein lone-pair electrons of the monovalent bromine atom
stabilize a trigonal carbocation. Although similar nucleophilic interactions of monovalent halogen atoms with non–carbon atom-centered
cations have long been predicted, we know of no experimental evidence of such an interaction. Here, we demonstrate a nucleophilic
interaction of neighboring monovalent halogen to stabilize an imino sp2 nitrogen cation. This interaction has an overwhelming impact on the reaction pathway, completely altering the migratory preference
under acid-catalyzed Beckmann rearrangement conditions. In sharp contrast to the general case of anti-migration, peri-chloro– and peri-bromo–substituted O-tosyl oximes of 1-tetralone substructures and their derivatives undergo syn-migration under Beckmann rearrangement conditions (i.e., migration of the group on the syn side of the leaving group). The peri-chloro or peri-bromo neighboring group turned out to provide strong anchimeric assistance for syn-migration via transient formation of a cyclic five-membered imino-halonium cation with dissociation of tosylic acid. Thus,
formation of the syn-migration products can be attributed to a reaction mechanism that is different from the conventional Beckmann rearrangement
mechanism. That is, the positively charged imino nitrogen atom can be stabilized by, or interact with, a chloro or bromo group
in close spatial proximity, and this interaction dramatically changes the reaction pathway, selectively affording regioisomeric
lactams from closely related starting materials.
Co-reporter:Kaoru Sato, Jun-ichi Kuriwaki, Kanako Takahashi, Yoshihiko Saito, Jun-ichiro Oka, Yuko Otani, Yu Sha, Ken Nakazawa, Yuko Sekino, and Tomohiko Ohwada
ACS Chemical Neuroscience 2012 Volume 3(Issue 2) pp:105
Publication Date(Web):November 14, 2011
DOI:10.1021/cn200091w
We recently found that tamoxifen suppresses l-glutamate transport activity of cultured astrocytes. Here, in an attempt to separate the l-glutamate transporter-inhibitory activity from the estrogen receptor-mediated genomic effects, we synthesized several compounds structurally related to tamoxifen. Among them, we identified two compounds, 1 (YAK01) and 3 (YAK037), which potently inhibited l-glutamate transporter activity. The inhibitory effect of 1 was found to be mediated through estrogen receptors and the mitogen-activated protein kinase (MAPK)/phosphatidylinositol 3-kinase (PI3K) pathway, though 1 showed greatly reduced transactivation activity compared with that of 17β-estradiol. On the other hand, compound 3 exerted its inhibitory effect through an estrogen receptor-independent and MAPK-independent, but PI3K-dependent pathway, and showed no transactivation activity. Compound 3 may represent a new platform for developing novel l-glutamate transporter inhibitors with higher brain transfer rates and reduced adverse effects.Keywords: astrocyte; ERα; l-glutamate transporter; nongenomic pathway; Tamoxifen; tetrasubstituted ethylene
Co-reporter:Hiroaki Kurouchi, Kyoko Kawamoto, Hiromichi Sugimoto, Satoshi Nakamura, Yuko Otani, and Tomohiko Ohwada
The Journal of Organic Chemistry 2012 Volume 77(Issue 20) pp:9313-9328
Publication Date(Web):October 1, 2012
DOI:10.1021/jo3020566
Although cations with three heteroatoms, such as monoprotonated guanidine and urea, are stabilized by Y-shaped conjugation and such Y-conjugated cations are sufficiently basic to be further protonated (or protosolvated) to dications in strongly acid media, only O-monoprotonated species have been detected in the case of carbamates even in magic acid. We found that the trifluoromethanesulfonic acid-catalyzed cyclization of arylethylcarbamates proceeds to afford dihydroisoquinolones in high yield. In strong acids, methyl carbamates are fully O-monoprotonated, and these monocations do not undergo cyclization even under heating. But, as the acidity of the reaction medium is further increased, the cyclization reaction of methyl phenethylcarbamates starts to proceed as a first-order reaction, with a linear relationship between rate and acidity. The sign and magnitude of the entropy of activation ΔS⧧ were found to be similar to those of other AAc1 reactions. These results strongly support the idea that further protonation of the O-protonated carbamates is involved in the cyclization, but the concentration of the dications is very low and suggests that the rate-determining step is dissociation of methanol from the diprotonated carbamate to generate protonated isocyanate, which reacts with the aromatic ring. Therefore, O-protonated carbamates are weak bases in sharp contrast to other Y-shaped monocations.
Co-reporter:Fumika Karaki;Yoji Kabasawa;Takahiro Yanagimoto;Dr. Nobuhiro Umeda;Firman;Dr. Yasuteru Urano;Dr. Tetsuo Nagano;Dr. Yuko Otani;Dr. Tomohiko Ohwada
Chemistry - A European Journal 2012 Volume 18( Issue 4) pp:1127-1141
Publication Date(Web):
DOI:10.1002/chem.201101427
Abstract
Although many organic/inorganic compounds that release nitric oxide (NO) upon photoirradiation (phototriggered caged-NOs) have been reported, their photoabsorption wavelengths mostly lie in the UV region, because XNO bonds (X=heteroatom and metal) generally have rather strong π-bond character. Thus, it is intrinsically difficult to generate organic compounds that release NO under visible light irradiation. Herein, the structures and properties of N-pyramidal nitrosamine derivatives of 7-azabicyclo[2.2.1]heptanes that release NO under visible light irradiation are described. Bathochromic shifts of the absorptions of these nitrosamines, attributed to HOMO (n)–LUMO (π*) transitions associated with the nonplanar structure of the NNO moiety, enable the molecules to absorb visible light, which results in NNO bond cleavage. Thus, these compounds are innate organic caged-NOs that are uncaged by visible light.
Co-reporter:Yuko Otani, Tetsuharu Hori, Masatoshi Kawahata, Kentaro Yamaguchi, Tomohiko Ohwada
Tetrahedron 2012 68(23) pp: 4418-4428
Publication Date(Web):
DOI:10.1016/j.tet.2012.01.018
Co-reporter:Tomohiko Ohwada, Satoko Ishikawa, Yusuke Mine, Keiko Inami, Takahiro Yanagimoto, Fumika Karaki, Yoji Kabasawa, Yuko Otani, Masataka Mochizuki
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 8) pp:2726-2741
Publication Date(Web):15 April 2011
DOI:10.1016/j.bmc.2011.02.049
Nitrosamines are potent carcinogens and toxicants in the rat and potential genotoxins in humans. They are metabolically activated by hydroxylation at an α-carbon atom with respect to the nitrosoamino group, catalyzed by cytochrome P450. However, there has been little systematic investigation of the structure–mutagenic activity relationship of N-nitrosamines. Herein, we evaluated the mutagenicity of a series of 7-azabicyclo[2.2.1]heptane N-nitrosamines and related monocyclic nitrosamines by using the Ames assay. Our results show that the N-nitrosamine functionality embedded in the bicyclic 7-azabicylo[2.2.1]heptane structure lacks mutagenicity, that is, it is inert to α-hydroxylation, which is the trigger of mutagenic events. Further, the calculated α-C–H bond dissociation energies of the bicyclic nitrosamines are larger in magnitude than those of the corresponding monocyclic nitrosamines and N-nitrosodimethylamine by as much as 20–30 kcal/mol. These results are consistent with lower α-C–H bond reactivity of the bicyclic nitrosamines. Thus, the 7-azabicyclo[2.2.1]heptane structural motif may be useful for the design of nongenotoxic nitrosamine compounds with potential biological/medicinal applications.
Co-reporter:Masahiro Hosoya ; Yuko Otani ; Masatoshi Kawahata ; Kentaro Yamaguchi
Journal of the American Chemical Society 2010 Volume 132(Issue 42) pp:14780-14789
Publication Date(Web):September 30, 2010
DOI:10.1021/ja1017877
Helical structures of oligomers of non-natural β-amino acids are significantly stabilized by intramolecular hydrogen bonding between main-chain amide moieties in many cases, but the structures are generally susceptible to the environment; that is, helices may unfold in protic solvents such as water. For the generation of non-hydrogen-bonded ordered structures of amides (tertiary amides in most cases), control of cis−trans isomerization is crucial, even though there is only a small sterical difference with respect to cis and trans orientations. We have established methods for synthesis of conformationally constrained β-proline mimics, that is, bridgehead-substituted 7-azabicyclo[2.2.1]heptane-2-endo-carboxylic acids. Our crystallographic, 1D- and 2D-NMR, and CD spectroscopic studies in solution revealed that a bridgehead methoxymethyl substituent completely biased the cis−trans equilibrium to the cis-amide structure along the main chain, and helical structures based on the cis-amide linkage were generated independently of the number of residues, from the minimalist dimer through the tetramer, hexamer, and up to the octamer, and irrespective of the solvent (e.g., water, alcohol, halogenated solvents, and cyclohexane). Generality of the control of the amide equilibrium by bridgehead substitution was also examined.
Co-reporter:Yong-Mei Cui, Eriko Yasutomi, Yuko Otani, Katsutoshi Ido, Takashi Yoshinaga, Kohei Sawada, Tomohiko Ohwada
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 24) pp:8642-8659
Publication Date(Web):15 December 2010
DOI:10.1016/j.bmc.2010.09.072
Oxime ether derivatives at the benzylic position of unsubstituted, dichloro, trichloro, and monobromo derivatives of the aromatic C-ring of dehydroabietic acid and podocarpic acid were synthesized and evaluated as BK channel openers in an assay system of CHO-K1 cells expressing hBKα channels. Detailed SAR analysis showed that the oximation was particularly effective in the cases of dehydroabietic acid derivatives, and some of these oxime derivatives showed more potent BK channel activities than the standard compound, NS1619. The present studies provide a new structural basis for development of efficient BK channel openers.Oxime ether derivatives at the benzylic position of unsubstituted, dichloro, trichloro, and monobromo derivatives of the aromatic C-ring of dehydroabietic acid and podocarpic acid were synthesized and evaluated as BK channel openers in an assay system of CHO-K1 cells expressing hBKα channels.
Co-reporter:Hiroaki Kurouchi ; Hiromichi Sugimoto ; Yuko Otani
Journal of the American Chemical Society 2009 Volume 132(Issue 2) pp:807-815
Publication Date(Web):December 17, 2009
DOI:10.1021/ja908749u
The chemical features, such as substrate stability, product distribution, and substrate generality, and the reaction mechanism of Brønsted superacid-catalyzed cyclization reactions of aromatic ring-containing acetoacetates (β-ketoesters) were examined in detail. While two types of carbonyl cyclization are possible, i.e., keto cyclization and ester cyclization, the former was found to take place exclusively. The reaction constitutes an efficient method to synthesize indene and 3,4-dihydronapthalene derivatives. Acid−base titration monitored with 13C NMR spectroscopy showed that the acetoacetates are fully O1,O3-diprotonated at H0 = −11. While the five-membered ring cyclization of the arylacetoacetates proceeded slowly at H0 = −11, a linear increase in the rate of the cyclization was found with increasing acidity in the high acidity region of H0 = −11.8 to −13.3. Therefore, the O1,O3-diprotonated acetoacetates exhibited some cyclizing reactivity, but they are not the reactive intermediates responsible for the acceleration of the cyclization in the high acidity region. The reactive cationic species might be formed by further protonation (or protosolvation) of the O1,O3-diprotonated acetoacetates; i.e., they may be tricationic species. Thermochemical data on the acid-catalyzed cyclization of the arylacetoacetates showed that the activation energy is decreased significantly as compared with that of the related acid-catalyzed cyclization reaction of a compound bearing a single functional group, such as a ketone. These findings indicate that intervention of the trication contributes to the activation of the cyclization of arylacetoacetates in strong acid, and the electron-withdrawing nature of the O-protonated ester functionality significantly increases the electrophilicity of the ketone moiety.
Co-reporter:Masazumi Iwashita ▽; Kumiko Makide ▽; Taro Nonomura ; Yoshimasa Misumi ; Yuko Otani ; Mayuko Ishida ; Ryo Taguchi ; Masafumi Tsujimoto ; Junken Aoki ; Hiroyuki Arai
Journal of Medicinal Chemistry 2009 Volume 52(Issue 19) pp:5837-5863
Publication Date(Web):September 10, 2009
DOI:10.1021/jm900598m
In response to various exogenous stimuli, mast cells (MCs) release a wide variety of inflammatory mediators stored in their cytoplasmic granules and this release initiates subsequent allergic reactions. Lysophosphatidylserine (lysoPS) has been known as an exogenous inducer to potentiate histamine release from MCs, though even at submicromolar concentrations. In this study, through SAR studies on lysoPS against MC degranulation, we identified lysoPT, a threonine-containing lysophospholipid and its 2-deoxy derivative as novel strong agonists. LysoPT and its 2-deoxy derivative induced histamine release from MCs both in vitro and in vivo at a concentration less than one-tenth that of lysoPS. Notably, lysoPT did not activate a recently proposed lysoPS receptor on MCs, GPR34, demonstrating the presence of another undefined receptor reactive to both lysoPS and lysoPT that is involved in MC degranulation. Thus, the present strong agonists, lysoPT and its 2-deoxy derivative, will be useful tools to understand the mechanisms of lysoPS-induced activation of degranulation of MCs.
Co-reporter:Toshiyuki Tsuji, Maki Onoda, Yuko Otani, Tomohiko Ohwada, Takahito Nakajima, Kimihiko Hirao
Chemical Physics Letters 2009 Volume 473(1–3) pp:196-200
Publication Date(Web):29 April 2009
DOI:10.1016/j.cplett.2009.03.066
We compare the absorption/emission energies of three heteroaromatic chromophores, indoles, benzofurazans and coumarins, both experimentally and theoretically. First we used TDDFT and SAC-CI methods to calculate S0 → S1 vertical excitation energies corresponding to the absorptions. The excitation energies obtained by the SAC-CI method were superior to those obtained by the TDDFT method, as well as being consistent with those experimentally obtained. We thus applied the SAC-CI method to estimate the emission energies of these chromophores on the basis of the CIS- or TDDFT-optimized excited-state structures. We found a good and consistent agreement of the CIS-structure-based values with the experimental values, whereas the TDDFT-structure based values showed less good agreement.The SAC-CI method (S) with the CIS-optimized structure (C) is generally superior to other approaches to calculate emission energies of heteroaromatic chromophores.
Co-reporter:Yong-Mei Cui, Eriko Yasutomi, Yuko Otani, Takashi Yoshinaga, Katsutoshi Ido, Kohei Sawada, Masatoshi Kawahata, Kentaro Yamaguchi, Tomohiko Ohwada
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 24) pp:6386-6389
Publication Date(Web):15 December 2008
DOI:10.1016/j.bmcl.2008.10.078
Oxime ether derivatives of the benzylic ketone of 12,14-dichlorodehydroabietic acid (diCl-DHAA, 4b) were synthesised, and their BK channel-opening activity was evaluated in an assay system of CHO-K1 cells expressing hBKα channels. Oxime ether structure on the B ring of diCl-DHAA significantly increased the BK channel-opening activity.The oxime ether structure, particularly when bearing O-short carbon chains, significantly increased the BK channel-opening activity of 12,14-dichlorodehydroabietic acid (4b).
Co-reporter:Yong-Mei Cui, Eriko Yasutomi, Yuko Otani, Takashi Yoshinaga, Katsutoshi Ido, Kohei Sawada, Tomohiko Ohwada
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 19) pp:5197-5200
Publication Date(Web):1 October 2008
DOI:10.1016/j.bmcl.2008.08.081
We found that the podocarpic acid structure provides a new scaffold for chemical modulators of large-conductance calcium-activated K+ channels (BK channels). Structure–activity analysis indicates the importance of both the arrangement (i.e., location and orientation) of the carboxylic acid functionality of ring A and the hydrophobic region of ring C for expression of BK channel-opening activity.We found that the podocarpic acid structure provides a new scaffold for chemical modulators of large-conductance calcium-activated K+ channels (BK channels).
Co-reporter:Yong-Mei Cui, Eriko Yasutomi, Yuko Otani, Takashi Yoshinaga, Katsutoshi Ido, Kohei Sawada, Tomohiko Ohwada
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 19) pp:5201-5205
Publication Date(Web):1 October 2008
DOI:10.1016/j.bmcl.2008.08.078
A series of dehydroabietic acid (DHAA, 2) derivatives was synthesized and evaluated as BK channel openers in an assay system of CHO-K1 cells expressing hBKα channels. Systematic modifications of the peripheral functionality of ring C of DHAA showed that the introduction of a nitro or (thio)urea group in ring C greatly enhanced the BK channel-opening activity.A series of dehydroabietic acid derivatives was synthesized and evaluated as BK channel openers in an assay system of CHO-K1 cells expressing hBKα channels.
Co-reporter:Hiromichi Sugimoto;Satoshi Nakamura
Advanced Synthesis & Catalysis 2007 Volume 349(Issue 4-5) pp:
Publication Date(Web):20 MAR 2007
DOI:10.1002/adsc.200600508
o-Quinone methides (o-QMs) are highly reactive, short-lived intermediates, which have potential synthetic applicability. However, few studies on the generation of o-QMs bearing an electron-withdrawing group have been reported. Herein we present a general method for the generation of o-QMs, particularly those substituted with an electrophilic substituent, from new precursors, 4H-1,2-benzoxazines 2. We have also studied systematically the Diels–Alder reactions of o-QMs with various dienophiles, such as vinyl ethers, enamines and imines. The reactions provide a versatile route to substituted chromans, phenols and 3,4-dihydro-2H-benzo[e][1,3]oxazines (3,4-dihydro-1,3-benzoxazines). Furthermore, we applied the new method to the derivatization of some natural products.
Co-reporter:Chie Gota, Seiichi Uchiyama and Tomohiko Ohwada
Analyst 2007 vol. 132(Issue 2) pp:121-126
Publication Date(Web):18 Dec 2006
DOI:10.1039/B615168J
Fluorescent polymeric thermometers consisting of only N-alkylacrylamide and fluorescent components show rather low temperature resolution in their functional ranges (ca. 15–50 °C) because of the occurrence of intermolecular aggregation, which causes hysteresis in their fluorescence response to changes in temperature. By adding an ionic component to prevent such intermolecular aggregation, we obtained four fluorescent polymeric thermometers that offer high temperature resolution (<0.2 °C). Each new fluorescent polymeric thermometer covered the temperature range, 9–33 °C, 30–51 °C, 49–66 °C or 4–38 °C.
Co-reporter:Toshihiko Tashima, Yoshimi Toriumi, Yumi Mochizuki, Taro Nonomura, Satoru Nagaoka, Katsuo Furukawa, Hiromichi Tsuru, Satomi Adachi-Akahane, Tomohiko Ohwada
Bioorganic & Medicinal Chemistry 2007 Volume 15(Issue 4) pp:1878
Publication Date(Web):15 February 2007
DOI:10.1016/j.bmc.2006.12.025
Co-reporter:Toshihiko Tashima, Yoshimi Toriumi, Yumi Mochizuki, Taro Nonomura, Satoru Nagaoka, Katsuo Furukawa, Hiromichi Tsuru, Satomi Adachi-Akahane, Tomohiko Ohwada
Bioorganic & Medicinal Chemistry 2006 Volume 14(Issue 23) pp:8014-8031
Publication Date(Web):1 December 2006
DOI:10.1016/j.bmc.2006.07.042
In order to explore new scaffolds for large-conductance Ca2+-activated K+ channel (BK channel) openers, we carried out molecular design and synthesis on the basis of the following two concepts: (1) introduction of a heteroatom into the dehydroabietic acid (BK channel opener) skeleton would allow easier introduction of substituents. (2) Because of the fourfold symmetrical structure of BK channels, dimeric compounds in which two pharmacophores are linked through a tether are expected to have a greater binding probability to the channels, resulting in increased channel-opening activity. Herein, we explore the usefulness of the hexahydrodibenzazepinone structure as a new scaffold for BK channel openers. The synthesized monomer compounds of hexahydrodibenzazepinone derivatives, which can be derived from dehydroabietic acid, were subjected to electrophysiological patch–clamp studies, followed by Magnus contraction–relaxation assay using rabbit urinary bladder smooth muscle strips to assess overall activities. Dimeric compounds were designed by linking the monomeric hexahydrodibenzazepinone derivatives through a diacetylenebenzene tether, and their channel-opening activities were evaluated by electrophysiological methods. Finally, we concluded that the critical structure for BK channel-opening activity is the hexahydrodibenzazepinone monomer substituted with a phenyl-bearing alkynyl substituent on the lactam amide.
Co-reporter:Tomohiko Ohwada Dr.;Daisuke Kojima;Tatsuto Kiwada;Shiroh Futaki Dr.;Yukio Sugiura Dr.;Kentaro Yamaguchi Dr.;Yoshinori Nishi;Yuji Kobayashi Dr.
Chemistry - A European Journal 2004 Volume 10(Issue 3) pp:
Publication Date(Web):2 FEB 2004
DOI:10.1002/chem.200305492
A method was developed for synthesizing α,α-disubstituted glycine residues bearing a large (more than 15-membered) hydrophobic ring. The ring-closing metathesis reactions of the dialkenylated malonate precursors proceed efficiently, particularly when long methylene chains tether both terminal olefin groups. Surprisingly, the amino groups of these α,α-disubstituted glycines are inert to conventional protective reactions (e.g., N-tert-butoxycarbonyl (Boc) protection: Boc2O/4-dimethylaminopyridine (DMAP)/CH2Cl2; N-benzyloxycarbonyl (Z) protection: Z-Cl/DMAP/CH2Cl2). Curtius rearrangement of the carboxylic acid functionality of the malonate derivative after ring-closing metathesis leads to formation of an amine functionality and can be catalyzed by diphenylphosphoryl azide. However, only the intermediate isocyanates can be isolated, even in the presence of alcohols such as benzyl alcohol. The isocyanates obtained by Curtius rearrangement in an aprotic solvent (benzene) were isolated in high yields and treated with 9-fluorenylmethanol in a high-boiling-point solvent (toluene) under reflux to give the N-9-fluorenylmethoxycarbonyl (Fmoc)-protected aminomalonate derivatives in high yield. These hydrophobic amino acids can be incorporated into a peptide by Fmoc solid-phase peptide synthesis and the acid fluoride activation method. The stability of the monomeric α-helical structure of a 17-amino-acid peptide was enhanced by replacement of two alanine residues with two hydrophobic amino acid residues bearing a cyclic 18-membered ring. The results of sedimentation equilibrium studies suggested that the peptide assembles into hexamers in the presence of 100 mM NaCl.
Co-reporter:Tomohiko Ohwada, Taro Nonomura, Keisuke Maki, Kazuho Sakamoto, Susumu Ohya, Katsuhiko Muraki, Yuji Imaizumi
Bioorganic & Medicinal Chemistry Letters 2003 Volume 13(Issue 22) pp:3971-3974
Publication Date(Web):17 November 2003
DOI:10.1016/j.bmcl.2003.08.072
We found that the dehydroabietic acid structure is a new scaffold for chemical modulators of large-conductance calcium-activated K+ channels (BK channels). Structure–activity relationship (SAR) studies of the dehydroabietic acid derivatives and related non-aromatic compounds such as pimaric acid revealed the importance of the carboxyl functionality and an appropriate hydrophobic moiety of the molecules for BK channel-opening ability.Dehydroabietic acid structure is a new scaffold for chemical activators of large-conductance calcium-activated K+ channels (BK channels).
Co-reporter:Tomonobu Yoshimura, Seiji Hasegawa, Naohide Hirashima, Mamoru Nakanishi, Tomohiko Ohwada
Bioorganic & Medicinal Chemistry Letters 2001 Volume 11(Issue 22) pp:2897-2901
Publication Date(Web):19 November 2001
DOI:10.1016/S0960-894X(01)00563-7
Novel cationic amphiphiles, based on lithocholic acid derivatives with two structural motifs, anchoring lipids and bola lipids, were designed and synthesized. Both bear extended hydrophobic space-filling substituents. A significant effect of the orientation and extension of hydrophobic regions around the ether linkage at the 3-position was found on the efficiency of DNA delivery.Graphic
Co-reporter:Siyuan Wang, Tohru Taniguchi, Kenji Monde, Masatoshi Kawahata, Kentaro Yamaguchi, Yuko Otani and Tomohiko Ohwada
Chemical Communications 2016 - vol. 52(Issue 21) pp:NaN4021-4021
Publication Date(Web):2016/02/18
DOI:10.1039/C6CC00284F
Nitrogen-pyramidalization of amide increases electron density on nitrogen and decreases that on carbonyl oxygen. We identified hydrogen-bonding to carbonyl of nitrogen-pyramidalized bicyclic β-proline derivatives by crystallography, and by NMR and vibrational circular dichroism (VCD) spectroscopy in solution. Such hydrogen-bonding can switch the preferred nitrogen-pyramidalization direction, as detected by VCD spectroscopy.
Co-reporter:Yingtang Ning, Tomoya Fukuda, Hirotaka Ikeda, Yuko Otani, Masatoshi Kawahata, Kentaro Yamaguchi and Tomohiko Ohwada
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 6) pp:NaN1392-1392
Publication Date(Web):2017/01/10
DOI:10.1039/C6OB02719A
Neighboring group participation is defined as the action of a substituent to stabilize a transition state or an intermediate by forming a bond or a partial bond with the reaction center. In addition to the primary interaction with the nearest neighboring group, secondary interactions involving another neighboring group(s) could also occur in principle. Here, we revisit this issue by examining the influence of secondary interactions on the stability and reactivity of the putative iminylium cation intermediates, formed by N–O bond cleavage of 1-tetralone oxime systems. A direct observation of a peri-bromo-iminylium intermediate in solution supported the involvement of iminylium cations and the stabilizing effect of secondary interactions arising from a distal tandem substituent. Both experimental and computational findings support the idea that secondary interactions of a tandem-neighboring group on the primary peri-heteroatom (Br, Cl, and O(Me))-iminylium bonding interaction, i.e., a weak halogen bonding interaction (ester (nitro) oxygen–halogen bonding) and an unprecedented hydrogen bonding interaction between a nitro oxygen atom and a CH3O hydrogen atom, are crucial determinants of the reaction pathway, leading to either overwhelmingly selective syn-migration of the oxime functionality or covalent bond formation under acid-catalyzed Beckmann rearrangement conditions.
Co-reporter:Akinari Sumita, Yuko Otani and Tomohiko Ohwada
Chemical Communications 2017 - vol. 53(Issue 9) pp:NaN1485-1485
Publication Date(Web):2017/01/13
DOI:10.1039/C6CC09618B
We describe the chemoselective conversion of carboxylic acids to functional aromatic ketones promoted by a tailored organophosphate ester in the presence of a Brønsted acid. The protonated phosphate ester reacts with the carboxylic acid to form acyl phosphate, which reacts with benzenes to give aromatic ketones, probably through the acylium ion or its equivalent. The reaction time is short even at room temperature, and the reaction is compatible with various other functional groups, including amines, olefins, esters, amides and nitriles.

![7-(tert-butyl) 1-methyl (1R,2R,4S)-2-(((R)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoyl)oxy)-7-azabicyclo[2.2.1]heptane-1,7-dicarboxylate](http://img.cochemist.com/ccimg/449200/449171-39-5.png)
![7-(tert-butyl) 1-methyl (1R,2R,4S)-2-(((R)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoyl)oxy)-7-azabicyclo[2.2.1]heptane-1,7-dicarboxylate](http://img.cochemist.com/ccimg/449200/449171-39-5_b.png)

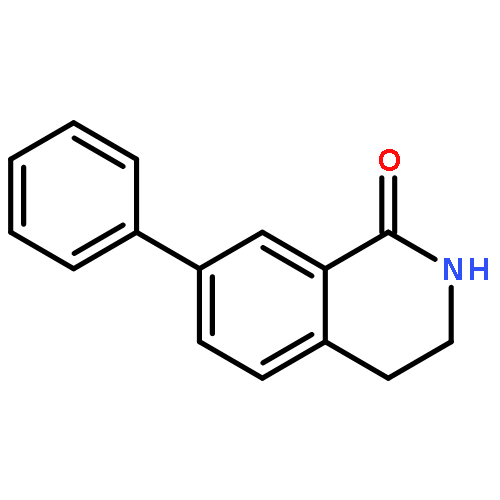
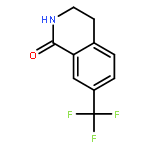
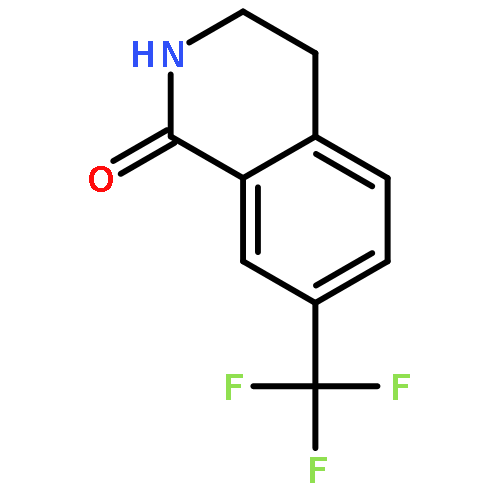
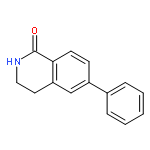
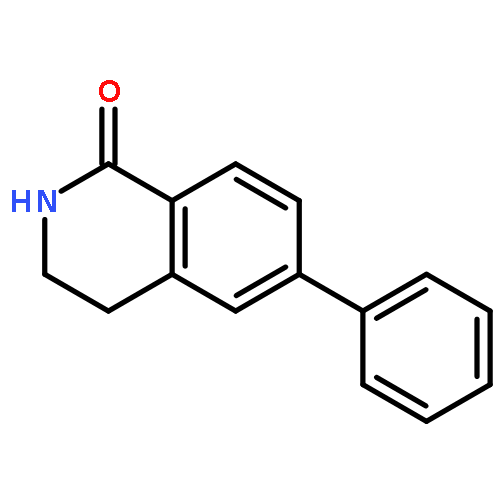
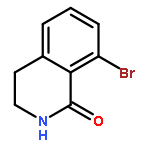
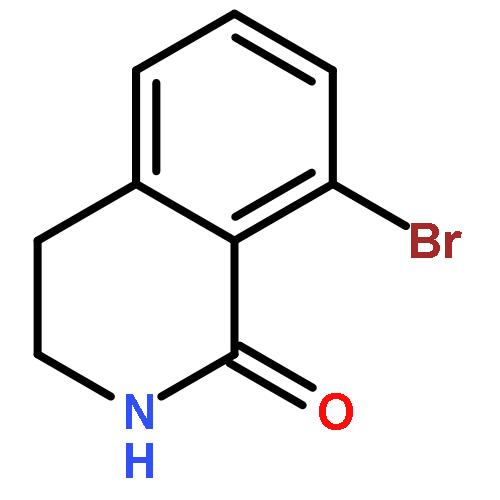

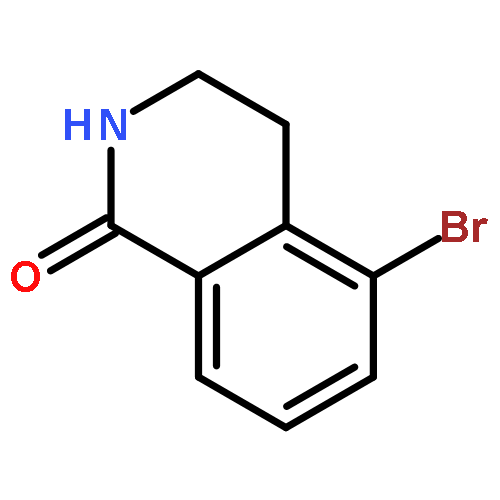
![BENZENEPROPANOIC ACID, 4-[(3-PHENOXYPHENYL)METHOXY]-](http://img.cochemist.com/ccimg/691900/691898-06-3.png)
![BENZENEPROPANOIC ACID, 4-[(3-PHENOXYPHENYL)METHOXY]-](http://img.cochemist.com/ccimg/691900/691898-06-3_b.png)


![Carbamic acid, [2-(4-bromophenyl)ethyl]-, methyl ester](http://img.cochemist.com/ccimg/305500/305447-65-8.png)
![Carbamic acid, [2-(4-bromophenyl)ethyl]-, methyl ester](http://img.cochemist.com/ccimg/305500/305447-65-8_b.png)




![[(1S)-2-[[(tert-Butyl)dimethylsilyl]oxy]-1-(hydroxymethyl)ethyl]-carbamic Acid tert-Butyl Ester](http://img.cochemist.com/ccimg/185700/185692-85-7.png)
![[(1S)-2-[[(tert-Butyl)dimethylsilyl]oxy]-1-(hydroxymethyl)ethyl]-carbamic Acid tert-Butyl Ester](http://img.cochemist.com/ccimg/185700/185692-85-7_b.png)


![1,5-Anhydro-2,3-dideoxy-6-O-[(1,1-dimethylethyl)dimethylsilyl]-D-erythro-Hex-2-enitol](http://img.cochemist.com/ccimg/132400/132375-36-1.png)
![1,5-Anhydro-2,3-dideoxy-6-O-[(1,1-dimethylethyl)dimethylsilyl]-D-erythro-Hex-2-enitol](http://img.cochemist.com/ccimg/132400/132375-36-1_b.png)




![Phenol, 4-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/126100/126070-20-0.png)
![Phenol, 4-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/126100/126070-20-0_b.png)


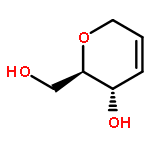
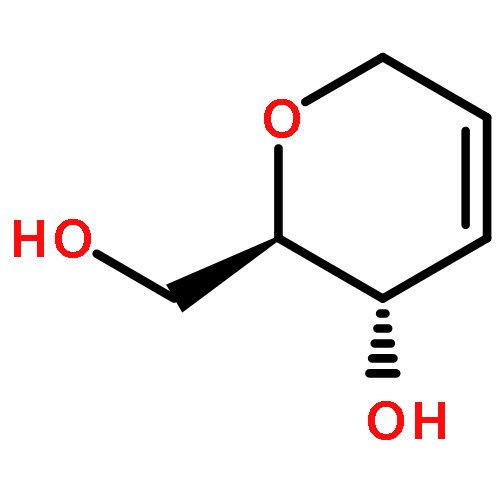
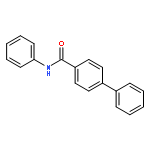
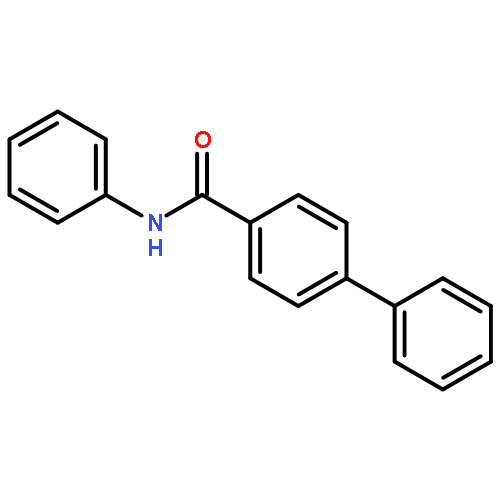
![D-Serine, N-[(1,1-dimethylethoxy)carbonyl]-O-[(1,1-dimethylethyl)dimethylsilyl]-, methyl ester](http://img.cochemist.com/ccimg/112500/112418-19-6.png)
![D-Serine, N-[(1,1-dimethylethoxy)carbonyl]-O-[(1,1-dimethylethyl)dimethylsilyl]-, methyl ester](http://img.cochemist.com/ccimg/112500/112418-19-6_b.png)

![Phenol, 3-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/96100/96013-78-4.png)
![Phenol, 3-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/96100/96013-78-4_b.png)









![[3-(4-CHLOROPHENOXY)PHENYL]METHANOL](http://img.cochemist.com/ccimg/72800/72714-63-7.png)
![[3-(4-CHLOROPHENOXY)PHENYL]METHANOL](http://img.cochemist.com/ccimg/72800/72714-63-7_b.png)



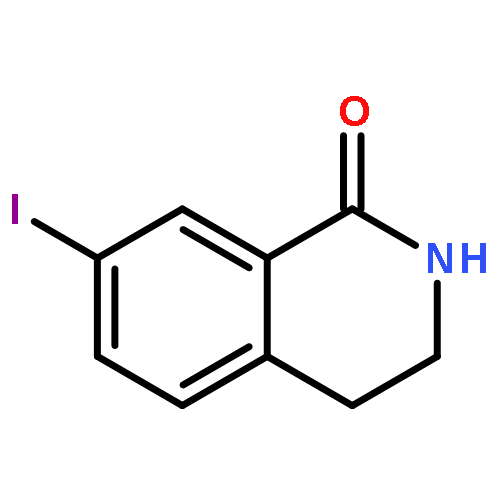




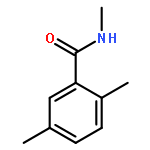
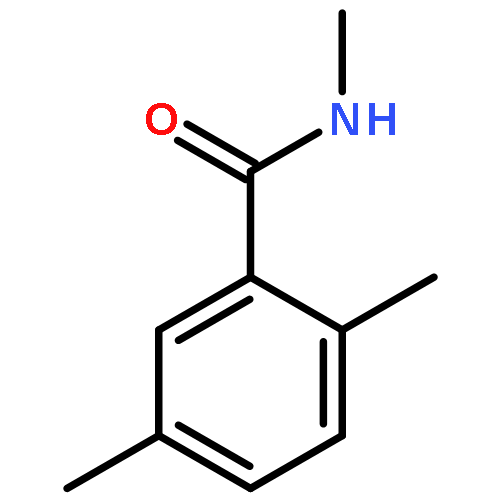










![Benzoic acid, 4-[[(dimethylamino)carbonyl]oxy]-, methyl ester](/data/chemimg/3742300/38491-27-9.png)
![Benzoic acid, 4-[[(dimethylamino)carbonyl]oxy]-, methyl ester](/data/chemimg/3742300/38491-27-9_b.png)


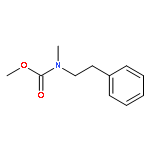
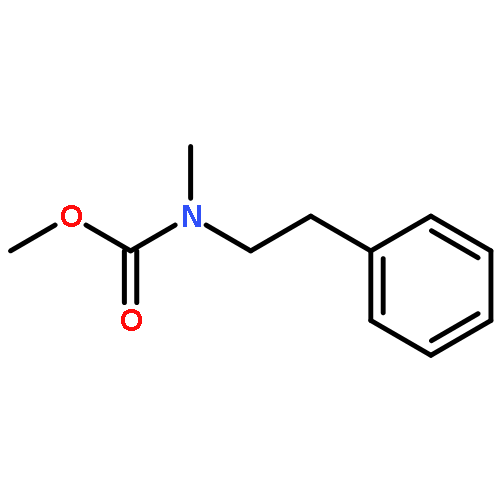
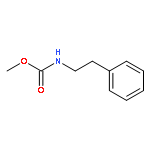
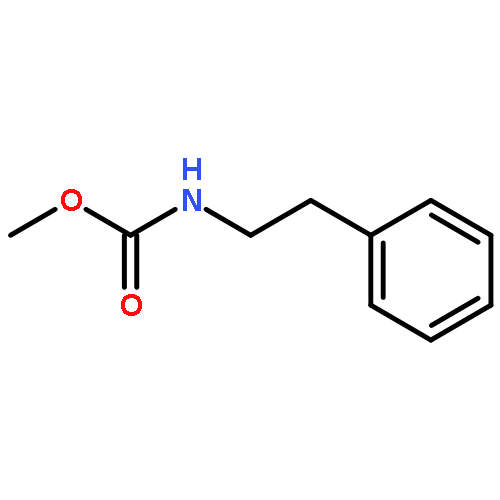
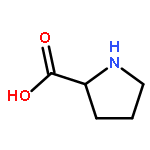







![1-Propanol, 3-[(9Z)-9-octadecenyloxy]-](http://img.cochemist.com/ccimg/17400/17367-37-2.png)
![1-Propanol, 3-[(9Z)-9-octadecenyloxy]-](http://img.cochemist.com/ccimg/17400/17367-37-2_b.png)
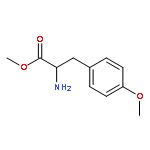
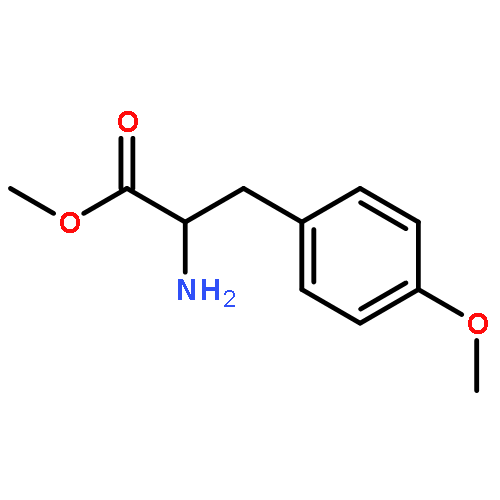
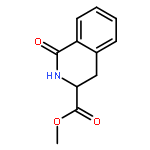
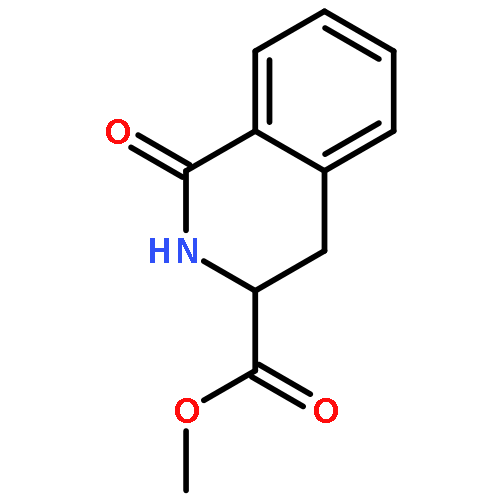
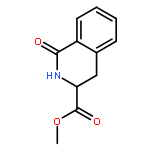
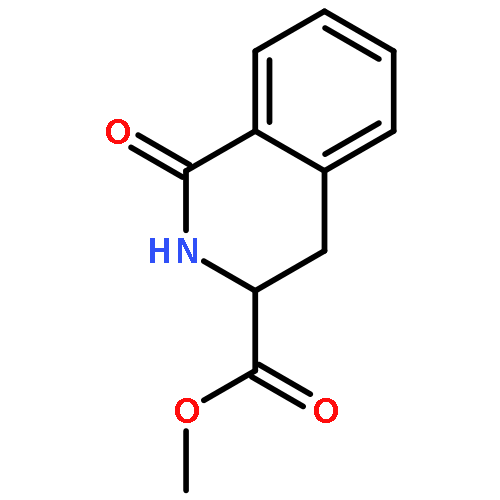
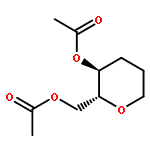
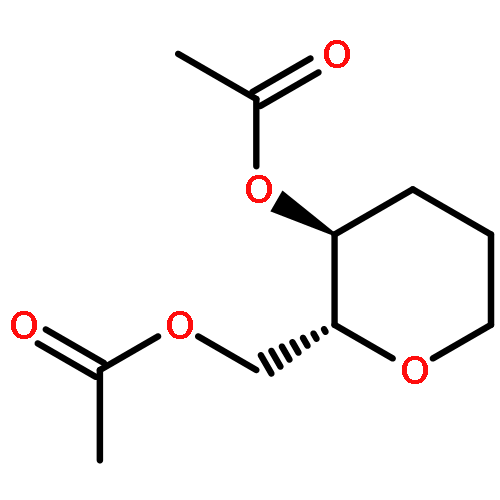
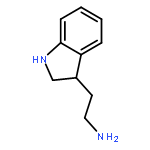

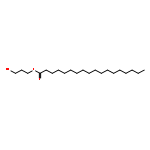


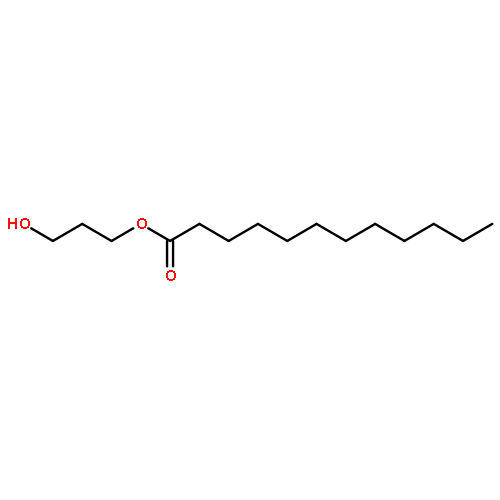
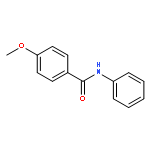
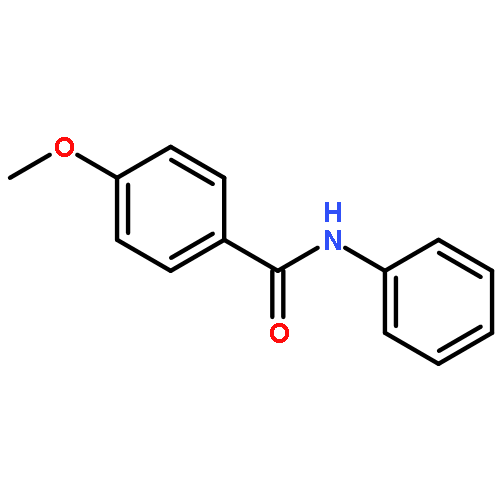
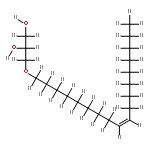
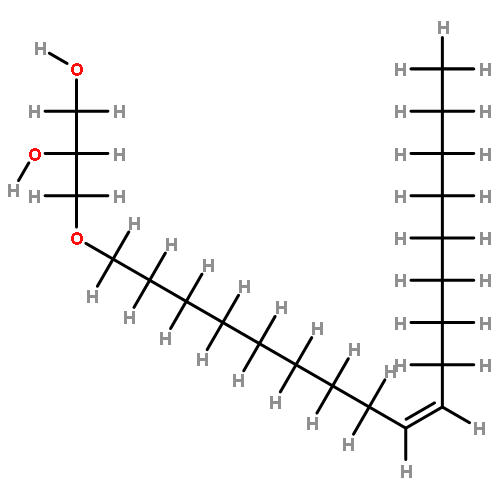








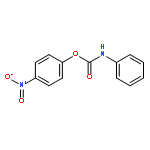







![Carbamic acid, [2-(4-methoxyphenyl)ethyl]-, methyl ester](http://img.cochemist.com/ccimg/91300/91247-71-1.png)
![Carbamic acid, [2-(4-methoxyphenyl)ethyl]-, methyl ester](http://img.cochemist.com/ccimg/91300/91247-71-1_b.png)

![Benzoic acid, 2,2'-[carbonylbis(oxy)]bis-, dimethyl ester](http://img.cochemist.com/ccimg/82100/82091-12-1.png)
![Benzoic acid, 2,2'-[carbonylbis(oxy)]bis-, dimethyl ester](http://img.cochemist.com/ccimg/82100/82091-12-1_b.png)