Co-reporter:Donglong Fu;Dr. Joel E. Schmidt;Dr. Zoran Ristanović;Dr. Abhishek Dutta Chowdhury;Dr. Florian Meirer; Dr. Bert M. Weckhuysen
Angewandte Chemie International Edition 2017 Volume 56(Issue 37) pp:11273-11273
Publication Date(Web):2017/09/04
DOI:10.1002/anie.201708258
Catalytically active aluminum can be incorporated into perfectly oriented zeolite films using economically viable alcohols. In their Communication on page 11217 ff., B. M. Weckhuysen and co-workers demonstrate this process, which is illustrated by the windmills that pump Al into the canals resulting in homogeneous incorporation, as confirmed by polarized confocal fluorescence microscopy and reactive fluorescent probe molecules, symbolized by the sun causing the “fluorescence of the canals”.
Co-reporter:R. Oord;I. C. ten Have;J. M. Arends;F. C. Hendriks;J. Schmidt;I. Lezcano-Gonzalez;B. M. Weckhuysen
Catalysis Science & Technology (2011-Present) 2017 vol. 7(Issue 17) pp:3851-3862
Publication Date(Web):2017/08/29
DOI:10.1039/C7CY00798A
Mesoporous Cu-SSZ-13 was created by first synthesizing zeolite H-SSZ-13 and subsequently desilicating the material by base leaching using NaOH in different concentrations. The catalyst materials were prepared by ion exchanging the leached samples back to their acidic form using NH4NO3, and to their active Cu form by ion exchanging them with CuSO4. For comparison, H- and Cu-SSZ-13 were steamed using a wide variety of different conditions. Using a 0.10 M NaOH solution for base leaching, it was found that Cu-SSZ-13 becomes more active in the selective catalytic reduction of NOx with NH3 (NH3-SCR) over the entire temperature region but especially in the low temperature region (<200 °C). This increase could be explained by a decrease in pore diffusion limitations due to the introduction of mesopores on the outside of the zeolite crystals but keeping the chemical environment of the catalyst nearly the same as that of the parent material. Higher base leaching concentrations do, however, lead to a decrease in the amount of Brønsted acid sites, pore volume and accessible surface area, accompanied by a decrease in NH3-SCR activity. Ar physisorption coupled with SEM and confocal fluorescence microscopy in combination with two differently sized fluorescent organic probe molecules (i.e., 4-(4-dimethyl-aminostyryl)-1-methyl-pyridinium-iodide and 4-(4-dicyclohexyl-aminostyryl)-1-methyl-pyridinium-iodide) show an increase in the external surface area due to the creation of mesopores. The development of mesoporosity starts from the crystal surface and continues into the crystal with increasing alkaline solution strength, but under our conditions it never reaches the center. On the other hand, zeolite steaming did not successfully introduce mesoporosity and mainly managed to deactivate the Cu-SSZ-13 zeolite catalysts.
Co-reporter:Donglong Fu;Katie Park;Guusje Delen;Özgün Attila;Florian Meirer;Derek Nowak;Sung Park;Joel E. Schmidt
Chemical Communications 2017 vol. 53(Issue 97) pp:13012-13014
Publication Date(Web):2017/12/05
DOI:10.1039/C7CC06832H
Characterizing the structures of zeolites and their catalytic performance with high-spatial-resolution is vital to developing new solid catalysts. We demonstrate the application of photoinduced force microscopy (PiFM), with nanometer scale resolution across the infrared spectral range, for the study of zeolite ZSM-5 thin-films with various Si/Al ratios after the methanol-to-hydrocarbons reaction. This first-of-its kind nanometer scale infrared imaging of zeolite materials demonstrates the possibility of PiFM for the study of functional porous materials.
Co-reporter:F. C. Hendriks;D. Valencia;P. C. A. Bruijnincx;B. M. Weckhuysen
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 3) pp:1857-1867
Publication Date(Web):2017/01/18
DOI:10.1039/C6CP07572J
A series of fluorescent probe molecules based on the commercially available trans-4-(4-(diethylamino)styryl)-N-methylpyridinium iodide (DAMPI) scaffold has been developed. The dynamic radii of these DAMPI-type probes covered a range of 5.8 to 10.1 Å and could be changed by varying the alkyl substituents on the amine donor group, with limited effect on the electronic properties. These probe molecules allow for the direct evaluation of the molecular accessibility into confined spaces, more specifically the micropore architecture of zeolite materials. Evaluation of industrially relevant zeolite materials with 8- (CHA), 10- (MFI) and 12-membered ring pores (FAU) showed that steric bulk influences the rate of adsorption, the amount of probe molecule taken up by the zeolite as well as the interaction of the probe molecule with the zeolite material. Furthermore, a positive linear correlation is found between the pore–probe size difference and total probe uptake by the zeolite. The absorption spectra of each probe molecule within the zeolites show that this DAMPI-type compound is chemically bound to the zeolite's acid sites. The new approach shows the general principle of determining size-accessibility relationships in microporous solids with a series of fluorescent probes of systematically tunable size.
Co-reporter:Donglong Fu;Dr. Joel E. Schmidt;Dr. Zoran Ristanović;Dr. Abhishek Dutta Chowdhury;Dr. Florian Meirer; Dr. Bert M. Weckhuysen
Angewandte Chemie 2017 Volume 129(Issue 37) pp:11427-11427
Publication Date(Web):2017/09/04
DOI:10.1002/ange.201708258
Katalytisch aktives Aluminium kann unter Einsatz kostengünstiger Alkohole in perfekt orientierte Zeolithfilme eingebaut werden. In ihrer Zuschrift auf S. 11369 demonstrieren B. M. Weckhuysen und Mitarbeiter diesen Prozess, der durch die Windmühlen veranschaulicht wird, die Al in die Kanäle pumpen. Der homogene Al-Einbau wird durch polarisierte Fluoreszenzmikroskopie mit reaktiven fluoreszierenden Sondenmolekülen bestätigt, symbolisiert durch die Sonne, die die “Fluoreszenz des Kanals” verursacht.
Co-reporter:Donglong Fu;Dr. Joel E. Schmidt;Dr. Zoran Ristanović;Dr. Abhishek Dutta Chowdhury;Dr. Florian Meirer; Dr. Bert M. Weckhuysen
Angewandte Chemie 2017 Volume 129(Issue 37) pp:11369-11373
Publication Date(Web):2017/09/04
DOI:10.1002/ange.201704846
AbstractHighly b-oriented zeolite ZSM-5 films are critical for applications in catalysis and separations and may serve as models to study diffusion and catalytic properties in single zeolite channels. However, the introduction of catalytically active Al3+ usually disrupts the orientation of zeolite films. Herein, using structure-directing agents with hydroxy groups, we demonstrate a new method to prepare highly b-oriented zeolite ZSM-5 films with a broad range of Si/Al ratios (Si/Al=45 to ∞). Fluorescence micro-(spectro)scopy was used to monitor misoriented microstructures, which are invisible to X-ray diffraction, and show Al3+ framework incorporation and illustrate the differences between misoriented and b-oriented films. The methanol-to-hydrocarbons process was studied by operando UV/Vis diffuse reflectance micro-spectroscopy with on-line mass spectrometry, showing that the b-oriented zeolite ZSM-5 films are active and stable under realistic process conditions.
Co-reporter:Dr. Haifeng Xiong;Dr. Sen Lin;Joris Goetze;Dr. Paul Pletcher; Dr. Hua Guo;Dr. Libor Kovarik;Dr. Kateryna Artyushkova; Dr. Bert M. Weckhuysen; Dr. Abhaya K. Datye
Angewandte Chemie 2017 Volume 129(Issue 31) pp:9114-9119
Publication Date(Web):2017/07/24
DOI:10.1002/ange.201701115
AbstractCeria (CeO2) supports are unique in their ability to trap ionic platinum (Pt), providing exceptional stability for isolated single atoms of Pt. The reactivity and stability of single-atom Pt species was explored for the industrially important light alkane dehydrogenation reaction. The single-atom Pt/CeO2 catalysts are stable during propane dehydrogenation, but are not selective for propylene. DFT calculations show strong adsorption of the olefin produced, leading to further unwanted reactions. In contrast, when tin (Sn) is added to CeO2, the single-atom Pt catalyst undergoes an activation phase where it transforms into Pt–Sn clusters under reaction conditions. Formation of small Pt–Sn clusters allows the catalyst to achieve high selectivity towards propylene because of facile desorption of the product. The CeO2-supported Pt–Sn clusters are very stable, even during extended reaction at 680 °C. Coke formation is almost completely suppressed by adding water vapor to the feed. Furthermore, upon oxidation the Pt–Sn clusters readily revert to the atomically dispersed species on CeO2, making Pt–Sn/CeO2 a fully regenerable catalyst.
Co-reporter:Donglong Fu;Dr. Joel E. Schmidt;Dr. Zoran Ristanović;Dr. Abhishek Dutta Chowdhury;Dr. Florian Meirer; Dr. Bert M. Weckhuysen
Angewandte Chemie International Edition 2017 Volume 56(Issue 37) pp:11217-11221
Publication Date(Web):2017/09/04
DOI:10.1002/anie.201704846
AbstractHighly b-oriented zeolite ZSM-5 films are critical for applications in catalysis and separations and may serve as models to study diffusion and catalytic properties in single zeolite channels. However, the introduction of catalytically active Al3+ usually disrupts the orientation of zeolite films. Herein, using structure-directing agents with hydroxy groups, we demonstrate a new method to prepare highly b-oriented zeolite ZSM-5 films with a broad range of Si/Al ratios (Si/Al=45 to ∞). Fluorescence micro-(spectro)scopy was used to monitor misoriented microstructures, which are invisible to X-ray diffraction, and show Al3+ framework incorporation and illustrate the differences between misoriented and b-oriented films. The methanol-to-hydrocarbons process was studied by operando UV/Vis diffuse reflectance micro-spectroscopy with on-line mass spectrometry, showing that the b-oriented zeolite ZSM-5 films are active and stable under realistic process conditions.
Co-reporter:Dr. Haifeng Xiong;Dr. Sen Lin;Joris Goetze;Dr. Paul Pletcher; Dr. Hua Guo;Dr. Libor Kovarik;Dr. Kateryna Artyushkova; Dr. Bert M. Weckhuysen; Dr. Abhaya K. Datye
Angewandte Chemie International Edition 2017 Volume 56(Issue 31) pp:8986-8991
Publication Date(Web):2017/07/24
DOI:10.1002/anie.201701115
AbstractCeria (CeO2) supports are unique in their ability to trap ionic platinum (Pt), providing exceptional stability for isolated single atoms of Pt. The reactivity and stability of single-atom Pt species was explored for the industrially important light alkane dehydrogenation reaction. The single-atom Pt/CeO2 catalysts are stable during propane dehydrogenation, but are not selective for propylene. DFT calculations show strong adsorption of the olefin produced, leading to further unwanted reactions. In contrast, when tin (Sn) is added to CeO2, the single-atom Pt catalyst undergoes an activation phase where it transforms into Pt–Sn clusters under reaction conditions. Formation of small Pt–Sn clusters allows the catalyst to achieve high selectivity towards propylene because of facile desorption of the product. The CeO2-supported Pt–Sn clusters are very stable, even during extended reaction at 680 °C. Coke formation is almost completely suppressed by adding water vapor to the feed. Furthermore, upon oxidation the Pt–Sn clusters readily revert to the atomically dispersed species on CeO2, making Pt–Sn/CeO2 a fully regenerable catalyst.
Co-reporter:Zoran Ristanović, Alexey V. Kubarev, Johan Hofkens, Maarten B. J. Roeffaers, and Bert M. Weckhuysen
Journal of the American Chemical Society 2016 Volume 138(Issue 41) pp:13586-13596
Publication Date(Web):October 6, 2016
DOI:10.1021/jacs.6b06083
Visualizing proton-transfer processes at the nanoscale is essential for understanding the reactivity of zeolite-based catalyst materials. In this work, the Brønsted-acid-catalyzed oligomerization of styrene derivatives was used for the first time as a single molecule probe reaction to study the reactivity of individual zeolite H-ZSM-5 crystals in different zeolite framework, reactant and solvent environments. This was accomplished via the formation of distinct dimeric and trimeric fluorescent carbocations, characterized by their different photostability, as detected by single molecule fluorescence microscopy. The oligomerization kinetics turned out to be very sensitive to the reaction conditions and the presence of the local structural defects in zeolite H-ZSM-5 crystals. The remarkably photostable trimeric carbocations were found to be formed predominantly near defect-rich crystalline regions. This spectroscopic marker offers clear prospects for nanoscale quality control of zeolite-based materials. Interestingly, replacing n-heptane with 1-butanol as a solvent led to a reactivity decrease of several orders and shorter survival times of fluorescent products due to the strong chemisorption of 1-butanol onto the Brønsted acid sites. A similar effect was achieved by changing the electrophilic character of the para-substituent of the styrene moiety. Based on the measured turnover rates we have established a quantitative, single turnover approach to evaluate substituent and solvent effects on the reactivity of individual zeolite H-ZSM-5 crystals.
Co-reporter:Anna M. Wise, Johanna Nelson Weker, Sam Kalirai, Maryam Farmand, David A. Shapiro, Florian Meirer, and Bert M. Weckhuysen
ACS Catalysis 2016 Volume 6(Issue 4) pp:2178
Publication Date(Web):February 26, 2016
DOI:10.1021/acscatal.6b00221
Understanding Fe deposition in fluid catalytic cracking (FCC) catalysis is critical for the mitigation of catalyst degradation. Here we employ soft X-ray ptychography to determine at the nanoscale the distribution and chemical state of Fe in an aged FCC catalyst particle. We show that both particle swelling due to colloidal Fe deposition and Fe penetration into the matrix as a result of precracking of large organic molecules occur. The application of ptychography allowed us to provide direct visual evidence for these two distinct Fe-based deactivation mechanisms, which have so far been proposed only on the basis of indirect evidence.Keywords: catalyst deactivation; chemical imaging; fluid catalytic cracking; iron and soft X-ray ptychography
Co-reporter:Hien N. Pham, Jesper J. H. B. Sattler, Bert M. Weckhuysen, and Abhaya K. Datye
ACS Catalysis 2016 Volume 6(Issue 4) pp:2257
Publication Date(Web):February 23, 2016
DOI:10.1021/acscatal.5b02917
Alumina-supported Pt is one of the major industrial catalysts for light alkane dehydrogenation. This catalyst loses activity during reaction, with coke formation often considered as the reason for deactivation. As we show in this study, the amount and nature of carbon deposits do not directly correlate with the loss of activity. Rather, it is the transformation of subnanometer Pt species into larger Pt nanoparticles that appears to be responsible for the loss of catalytic activity. Surprisingly, a portion of the Sn remains atomically dispersed on the alumina surface in the spent catalyst and helps in the redispersion of the Pt. In the absence of Sn on the alumina support, the larger Pt nanoparticles formed during reaction are not redispersed during oxidative regeneration. It is known that Sn is added as a promoter in the industrial catalyst to help in achieving high propene selectivity and to minimize coke formation. This work shows that an important role of Sn is to help in the regeneration of Pt, by providing nucleation sites on the alumina surface. Aberration-corrected scanning transmission electron microscopy helps to provide unique insights into the operating characteristics of an industrially important catalyst by demonstrating the role of promoter elements, such as Sn, in the oxidative regeneration of Pt on γ-Al2O3.Keywords: coking; nucleation sites; oxidative regeneration; propane dehydrogenation; Pt-Sn/γ-Al2O3; Pt/γ-Al2O3; sintering; STEM/HRTEM
Co-reporter:D. A. Matthijs de Winter, Florian Meirer, and Bert M. Weckhuysen
ACS Catalysis 2016 Volume 6(Issue 5) pp:3158
Publication Date(Web):April 18, 2016
DOI:10.1021/acscatal.6b00302
The overall performance of a catalyst particle strongly depends on the ability of mass transport through its pore space. Characterizing the three-dimensional structure of the macro- and mesopore space of a catalyst particle and establishing a correlation with transport efficiency is an essential step toward designing highly effective catalyst particles. In this work, a generally applicable workflow is presented to characterize the transport efficiency of individual catalyst particles. The developed workflow involves a multiscale characterization approach making use of a focused ion beam-scanning electron microscope (FIB-SEM). SEM imaging is performed on cross sections of 10.000 μm2, visualizing a set of catalyst particles, while FIB-SEM tomography visualized the pore space of a large number of 8 μm3 cubes (subvolumes) of individual catalyst particles. Geometrical parameters (porosity, pore connectivity, and heterogeneity) of the material were used to generate large numbers of virtual 3D volumes resembling the sample’s pore space characteristics, while being suitable for computationally demanding transport simulations. The transport ability, defined as the ratio of unhindered flow over hindered flow, is then determined via transport simulations through the virtual volumes. The simulation results are used as input for an upscaling routine based on an analogy with electrical networks, taking into account the spatial heterogeneity of the pore space over greater length scales. This novel approach is demonstrated for two distinct types of industrially manufactured fluid catalytic cracking (FCC) particles with zeolite Y as the active cracking component. Differences in physicochemical and catalytic properties were found to relate to differences in heterogeneities in the spatial porosity distribution. In addition to the characterization of existing FCC particles, our method of correlating pore space with transport efficiency does also allow for an up-front evaluation of the transport efficiency of new designs of FCC catalyst particles.Keywords: diffusion simulation; fluid catalytic cracking; focused ion beam-scanning electron microscopy; porous media; transport ability; upscaling
Co-reporter:Sang-Ho Chung, Carlo Angelici, Stijn O.M. Hinterding, Markus Weingarth, Marc Baldus, Klaartje Houben, Bert M. Weckhuysen, and Pieter C.A. Bruijnincx
ACS Catalysis 2016 Volume 6(Issue 6) pp:4034
Publication Date(Web):May 26, 2016
DOI:10.1021/acscatal.5b02972
Wet-kneading is a technique commonly used for the synthesis of SiO2–MgO catalysts for the Lebedev ethanol-to-butadiene process, with catalyst performance known to depend heavily on the preparation parameters used in this method. Here, the large influence of Mg precursor and MgO content on morphology, chemical structure (as determined by TEM(-EDX), FT-IR, XRD and solid-state 1H–29Si cross-polarized MAS NMR), and on catalyst performance is demonstrated. The Mg precursor used is found to influence the extent of magnesium silicate formation during wet-kneading, as estimated from TEM and FT-IR, which, in turn, was found to correlate with catalyst performance. Accordingly, the catalyst synthesized from a nanosized Mg(OH)2 precursor (SiO2–MgO (III)nano), showing the highest degree of chemical contact between the SiO2 and MgO components, gave the highest butadiene yield. Variation of the Mg/Si ratio in a series of SiO2–MgO (III)nano materials showed a volcano-type dependence of the butadiene yield on MgO content. 1H–29Si CP-MAS NMR studies allowed for the identification of the type and an estimation of the amount of magnesium silicates formed during wet-kneading. Here, we argue that the structural characteristics of the hydrous magnesium silicates, lizardite and talc, formed during catalyst preparation, together with the ratio of the magnesium silicates to MgO, determine the overall acid/base properties of the SiO2–MgO (III)nano catalyst materials and as a result, catalyst performance.Keywords: 29Si NMR; ethanol-to-butadiene; magnesium silicates; silica-magnesia; wet-kneading
Co-reporter:Jamal Ftouni, Ara Muñoz-Murillo, Andrey Goryachev, Jan P. Hofmann, Emiel J. M. Hensen, Li Lu, Christopher J. Kiely, Pieter C. A. Bruijnincx, and Bert M. Weckhuysen
ACS Catalysis 2016 Volume 6(Issue 8) pp:5462
Publication Date(Web):July 12, 2016
DOI:10.1021/acscatal.6b00730
Catalyst stability in the liquid phase under polar conditions, typically required for the catalytic conversion of renewable platform molecules, is a major concern but has been only sparsely studied. Here, the activity, selectivity, and stability of Ru-based catalysts supported on TiO2, ZrO2, and C in the conversion of levulinic acid (LA) to γ-valerolactone (GVL) has been studied at 30 bar of H2 and 423 K in dioxane as solvent. All catalysts showed excellent yields of GVL when used fresh, but only the Ru/ZrO2 catalyst could maintain these high yields upon multiple recycling. Surprisingly, the widely used Ru/TiO2 catalyst showed quick signs of deactivation already after the first catalytic test. XPS, CO/FT-IR, TGA, AC-STEM, and physisorption data showed that the partial deactivation is not due to Ru sintering or coking but rather due to reduction of the titania support in combination with partial coverage of the Ru nanoparticles, i.e. due to a detrimental strong metal–support interaction. In contrast, the zirconia support showed no signs of reduction and displayed high morphological and structural stability even after five recycling tests. Remarkably, in the fresh Ru/ZrO2 catalyst, Ru was found to be fully atomically dispersed on the fresh catalyst even at 1 wt % Ru loading, with some genesis of Ru nanoparticles being observed upon recycling. Further studies with the Ru/ZrO2 catalyst showed that dioxane can be readily replaced by more benign solvents, including GVL itself. The addition of water to the reaction mixture was furthermore shown to promote the selective hydrogenation reaction.Keywords: biomass; catalyst stability; levulinic acid; single-atom catalysis; strong metal−support interaction
Co-reporter:Sang-Ho Chung, Carlo Angelici, Stijn O.M. Hinterding, Markus Weingarth, Marc Baldus, Klaartje Houben, Bert M. Weckhuysen, and Pieter C.A. Bruijnincx
ACS Catalysis 2016 Volume 6(Issue 11) pp:7685
Publication Date(Web):October 12, 2016
DOI:10.1021/acscatal.6b02795
Co-reporter:Kristof De Wispelaere, Caterina S. Wondergem, Bernd Ensing, Karen Hemelsoet, Evert Jan Meijer, Bert M. Weckhuysen, Veronique Van Speybroeck, and Javier Ruiz-Martı́nez
ACS Catalysis 2016 Volume 6(Issue 3) pp:1991
Publication Date(Web):February 9, 2016
DOI:10.1021/acscatal.5b02139
The role of water in the methanol-to-olefins (MTO) process over H-SAPO-34 has been elucidated by a combined theoretical and experimental approach, encompassing advanced molecular dynamics simulations and in situ microspectroscopy. First-principles calculations at the molecular level point out that water competes with methanol and propene for direct access to the Brønsted acid sites. This results in less efficient activation of these molecules, which are crucial for the formation of the hydrocarbon pool. Furthermore, lower intrinsic methanol reactivity toward methoxide formation has been observed. These observations are in line with a longer induction period observed from in situ UV–vis microspectroscopy experiments. These experiments revealed a slower and more homogeneous discoloration of H-SAPO-34, while in situ confocal fluorescence microscopy confirmed the more homogeneous distribution and larger amount of MTO intermediates when cofeeding water. As such, it is shown that water induces a more efficient use of the H-SAPO-34 catalyst crystals at the microscopic level. The combined experimental–theoretical approach gives a profound insight into the role of water in the catalytic process at the molecular and single-particle level.Keywords: confocal fluorescence microscopy; metadynamics; methanol-to-olefins; molecular dynamics; UV−vis spectroscopy; water; zeolites
Co-reporter:Dilek A. Boga, Fang Liu, Pieter C. A. Bruijnincx and Bert M. Weckhuysen
Catalysis Science & Technology 2016 vol. 6(Issue 1) pp:134-143
Publication Date(Web):09 Sep 2015
DOI:10.1039/C4CY01711K
The aqueous-phase reforming (APR) of a crude glycerol that originates from an industrial process and the effect of the individual components of crude glycerol on APR activity have been studied over 1 wt% Pt/Mg(Al)O, 1 wt% Pt/Al2O3, 5 wt% Pt/Al2O3 and 5 wt% Pt/C catalysts at 29 bar and 225 °C. The use of a 10 wt% alkaline crude glycerol solution in water, containing 6.85 wt% glycerol, 1.62 wt% soaps, 1.55 wt% methanol, and 0.07 wt% ester, resulted in a dramatic drop in APR activity compared to the corresponding 6.85 wt% solution of pure glycerol in water. Catalytic performance in crude glycerol APR increased in the order: 1 wt% Pt/Al2O3 < 5 wt% Pt/Al2O3 < 1 wt% Pt/Mg(Al)O < 5 wt% Pt/C. A H2 selectivity of only 1% was obtained with crude glycerol over a 1 wt% Pt/Al2O3 catalyst, while the same catalyst material under identical reaction conditions gave 64% H2 with pure glycerol. The cause of deactivation was investigated with synthetic mixtures that mimicked the composition of the crude glycerol and contained glycerol, methanol and varying amounts of NaCl and sodium oleate. The results showed that Na salts of fatty acids have a much more pronounced negative influence on APR activity than NaCl does and greatly inhibit H2 formation. Stearic acid and long chain aliphatics and olefins were shown to be formed and to be involved in the deactivation of the catalyst. The relatively high activity/selectivity of the 1 wt% Pt/Mg(Al)O could be attributed to intercalation of oleate/stearate in the sheets of the layered double hydroxide that is formed under reaction conditions.
Co-reporter:Leila Negahdar, Arturo Gonzalez-Quiroga, Daria Otyuskaya, Hilal E. Toraman, Li Liu, Johann T. B. H. Jastrzebski, Kevin. M. Van Geem, Guy B. Marin, Joris W. Thybaut, and Bert M. Weckhuysen
ACS Sustainable Chemistry & Engineering 2016 Volume 4(Issue 9) pp:4974
Publication Date(Web):July 29, 2016
DOI:10.1021/acssuschemeng.6b01329
Fast pyrolysis bio-oils are feasible energy carriers and a potential source of chemicals. Detailed characterization of bio-oils is essential to further develop its potential use. In this study, quantitative 13C nuclear magnetic resonance (13C NMR) combined with comprehensive two-dimensional gas chromatography (GC × GC) was used to characterize fast pyrolysis bio-oils originated from pinewood, wheat straw, and rapeseed cake. The combination of both techniques provided new information on the chemical composition of bio-oils for further upgrading. 13C NMR analysis indicated that pinewood-based bio-oil contained mostly methoxy/hydroxyl (≈30%) and carbohydrate (≈27%) carbons; wheat straw bio-oil showed to have high amount of alkyl (≈35%) and aromatic (≈30%) carbons, while rapeseed cake-based bio-oil had great portions of alkyl carbons (≈82%). More than 200 compounds were identified and quantified using GC × GC coupled to a flame ionization detector (FID) and a time of flight mass spectrometer (TOF-MS). Nonaromatics were the most abundant and comprised about 50% of the total mass of compounds identified and quantified via GC × GC. In addition, this analytical approach allowed the quantification of high value-added phenolic compounds, as well as of low molecular weight carboxylic acids and aldehydes, which exacerbate the unstable and corrosive character of the bio-oil.Keywords: Agricultural residue; Aromatics; Bio-oil stability; Chemical shift; Compositional analysis; Light oxygenates; Pyrolysis oil; Softwood
Co-reporter:Meenakshisundaram Sankar, Qian He, Simon Dawson, Ewa Nowicka, Li Lu, Pieter C. A. Bruijnincx, Andrew M. Beale, Christopher J. Kiely and Bert M. Weckhuysen
Catalysis Science & Technology 2016 vol. 6(Issue 14) pp:5473-5482
Publication Date(Web):23 Mar 2016
DOI:10.1039/C6CY00425C
The synthesis and functionalization of imines and amines are key steps in the preparation of many fine chemicals and for pharmaceuticals in particular. Traditionally, metal complexes are used as homogeneous catalysts for these organic transformations. Here we report gold–palladium and ruthenium–palladium nano-alloys supported on TiO2 acting as highly efficient heterogeneous catalysts for the one-pot synthesis of the imine N-benzylideneaniline and the secondary amine N-benzylaniline directly from the easily available and stable nitrobenzene and benzyl alcohol precursors using a hydrogen auto-transfer strategy. These reactions were carried out without any added external hydrogen, sacrificial hydrogen donor or a homogeneous base. The bimetallic catalysts were prepared by the recently developed modified impregnation strategy, giving efficient control of size and nano-alloy composition. Both bimetallic catalysts were found to be far more active than their monometallic analogues due to a synergistic effect. Based on the turnover numbers the catalytic activities follow the order Ru < Pd < Au ≪ Au–Pd < Ru–Pd. Aberration corrected scanning transmission electron microscopy (AC-STEM) and X-ray absorption spectroscopy (XAFS) studies of these catalysts revealed that the reason for the observed synergistic effect is the electronic modification of the metal sites in the case of the Au–Pd system and a size stabilisation effect in the case of the Ru–Pd catalyst.
Co-reporter:K. H. Cats, J. C. Andrews, O. Stéphan, K. March, C. Karunakaran, F. Meirer, F. M. F. de Groot and B. M. Weckhuysen
Catalysis Science & Technology 2016 vol. 6(Issue 12) pp:4438-4449
Publication Date(Web):16 Feb 2016
DOI:10.1039/C5CY01524C
The Fischer–Tropsch synthesis (FTS) reaction is one of the most promising processes to convert alternative energy sources, such as natural gas, coal or biomass, into liquid fuels and other high-value products. Despite its commercial implementation, we still lack fundamental insights into the various deactivation processes taking place during FTS. In this work, a combination of three methods for studying single catalyst particles at different length scales has been developed and applied to study the deactivation of Co/TiO2 Fischer–Tropsch synthesis (FTS) catalysts. By combining transmission X-ray microscopy (TXM), scanning transmission X-ray microscopy (STXM) and scanning transmission electron microscopy-electron energy loss spectroscopy (STEM-EELS) we visualized changes in the structure, aggregate size and distribution of supported Co nanoparticles that occur during FTS. At the microscale, Co nanoparticle aggregates are transported over several μm leading to a more homogeneous Co distribution, while at the nanoscale Co forms a thin layer of ∼1–2 nm around the TiO2 support. The formation of the Co layer is the opposite case to the “classical” strong metal–support interaction (SMSI) in which TiO2 surrounds the Co, and is possibly related to the surface oxidation of Co metal nanoparticles in combination with coke formation. In other words, the observed migration and formation of a thin CoOx layer are similar to a previously discussed reaction-induced spreading of metal oxides across a TiO2 surface.
Co-reporter:D. Cicmil, I. K. van Ravenhorst, J. Meeuwissen, A. Vantomme and B. M. Weckhuysen
Catalysis Science & Technology 2016 vol. 6(Issue 3) pp:731-743
Publication Date(Web):18 Nov 2015
DOI:10.1039/C5CY01512J
A study on the influence of H2 and the degree of titanation of a Cr/SiO2 Phillips polymerisation catalyst on the selective oligomerisation of ethylene induced by pre-contacting the catalyst with triethylaluminium (TEAl) is presented. Ethylene oligomerisation reactions were performed at 373 K and 1 bar, inside a quartz reactor of a specially designed operando experimental setup, which allowed examination of the catalysts by UV-vis-NIR diffuse reflectance spectroscopy, while the gas phase was continuously monitored by on-line mass spectroscopy and subsequently analysed by gas chromatography. With this combination of techniques, it was possible to acquire detailed insight into the different distributions of produced oligomers, which were highly dependent on the catalyst structure. The addition of small amounts of Ti significantly changes the electronic environment of Cr oligomerisation sites by the formation of Cr–O–Ti–O–Si linkages, favouring β-H transfer and increasing the selectivity towards butene at the expense of 1-hexene. Moreover, we show that ethylene oligo-/polymerisation reactions follow at least two different pathways, i.e. metallacycle for olefinic species with a broken Schulz-Flory distribution, and Cossee–Arlman for other hydrocarbon species.
Co-reporter:Diego Valencia, Gareth T. Whiting, Rosa E. Bulo and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 3) pp:2080-2086
Publication Date(Web):21 Dec 2015
DOI:10.1039/C5CP06477E
In an earlier work, protonated thiophene-based oligomers were identified inside ZSM-5 zeolites. The novel compounds exhibited π–π* absorption wavelengths deep within the visible region, earmarking them for possible use as chromophores in a variety of applications. In this computational study, we determine the factors that cause such low-energy transitions, and describe the electronic structure of these remarkable compounds. DFT calculations of conjugated thiophene-based oligomers with up to five monomer units reveal that the main absorption band of each protonated oligomer is strongly red-shifted compared to the unprotonated form. This effect is counterintuitive, since protonation is expected to diminish aromaticity, and thereby increase the HOMO–LUMO gap. We find that upon protonation the π-electrons remain delocalized over the entire π-conjugated molecule, but the positive charge is localized predominantly on the protonated side of the molecule. A possible explanation for this ground-state charge localization is the participation of the C–H bond in the π-system of the protonated ring, locally providing aromatic stabilization for the positive charge. The addition of the proton stabilizes all electronic orbitals, but due to the ground state π-electron distribution away from the added nucleus, the HOMO is stabilized less than the LUMO. The main absorption peak upon protonation corresponds to the charge transfer excitation involving the frontier orbitals, and the small band gap explains the observed red shift. Analogue calculations on thiophene within a ZSM-5 zeolite cluster model confirm the same trends upon protonation as observed in the non-interacting compounds. Understanding the electronic structure of these compounds is very relevant to correlate UV-Vis bands with acidic strength and possibly environment in zeolites and to improve their performance in catalytic and energy related applications.
Co-reporter:Dr. Inés Lezcano-González;Ramon Oord;Dr. Mauro Rovezzi;Dr. Pieter Glatzel;Dr. Stanley W. Botchway;Dr. Bert M. Weckhuysen;Dr. Andrew M. Beale
Angewandte Chemie International Edition 2016 Volume 55( Issue 17) pp:5215-5219
Publication Date(Web):
DOI:10.1002/anie.201601357
Abstract
Combined high-resolution fluorescence detection X-ray absorption near-edge spectroscopy, X-ray diffraction, and X-ray emission spectroscopy have been employed under operando conditions to obtain detailed new insight into the nature of the Mo species on zeolite ZSM-5 during methane dehydroaromatization. The results show that isolated Mo–oxo species present after calcination are converted by CH4 into metastable MoCxOy species, which are primarily responsible for C2Hx/C3Hx formation. Further carburization leads to MoC3 clusters, whose presence coincides with benzene formation. Both sintering of MoC3 and accumulation of large hydrocarbons on the external surface, evidenced by fluorescence-lifetime imaging microscopy, are principally responsible for the decrease in catalytic performance. These results show the importance of controlling Mo speciation to achieve the desired product formation, which has important implications for realizing the impact of CH4 as a source for platform chemicals.
Co-reporter:Dr. Inés Lezcano-González;Ramon Oord;Dr. Mauro Rovezzi;Dr. Pieter Glatzel;Dr. Stanley W. Botchway;Dr. Bert M. Weckhuysen;Dr. Andrew M. Beale
Angewandte Chemie International Edition 2016 Volume 55( Issue 17) pp:
Publication Date(Web):
DOI:10.1002/anie.201602430
Co-reporter:Zoran Ristanovi&x107;;Dr. Jan P. Hofmann;Dr. Marie-Ingrid Richard;Dr. Tao Jiang;Dr. Gilbert A. Chahine;Dr. Tobias U. Schülli;Dr. Florian Meirer;Dr. Bert M. Weckhuysen
Angewandte Chemie International Edition 2016 Volume 55( Issue 26) pp:7496-7500
Publication Date(Web):
DOI:10.1002/anie.201601796
Abstract
Structure–activity relationships in heterogeneous catalysis are challenging to be measured on a single-particle level. For the first time, one X-ray beam is used to determine the crystallographic structure and reactivity of a single zeolite crystal. The method generates μm-resolved X-ray diffraction (μ-XRD) and X-ray excited optical fluorescence (μ-XEOF) maps of the crystallinity and Brønsted reactivity of a zeolite crystal previously reacted with a styrene probe molecule. The local gradients in chemical reactivity (derived from μ-XEOF) were correlated with local crystallinity and framework Al content, determined by μ-XRD. Two distinctly different types of fluorescent species formed selectively, depending on the local zeolite crystallinity. The results illustrate the potential of this approach to resolve the crystallographic structure of a porous material and its reactivity in one experiment via X-ray induced fluorescence of organic molecules formed at the reactive centers.
Co-reporter:Sophie C. C. Wiedemann;Zoran Ristanovi&x107;;Dr. Gareth T. Whiting;Dr. V. R. ReddyMarthala;Dr. Jörg Kärger;Dr. Jens Weitkamp;Dr. Bas Wels;Dr. Pieter C. A. Bruijnincx;Dr. Bert M. Weckhuysen
Chemistry - A European Journal 2016 Volume 22( Issue 1) pp:199-210
Publication Date(Web):
DOI:10.1002/chem.201503551
Abstract
Large zeolite crystals of ferrierite have been used to study the deactivation, at the single particle level, of the alkyl isomerisation catalysis of oleic acid and elaidic acid by a combination of visible micro-spectroscopy and fluorescence microscopy (both polarised wide-field and confocal modes). The large crystals did show the desired activity, albeit only traces of the isomerisation product were obtained and low conversions were achieved compared to commercial ferrierite powders. This limited activity is in line with their lower external non-basal surface area, supporting the hypothesis of pore mouth catalysis. Further evidence for the latter comes from visible micro-spectroscopy, which shows that the accumulation of aromatic species is limited to the crystal edges, while fluorescence microscopy strongly suggests the presence of polyenylic carbocations. Light polarisation associated with the spatial resolution of fluorescence microscopy reveals that these carbonaceous deposits are aligned only in the larger 10-MR channels of ferrierite at all crystal edges. The reaction is hence further limited to these specific pore mouths.
Co-reporter:Korneel H. Cats ;Dr. Bert M. Weckhuysen
ChemCatChem 2016 Volume 8( Issue 8) pp:1531-1542
Publication Date(Web):
DOI:10.1002/cctc.201600074
Abstract
A novel laboratory setup for combined operando X-ray diffraction and Raman spectroscopy of catalytic solids with online product analysis by gas chromatography is presented. The setup can be used with a laboratory-based X-ray source, which results in important advantages in terms of time-on-stream that can be measured, compared to synchrotron-based experiments. The data quality was much improved by the use of a relatively high-energy MoKα radiation instead of the more conventional CuKα radiation. We have applied the instrument to study the long-term deactivation of Co/TiO2 Fischer–Tropsch synthesis (FTS) catalysts. No sign of Co sintering or bulk oxidation was found during the experiments. However, part of the metallic Co was converted into cobalt carbide (Co2C), at elevated pressure (10 bar). Furthermore, graphitic-like coke species are clearly formed during FTS at atmospheric pressure, whereas at elevated pressure fluorescence hampered the interpretation of the measured Raman spectra.
Co-reporter:Dimitrije Cicmil;Dr. Jurjen Meeuwissen;Dr. Aurélien Vantomme;Dr.Ir. Bert M. Weckhuysen
ChemCatChem 2016 Volume 8( Issue 11) pp:1937-1944
Publication Date(Web):
DOI:10.1002/cctc.201600200
Abstract
A diffuse reflectance infrared Fourier-transform (DRIFT) study has been conducted at 373 K and 1 bar on an industrial Cr/Ti/SiO2 Phillips-type catalyst modified with, and without, triethylaluminium (TEAl) as co-catalyst. The reaction rate of the polymerization of ethylene, as monitored by the increase in the methylene stretching band of the growing polyethylene (PE), has been investigated as a function of the titanium content. After an initial period of mixed kinetics, with the reaction rate significantly higher for the TEAl-modified catalysts compared with the non-modified catalysts, the polymerization proceeded as a pseudo-zero-order reaction with a reaction rate that increased as a function of titanium loading. Furthermore, it was found that the higher Ti loading caused the appearance of more acidic hydroxyl groups and modified the Cr sites by making them more Lewis acidic, ultimately shortening the induction time and increasing the initial polymerization rate.
Co-reporter:Bert M. Weckhuysen;Pieter C. A. Bruijnincx;Monique F. A. Lamers
Topics in Catalysis 2016 Volume 59( Issue 19-20) pp:1669-1670
Publication Date(Web):2016 December
DOI:10.1007/s11244-016-0692-x
Co-reporter:Dr. Inés Lezcano-González;Ramon Oord;Dr. Mauro Rovezzi;Dr. Pieter Glatzel;Dr. Stanley W. Botchway;Dr. Bert M. Weckhuysen;Dr. Andrew M. Beale
Angewandte Chemie 2016 Volume 128( Issue 17) pp:5301-5305
Publication Date(Web):
DOI:10.1002/ange.201601357
Abstract
Combined high-resolution fluorescence detection X-ray absorption near-edge spectroscopy, X-ray diffraction, and X-ray emission spectroscopy have been employed under operando conditions to obtain detailed new insight into the nature of the Mo species on zeolite ZSM-5 during methane dehydroaromatization. The results show that isolated Mo–oxo species present after calcination are converted by CH4 into metastable MoCxOy species, which are primarily responsible for C2Hx/C3Hx formation. Further carburization leads to MoC3 clusters, whose presence coincides with benzene formation. Both sintering of MoC3 and accumulation of large hydrocarbons on the external surface, evidenced by fluorescence-lifetime imaging microscopy, are principally responsible for the decrease in catalytic performance. These results show the importance of controlling Mo speciation to achieve the desired product formation, which has important implications for realizing the impact of CH4 as a source for platform chemicals.
Co-reporter:Dr. Inés Lezcano-González;Ramon Oord;Dr. Mauro Rovezzi;Dr. Pieter Glatzel;Dr. Stanley W. Botchway;Dr. Bert M. Weckhuysen;Dr. Andrew M. Beale
Angewandte Chemie 2016 Volume 128( Issue 17) pp:
Publication Date(Web):
DOI:10.1002/ange.201602430
Co-reporter:Zoran Ristanovi&x107;;Dr. Jan P. Hofmann;Dr. Marie-Ingrid Richard;Dr. Tao Jiang;Dr. Gilbert A. Chahine;Dr. Tobias U. Schülli;Dr. Florian Meirer;Dr. Bert M. Weckhuysen
Angewandte Chemie 2016 Volume 128( Issue 26) pp:7622-7626
Publication Date(Web):
DOI:10.1002/ange.201601796
Abstract
Structure–activity relationships in heterogeneous catalysis are challenging to be measured on a single-particle level. For the first time, one X-ray beam is used to determine the crystallographic structure and reactivity of a single zeolite crystal. The method generates μm-resolved X-ray diffraction (μ-XRD) and X-ray excited optical fluorescence (μ-XEOF) maps of the crystallinity and Brønsted reactivity of a zeolite crystal previously reacted with a styrene probe molecule. The local gradients in chemical reactivity (derived from μ-XEOF) were correlated with local crystallinity and framework Al content, determined by μ-XRD. Two distinctly different types of fluorescent species formed selectively, depending on the local zeolite crystallinity. The results illustrate the potential of this approach to resolve the crystallographic structure of a porous material and its reactivity in one experiment via X-ray induced fluorescence of organic molecules formed at the reactive centers.
Co-reporter:Thomas Hartman; Caterina S. Wondergem; Naresh Kumar; Albert van den Berg
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 8) pp:1570-1584
Publication Date(Web):April 14, 2016
DOI:10.1021/acs.jpclett.6b00147
Surface- and tip-enhanced Raman spectroscopy (SERS and TERS) techniques exhibit highly localized chemical sensitivity, making them ideal for studying chemical reactions, including processes at catalytic surfaces. Catalyst structures, adsorbates, and reaction intermediates can be observed in low quantities at hot spots where electromagnetic fields are the strongest, providing ample opportunities to elucidate reaction mechanisms. Moreover, under ideal measurement conditions, it can even be used to trigger chemical reactions. However, factors such as substrate instability and insufficient signal enhancement still limit the applicability of SERS and TERS in the field of catalysis. By the use of sophisticated colloidal synthesis methods and advanced techniques, such as shell-isolated nanoparticle-enhanced Raman spectroscopy, these challenges could be overcome.
Co-reporter:E. T. C. Vogt and B. M. Weckhuysen
Chemical Society Reviews 2015 vol. 44(Issue 20) pp:7342-7370
Publication Date(Web):18 Sep 2015
DOI:10.1039/C5CS00376H
Fluid catalytic cracking (FCC) is one of the major conversion technologies in the oil refinery industry. FCC currently produces the majority of the world's gasoline, as well as an important fraction of propylene for the polymer industry. In this critical review, we give an overview of the latest trends in this field of research. These trends include ways to make it possible to process either very heavy or very light crude oil fractions as well as to co-process biomass-based oxygenates with regular crude oil fractions, and convert these more complex feedstocks in an increasing amount of propylene and diesel-range fuels. After providing some general background of the FCC process, including a short history as well as details on the process, reactor design, chemical reactions involved and catalyst material, we will discuss several trends in FCC catalysis research by focusing on ways to improve the zeolite structure stability, propylene selectivity and the overall catalyst accessibility by (a) the addition of rare earth elements and phosphorus, (b) constructing hierarchical pores systems and (c) the introduction of new zeolite structures. In addition, we present an overview of the state-of-the-art micro-spectroscopy methods for characterizing FCC catalysts at the single particle level. These new characterization tools are able to explain the influence of the harsh FCC processing conditions (e.g. steam) and the presence of various metal poisons (e.g. V, Fe and Ni) in the crude oil feedstocks on the 3-D structure and accessibility of FCC catalyst materials.
Co-reporter:Bert M. Weckhuysen and Jihong Yu
Chemical Society Reviews 2015 vol. 44(Issue 20) pp:7022-7024
Publication Date(Web):24 Sep 2015
DOI:10.1039/C5CS90100F
A graphical abstract is available for this content
Co-reporter:Hendrik E. van der Bij and Bert M. Weckhuysen
Chemical Society Reviews 2015 vol. 44(Issue 20) pp:7406-7428
Publication Date(Web):05 Jun 2015
DOI:10.1039/C5CS00109A
Phosphorus and microporous aluminosilicates, better known as zeolites, have a unique but poorly understood relationship. For example, phosphatation of the industrially important zeolite H-ZSM-5 is a well-known, relatively inexpensive and seemingly straightforward post-synthetic modification applied by the chemical industry not only to alter its hydrothermal stability and acidity, but also to increase its selectivity towards light olefins in hydrocarbon catalysis. On the other hand, phosphorus poisoning of zeolite-based catalysts, which are used for removing nitrogen oxides from exhaust fuels, poses a problem for their use in diesel engine catalysts. Despite the wide impact of phosphorus–zeolite chemistry, the exact physicochemical processes that take place require a more profound understanding. This review article provides the reader with a comprehensive and state-of-the-art overview of the academic literature, from the first reports in the late 1970s until the most recent studies. In the first part an in-depth analysis is undertaken, which will reveal universal physicochemical and structural effects of phosphorus–zeolite chemistry on the framework structure, accessibility, and strength of acid sites. The second part discusses the hydrothermal stability of zeolites and clarifies the promotional role that phosphorus plays. The third part of the review paper links the structural and physicochemical effects of phosphorus on zeolite materials with their catalytic performance in a variety of catalytic processes, including alkylation of aromatics, catalytic cracking, methanol-to-hydrocarbon processing, dehydration of bioalcohol, and ammonia selective catalytic reduction (SCR) of NOx. Based on these insights, we discuss potential applications and important directions for further research.
Co-reporter:Christoph Sprung
Journal of the American Chemical Society 2015 Volume 137(Issue 5) pp:1916-1928
Publication Date(Web):January 15, 2015
DOI:10.1021/ja511381f
A detailed and systematic polarized confocal fluorescence microscopy investigation is presented on three batches of large coffin-shaped ZSM-5 crystals (i.e., parent, steamed at 500 °C, and steamed at 700 °C). In total, six laser lines of different wavelength in the visible region are employed on two crystal positions and three orientations with respect to the polarization plane of the excitation laser light. A fluorescent probe molecule is generated inside the zeolite pores, originating from the acid-catalyzed oligomerization of 4-fluorostyrene. A thorough analysis of the polarization plane of emitting fluorescent light reveals insight into the orientation of the fluorescent probe molecule restricted by the highly ordered zeolite channel framework, thereby visualizing pore accessibility and clearly distinguishing the occupation of straight and sinusoidal channels by the probe molecule. Spectral features are, furthermore, observed to tell apart molecules situated in one or the other pore. Special focus was given on the rim and tip regions of the zeolite ZSM-5 crystals. On the basis of the confocal approach of the investigation, the aforementioned features are evaluated in three dimensions, while the degradation of the zeolite framework upon postsynthesis steam treatment could be visualized by occupation of the sinusoidal pores.
Co-reporter:Zoran Ristanović; Jan P. Hofmann; Gert De Cremer; Alexey V. Kubarev; Marcus Rohnke; Florian Meirer; Johan Hofkens; Maarten B. J. Roeffaers
Journal of the American Chemical Society 2015 Volume 137(Issue 20) pp:6559-6568
Publication Date(Web):April 13, 2015
DOI:10.1021/jacs.5b01698
Optimizing the number, distribution, and accessibility of Brønsted acid sites in zeolite-based catalysts is of a paramount importance to further improve their catalytic performance. However, it remains challenging to measure real-time changes in reactivity of single zeolite catalyst particles by ensemble-averaging characterization methods. In this work, a detailed 3D single molecule, single turnover sensitive fluorescence microscopy study is presented to quantify the reactivity of Brønsted acid sites in zeolite H-ZSM-5 crystals upon steaming. This approach, in combination with the oligomerization of furfuryl alcohol as a probe reaction, allowed the stochastic behavior of single catalytic turnovers and temporally resolved turnover frequencies of zeolite domains smaller than the diffraction limited resolution to be investigated with great precision. It was found that the single turnover kinetics of the parent zeolite crystal proceeds with significant spatial differences in turnover frequencies on the nanoscale and noncorrelated temporal fluctuations. Mild steaming of zeolite H-ZSM-5 crystals at 500 °C led to an enhanced surface reactivity, with up to 4 times higher local turnover rates than those of the parent H-ZSM-5 crystals, and revealed remarkable heterogeneities in surface reactivity. In strong contrast, severe steaming at 700 °C significantly dealuminated the zeolite H-ZSM-5 material, leading to a 460 times lower turnover rate. The differences in measured turnover activities are explained by changes in the 3D aluminum distribution due to migration of extraframework Al-species and their subsequent effect on pore accessibility, as corroborated by time-of-flight secondary ion mass spectrometry (TOF-SIMS) sputter depth profiling data.
Co-reporter:Florian Meirer; Darius T. Morris; Sam Kalirai; Yijin Liu; Joy C. Andrews
Journal of the American Chemical Society 2015 Volume 137(Issue 1) pp:102-105
Publication Date(Web):January 2, 2015
DOI:10.1021/ja511503d
Full-field transmission X-ray microscopy has been used to determine the 3D structure of a whole individual fluid catalytic cracking (FCC) particle at high spatial resolution and in a fast, noninvasive manner, maintaining the full integrity of the particle. Using X-ray absorption mosaic imaging to combine multiple fields of view, computed tomography was performed to visualize the macropore structure of the catalyst and its availability for mass transport. We mapped the relative spatial distributions of Ni and Fe using multiple-energy tomography at the respective X-ray absorption K-edges and correlated these distributions with porosity and permeability of an equilibrated catalyst (E-cat) particle. Both metals were found to accumulate in outer layers of the particle, effectively decreasing porosity by clogging of pores and eventually restricting access into the FCC particle.
Co-reporter:E. Borodina, F. Meirer, I. Lezcano-González, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, J. Ruiz-Martinez, and B. M. Weckhuysen
ACS Catalysis 2015 Volume 5(Issue 2) pp:992
Publication Date(Web):December 23, 2014
DOI:10.1021/cs501345g
The formation of hydrocarbon species during the methanol to olefins (MTO) reaction over zeolite H-SSZ-13 has been systematically studied at reaction temperatures between 573 and 723 K with a combination of operando UV–vis spectroscopy and online gas chromatography. It was found that the applied reaction temperature influences the rate and nature of coke formation as well as the catalyst stability. Correlation between all hydrocarbon species formed inside the catalyst pores with the activity and deactivation of H-SSZ-13 zeolite material was established. By using a multivariate analysis, we found that the nature of the active and deactivating species varies with the reaction temperature. The majority of active hydrocarbon pool species at low reaction temperatures (i.e., 573–598 K) are methylated benzene carbocations, while at high reaction temperatures (i.e., 623–723 K) methylated naphthalene carbocations become dominant. At low reaction temperatures the deactivation occurs because of the pore filling with methylated naphthalene carbocations. In contrast, at higher reaction temperatures the formation of phenanthrene, pyrene carbocations, and highly conjugated polyaromatics during the deactivation increases. This suggests that the formation of highly conjugated polyaromatics on the external surface can play a role in the deactivation of the material by pore blockage.Keywords: active species; catalyst deactivation; in situ spectroscopy; methanol to olefins; UV−vis spectroscopy
Co-reporter:Carlo Angelici, Florian Meirer, Ad M. J. van der Eerden, Herrick L. Schaink, Andrey Goryachev, Jan P. Hofmann, Emiel J. M. Hensen, Bert M. Weckhuysen, and Pieter C. A. Bruijnincx
ACS Catalysis 2015 Volume 5(Issue 10) pp:6005
Publication Date(Web):September 9, 2015
DOI:10.1021/acscatal.5b00755
Dehydrogenation promoters greatly enhance the performance of SiO2–MgO catalysts in the Lebedev process. Here, the effect of preparation method and order of addition of Cu on the structure and performance of Cu-promoted SiO2–MgO materials is detailed. Addition of Cu to MgO via incipient wetness impregnation (IWI) or coprecipitation (CP) prior to wet-kneading with SiO2 gave similar butadiene yields (∼40%) as when Cu was added to the already wet-kneaded catalyst. In contrast, the catalyst prepared by impregnation of Cu on SiO2 first proved to be the worst catalyst of the series. TEM, XRD, and XPS analyses suggested that, for all catalyst materials, Cu2+ forms a solid solution with MgO. This was confirmed by UV–vis, XANES, and EXAFS data, with Cu being found in a distorted octahedral geometry. As a result, the acid–base properties, as determined by Pyridine- and CDCl3–IR as well as NH3-TPD, are modified, contributing to the improved performance. Operando XANES and EXAFS studies of the evolution of the copper species showed that Cu2+, the only species initially present, is extensively reduced to a mixture of Cu0 and Cu+, leaving only a limited amount of unreduced Cu2+. This formation of Cu0 is the result of the reducing environment of the Lebedev process and is thought to be mainly responsible for the improved performance of the Cu-promoted catalysts.Keywords: butadiene; ethanol; operando; SiO2−MgO catalysts; solid solution
Co-reporter:Ilona van Zandvoort, Eline J. Koers, Markus Weingarth, Pieter C. A. Bruijnincx, Marc Baldus and Bert M. Weckhuysen
Green Chemistry 2015 vol. 17(Issue 8) pp:4383-4392
Publication Date(Web):09 Jul 2015
DOI:10.1039/C5GC00327J
Humin by-products are formed during the acid-catalyzed dehydration of carbohydrates to bio-based platform molecules, such as hydroxymethylfurfural and levulinic acid. The molecular structure of these humins has not yet been unequivocally established. 1D 13C solid-state NMR data reported have, for example, provided considerable insight, but do not allow for the unambiguous assignment of key structural motifs. Complementary (2D) techniques are needed to gain additional insight into the molecular structure of humins. Here, the preparation of 13C-enriched humins is reported, together with the reactive solubilization of these labeled humins and their characterization with complementary 1D and 2D solid-state NMR techniques. 1D cross polarization (CP) and direct excitation (DE) 13C solid-state NMR spectra, 2D 13C-detected double-quantum single-quantum (DQSQ) as well as 2D 1H-detected heteronuclear correlation (HETCOR) were recorded with different excitation schemes. These experiments unambiguously established that the original humins have a furan-rich structure with aliphatic linkers and allowed for a refinement of the molecular structure proposed previously. Solid-state NMR data of alkali-treated 13C-labeled humins showed that an arene-rich structure is formed at the expense of the furanic network during alkaline pretreatment.
Co-reporter:F. Meirer, S. Kalirai, J. Nelson Weker, Y. Liu, J. C. Andrews and B. M. Weckhuysen
Chemical Communications 2015 vol. 51(Issue 38) pp:8097-8100
Publication Date(Web):14 Apr 2015
DOI:10.1039/C5CC00401B
Metal accumulation at the catalyst particle surface plays a role in particle agglutination during fluid catalytic cracking.
Co-reporter:Carlo Angelici, Marjolein E. Z. Velthoen, Bert M. Weckhuysen and Pieter C. A. Bruijnincx
Catalysis Science & Technology 2015 vol. 5(Issue 5) pp:2869-2879
Publication Date(Web):25 Mar 2015
DOI:10.1039/C5CY00200A
The Lebedev ethanol-to-butadiene process entails a complex chain of reactions that require catalysts to possess a subtle balance in the number and strength of acidic and basic sites. SiO2–MgO materials can be excellent Lebedev catalysts if properly prepared, as catalyst performance has been found to depend significantly on the synthesis method. To assess the specific requirements for butadiene production in terms of active sites and to link their presence to the specific preparation method applied, five distinct SiO2–MgO catalysts, prepared by wet-kneading and co-precipitation methods, were thoroughly characterized. The amount and strength of the acidic (pyridine-IR and NH3-TPD) and basic (CDCl3-IR and CO2-TPD) sites of the materials as well as the overall acid/base properties in the liquid phase (Hammett indicators) were determined. The number of acidic and strong basic sites could be correlated with the extent of ethylene and diethyl ether by-product formation. The best performing catalysts are those containing a small amount of strong basic sites, combined with an intermediate amount of acidic sites and weak basic ones. These results thus provide further insight into the relation between the amount and strength of acidic/basic sites, preparation method and catalytic performance.
Co-reporter:Ilona van Zandvoort, Ernst R. H. van Eck, Peter de Peinder, Hero J. Heeres, Pieter C. A. Bruijnincx, and Bert M. Weckhuysen
ACS Sustainable Chemistry & Engineering 2015 Volume 3(Issue 3) pp:533
Publication Date(Web):January 30, 2015
DOI:10.1021/sc500772w
The valorization of the humin byproducts that are formed during hydrothermal, acid-catalyzed dehydration of carbohydrates is hampered by the insolubility of these byproducts. Here, we report on an alkaline pretreatment method that allows for the insolubility of this highly recalcitrant and structurally complex feed to be overcome. The reactive solubilization of glucose-derived humins was found to require a treatment at 200 °C in 0.5 M NaOH for 3.5 h. Fructose- and xylose-derived humins were found to be more recalcitrant, and complete dissolution required raising the temperature to 240 °C. Gel permeation chromatographic analyses show the relative average molecular weight of the now soluble humins to decrease with increasing temperature and reaction time. The alkali-treated humins are soluble in water of pH ≥ 7. Elemental analysis, IR, 2D PASS 13C solid-state NMR and pyrolysis-GC–MS (gas chromatography–mass spectrometry) data indicate that the alkaline pretreatment leads to considerable changes in the molecular structure of the humins. Cleavage of C–O–C bonds and further aromatization of the originally highly furanic humins result in the formation of (polycyclic) aromatic structures decorated with carboxylic acids. The combination of the reduction in Mw and the formation of polar functional groups are thought to be the reasons behind the improved solubility.Keywords: alkaline treatment; biorefinery; carbohydrates biomass; Humins; hydroxymethylfurfural
Co-reporter:Pushkar Singh, Tanja Deckert-Gaudig, Henrik Schneidewind, Konstantin Kirsch, Evelien M. van Schrojenstein Lantman, Bert M. Weckhuysen and Volker Deckert
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 5) pp:2991-2995
Publication Date(Web):10 Dec 2014
DOI:10.1039/C4CP04850D
Vibrational spectroscopy usually provides structural information averaged over many molecules. We report a larger peak position variation and reproducibly smaller FWHM of TERS spectra compared to SERS spectra indicating that the number of molecules excited in a TERS experiment is extremely low. Thus, orientational averaging effects are suppressed and micro ensembles are investigated. This is shown for a thiophenol molecule adsorbed on Au nanoplates and nanoparticles.
Co-reporter:Zafer Öztürk;Jan P. Hofmann;Martin Lutz;Matja&x17e; Mazaj;Nata&x161;a Zabukovec Logar
European Journal of Inorganic Chemistry 2015 Volume 2015( Issue 9) pp:1625-1630
Publication Date(Web):
DOI:10.1002/ejic.201403077
Abstract
The synthesis of phase-pure Co-ZIF-9, an important cobalt-based zeolitic imidazolate framework, could be achieved by modification of the reported synthesis procedure through pH adjustment of the starting synthesis mixture. The phase-pure Co-ZIF-9 material obtained has been characterized by a combination of UV/Vis, FTIR, and Raman spectroscopy as well as by thermogravimetric analysis (TGA) and XRD and possesses a lower overall crystallinity. This can be explained by the addition of the base for the pH adjustment method. On the basis of these findings, a synthesis pathway for the formation of the secondary phase, cobalt formate, is proposed along with its relationship to the flexibility of the coordination environment of cobalt ions. The crystal structures of both phases have been determined by single-crystal X-ray crystallography, and the resolved structures also reflect the coordination flexibility of the framework cobalt ions.
Co-reporter:Dimitrije Cicmil;Dr. Jurjen Meeuwissen;Dr. Aurélien Vantomme;Dr. Jian Wang;Ilse K. vanRavenhorst;Dr. Hendrik E. vanderBij;Dr. Ara Muñoz-Murillo;Dr. Bert M. Weckhuysen
Angewandte Chemie International Edition 2015 Volume 54( Issue 44) pp:13073-13079
Publication Date(Web):
DOI:10.1002/anie.201506718
Abstract
A triethylaluminium(TEAl)-modified Phillips ethylene polymerisation Cr/Ti/SiO2 catalyst has been developed with two distinct active regions positioned respectively in the inner core and outer shell of the catalyst particle. DRIFTS, EPR, UV-Vis-NIR DRS, STXM, SEM-EDX and GPC-IR studies revealed that the catalyst produces simultaneously two different polymers, i.e., low molecular weight linear-chain polyethylene in the Ti-abundant catalyst particle shell and high molecular weight short-chain branched polyethylene in the Ti-scarce catalyst particle core. Co-monomers for the short-chain branched polymer were generated in situ within the TEAl-impregnated confined space of the Ti-scarce catalyst particle core in close proximity to the active sites that produced the high molecular weight polymer. These results demonstrate that the catalyst particle architecture directly affects polymer composition, offering the perspective of making high-performance polyethylene from a single reactor system using this modified Phillips catalyst.
Co-reporter:Dr. Gareth T. Whiting;Dr. Florian Meirer;Dr. Machteld M. Mertens;Dr. Anton-Jan Bons;Dr. Brian M. Weiss;Dr. Paul A. Stevens;Dr. Emiel deSmit; Bert M. Weckhuysen
ChemCatChem 2015 Volume 7( Issue 8) pp:1312-1321
Publication Date(Web):
DOI:10.1002/cctc.201402897
Abstract
Microspectroscopic methods were explored to investigate binder effects occurring in ZSM-5-containing SiO2- and Al2O3-bound millimetre-sized extrudates. Using thiophene as a selective probe for Brønsted acidity, coupled with time-resolved in situ UV/Vis and confocal fluorescence microspectroscopy, variations in reactivity and selectivity between the two distinct binder types were established. It was found that aluminium migration occurs in ZSM-5-containing Al2O3-bound extrudates, forming additional Brønsted acid sites. These sites strongly influence the oligomer selectivity, favouring the formation of thiol-like species (i.e., ring-opened species) in contrast to higher oligomers, predominantly formed on SiO2-bound ZSM-5-containing extrudates. Not only were the location and distribution of these oligomers visualised by 3 D analysis, it was also observed that more conjugated species appeared to grow off the surface of the zeolite ZSM-5 crystals (containing less conjugated species) into the surrounding binder material. Furthermore, a higher binder content resulted in an increasing overall reactivity owing to the greater number of stored thiophene monomers available per Brønsted acid site.
Co-reporter:Dr. Fiona Kirby;Anne-Eva Nieuwelink;Bonny W. M. Kuipers;Dr. Anton Kaiser;Dr. Pieter C. A. Bruijnincx;Dr. Bert M. Weckhuysen
Chemistry - A European Journal 2015 Volume 21( Issue 13) pp:5101-5109
Publication Date(Web):
DOI:10.1002/chem.201405906
Abstract
Glycerol is an attractive renewable building block for the synthesis of polyglycerols, which find application in the cosmetic and pharmaceutical industries. The selective etherification of glycerol to higher oligomers was studied in the presence of CaO colloids and the data are compared with those obtained from NaOH and CaO. The materials were prepared by dispersing CaO, CaCO3, or Ca(OH)2 onto a carbon nanofiber (CNF) support. Colloidal nanoparticles were subsequently dispensed from the CNF into the reaction mixture to give CaO colloids that have a higher activity than equimolar amounts of bulk CaO and NaOH. Optimization of the reaction conditions allowed us to obtain a product with Gardner color number <2, containing no acrolein and minimal cyclic byproducts. The differences in the CaO colloids originating from CNF and bulk CaO were probed using light scattering and conductivity measurements. The results confirmed that the higher activity of the colloids originating from CaO/CNF was due to their more rapid formation and smaller size compared with colloids from bulk CaO. We thus have developed a practical method for the synthesis of polyglycerols containing low amounts of Ca.
Co-reporter:E. M. van Schrojenstein Lantman;Dr. P. de Peinder;Dr. A. J. G. Mank; Dr. B. M. Weckhuysen
ChemPhysChem 2015 Volume 16( Issue 3) pp:547-554
Publication Date(Web):
DOI:10.1002/cphc.201402709
Abstract
Straightforward analysis of chemical processes on the nanoscale is difficult, as the measurement volume is linked to a discrete number of molecules, ruling out any ensemble averaging over rotation and diffusion processes. Raman spectroscopy is sufficiently selective for monitoring chemical changes, but is not sufficiently sensitive to be applied directly. Surface-enhanced Raman spectroscopy (SERS) can be applied for studying reaction kinetics, but adds additional variability in the signal as the enhancement factor is not the same for every location. A novel chemometric method described here separates reaction kinetics from short-term variability, based on the lack of fit in a principal-component analysis. We show that it is possible to study effects that occur on different time scales independently without data reduction using the photocatalytic reduction of p-nitrothiophenol as a showcase system. Using this approach a better description of the nanoscale reaction kinetics becomes available, while the short-term variations can be examined separately to examine reorientation and/or diffusion effects. It may even be possible to identify reaction intermediates through this approach. With only a limited number of reactive molecules in the studied volume, an intermediate on a SERS hot spot may temporarily dominate the spectrum. Now such events can be easily separated from the bulk conversion process by making use of this chemometric method.
Co-reporter:E. M. van Schrojenstein Lantman;Dr. P. de Peinder;Dr. A. J. G. Mank; Dr. B. M. Weckhuysen
ChemPhysChem 2015 Volume 16( Issue 3) pp:
Publication Date(Web):
DOI:10.1002/cphc.201500027
Abstract
The front cover artwork is provided by Prof. Bert Weckhuysen (Utrecht University, The Netherlands). The image outlines the experimental approach taken. Single Ag nanoparticles (grey) are deposited on a self-assembled monolayer of p-nitrothiophenol (pNTP, red) assembled on a flat Au substrate (yellow). The monolayer of pNTP can be reduced to p,p’-dimercaptoazobisbenzene (DMAB) through photocatalysis over plasmonic nanoparticles. By using a laser excitation of 785 nm (yellow arrow) the coupled plasmons of Au and Ag are excited and give sufficient surface enhanced Raman spectroscopy (SERS) enhancement effect to observe dynamics and kinetics over single nanoparticle hotspots. Read the full text of the article at 10.1002/cphc.201402709.
Co-reporter:E. M. van Schrojenstein Lantman;Dr. P. de Peinder;Dr. A. J. G. Mank; Dr. B. M. Weckhuysen
ChemPhysChem 2015 Volume 16( Issue 3) pp:
Publication Date(Web):
DOI:10.1002/cphc.201402775
Co-reporter:Dr. Emily C. Nordvang;Dr. Elena Borodina;Dr. Javier Ruiz-Martínez;Dr. Rasmus Fehrmann;Dr. Bert M. Weckhuysen
Chemistry - A European Journal 2015 Volume 21( Issue 48) pp:17324-17335
Publication Date(Web):
DOI:10.1002/chem.201503136
Abstract
The catalytic activity of large zeolite H-ZSM-5 crystals in methanol (MTO) and ethanol-to-olefins (ETO) conversions was investigated and, using operando UV/Vis measurements, the catalytic activity and deactivation was correlated with the formation of coke. These findings were related to in situ single crystal UV/Vis and confocal fluorescence micro-spectroscopy, allowing the observation of the spatiotemporal formation of intermediates and coke species during the MTO and ETO conversions. It was observed that rapid deactivation at elevated temperatures was due to the fast formation of aromatics at the periphery of the H-ZSM-5 crystals, which are transformed into more poly-aromatic coke species at the external surface, preventing the diffusion of reactants and products into and out of the H-ZSM-5 crystal. Furthermore, we were able to correlate the operando UV/Vis spectroscopy results observed during catalytic testing with the single crystal in situ results.
Co-reporter:Dimitrije Cicmil;Dr. Jurjen Meeuwissen;Dr. Aurélien Vantomme;Dr. Jian Wang;Ilse K. vanRavenhorst;Dr. Hendrik E. vanderBij;Dr. Ara Muñoz-Murillo;Dr. Bert M. Weckhuysen
Angewandte Chemie 2015 Volume 127( Issue 44) pp:13265-13271
Publication Date(Web):
DOI:10.1002/ange.201506718
Abstract
A triethylaluminium(TEAl)-modified Phillips ethylene polymerisation Cr/Ti/SiO2 catalyst has been developed with two distinct active regions positioned respectively in the inner core and outer shell of the catalyst particle. DRIFTS, EPR, UV-Vis-NIR DRS, STXM, SEM-EDX and GPC-IR studies revealed that the catalyst produces simultaneously two different polymers, i.e., low molecular weight linear-chain polyethylene in the Ti-abundant catalyst particle shell and high molecular weight short-chain branched polyethylene in the Ti-scarce catalyst particle core. Co-monomers for the short-chain branched polymer were generated in situ within the TEAl-impregnated confined space of the Ti-scarce catalyst particle core in close proximity to the active sites that produced the high molecular weight polymer. These results demonstrate that the catalyst particle architecture directly affects polymer composition, offering the perspective of making high-performance polyethylene from a single reactor system using this modified Phillips catalyst.
Co-reporter:Florian Meirer;Darius Morris;Santosh Soparawalla;Sam Kalirai;Gerbrand Mesu;Yijin Liu;Joy C. Andrews
Science Advances 2015 Volume 1(Issue 3) pp:e1400199
Publication Date(Web):03 Apr 2015
DOI:10.1126/sciadv.1400199
Macropore blocking through metal deposition and intrusion of particles is a major deactivation mechanism in FCC catalysts essential to gasoline production.
Co-reporter:Clare E. Harvey
Catalysis Letters 2015 Volume 145( Issue 1) pp:40-57
Publication Date(Web):2015 January
DOI:10.1007/s10562-014-1420-4
Surface-enhanced Raman spectroscopy (SERS) and tip-enhanced Raman spectroscopy (TERS) were until recently limited in their applicability to the majority of heterogeneous catalytic reactions. Recent developments begin to resolve the conflicting experimental requirements for SERS and TERS on the one hand, and heterogeneous catalysis on the other hand. This article discusses the development and use of SERS and TERS to study heterogeneous catalytic reactions, and the exciting possibilities that may now be within reach thanks to the latest technical developments. This will be illustrated with showcase examples from photo- and electrocatalysis.
Co-reporter:Jesper J. H. B. Sattler, Javier Ruiz-Martinez, Eduardo Santillan-Jimenez, and Bert M. Weckhuysen
Chemical Reviews 2014 Volume 114(Issue 20) pp:10613
Publication Date(Web):August 27, 2014
DOI:10.1021/cr5002436
Co-reporter:Hendrik E. van der Bij ; Dimitrije Cicmil ; Jian Wang ; Florian Meirer ; Frank M. F. de Groot
Journal of the American Chemical Society 2014 Volume 136(Issue 51) pp:17774-17787
Publication Date(Web):November 21, 2014
DOI:10.1021/ja508545m
In this work, three industrially relevant zeolites with framework topologies of MOR, FAU and FER have been explored on their ability to form an AlPO4 phase by reaction of a phosphate precursor with expelled framework aluminum. A detailed study was performed on zeolite H-mordenite, using in situ STXM and soft X-ray absorption tomography, complemented with 27Al and 31P magic angle spinning nuclear magnetic resonance (MAS NMR) spectroscopy, XRD, FT-IR spectroscopy, and N2 physisorption. Extraframework aluminum was extracted from steam-dealuminated H-mordenite and shown to dominantly consist of amorphous AlO(OH). It was found that phosphoric acid readily reacts with the AlO(OH) phase in dealuminated H-mordenite and forms an extraframework amorphous AlPO4 phase. It was found that while AlPO4 crystallizes outside of the zeolitic channel system forming AlPO4 islands, AlPO4 that remains inside tends to stay more amorphous. In the case of ultrastable zeolite Y the FAU framework collapsed during phosphatation, due to extraction of framework aluminum from the lattice. However, using milder phosphatation conditions an extraframework AlPO4 α-cristobalite/tridymite phase could also be produced within the FAU framework. Finally, in steamed zeolite ferrierite with FER topology the extraframework aluminum species were trapped and therefore not accessible for phosphoric acid; hence, no AlPO4 phase could be formed within the structure. Therefore, the parameters to be taken into account in AlPO4 synthesis are the framework Si/Al ratio, stability of framework aluminum, pore dimensionality and accessibility of extraframework aluminum species.
Co-reporter:Gareth T. Whiting, Florian Meirer, Diego Valencia, Machteld M. Mertens, Anton-Jan Bons, Brian M. Weiss, Paul A. Stevens, Emiel de Smit and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 39) pp:21531-21542
Publication Date(Web):2014/08/29
DOI:10.1039/C4CP03649B
Optical absorption and confocal fluorescence micro-spectroscopy were applied to investigate Brønsted acidity in millimetre-sized extrudates of Na(H)-ZSM-5 and SiO2 with varying ZSM-5 content. Partially (residual Na present) and fully proton-exchanged extrudates were employed, using thiophene oligomerization as a probe reaction. Time-resolved in situ optical absorption spectra and time dependent DFT calculations revealed several initial reaction pathways during the oligomerization reaction. In particular, it was found that protonated thiophene monomers reacted by either oligomerization (via reaction with un-reacted thiophene monomers) or ring-opening, depending on the Brønsted acid site density in each sample. Moreover, fully-exchanged extrudates not only have significantly higher reactivity than partially-exchanged samples, but they also favour the formation of ring-opening products, that are not formed on the partially-exchanged samples. Confocal fluorescence microscopy was employed to visualise non-invasively in 3D, the heterogeneity and homogeneity of thiophene oligomers on partially- and fully-exchanged extrudates, respectively. Furthermore, it was observed that extrudates with high binder content produce a higher relative amount of conjugated species, related with a higher quantity of available monomer in the binder, which is able to react further with intermediates adsorbed on active sites. Moreover, these conjugated species appear to form near the external surface of ZSM-5 crystals/agglomerates.
Co-reporter:Hendrik E. van der Bij and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 21) pp:9892-9903
Publication Date(Web):18 Dec 2013
DOI:10.1039/C3CP54791D
In order to elucidate phosphorus–zeolite H-ZSM-5 interactions, a variety of phosphorus-modified zeolite H-ZSM-5 materials were studied in a multi-spectroscopic manner. By deploying single pulse 27Al, 31P MAS NMR, 2D heteronuclear 27Al–31P correlation (HETCOR), 27Al MQ MAS NMR spectroscopy, TPD of pyridine monitored by FT-IR spectroscopy, and Scanning Transmission X-ray Microscopy (STXM) the interplay and influence of acidity, thermal treatment and phosphorus on the structure and acidity of H-ZSM-5 were established. It was found that while acid treatment did not affect the zeolite structure, thermal treatment leads to the breaking of Si–OH–Al bonds, a decrease in the strong acid site number and the formation of terminal Al–OH groups. No extra-framework aluminium species was observed. Phosphorus precursors interact with the zeolitic framework through hydrogen bonds and physical coordination, as phosphorus species can be simply washed out with hot water. After phosphatation and thermal treatment two effects occur simultaneously, namely (i) phosphorus species transform into water insoluble condensed poly-phosphoric acid and (ii) phosphoric acid binds irreversibly to the terminal Al–OH groups of partially dislodged four-coordinated framework aluminium, forming local silico-aluminophosphate interfaces. These interfaces are potentially the promoters of hydrothermal stability in phosphated zeolite H-ZSM-5.
Co-reporter:I. Lezcano-Gonzalez, U. Deka, B. Arstad, A. Van Yperen-De Deyne, K. Hemelsoet, M. Waroquier, V. Van Speybroeck, B. M. Weckhuysen and A. M. Beale
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 4) pp:1639-1650
Publication Date(Web):28 Nov 2013
DOI:10.1039/C3CP54132K
Three different types of NH3 species can be simultaneously present on Cu2+-exchanged CHA-type zeolites, commonly used in Ammonia Selective Catalytic Reduction (NH3-SCR) systems. These include ammonium ions (NH4+), formed on the Brønsted acid sites, [Cu(NH3)4]2+ complexes, resulting from NH3 coordination with the Cu2+ Lewis sites, and NH3 adsorbed on extra-framework Al (EFAl) species, in contrast to the only two reacting NH3 species recently reported on Cu-SSZ-13 zeolite. The NH4+ ions react very slowly in comparison to NH3 coordinated to Cu2+ ions and are likely to contribute little to the standard NH3-SCR process, with the Brønsted groups acting primarily as NH3 storage sites. The availability/reactivity of NH4+ ions can be however, notably improved by submitting the zeolite to repeated exchanges with Cu2+, accompanied by a remarkable enhancement in the low temperature activity. Moreover, the presence of EFAl species could also have a positive influence on the reaction rate of the available NH4+ ions. These results have important implications for NH3 storage and availability in Cu-Chabazite-based NH3-SCR systems.
Co-reporter:Marleen M. Kerssens, Christoph Sprung, Gareth T. Whiting, Bert M. Weckhuysen
Microporous and Mesoporous Materials 2014 Volume 189() pp:136-143
Publication Date(Web):1 May 2014
DOI:10.1016/j.micromeso.2013.10.015
•We provide a methodical overview of probing acidity in zeolites.•Confocal fluorescence microscopy is introduced in detail.•Developed methods on model systems are transferred to industrially relevant catalysts.•Selective staining of zeolites, FCC particles, and extrudates.•Perspective outlook on future acidity probing in zeolites.This perspective article highlights recent methodical approaches for probing acid sites in zeolites using selective chemical staining methods. Research and method development on model systems (large zeolite crystals) is presented in close relation to the investigation of industrially relevant catalyst particles for Fluid Catalytic Cracking (FCC) and zeolite-based extrudates. The article begins with an (1) introduction on characteristics of zeolites and industrial catalyst particles, followed by a methodical overview on (2) probing acidity in zeolites, including temperature programmed desorption (TPD), solid state nuclear magnetic resonance (NMR) spectroscopy, and infrared (IR) spectroscopy. Instrumental details on (3) fluorescence microscopy are provided to prepare for the focus on (4) selective staining by probe molecules and oligomerisation reactions to highlight or distinguish materials (i.e. zeolite vs. binder or matrix components) and visualise acid sites within zeolites. Confocal fluorescence yields, in contrast to the other discussed techniques, a high spatiotemporal resolution giving way to exciting prospects such as probing single (fluorescent) reaction products. (5) Concluding remarks and future perspectives envision how Brønsted and Lewis acid sites can be investigated selectively.Sections: (1) Introduction (2) Probing acidity – methodical overview (3) Confocal fluorescence microscopy – instrumentation (4) Selective staining (5) Concluding Remarks and Future Perspectives.
Co-reporter:Zoran Ristanovi&x107; ;Dr. Bert M. Weckhuysen
Angewandte Chemie 2014 Volume 126( Issue 33) pp:8696-8698
Publication Date(Web):
DOI:10.1002/ange.201404463
Co-reporter:Jesper J. H. B. Sattler;Dr. Ines D. Gonzalez-Jimenez;Dr. Lin Luo;Brien A. Stears;Dr. Andrzej Malek;Dr. David G. Barton;Dr. Beata A. Kilos;Dr. Mark P. Kaminsky;Tiny W. G. M. Verhoeven;Eline J. Koers;Dr. Marc Baldus;Dr. Bert M. Weckhuysen
Angewandte Chemie 2014 Volume 126( Issue 35) pp:9405-9410
Publication Date(Web):
DOI:10.1002/ange.201404460
Abstract
A novel catalyst material for the selective dehydrogenation of propane is presented. The catalyst consists of 1000 ppm Pt, 3 wt % Ga, and 0.25 wt % K supported on alumina. We observed a synergy between Ga and Pt, resulting in a highly active and stable catalyst. Additionally, we propose a bifunctional active phase, in which coordinately unsaturated Ga3+ species are the active species and where Pt functions as a promoter.
Co-reporter:Zoran Ristanovi&x107; ;Dr. Bert M. Weckhuysen
Angewandte Chemie International Edition 2014 Volume 53( Issue 33) pp:8556-8558
Publication Date(Web):
DOI:10.1002/anie.201404463
Co-reporter:Jesper J. H. B. Sattler;Dr. Ines D. Gonzalez-Jimenez;Dr. Lin Luo;Brien A. Stears;Dr. Andrzej Malek;Dr. David G. Barton;Dr. Beata A. Kilos;Dr. Mark P. Kaminsky;Tiny W. G. M. Verhoeven;Eline J. Koers;Dr. Marc Baldus;Dr. Bert M. Weckhuysen
Angewandte Chemie International Edition 2014 Volume 53( Issue 35) pp:9251-9256
Publication Date(Web):
DOI:10.1002/anie.201404460
Abstract
A novel catalyst material for the selective dehydrogenation of propane is presented. The catalyst consists of 1000 ppm Pt, 3 wt % Ga, and 0.25 wt % K supported on alumina. We observed a synergy between Ga and Pt, resulting in a highly active and stable catalyst. Additionally, we propose a bifunctional active phase, in which coordinately unsaturated Ga3+ species are the active species and where Pt functions as a promoter.
Co-reporter:Dr. Matthijs Ruitenbeek;Dr. Bert M. Weckhuysen
Angewandte Chemie International Edition 2014 Volume 53( Issue 42) pp:11137-11139
Publication Date(Web):
DOI:10.1002/anie.201407109
Co-reporter:Hendrik E. van der Bij;Dr. Luis R. Aramburo;Dr. Bjørnar Arstad;Dr. James J. Dynes;Dr. Jian Wang; Dr. Bert M. Weckhuysen
ChemPhysChem 2014 Volume 15( Issue 2) pp:283-292
Publication Date(Web):
DOI:10.1002/cphc.201300910
Abstract
A variety of phosphated zeolite H-ZSM-5 samples are investigated by using a combination of Fourier transfer infrared (FTIR) spectroscopy, single pulse 27Al, 29Si, 31P, 1H-31P cross polarization (CP), 27Al-31P CP, and 27Al 3Q magic angle spinning nuclear magnetic resonance (MAS NMR) spectroscopy, scanning transmission X-ray microscopy (STXM) and N2 physisorption. This approach leads to insights into the physicochemical processes that take place during phosphatation. Direct phosphatation of H-ZSM-5 promotes zeolite aggregation, as phosphorus does not penetrate deep into the zeolite material and is mostly found on and close to the outer surface of the zeolite, acting as a glue. Phosphatation of pre-steamed H-ZSM-5 gives rise to the formation of a crystalline tridymite AlPO4 phase, which is found in the mesopores of dealuminated H-ZSM-5. Framework aluminum species interacting with phosphorus are not affected by hydrothermal treatment. Dealuminated H-ZSM-5, containing AlPO4, retains relatively more framework Al atoms and acid sites during hydrothermal treatment than directly phosphated H-ZSM-5.
Co-reporter:Dr. Christoph Sprung ;Dr. Bert M. Weckhuysen
Chemistry - A European Journal 2014 Volume 20( Issue 13) pp:3667-3677
Publication Date(Web):
DOI:10.1002/chem.201303549
Abstract
Confocal fluorescence microscopy was employed to selectively visualize the dispersion and orientation of zeolite ZSM-5 domains inside a single industrially applied fluid catalytic cracking (FCC) catalyst particle. Large ZSM-5 crystals served as a model system together with the acid-catalyzed fluorostyrene oligomerization reaction to study the interaction of plane-polarized light with these anisotropic zeolite crystals. The distinction between zeolite and binder material, such as alumina, silica, and clay, within an individual FCC particle was achieved by utilizing the anisotropic nature of emitted fluorescence light arising from the entrapped fluorostyrene-derived carbocations inside the zeolite channels. This characterization approach provides a non-invasive way for post-synthesis characterization of an individual FCC catalyst particle in which the size, distribution, orientation, and amount of zeolite ZSM-5 aggregates can be determined. It was found that the amount of detected fluorescence light originating from the stained ZSM-5 aggregates corresponds to about 15 wt %. Furthermore, a statistical analysis of the emitted fluorescence light indicated that a large number of the ZSM-5 domains appeared in small sizes of about 0.015–0.25 μm2, representing single zeolite crystallites or small aggregates thereof. This observation illustrated a fairly high degree of zeolite dispersion within the FCC binder material. However, the highest amount of crystalline material was aggregated into larger domains (ca. 1–5 μm2) with more or less similarly oriented zeolite crystallites. It is clear that this visualization approach may serve as a post-synthesis quality control on the dispersion of zeolite ZSM-5 crystallites within FCC particles.
Co-reporter:Hendrik E. vanderBij;Dr. Florian Meirer;Sam Kalirai;Dr. Jian Wang;Dr. Bert M. Weckhuysen
Chemistry - A European Journal 2014 Volume 20( Issue 51) pp:
Publication Date(Web):
DOI:10.1002/chem.201490213
Co-reporter:Qingyun Qian;Dr. Javier Ruiz-Martínez; Mohamed Mokhtar; Abdullah M. Asiri; Shaeel A. Al-Thabaiti; Suliman N. Basahel;Dr. Bert M. Weckhuysen
ChemCatChem 2014 Volume 6( Issue 3) pp:772-783
Publication Date(Web):
DOI:10.1002/cctc.201300962
Abstract
In situ synchrotron-based IR and UV/Vis micro-spectroscopy combined with isotopically labeled reactants have been used to identify the different hydrocarbon species formed as well as to assess the activity and accessibility of individual 50 μm-sized SAPO-34 crystals. For the methanol-to-olefins process, two reaction stages can be distinguished. The first involves the formation of methoxy species, protonated dimethyl ether, and polyalkylated benzene (PAB) carbocations, which do not affect the accessibility of the SAPO-34 crystal. In addition, methoxy species are very dynamic during this stage. The second stage is related to the formation of polyaromatic (PA) species concentrated in the outer rim of the crystal, which are bulky and interact with the acid sites and thus alter the overall accessibility of the crystal. In contrast, the ethanol-to-olefins process only consists of one major stage, as the formation of PAB and PA species cannot be separated. Furthermore, the formation of these species is more internal, and coke formation is mainly concentrated in a layer located in the inner part of the SAPO-34 crystal.
Co-reporter:Qingyun Qian;Dr. Javier Ruiz-Martínez; Mohamed Mokhtar; Abdullah M. Asiri; Shaeel A. Al-Thabaiti; Suliman N. Basahel;Dr. Bert M. Weckhuysen
ChemCatChem 2014 Volume 6( Issue 3) pp:
Publication Date(Web):
DOI:10.1002/cctc.201400102
Abstract
The front cover artwork for Issue 3/2014 is provided by the Inorganic Chemistry and Catalysis group of Utrecht University. The image shows the concept of synchrotron-based IR and UV/Vis microspectroscopy monitoring a catalytic reaction on a single SAPO-34 crystal and the obtained spatially resolved information on the retained hydrocarbon species and crystal accessibility. See the Full Paper itself on http://dx.doi.org/10.1002/cctc.201300962.
Co-reporter:Qingyun Qian;Dr. Javier Ruiz-Martínez; Mohamed Mokhtar; Abdullah M. Asiri; Shaeel A. Al-Thabaiti; Suliman N. Basahel;Dr. Bert M. Weckhuysen
ChemCatChem 2014 Volume 6( Issue 3) pp:
Publication Date(Web):
DOI:10.1002/cctc.201490014
Co-reporter:Jesper J. H. B. Sattler;Ad M. Mens ;Dr. Bert M. Weckhuysen
ChemCatChem 2014 Volume 6( Issue 11) pp:3139-3145
Publication Date(Web):
DOI:10.1002/cctc.201402649
Abstract
Combined operando UV/vis–Raman spectroscopy has been used to study the deactivation of CrOx/Al2O3 catalyst extrudates in a pilot scale propane dehydrogenation reactor. For this purpose, UV/vis and Raman optical fiber probes have been designed, constructed and tested. The light absorption measured by operando UV/vis spectroscopy was used to correct for the effect of catalyst darkening. This makes operando Raman spectroscopy a quantitative technique to follow the formation of coke deposits on-line during the first hour of catalytic propane dehydrogenation process. The probe was used to study the coke deposition at the top and bottom of the reactor. Differences in the rate of coke deposition were observed, which were related to the local temperature of the catalyst bed. The chemical nature of the coke deposits formed on a catalyst extrudate, as expressed by the D1/G ratio, was independent of reaction time as well as of the position of the catalyst extrudate within the reactor bed.
Co-reporter:Dr. Qingyun Qian;Charlotte Vogt;Dr. Mohamed Mokhtar;Dr. Abdullah M. Asiri;Dr. Shaeel A. Al-Thabaiti;Dr. Suliman N. Basahel;Dr. Javier Ruiz-Martínez;Dr. Bert M. Weckhuysen
ChemCatChem 2014 Volume 6( Issue 12) pp:3396-3408
Publication Date(Web):
DOI:10.1002/cctc.201402714
Abstract
The methanol-to-olefins (MTO) process over H-SAPO-34 is investigated by using an operando approach combining UV/Vis and IR spectroscopies with on-line mass spectrometry. Methanol, methoxy, and protonated dimethyl ether are the major species during the induction period, whereas polyalkylated benzenes and polyaromatic species are encountered in the active stage of the MTO process. The accessibility of SAPO-34 is linked with the amount of methoxy species, whereas the formation of polyaromatic species that block the pores is the main cause of deactivation. Furthermore, the reaction pathways responsible for the formation of olefins and polyaromatics co-exist and compete during the whole MTO process, and both routes are directly related to the amount of surface polyalkylated benzene carbocations and methoxy species. Hence, a first-order kinetic model is proposed and comparable activation energies for both processes are obtained.
Co-reporter:Evelien M. vanSchrojensteinLantman;Dr. Onno L. J. Gijzeman;Dr. Arjan J. G. Mank;Dr. Bert M. Weckhuysen
ChemCatChem 2014 Volume 6( Issue 12) pp:3342-3346
Publication Date(Web):
DOI:10.1002/cctc.201402647
Abstract
Heterogeneous catalysis is a surface phenomenon. Yet, though the catalysis itself takes place on surfaces, the reactants and products rapidly take the form of another physical state, as either a liquid or a gas. Catalytic reactions within a self-assembled monolayer are confined within two dimensions, as the molecules involved do not leave the surface. Surface-enhanced Raman spectroscopy is an ideal technique to probe these self-assembled monolayers as it gives molecular information in a measured volume limited to the surface. We show how surface-enhanced Raman spectroscopy can be used to determine the reaction kinetics of a two-dimensional reaction. As a proof of principle, we study the photocatalytic reduction of p-nitrothiophenol. A study of the reaction rate and dilution effects leads to the conclusion that a dimerization must take place as one of the reaction steps.
Co-reporter:Hendrik E. vanderBij;Dr. Florian Meirer;Sam Kalirai;Dr. Jian Wang;Dr. Bert M. Weckhuysen
Chemistry - A European Journal 2014 Volume 20( Issue 51) pp:16922-16932
Publication Date(Web):
DOI:10.1002/chem.201404924
Abstract
The nature behind the promotional effect of phosphorus on the catalytic performance and hydrothermal stability of zeolite H-ZSM-5 has been studied using a combination of 27Al and 31P MAS NMR spectroscopy, soft X-ray absorption tomography and n-hexane catalytic cracking, complemented with NH3 temperature-programmed desorption and N2 physisorption. Phosphated H-ZSM-5 retains more acid sites and catalytic cracking activity after steam treatment than its non-phosphated counterpart, while the selectivity towards propylene is improved. It was established that the stabilization effect is twofold. First, the local framework silico-aluminophosphate (SAPO) interfaces, which form after phosphatation, are not affected by steam and hold aluminum atoms fixed in the zeolite lattice, preserving the pore structure of zeolite H-ZSM-5. Second, the four-coordinate framework aluminum can be forced into a reversible sixfold coordination by phosphate. These species remain stationary in the framework under hydrothermal conditions as well. Removal of physically coordinated phosphate after steam-treatment leads to an increase in the number of strong acid sites and increased catalytic activity. We propose that the improved selectivity towards propylene during catalytic cracking can be attributed to local SAPO interfaces located at channel intersections, where they act as impediments in the formation of bulky carbenium ions and therefore suppress the bimolecular cracking mechanism.
Co-reporter:Carlo Angelici;Marjolein E. Z. Velthoen; Bert M. Weckhuysen;Dr. Pieter C. A. Bruijnincx
ChemSusChem 2014 Volume 7( Issue 9) pp:2505-2515
Publication Date(Web):
DOI:10.1002/cssc.201402361
Abstract
Silica–magnesia (Si/Mg=1:1) catalysts were studied in the one-pot conversion of ethanol to butadiene. The catalyst synthesis method was found to greatly influence morphology and performance, with materials prepared through wet-kneading performing best both in terms of ethanol conversion and butadiene yield. Detailed characterization of the catalysts synthesized through co-precipitation or wet-kneading allowed correlation of activity and selectivity with morphology, textural properties, crystallinity, and acidity/basicity. The higher yields achieved with the wet-kneaded catalysts were attributed to a morphology consisting of SiO2 spheres embedded in a thin layer of MgO. The particle size of the SiO2 catalysts also influenced performance, with catalysts with smaller SiO2 spheres showing higher activity. Temperature-programmed desorption (TPD) measurements showed that best butadiene yields were obtained with SiO2–MgO catalysts characterized by an intermediate amount of acidic and basic sites. A Hammett indicator study showed the catalysts’ pKa value to be inversely correlated with the amount of dehydration by-products formed. Butadiene yields could be further improved by the addition of 1 wt % of CuO as promoter to give butadiene yields and selectivities as high as 40 % and 53 %, respectively. The copper promoter boosts the production of the acetaldehyde intermediate changing the rate-determining step of the process. TEM-energy-dispersive X-ray (EDX) analyses showed CuO to be present on both the SiO2 and MgO components. UV/Vis spectra of promoted catalysts in turn pointed at the presence of cluster-like CuO species, which are proposed to be responsible for the increased butadiene production.
Co-reporter:Anna L. Jongerius, John R. Copeland, Guo Shiou Foo, Jan P. Hofmann, Pieter C. A. Bruijnincx, Carsten Sievers, and Bert M. Weckhuysen
ACS Catalysis 2013 Volume 3(Issue 3) pp:464
Publication Date(Web):February 8, 2013
DOI:10.1021/cs300684y
The stability of a 1 wt % Pt/γ-Al2O3 catalyst was tested in an ethanol/water mixture at 225 °C and autogenic pressure, conditions at which it is possible to dissolve and depolymerize various kinds of lignin, and structural changes to the catalysts were studied by means of X-ray diffraction (XRD), 27Al MAS NMR, N2 physisorption, transmission electron microscopy (TEM), H2 chemisorption, elemental analysis, thermogravimetric analysis-mass spectrometry (TGA-MS), and IR. In the absence of reactants the alumina support is found to transform into boehmite within 4 h, leading to a reduction in support surface area, sintering of the supported Pt nanoparticles, and a reduction of active metal surface area. Addition of aromatic oxygenates to mimic the compounds typically obtained by lignin depolymerization leads to a slower transformation of the support oxide. These compounds, however, were not able to slow down the decrease in dispersion of the Pt nanoparticles. Vanillin and guaiacol stabilize the aluminum oxide more than phenol, anisole, and benzaldehyde because of the larger number of oxygen functionalities that can interact with the alumina. Interestingly, catalyst samples treated in the presence of lignin showed almost no formation of boehmite, no reduction in support or active metal surface area, and no Pt nanoparticle sintering. Furthermore, in the absence of lignin-derived aromatic oxygenates, ethanol forms a coke-like layer on the catalyst, while oxygenates prevent this by adsorption on the support by coordination via the oxygen functionalities.Keywords: alumina; biomass; boehmite; hydration; hydrothermal stability
Co-reporter:Emma K. Gibson, Mathijs W. Zandbergen, Simon D. M. Jacques, Cai Biao, Robert J. Cernik, Matthew G. O’Brien, Marco Di Michiel, Bert M. Weckhuysen, and Andrew M. Beale
ACS Catalysis 2013 Volume 3(Issue 3) pp:339
Publication Date(Web):January 15, 2013
DOI:10.1021/cs300746a
A combination of X-ray absorption microcomputed tomography (μ-CT) and diagonal offset raman spectroscopy (DORS) have been used to follow in real time the 2-D and 3-D evolution of Mo species within 3 mm γ-Al2O3 extrudates during catalyst impregnation and drying processes. In a first set of experiments, we have followed the real-time incipient wetness impregnation process using an aqueous solution of ammonium heptamolybdate (AHM). We observed that during the equilibration period, singly impregnated samples formed Al(OH)6Mo6O183– (Al–Mo) hot spots distributed over the entire sample volume and that these heterogeneities grow in number and size as a function of time. A second set of measurements focused on the coimpregnation of AHM with H3PO4 and the subsequent equilibration and drying stages. It was found that the presence of phosphorus in the impregnating solution prevented the formation of the hot spots via the formation of weakly bound HxP2Mo5O23(6–x)– species that were uniformly distributed over the sample after 70 min of equilibration. During drying, however, these species migrated to the periphery of the sample, resulting in an egg shell distribution of HxP2Mo5O23(6–x)–. We show that by performing these studies noninvasively with a sufficiently high time resolution, the behavior and evolution of the Mo species were reproduced more faithfully than by using more conventional and invasive cut-and-measure approaches.Keywords: catalyst bodies; in situ; incipient wetness impregnation; spatially resolved
Co-reporter:Upakul Deka, Ines Lezcano-Gonzalez, Bert M. Weckhuysen, and Andrew M. Beale
ACS Catalysis 2013 Volume 3(Issue 3) pp:413
Publication Date(Web):January 22, 2013
DOI:10.1021/cs300794s
Cu-exchanged zeolites have demonstrated widespread use as catalyst materials in the abatement of NOx, especially from mobile sources. Recent studies focusing on Cu-exchanged zeolites with the CHA structure have demonstrated them to be excellent catalysts in the ammonia-assisted selective catalytic reduction (NH3-SCR) of NOx. Thorough characterization of these materials using state-of-the-art techniques has led to a significant improvement in the understanding of active sites present, which contributes toward a fundamental understanding of the catalytic processes and the rational design of new materials; however, the availability of multiple techniques at our disposal has led to various observations and conclusions on the nature of the active sites. This article begins with a brief introduction to exhaust emission control in the mobile sector, followed by an overview of hydrocarbon-SCR and NH3-SCR; the former technology having found common use in light duty passenger vehicles, whereas the latter are applied for medium (or heavy) duty vehicles, such as trucks and busses. This is followed by an overview of zeolite-based catalysts, especially for NH3-SCR reaction with a focus toward zeolites known to possess high activity. They include zeolites Y (FAU framework), ZSM-5 (MFI framework), SSZ-13 (CHA framework), and (briefly) zeolite Beta (BEA framework). A few common techniques used for the characterization of zeolites and the information that they bring to help determine the salient structural and mechanistic aspects of the NH3-SCR process are introduced. The combination and comparison of the information obtained from the approaches have resulted in an accurate elucidation of the local geometry and environment of Cu within zeolites, thus forming the active site. The article further focuses on three main aspects: (a) the crystallographic cation location of Cu within the structures as compared to results from techniques more sensitive to the local environment; (b) the interaction of Cu at these sites with reactant or probe molecules, which illustrates their (potential) mobility and accessibility; and (c) the proposed active sites within the zeolites ZSM-5, Y, and SSZ-13 as evident in literature. The discussion is focused toward the influence of the zeolite structure, from both a long-range perspective and that of the local structure around the active Cu species, on the thus formed active sites and their implications toward the NH3-SCR reaction.Keywords: active sites; Cu-zeolites; NH3-SCR
Co-reporter:Anna L. Jongerius, Pieter C. A. Bruijnincx and Bert M. Weckhuysen
Green Chemistry 2013 vol. 15(Issue 11) pp:3049-3056
Publication Date(Web):31 Jul 2013
DOI:10.1039/C3GC41150H
A two-step approach to the conversion of organosolv, kraft and sugarcane bagasse lignin to monoaromatic compounds of low oxygen content is presented. The first step consists of lignin depolymerization in a liquid phase reforming (LPR) reaction over a 1 wt% Pt/γ-Al2O3 catalyst at 225 °C in alkaline ethanol–water. The first LPR step resulted in a decrease in lignin molecular weight of 32%, 57% and 27% for organosolv, kraft and bagasse lignin, respectively. GC analysis of the depolymerized lignin reaction mixture furthermore showed the formation of alkylated phenol, guaiacol and syringol-type products in 11%, 9% and 5% yields from organosolv, kraft and bagasse lignin, respectively. The lignin-oil that was isolated by extraction of the ethanol–water solution was subjected to a subsequent hydrodeoxygenation (HDO) reaction in the second conversion step. HDO of the lignin-oil was performed in dodecane at 300 °C under 50 bar hydrogen pressure over CoMo/Al2O3 and Mo2C/CNF catalysts. GC analysis of the product mixture obtained after the two-step LPR–HDO process revealed the formation of, amongst others, benzene, toluene, xylenes and ethylmethylbenzenes. Of the total observed monomeric products (9%), 25% consisted of these oxygen-free products. Notably, such products cannot be obtained by direct HDO of lignin. HDO of the lignin-oil at 350 °C resulted in the conversion of all tris-oxygenated products, with 57% of the observed monomeric products now identified as mono-oxygenated phenolics.
Co-reporter:Korneel H. Cats, Ines D. Gonzalez-Jimenez, Yijin Liu, Johanna Nelson, Douglas van Campen, Florian Meirer, Ad M. J. van der Eerden, Frank M. F. de Groot, Joy C. Andrews and Bert M. Weckhuysen
Chemical Communications 2013 vol. 49(Issue 41) pp:4622-4624
Publication Date(Web):03 Apr 2013
DOI:10.1039/C3CC00160A
Transmission X-ray microscopy has been used to investigate individual Co/TiO2 Fischer–Tropsch (FT) catalyst particles in 2-D and 3-D with 30 nm spatial resolution. Tomographic elemental mapping showed that Co is heterogeneously concentrated in the centre of the catalyst particles. In addition, it was found that Co is mostly metallic during FT at 250 °C and 10 bar. No evidence for Co oxidation was found.
Co-reporter:J. J. H. B. Sattler, I. D. González-Jiménez, A. M. Mens, M. Arias, T. Visser and B. M. Weckhuysen
Chemical Communications 2013 vol. 49(Issue 15) pp:1518-1520
Publication Date(Web):04 Jan 2013
DOI:10.1039/C2CC38978A
A novel operando UV-Vis spectroscopic set-up has been constructed and tested for the investigation of catalyst bodies loaded in a pilot-scale reactor under relevant reaction conditions. Spatiotemporal insight into the formation and burning of coke deposits on an industrial CrOx/Al2O3 catalyst during propane dehydrogenation has been obtained.
Co-reporter:Peter J. C. Hausoul, Sinedu D. Tefera, Jelle Blekxtoon, Pieter C. A. Bruijnincx, Robertus J. M. Klein Gebbink and Bert M. Weckhuysen
Catalysis Science & Technology 2013 vol. 3(Issue 5) pp:1215-1223
Publication Date(Web):01 Nov 2012
DOI:10.1039/C2CY20522J
The Pd/TOMPP-catalysed (TOMPP = tris(2-methoxyphenyl)phosphine) telomerisation of 1,3-butadiene was studied under solvent- and base-free conditions with phenolic substrates that can be potentially derived from lignin. Large differences in catalytic activity were observed, with reactivity increasing in the order of phenol, p-cresol, guaiacol, creosol and syringol. This reactivity trend can be attributed to the substrates' relative nucleophilicities, as induced by the donating effects of the p-methyl and o-methoxy substituents. The chosen reaction conditions, i.e. temperature, ligand/metal and butadiene/substrate ratios, strongly influenced both the conversion and selectivity of the reaction. Remarkably, the composition of the reaction medium, i.e. the butadiene/substrate ratio, exerted a strong influence on the linear/branched ratio. High conversions and selectivities to the linear products are obtained when excess butadiene is used. The linear telomer products could be readily converted from O-alkylated to C-alkylated phenolics via the thermal Claisen rearrangement. High conversions and selectivities were observed after 2 hours at 200 °C. Branched o-octadienyl phenols were obtained in all cases except for the syringol telomer which gave the linear p-octadienyl product exclusively.
Co-reporter:Luis R. Aramburo, Javier Ruiz-Martínez, Jan P. Hofmann and Bert M. Weckhuysen
Catalysis Science & Technology 2013 vol. 3(Issue 5) pp:1208-1214
Publication Date(Web):01 Nov 2012
DOI:10.1039/C2CY20661G
Confocal fluorescence microscopy has been used in combination with bulky non-reactive dyes (i.e. proflavine, stilbene and nile blue A) and two staining reactions (i.e. fluorescein synthesis and 4-fluorostyrene oligomerisation) to study the effect of steaming on pore accessibility and acidity of large ZSM-5 zeolite crystals. This approach enabled the 3-D visualization of cracks and mesopores connected to the outer zeolite surface as well as mesoporous “cavities” within steamed ZSM-5 zeolite crystals. It has been found that besides the generation of mesoporosity steaming makes the boundaries between the different crystal sub-units accessible for bulky molecules. Additionally, the fluorescein staining reaction reveals prominent formation of structural defects that are connected to the surface of the crystal via the microporous ZSM-5 system and which contain either Brønsted or Lewis acid sites. On the other hand, the 4-fluorostyrene staining reaction shows how mild steaming conditions increase the accessibility towards the Brønsted acid sites, while under severe steaming conditions the Brønsted acidity contained in the internal crystal sub-units is more accessible, although it is preferentially removed close to the surface of the lateral sub-units of ZSM-5 zeolite crystals.
Co-reporter:Jesper J. H. B. Sattler, Andrew M. Beale and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 29) pp:12095-12103
Publication Date(Web):11 Apr 2013
DOI:10.1039/C3CP50646K
The deactivation of 0.5 wt% Pt/Al2O3 and 0.5 wt% Pt–1.5 wt% Sn/Al2O3 catalysts has been studied by operando Raman spectroscopy during the dehydrogenation of propane and subsequent regeneration in air for 10 successive dehydrogenation–regeneration cycles. Furthermore, the reaction feed was altered by using different propane/propene/hydrogen ratios. It was found that the addition of hydrogen to the feed increases the catalyst performance and decreases the formation of coke deposits, as was revealed by thermogravimetrical analysis. The positive effect of hydrogen on the catalyst performance is comparable to the addition of Sn, a promoter element which increases both the propane conversion and propene selectivity. Operando Raman spectroscopy showed that hydrogen altered the nature of the coke deposits formed during propane dehydrogenation. Due to this approach it was possible to perform a systematic deconvolution procedure on the Raman spectra. By analysing the related intensity, band position and bandwidth of the different Raman features, it was determined that smaller graphite crystallites, which have less defects, are formed when the partial pressure of hydrogen in the feed was increased.
Co-reporter:Inge L.C. Buurmans, Fouad Soulimani, Javier Ruiz-Martínez, Hendrik E. van der Bij, Bert M. Weckhuysen
Microporous and Mesoporous Materials 2013 Volume 166() pp:86-92
Publication Date(Web):15 January 2013
DOI:10.1016/j.micromeso.2012.08.007
A synchrotron-based infrared micro-spectroscopy study has been conducted to investigate the structure as well as the Brønsted and Lewis acidity of Fluid Catalytic Cracking (FCC) catalyst particles at the individual particle level. Both fresh and laboratory-deactivated catalyst particles have been studied. The applied deactivation protocols were steaming (ST), two-step cyclic deactivation (CD) and Mitchell impregnation-steam deactivation (MI). In addition, an equilibrium catalyst (Ecat) taken from a real cracking unit has been investigated. From the infrared spectra of the fresh and laboratory-deactivated samples it was clear that the zeolite component experiences partial collapse upon deactivation. Furthermore, it was found that characteristic bands, caused by the presence of clay material, are lost upon deactivation. After pyridine adsorption, the acidity of the samples could be monitored. Both Brønsted and Lewis acidity decreased in the following order: Fresh > ST > CD > MI. The Ecat sample was found to display acidity in between those of CD and MI samples. These findings are in line with earlier bulk transmission infrared as well as ammonia temperature programmed desorption measurements, which confirms the validity of acidity measurements at the single particle level. However, additional information about the distribution of Brønsted and Lewis acidity within individual catalyst particles becomes available. The developed approach reveals a larger variety in the amount of Brønsted acid sites for individual Ecat particles as compared to CD and MI particles. This observation can be attributed to the wide age distribution within industrial equilibrium catalysts and directly shows the added value of micro-spectroscopy approaches in the investigation of interparticle heterogeneities.Graphical abstractHighlights► Infrared micro-spectroscopy enables the study of individual cracking catalyst particles. ► Structural changes in the zeolite and clay component are observed upon deactivation. ► Industrial deactivation protocols lead to a decrease in Brønsted and Lewis acidity. ► Equilibrium catalysts display large heterogeneities in acidity.
Co-reporter:Dr. Javier Ruiz-Martínez;Dr. Andrew M. Beale;Dr. Upakul Deka;Dr. Mathew G. O'Brien;Dr. Paul D. Quinn;Dr. J. Fred W. Mosselmans;Dr. Bert M. Weckhuysen
Angewandte Chemie 2013 Volume 125( Issue 23) pp:6099-6103
Publication Date(Web):
DOI:10.1002/ange.201210030
Co-reporter:Zoran Ristanovi&x107;;Dr. Jan P. Hofmann;Dr. Upakul Deka;Dr. Tobias U. Schülli;Dr. Marcus Rohnke;Dr. Andrew M. Beale;Dr.Ir. Bert M. Weckhuysen
Angewandte Chemie 2013 Volume 125( Issue 50) pp:13624-13628
Publication Date(Web):
DOI:10.1002/ange.201306370
Co-reporter:Dr. Javier Ruiz-Martínez;Dr. Andrew M. Beale;Dr. Upakul Deka;Dr. Mathew G. O'Brien;Dr. Paul D. Quinn;Dr. J. Fred W. Mosselmans;Dr. Bert M. Weckhuysen
Angewandte Chemie International Edition 2013 Volume 52( Issue 23) pp:5983-5987
Publication Date(Web):
DOI:10.1002/anie.201210030
Co-reporter:Zoran Ristanovi&x107;;Dr. Jan P. Hofmann;Dr. Upakul Deka;Dr. Tobias U. Schülli;Dr. Marcus Rohnke;Dr. Andrew M. Beale;Dr.Ir. Bert M. Weckhuysen
Angewandte Chemie International Edition 2013 Volume 52( Issue 50) pp:13382-13386
Publication Date(Web):
DOI:10.1002/anie.201306370
Co-reporter:Dr. Bert M. Weckhuysen
ChemSusChem 2013 Volume 6( Issue 9) pp:1559-1563
Publication Date(Web):
DOI:10.1002/cssc.201300602
Co-reporter:Dr. Bert M. Weckhuysen
ChemSusChem 2013 Volume 6( Issue 9) pp:
Publication Date(Web):
DOI:10.1002/cssc.201300884
Co-reporter:Veronique VanSpeybroeck;Karen Hemelsoet;Kristof DeWispelaere;Qingyun Qian;Jeroen VanderMynsbrugge;Bart DeSterck;Michel Waroquier
ChemCatChem 2013 Volume 5( Issue 1) pp:173-184
Publication Date(Web):
DOI:10.1002/cctc.201200580
Abstract
The formation and nature of active sites for methanol conversion over solid acid catalyst materials are studied by using a unique combined spectroscopic and theoretical approach. A working catalyst for the methanol-to-olefin conversion has a hybrid organic–inorganic nature in which a cocatalytic organic species is trapped in zeolite pores. As a case study, microporous materials with the chabazite topology, namely, H-SAPO-34 and H-SSZ-13, are considered with trapped (poly)aromatic species. First-principle rate calculations on methylation reactions and in situ UV/Vis spectroscopy measurements are performed. The theoretical results show that the structure of the organic compound and zeolite composition determine the methylation rates: 1) the rate increases by 6 orders of magnitude if more methyl groups are added on benzenic species, 2) transition state selectivity occurs for organic species with more than one aromatic core and bearing more than three methyl groups, 3) methylation rates for H-SSZ-13 are approximately 3 orders of magnitude higher than on H-SAPO-34 owing to its higher acidity. The formation of (poly)aromatic cationic compounds can be followed by using in situ UV/Vis spectroscopy because these species yield characteristic absorption bands in the visible region of the spectrum. We have monitored the growth of characteristic peaks and derived activation energies of formation for various sets of (poly)aromatic compounds trapped in the zeolite host. The formation–activation barriers deduced by using UV/Vis microspectroscopy correlate well with the activation energies for the methylation of the benzenic species and the lower methylated naphthalenic species. This study shows that a fundamental insight at the molecular level can be obtained by using a combined in situ spectroscopic and theoretical approach for a complex catalyst of industrial relevance.
Co-reporter:Veronique VanSpeybroeck;Karen Hemelsoet;Kristof DeWispelaere;Qingyun Qian;Jeroen VanderMynsbrugge;Bart DeSterck;Michel Waroquier
ChemCatChem 2013 Volume 5( Issue 1) pp:
Publication Date(Web):
DOI:10.1002/cctc.201290053
Co-reporter:Dilek A. Boga;Ramon Oord;Dr. Andrew M. Beale;Dr. Young-Min Chung;Dr. Pieter C. A. Bruijnincx;Dr. Bert M. Weckhuysen
ChemCatChem 2013 Volume 5( Issue 2) pp:529-537
Publication Date(Web):
DOI:10.1002/cctc.201200112
Abstract
Monometallic Pt and bimetallic Pt-Cu catalysts supported on Mg(Al)O mixed oxides, obtained by calcination of the corresponding layered double hydroxides (LDHs), were prepared and tested in the aqueous-phase reforming (APR) of glycerol. The effect of the Mg/Al ratio and calcination temperature of the LDH support, as well as the effect of varying Pt and Cu amounts on glycerol reforming, was investigated. The use of a basic support increases the selectivity to hydrogen and the use of a Pt-Cu bimetallic catalyst results in a decrease in alkane formation. The 0.9 wt. % Pt-0.4 wt. % Cu/Mg(Al)O_2.95 catalyst system with an Mg(Al)O mixed oxide support obtained by the calcination of the corresponding LDH material with Mg/Al ratio of 2.95 at 673 K, showed higher hydrogen selectivity (55.3 %) and lower methane production (1.9 %) after 5 h reaction than the benchmark Pt/Al2O3 catalyst (49.4 % and 5.6 %, respectively). Catalyst characterization by extended X-ray absorption fine structure (EXAFS) spectroscopy showed a bimetallic interaction between Pt and Cu. The bimetallic interaction is thought to be responsible for the lowered methane formation and, ultimately, the high hydrogen selectivity observed.
Co-reporter:Dr. Luis R. Aramburo;Dr. Javier Ruiz-Martínez;Dr. Linn Sommer;Dr. Bjørnar Arstad;Robison Buitrago-Sierra;Dr. Antonio Sepúlveda-Escribano;Dr. Henny W. Zbergen;Dr. Unni Olsbye;Dr. Frank M. F. deGroot;Dr. Bert M. Weckhuysen
ChemCatChem 2013 Volume 5( Issue 6) pp:1386-1394
Publication Date(Web):
DOI:10.1002/cctc.201200670
Abstract
The effect of a severe steaming treatment on the physicochemical properties and catalytic performance of H-SAPO-34 molecular sieves during the methanol-to-hydrocarbons (MTH) reaction has been investigated with a combination of scanning transmission X-ray microscopy (STXM), catalytic testing, and bulk characterization techniques, including ammonia temperature programmed desorption and 27Al and 29Si magic angle spinning nuclear magnetic resonance. For this purpose, two samples, namely a calcined and a steamed H-SAPO-34 catalyst powder, have been compared. It has been found that calcined H-SAPO-34 displays a high selectivity towards light olefins, yet shows a poor stability as compared to a zeolite H-ZSM-5 catalyst. Moreover, in situ STXM at the carbon K-edge during the MTH reaction allows construction of nanoscale chemical maps of the hydrocarbon species formed within the H-SAPO-34 aggregates as a function of reaction time and steam post-treatment. It was found that there is an initial preferential formation of coke precursor species within the core of the H-SAPO-34 aggregates. For longer times on stream the formation of the coke precursor species is extended to the outer regions, progressively filling the entire H-SAPO-34 catalyst particle. In contrast, the hydrothermally treated H-SAPO-34 showed similar reaction selectivity, but decreased activity and catalyst stability with respect to its calcined counterpart. These variations in MTH performance are related to a faster and more homogeneous formation of coke precursor species filling up the entire steamed H-SAPO-34 catalyst particle. Finally, the chemical imaging capabilities of the STXM method at the Al and Si K-edge are illustrated by visualizing the silicon islands at the nanoscale before and after steaming H-SAPO-34.
Co-reporter:Anna L. Jongerius;Robert W. Gosselink;Jelmer Dijkstra;Dr. Johannes H. Bitter;Dr. Pieter C. A. Bruijnincx ; Bert M. Weckhuysen
ChemCatChem 2013 Volume 5( Issue 10) pp:2964-2972
Publication Date(Web):
DOI:10.1002/cctc.201300280
Abstract
Hydrodeoxygenation (HDO) studies over carbon nanofiber-supported (CNF) W2C and Mo2C catalysts were performed on guaiacol, a prototypical substrate to evaluate the potential of a catalyst for valorization of depolymerized lignin streams. Typical reactions were executed at 55 bar hydrogen pressure over a temperature range of 300–375 °C for 4 h in dodecane, using a batch autoclave system. Combined selectivities of up to 87 and 69 % to phenol and methylated phenolics were obtained at 375 °C for W2C/CNF and Mo2C/CNF at >99 % conversion, respectively. The molybdenum carbide-based catalyst showed a higher activity than W2C/CNF and yielded more completely deoxygenated aromatic products, such as benzene and toluene. Catalyst recycling experiments, performed with and without regeneration of the carbide phase, showed that the Mo2C/CNF catalyst was stable during reusability experiments. The most promising results were obtained with the Mo2C/CNF catalyst, as it showed a much higher activity and higher selectivity to phenolics compared to W2C/CNF.
Co-reporter:Dr. Pedro Castaño;Dr. Javier Ruiz-Martínez;Eva Epelde;Dr. Ana G. Gayubo;Dr. Bert M. Weckhuysen
ChemCatChem 2013 Volume 5( Issue 10) pp:2827-2831
Publication Date(Web):
DOI:10.1002/cctc.201300218
Co-reporter:Matthia A. Karreman;Dr. Inge L. C. Buurmans;Dr. Alexra V. Agronskaia;Dr. John W. Geus;Dr. Hans C. Gerritsen;Dr. Bert M. Weckhuysen
Chemistry - A European Journal 2013 Volume 19( Issue 12) pp:3846-3859
Publication Date(Web):
DOI:10.1002/chem.201203491
Abstract
While cycling through a fluid catalytic cracking (FCC) unit, the structure and performance of FCC catalyst particles are severely affected. In this study, we set out to characterize the damage to commercial equilibrium catalyst particles, further denoted as ECat samples, and map the different pathways involved in their deactivation in a practical unit. The degradation was studied on a structural and a functional level. Transmission electron microscopy (TEM) of ECat samples revealed several structural features; including zeolite crystals that were partly or fully severed, mesoporous, macroporous, and/or amorphous. These defects were then correlated to structural features observed in FCC particles that were treated with different levels of hydrothermal deactivation. This allowed us not only to identify which features observed in ECat samples were a result of hydrothermal deactivation, but also to determine the severity of treatments resulting in these defects. For functional characterization of the ECat sample, the Brønsted acidity within individual FCC particles was studied by a selective fluorescent probe reaction with 4-fluorostyrene. Integrated laser and electron microscopy (iLEM) allowed correlating this Brønsted acidity to structural features by combining a fluorescence and a transmission electron microscope in a single set-up. Together, these analyses allowed us to postulate a plausible model for the degradation of zeolite crystals in FCC particles in the ECat sample. Furthermore, the distribution of the various deactivation processes within particles of different ages was studied. A rim of completely deactivated zeolites surrounding each particle in the ECat sample was identified by using iLEM. These zeolites, which were never observed in fresh or steam-deactivated samples, contained clots of dense structures. The structures are proposed to be carbonaceous deposits formed during the cracking process, and seem resistant towards burning off during catalyst regeneration.
Co-reporter:Dr. Jan P. Hofmann;Dr. Davide Mores;Luis R. Aramburo;Shewangizaw Teketel;Dr. Marcus Rohnke;Dr. Jürgen Janek;Dr. Unni Olsbye;Dr. Bert M. Weckhuysen
Chemistry - A European Journal 2013 Volume 19( Issue 26) pp:8533-8542
Publication Date(Web):
DOI:10.1002/chem.201203351
Abstract
The catalytic, deactivation, and regeneration characteristics of large coffin-shaped H-ZSM-5 crystals were investigated during the methanol-to-hydrocarbons (MTH) reaction at 350 and 500 °C. Online gas-phase effluent analysis and examination of retained material thereof were used to explore the bulk properties of large coffin-shaped zeolite H-ZSM-5 crystals in a fixed-bed reactor to introduce them as model catalysts for the MTH reaction. These findings were related to observations made at the individual particle level by using polarization-dependent UV-visible microspectroscopy and mass spectrometric techniques after reaction in an in situ microspectroscopy reaction cell. Excellent agreement between the spectroscopic measurements and the analysis of hydrocarbon deposits by means of retained hydrocarbon analysis and time-of-flight secondary-ion mass spectrometry of spent catalyst materials was observed. The obtained data reveal a shift towards more condensed coke deposits on the outer zeolite surface at higher reaction temperatures. Zeolites in the fixed-bed reactor setup underwent more coke deposition than those reacted in the in situ microspectroscopy reaction cell. Regeneration studies of the large zeolite crystals were performed by oxidation in O2/inert gas mixtures at 550 °C. UV-visible microspectroscopic measurements using the oligomerization of styrene derivatives as probe reaction indicated that the fraction of strong acid sites decreased during regeneration. This change was accompanied by a slight decrease in the initial conversion obtained after regeneration. H-ZSM-5 deactivated more rapidly at higher reaction temperature.
Co-reporter:Luis R. Aramburo;Dr. Yijin Liu;Dr. Tolek Tyliszczak; Dr. Frank M. F. de Groot; Dr. Joy C. Andrews; Dr. Bert M. Weckhuysen
ChemPhysChem 2013 Volume 14( Issue 3) pp:496-499
Publication Date(Web):
DOI:10.1002/cphc.201201015
Co-reporter:Luis R. Aramburo;Dr. Yijin Liu;Dr. Tolek Tyliszczak; Dr. Frank M. F. de Groot; Dr. Joy C. Andrews; Dr. Bert M. Weckhuysen
ChemPhysChem 2013 Volume 14( Issue 3) pp:
Publication Date(Web):
DOI:10.1002/cphc.201390011
Co-reporter:Dr. Joy C. Andrews; Dr. Bert M. Weckhuysen
ChemPhysChem 2013 Volume 14( Issue 16) pp:3655-3666
Publication Date(Web):
DOI:10.1002/cphc.201300529
Abstract
Heterogeneous catalysts often consist of an active metal (oxide) in close contact with a support material and various promoter elements. Although macroscopic properties, such as activity, selectivity and stability, can be assessed with catalyst performance testing, the development of relevant, preferably quantitative structure–performance relationships require the use of advanced characterisation methods. Spectroscopic imaging in the hard X-ray region with nanometer-scale resolution has very recently emerged as a powerful approach to elucidate the hierarchical structure and related chemistry of catalytic solids in action under realistic reaction conditions. This X-ray-based chemical imaging method benefits from the combination of high resolution (∼30 nm) with large X-ray penetration and depth of focus, and the possibility for probing large areas with mosaic imaging. These capabilities make it possible to obtain spatial and temporal information on chemical changes in catalytic solids as well as a wide variety of other functional materials, such as fuel cells and batteries, in their full complexity and integrity. In this concept article we provide details on the method and setup of full-field hard X-ray spectroscopic imaging, illustrate its potential for spatiotemporal chemical imaging by making use of recent showcases, outline the pros and cons of this experimental approach and discuss some future directions for hierarchical functional materials research.
Co-reporter:Ilona vanZvoort;Yuehu Wang;Carolus B. Rasrendra;Dr. Ernst R. H. vanEck;Dr. Pieter C. A. Bruijnincx;Dr. Hero J. Heeres;Dr. Bert M. Weckhuysen
ChemSusChem 2013 Volume 6( Issue 9) pp:1745-1758
Publication Date(Web):
DOI:10.1002/cssc.201300332
Abstract
Neither the routes through which humin byproducts are formed, nor their molecular structure have yet been unequivocally established. A better understanding of the formation and physicochemical properties of humins, however, would aid in making biomass conversion processes more efficient. Here, an extensive multiple-technique-based study of the formation, molecular structure, and morphology of humins is presented as a function of sugar feed, the presence of additives (e.g., 1,2,4-trihydroxybenzene), and the applied processing conditions. Elemental analyses indicate that humins are formed through a dehydration pathway, with humin formation and levulinic acid yields strongly depending on the processing parameters. The addition of implied intermediates to the feedstocks showed that furan and phenol compounds formed during the acid-catalyzed dehydration of sugars are indeed included in the humin structure. IR spectra, sheared sum projections of solid-state 2DPASS 13C NMR spectra, and pyrolysis GC–MS data indicate that humins consist of a furan-rich polymer network containing different oxygen functional groups. The structure is furthermore found to strongly depend on the type of feedstock. A model for the molecular structure of humins is proposed based on the data presented.
Co-reporter:Carlo Angelici; Bert M. Weckhuysen;Dr. Pieter C. A. Bruijnincx
ChemSusChem 2013 Volume 6( Issue 9) pp:1595-1614
Publication Date(Web):
DOI:10.1002/cssc.201300214
Abstract
The development of new and improved processes for the synthesis of bio-based chemicals is one of the scientific challenges of our time. These new discoveries are not only important from an environmental point of view, but also represent an important economic opportunity, provided that the developed processes are selective and efficient. Bioethanol is currently produced from renewable resources in large amounts and, in addition to its use as biofuel, holds considerable promise as a building block for the chemical industry. Indeed, further improvements in production, both in terms of efficiency and feedstock selection, will guarantee availability at competitive prices. The conversion of bioethanol into commodity chemicals, in particular direct ‘drop-in’ replacements is, therefore, becoming increasingly attractive, provided that the appropriate (catalytic) technology is in place. The production of green and renewable 1,3-butadiene is a clear example of this approach. The Lebedev process for the one-step catalytic conversion of ethanol to butadiene has been known since the 1930s and has been applied on an industrial scale to produce synthetic rubber. Later, the availability of low-cost oil made it more convenient to obtain butadiene from petrochemical sources. The desire to produce bulk chemicals in a sustainable way and the availability of low-cost bioethanol in large volumes has, however, resulted in a renaissance of this old butadiene production process. This paper reviews the catalytic aspects associated with the synthesis of butadiene via the Lebedev process, as well as the production of other, mechanistically related bulk chemicals that can be obtained from (bio)ethanol.
Co-reporter:Qingyun Qian;Dr. Javier Ruiz-Martínez;Dr. Mohamed Mokhtar;Dr. Abdullah M. Asiri;Dr. Shaeel A. Al-Thabaiti;Dr. Suliman N. Basahel;Hendrik E. vanderBij;Dr. Jan Kornatowski;Dr. Bert M. Weckhuysen
Chemistry - A European Journal 2013 Volume 19( Issue 34) pp:11204-11215
Publication Date(Web):
DOI:10.1002/chem.201300540
Abstract
The formation of hydrocarbon pool (HCP) species during methanol-to-olefin (MTO) and ethanol-to-olefin (ETO) processes have been studied on individual micron-sized SAPO-34 crystals with a combination of in situ UV/Vis, confocal fluorescence, and synchrotron-based IR microspectroscopic techniques. With in situ UV/Vis microspectroscopy, the intensity changes of the λ=400 nm absorption band, ascribed to polyalkylated benzene (PAB) carbocations, have been monitored and fitted with a first-order kinetics at low reaction temperatures. The calculated activation energy (Ea) for MTO, approximately 98 kJ mol−1, shows a strong correlation with the theoretical values for the methylation of aromatics. This provides evidence that methylation reactions are the rate-determining steps for the formation of PAB. In contrast for ETO, the Ea value is approximately 60 kJ mol−1, which is comparable to the Ea values for the condensation of light olefins into aromatics. Confocal fluorescence microscopy demonstrates that during MTO the formation of the initial HCP species are concentrated in the outer rim of the SAPO-34 crystal when the reaction temperature is at 600 K or lower, whereas larger HCP species are gradually formed inwards the crystal at higher temperatures. In the case of ETO, the observed egg-white distribution of HCP at 509 K suggests that the ETO process is kinetically controlled, whereas the square-shaped HCP distribution at 650 K is indicative of a diffusion-controlled process. Finally, synchrotron-based IR microspectroscopy revealed a higher degree of alkylation for aromatics for MTO as compared to ETO, whereas high reaction temperatures favor dealkylation processes for both the MTO and ETO processes.
Co-reporter:Matthew G. O'Brien, Simon D. M. Jacques, Marco Di Michiel, Paul Barnes, Bert M. Weckhuysen and Andrew M. Beale
Chemical Science 2012 vol. 3(Issue 2) pp:509-523
Publication Date(Web):09 Nov 2011
DOI:10.1039/C1SC00637A
A combination of synchrotron μ-XRD-CT and μ-absorption-CT (CT = computed tomography) is demonstrated, providing a unique insight into the solid state changes occurring from within crystalline materials. Specifically, we examine here the solid state changes that occur in a millimetre-sized Ni/γ-Al2O3 catalyst body in both 2D and 3D during calcination and CO methanation for the first time. The combination provides a unique insight into the spatial phase distribution of these materials and how these evolve via a series of solid state transformation processes. For example, initially, two Ni-ethylenediamine (en) complexes were observed on the impregnated and dried body; a hydrated and non-hydrated form, which 2D scans reveal possess an egg-shell and egg-yolk distribution, respectively. Furthermore, the μ-XRD data were of sufficient quality so as to be able to reveal that the particles within the ‘egg-shell’ were larger (∼35 nm) than those of the ‘egg-yolk’ (∼19 nm) and that there were more of them. On calcination, both precursors collapsed, yielding metallic fcc Ni particles with a surprisingly uniform average size distribution over the catalyst (∼4 nm). However, a comparison of the scattering at different stages of the experiment suggested that the crystalline structure of some of the Ni remained diffraction ‘silent’. Calcination in oxygen lead to both Ni oxidation and particle sintering, mainly at the exterior, which on pre-reaction reduction (in H2) yielded again fcc Ni particles (∼4 nm interior, ∼6 nm exterior) with a significant reduction in the amorphous Ni component. The catalyst proved active for CO methanation and, during 2 h time on-stream, no change in the structure composition or shape was observed, leading us to conclude that nano-sized fcc Ni particles on γ-Al2O3 are the active component in CO methanation. This work therefore demonstrates both the power of spatially resolved μ-XRD-CT/μ-absorption-CT measurement of catalytic systems and its advantage over more ‘traditional’ single point studies on small sieve fractions.
Co-reporter:Clare E. Harvey, Evelien M. van Schrojenstein Lantman, Arjan J. G. Mank and Bert M. Weckhuysen
Chemical Communications 2012 vol. 48(Issue 12) pp:1742-1744
Publication Date(Web):04 Jan 2012
DOI:10.1039/C2CC15939B
The integration of Atomic Force Microscopy and Raman spectroscopy is tested for use in heterogeneous catalysis research by a preliminary investigation, the photo-oxidation of rhodamine-6G. Temperature and atmosphere were varied in an in situcell to show compatibility with realistic reaction conditions.
Co-reporter:Bart P. C. Hereijgers, Rudy F. Parton and Bert M. Weckhuysen
Catalysis Science & Technology 2012 vol. 2(Issue 5) pp:951-960
Publication Date(Web):06 Jan 2012
DOI:10.1039/C2CY00455K
Olefin epoxidation with cyclohexyl hydroperoxide offers great perspective in increasing the yield from industrial cyclohexane oxidation and the production of epoxides in an apolar medium. Two competing hydroperoxide conversion routes, namely direct epoxidation and thermal decomposition, were identified. The formation of radicals seemed to play a role in both mechanisms. However, olefin epoxidation was found to solely take place at the catalyst. Allylic oxidation of cyclohexene occurs under reaction conditions primarily by molecular oxygen and only constitutes a minor route. The presence of molecular oxygen was found to increase the overall yield of the process by solvent oxidation yielding new cyclohexyl hydroperoxide. Hydrolysis and isomerization of the epoxide were found to be negligible reactions, although the epoxide gets converted at higher concentrations, presumably by the radical initiated polymerization. UV-Vis spectroscopy provided proof for the formation of titanium-hydroperoxide species as the active catalytic site in the direct epoxidation reaction.
Co-reporter:Luis R. Aramburo, Sue Wirick, Piter S. Miedema, Inge L. C. Buurmans, Frank M. F. de Groot and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 19) pp:6967-6973
Publication Date(Web):25 Jan 2012
DOI:10.1039/C2CP22848C
The Brønsted acid-catalyzed oligomerization of 4-fluorostyrene has been studied on a series of H-ZSM-5 zeolite powders, steamed under different conditions, with a combination of UV-Vis micro-spectroscopy and Scanning Transmission X-ray Microscopy (STXM). UV-Vis micro-spectroscopy and STXM have been used to monitor the relative formation of cyclic and linear dimeric carbocations as a function of the steaming post-treatment (i.e., parent vs. steaming at 600, 700 and 800 °C). It was found that the UV-Vis band intensity ratios of linear to cyclic dimeric species increase from 0.79 (parent H-ZSM-5) over 1.41 (H-ZSM-5 steamed at 600 °C) and 1.88 (H-ZSM-5 steamed at 700 °C) to 2.33 (H-ZSM-5 steamed at 800 °C). STXM confirms this trend in reaction product selectivity, as the relative intensities of the transitions attributed to the presence of the cyclic dimer in the carbon K-edge spectra decrease with increasing severity of the steaming post-treatment. Furthermore, STXM reveals spatial heterogeneities in reaction product formation within the H-ZSM-5 zeolite powders at the nanoscale. More specifically, a shrinking carbon core–shell distribution was detected within the zeolite aggregates, in which the relative amount of cyclic dimeric species is higher in the core relative to the shell of the zeolite aggregate and the relative amount of cyclic dimeric species in the zeolite core gradually decreases with increasing severity of the steaming post-treatment. These differences are rationalized in terms of spatial differences in Brønsted acidity within H-ZSM-5 zeolite powders as well as by changes in the formation process of linear and dimeric carbocations within H-ZSM-5 micro- and mesopores.
Co-reporter:Ana Mijovilovich;Hisashi Hayashi;Naomi Kawamura;Hitoshi Osawa;Pieter C. A. Bruijnincx;Robertus J. M. Klein Gebbink;Frank M. F. de Groot
European Journal of Inorganic Chemistry 2012 Volume 2012( Issue 10) pp:1589-1597
Publication Date(Web):
DOI:10.1002/ejic.201101075
Abstract
Proteins sharing the same “2-His-1-carboxylate” structural motif have little amino acid sequence similarity and are able to perform many different reactions. Many factors have been cited to explain their different specificity and turnover rates, like protein environment, coordinated ligand geometry, electronic structure of the active site, etc. In this paper, we present a combined approach applying high-resolution XANES spectroscopy and theory simulations to different model complexes that mimic the binding modes of the amino acids to the metal site. Experiments were performed on three compounds showing three metal sites: ferrous hexacoordinate, ferric pentacoordinate and ferrous pentacoordinate. The first two compounds bear an N,N,O-tridentate 3,3-bis(1-alkylimidazol-2-yl)propionate ligand that features a monodentate carboxylate group. These complexes mimic the activity of extradiol dioxygenases but also exhibit intradiol cleavage activity. The third compound features a bidentate terphenylcarboxylate ligand and a sterically hindered bidentate N,N-donor, thus providing a good structural mimic of the ternary enzyme-tetrahydrobiopterin-substrate complex in pterin-dependent phenylalanine hydroxylase, which also contains a bidentate carboxylate. Modeling of high-resolution XANES on well-defined model complexes of different geometry can aid in protein structure elucidation. XANES gives the oxidation state and coordination number of the metal in the non-crystallized protein at natural pH. The accuracy of the results is limited by the core-hole and experimental broadenings. We found that high-resolution XANES experiments give increased resolution at the pre-edges, but limited improvement at the main edge. These high-resolution pre-edges can be accurately simulated by using crystal field multiplet theory (CFM). We show that by combining modelling and XANES simulations with FEFF8, detailed structural and chemical information can be obtained. We found that a short O2–metal distance for the carboxylate oxygen atom not bound to the metal causes a higher white line in FeII, which is similar to the results obtained for the pterin-dependent hydroxylase, tyrosine hydroxylase (TYH). Full-potential FDMNES simulations for each sample confirm the accuracy of the main results with muffin-tin approximation (FEFF8).
Co-reporter:Ana Mijovilovich;Hisashi Hayashi;Naomi Kawamura;Hitoshi Osawa;Pieter C. A. Bruijnincx;Robertus J. M. Klein Gebbink;Frank M. F. de Groot
European Journal of Inorganic Chemistry 2012 Volume 2012( Issue 10) pp:
Publication Date(Web):
DOI:10.1002/ejic.201290026
Co-reporter:Inge L. C. Buurmans;Dr. Javier Ruiz-Martínez;Sanne L. vanLeeuwen;Dr. David vanderBeek;Dr. Jaap A. Bergwerff;Dr. William V. Knowles;Dr. Eelco T. C. Vogt;Dr.Ir. Bert M. Weckhuysen
Chemistry - A European Journal 2012 Volume 18( Issue 4) pp:1094-1101
Publication Date(Web):
DOI:10.1002/chem.201102949
Abstract
A time-resolved in situ micro-spectroscopic approach has been used to investigate the Brønsted acidic properties of fluid-catalytic-cracking (FCC) catalysts at the single particle level by applying the acid-catalysed styrene oligomerisation probe reaction. The reactivity of individual FCC components (zeolite, clay, alumina and silica) was monitored by UV/Vis micro-spectroscopy and showed that only clay and zeolites (Y and ZSM-5) contain Brønsted acid sites that are strong enough to catalyse the conversion of 4-fluorostyrene into carbocationic species. By applying the same approach to complete FCC catalyst particles, it has been found that the fingerprint of the zeolitic UV/Vis spectra is clearly recognisable. This almost exclusive zeolitic activity is confirmed by the fact that hardly any reactivity is observed for FCC particles that contain no zeolite. Confocal fluorescence microscopy images of FCC catalyst particles reveal inhomogeneously distributed micron-sized zeolite domains with a highly fluorescent signal upon reaction. By examining laboratory deactivated FCC catalyst particles in a statistical approach, a clear trend of decreasing fluorescence intensity, and thus Brønsted acidity, of the zeolite domains is observed with increasing severity of the deactivation method. By comparing the average fluorescence intensities obtained with two styrenes that differ in reactivity, it has been found that the Brønsted acid site strength within FCC catalyst particles containing ZSM-5 is more uniform than within those containing zeolite Y, as confirmed with temperature-programmed desorption of ammonia.
Co-reporter:Inge L. C. Buurmans;Dr. Javier Ruiz-Martínez;Sanne L. vanLeeuwen;Dr. David vanderBeek;Dr. Jaap A. Bergwerff;Dr. William V. Knowles;Dr. Eelco T. C. Vogt;Dr.Ir. Bert M. Weckhuysen
Chemistry - A European Journal 2012 Volume 18( Issue 4) pp:
Publication Date(Web):
DOI:10.1002/chem.201290008
Co-reporter:Dr. Mathijs W. Zbergen;Dr. Andrew M. Beale ;Dr. ir. Bert M. Weckhuysen
ChemCatChem 2012 Volume 4( Issue 2) pp:217-227
Publication Date(Web):
DOI:10.1002/cctc.201100333
Abstract
The incipient wetness impregnation steps of 3 mm cylindrical γ-Al2O3 pellets with CrIII-nitrate and CrVI-oxide as precursors was investigated by using UV/Vis and Raman microspectroscopy. The speciation and microdistributions of metal-ion complexes in the pellets were followed in time after impregnation. Measurements on the catalyst bodies were performed by applying a scan-line on the surface of bisected pellets. The microdistributions of the Cr-ion complexes were altered by varying the pH, the ratio of the CrIII-nitrate and CrVI-oxide and by including either citric acid or phosphoric acid to the impregnation solution. Three factors were found to be most influential: i) Hydrolysis of CrIII complexes to yield solid Cr(OH)3 near the exterior of the pellets, ii) electrostatic interaction between the support and the respective CrIII and CrVI complexes, and iii) competitive adsorption of the additives. The opposite charge of the CrIII and CrVI complexes was exploited to obtain differences in the overall Cr-ion complex microdistributions, for example, egg-shell, egg-yolk, egg-white, and uniform type of distributions.
Co-reporter:Dr. Joseph Zakzeski;Dr. Ruud J. H. Grisel;Arjan T. Smit;Dr. Bert M. Weckhuysen
ChemSusChem 2012 Volume 5( Issue 2) pp:430-437
Publication Date(Web):
DOI:10.1002/cssc.201100631
Abstract
The solid acid-catalyzed hydrolysis of cellulose was studied under elevated temperatures and autogenous pressures using in situ ATR-IR spectroscopy. Standards of cellulose and pure reaction products, which include glucose, fructose, hydroxymethylfurfural (HMF), levulinic acid (LA), formic acid, and other compounds, were measured in water under ambient and elevated temperatures. A combination of spectroscopic and HPLC analysis revealed that the cellulose hydrolysis proceeds first through the disruption of the glycosidic linkages of cellulose to form smaller cellulose molecules, which are readily observed by their distinctive CO vibrational stretches. The continued disruption of the linkages in these oligomers eventually results in the formation and accumulation of monomeric glucose. The solid-acid catalyst accelerated the isomerization of glucose to fructose, which then rapidly reacted under hydrothermal conditions to form degradation products, which included HMF, LA, formic acid, and acetic acid. The formation of these species could be suppressed by decreasing the residence time of glucose in the reactor, reaction temperature, and contact with the metal reactor. The hydrolysis of regenerated cellulose proceeded faster and under milder conditions than microcrystalline cellulose, which resulted in increased glucose yield and selectivity.
Co-reporter:Dr. Joseph Zakzeski;Anna L. Jongerius;Dr. Pieter C. A. Bruijnincx ; Bert M. Weckhuysen
ChemSusChem 2012 Volume 5( Issue 8) pp:1602-1609
Publication Date(Web):
DOI:10.1002/cssc.201100699
Abstract
With dwindling reserves of fossil feedstock as a resource for chemicals production, the fraction of chemicals and energy supplied by alternative, renewable resources, such as lignin, can be expected to increase in the foreseeable future. Here, we demonstrate a catalytic process to valorize lignin (exemplified with kraft, organosolv, and sugarcane bagasse lignin) using a mixture of cheap, bio-renewable ethanol and water as solvent. Ethanol/water mixtures readily solubilize lignin under moderate temperatures and pressures with little residual solids. The molecular weight of the dissolved lignins was shown to be reduced by gel permeation chromatography and quantitative HSQC NMR methods. The use of liquid-phase reforming of the solubilized lignin over a Pt/Al2O3 catalyst at 498 K and 58 bar is introduced to yield up to 17 % combined yield of monomeric aromatic oxygenates such as guaiacol and substituted guaiacols generating hydrogen as a useful by-product. Reduction of the lignin dissolved in ethanol/water using a supported transition metal catalyst at 473 K and 30 bar hydrogen yields up to 6 % of cyclic hydrocarbons and aromatics.
Co-reporter:Dr. Ines D. Gonzalez-Jimenez;Korneel Cats;Dr. Thomas Davidian;Dr. Matthijs Ruitenbeek;Dr. Florian Meirer;Dr. Yijin Liu;Dr. Johanna Nelson;Dr. Joy C. Andrews;Dr. Piero Pianetta;Dr. Frank M. F. deGroot;Dr. Bert M. Weckhuysen
Angewandte Chemie 2012 Volume 124( Issue 48) pp:12152-12156
Publication Date(Web):
DOI:10.1002/ange.201204930
Co-reporter:Luis R. Aramburo;Dr. Emiel deSmit;Dr. Bjørnar Arstad;Matti M. vanSchooneveld;Dr. Linn Sommer;Dr. Amélie Juhin;Dr. Tadahiro Yokosawa;Dr. Henny W. Zbergen;Dr. Unni Olsbye;Dr. Frank M. F. deGroot;Dr. Bert M. Weckhuysen
Angewandte Chemie 2012 Volume 124( Issue 15) pp:3676-3679
Publication Date(Web):
DOI:10.1002/ange.201109026
Co-reporter:Matthia A. Karreman;Inge L. C. Buurmans;Dr. John W. Geus;Dr. Alexra V. Agronskaia;Dr. Javier Ruiz-Martínez;Dr. Hans C. Gerritsen;Dr. Bert M. Weckhuysen
Angewandte Chemie 2012 Volume 124( Issue 6) pp:1457-1460
Publication Date(Web):
DOI:10.1002/ange.201106651
Co-reporter:Dr. Mathijs W. Zbergen;Dr. Simon D. M. Jacques;Dr. Bert M. Weckhuysen;Dr. Andrew M. Beale
Angewandte Chemie International Edition 2012 Volume 51( Issue 4) pp:957-960
Publication Date(Web):
DOI:10.1002/anie.201107175
Co-reporter:Matthia A. Karreman;Inge L. C. Buurmans;Dr. John W. Geus;Dr. Alexra V. Agronskaia;Dr. Javier Ruiz-Martínez;Dr. Hans C. Gerritsen;Dr. Bert M. Weckhuysen
Angewandte Chemie International Edition 2012 Volume 51( Issue 6) pp:1428-1431
Publication Date(Web):
DOI:10.1002/anie.201106651
Co-reporter:Luis R. Aramburo;Dr. Emiel deSmit;Dr. Bjørnar Arstad;Matti M. vanSchooneveld;Dr. Linn Sommer;Dr. Amélie Juhin;Dr. Tadahiro Yokosawa;Dr. Henny W. Zbergen;Dr. Unni Olsbye;Dr. Frank M. F. deGroot;Dr. Bert M. Weckhuysen
Angewandte Chemie International Edition 2012 Volume 51( Issue 15) pp:3616-3619
Publication Date(Web):
DOI:10.1002/anie.201109026
Co-reporter:Dr. Ines D. Gonzalez-Jimenez;Korneel Cats;Dr. Thomas Davidian;Dr. Matthijs Ruitenbeek;Dr. Florian Meirer;Dr. Yijin Liu;Dr. Johanna Nelson;Dr. Joy C. Andrews;Dr. Piero Pianetta;Dr. Frank M. F. deGroot;Dr. Bert M. Weckhuysen
Angewandte Chemie International Edition 2012 Volume 51( Issue 48) pp:11986-11990
Publication Date(Web):
DOI:10.1002/anie.201204930
Co-reporter:Dr. Mathijs W. Zbergen;Dr. Simon D. M. Jacques;Dr. Bert M. Weckhuysen;Dr. Andrew M. Beale
Angewandte Chemie 2012 Volume 124( Issue 4) pp:981-984
Publication Date(Web):
DOI:10.1002/ange.201107175
Co-reporter:A. N. Parvulescu, P. J. C. Hausoul, P. C. A. Bruijnincx, S. T. Korhonen, C. Teodorescu, R. J. M. Klein Gebbink, and B. M. Weckhuysen
ACS Catalysis 2011 Volume 1(Issue 5) pp:526
Publication Date(Web):March 23, 2011
DOI:10.1021/cs1001477
The telomerization of 1,3-butadiene with homogeneous palladium/phosphine catalysts is an efficient method to transform biomass-based oxygenates into useful fine and bulk chemicals, e.g. surfactants. Recovery and reuse of the expensive noble metal-based catalyst is highly desired for these systems and therefore a heterogeneous telomerization catalyst would be preferred. Layered double hydroxides (LDH) were investigated as supports for the heterogenization of the Pd/TPPTS (trisodium salt of 3, 3′,3′′-phosphanetriyl benzene sulfonic acid) telomerization catalyst. Turn over numbers (TONs) up to 1300 were obtained with heterogeneous (immobilized palladium and ligand) and pseudo-heterogeneous (immobilized ligand) catalysts in the telomerization of 1,3-butadiene with ethylene glycol (EG) and methanol (MeOH) under solvent- and base-free conditions; TONs are comparable to those obtained with the homogeneous catalysts under similar conditions. Importantly, the LDH support was found to induce a change in product selectivity. In addition to the expected C8 telomers, higher telomers with C16 or C24 chains were observed with rather high selectivities (up to 48% for EG and 77% for MeOH).Keywords: biomass; heterogeneous catalysis; layered double hydroxides; palladium; phosphines; telomerization
Co-reporter:Bart P. C. Hereijgers, Rudy F. Parton, and Bert M. Weckhuysen
ACS Catalysis 2011 Volume 1(Issue 10) pp:1183
Publication Date(Web):August 15, 2011
DOI:10.1021/cs200354c
The industrially important deperoxidation reaction of cyclohexyl hydroperoxide was combined with the epoxidation of cyclohexene over a series of mesoporous titanium silicates. The process was found to proceed with high selectivity, forming cyclohexanol, cyclohexanone, and epoxy-cyclohexane. The deperoxidation and epoxidation reactions were found to compete. However, by changing the surface hydrophobicity of the catalysts or the applied olefin/peroxide ratio, the overall mechanism could be directed in favor of the epoxidation. In this way, the combined selectivity toward valuable alicyclic oxygenates from cyclohexane oxidation based on peroxide conversion could be increased up to 170%. The catalysts where found to be stable from recycling and filtration experiments.Keywords: heterogeneous catalysis; olefin; selective oxidation; titanium; TS-1;
Co-reporter:Joseph Zakzeski, Pieter C. A. Bruijnincx and Bert M. Weckhuysen
Green Chemistry 2011 vol. 13(Issue 3) pp:671-680
Publication Date(Web):04 Feb 2011
DOI:10.1039/C0GC00437E
The cobalt-catalyzed oxidation of lignin and lignin model compounds using molecular oxygen in ionic liquids proceeds readily under mild conditions, but mechanistic insight and evidence for the species involved in the catalytic cycle is lacking. In this study, a spectroscopic investigation of the complexes involved during this process was conducted using in situ ATR-IR, Raman, and UV-Vis spectroscopy. A plausible mechanism was proposed that explains the role of the ionic liquid and the other reaction conditions necessary to achieve high catalytic activity. Direct spectroscopic evidence for the species involved in the catalytic cycle was obtained. In addition, substrate consumption and product formation during the oxidation of several lignin model compounds, such as veratryl alcohol, cinnamyl alcohol, and a model compound with a β-O-4 linkage, was directly monitored. The reaction proceeds via the coordination of alcohol-containing substrates to the Co followed by formation of a Co-superoxo species. The presence of hydroxide is necessary for coordination of the alcohol to occur. Hydrogen peroxide that forms as a reaction by-product underwent rapid disproportionation to yield water and molecular oxygen. Involvement of the various intermediates was further confirmed by 18O2 labeling studies. The properties of the ionic liquid greatly influence the catalytic activity both by stabilizing reactive intermediates and by favoring the coordination of the substrate to the cobalt over the direct oxidation of the cobalt without substrate.
Co-reporter:Davide Mores, Eli Stavitski, Suzanna P. Verkleij, Antoinette Lombard, Amandine Cabiac, Loïc Rouleau, Joël Patarin, Angélique Simon-Masseron and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 35) pp:15985-15994
Publication Date(Web):03 Aug 2011
DOI:10.1039/C1CP21324E
A combination of in situUV-Vis and confocal fluorescence micro-spectroscopy is applied to investigate the influence of an external silicalite-1 shell on the Brønsted acidity and coke formation process of individual H-ZSM-5 zeolite crystals. Three probe reactions were used: oligomerization of styrene, methanol-to-olefin (MTO) conversion and aromatization of light naphtha (LNA) derivatives. Oligomerization of styrene leads to the formation of optically active carbocationic oligomers. Different styrene substitutions indicate the conversion ability of the catalyst acid core, a preferred alignment of the oligomers within the straight zeolite channels and a Brønsted acidity gradient throughout the zeolite crystal. Both the MTO conversion and the LNA process lead to limited carbonaceous deposition within the external silicalite-1 layer. This outer shell furthermore prevents the growth of extended coke species at the zeolite external surface. During MTO, the formation of carbonaceous compounds initiates at the center of the H-ZSM-5 zeolite core and expands towards the zeolite exterior. This coke build-up starts with a 420 nm UV-Vis absorption band, assigned to methyl-substituted aromatic carbocations, and a second band around 550 nm, which is indicative of their growth towards larger conjugated systems. Aromatization of linear and branched C5 paraffins causes negligible darkening of the zeolite crystals though it forms fluorescent coke deposits and their precursors within the H-ZSM-5 catalyst. Olefin homologues on the contrary cause pronounced darkening of the zeolite composite. Methyl-branching of these reactants slows down the coke formation rate and produces carbonaceous species that are more restricted in their molecular size.
Co-reporter:Ana Mijovilovich, Sylvain Hamman, Fabrice Thomas, Frank M. F. de Groot and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 13) pp:5600-5604
Publication Date(Web):31 Jan 2011
DOI:10.1039/C0CP01144D
X-ray Emission Spectroscopy (XES) crossover peaks were shown to be sensitive to the protonation state of solvent molecules in the Zn protein carbonic anhydrase and its model compounds. Here we extend such studies to galactose oxidase models i.e.Cu(II) open d-shell systems, illustrating that XES combined with FEFF8 simulations reflect changes in the protonation state of the phenolate ligand for the copper center.
Co-reporter:Lukasz Karwacki and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 9) pp:3681-3685
Publication Date(Web):22 Dec 2010
DOI:10.1039/C0CP02220A
A combination of in situUV-Vis and confocal fluorescence micro-spectroscopy was used to study the template decomposition process in large zeolite ZSM-5 crystals. Correlation of polarized light dependent UV-Vis absorption spectra with confocal fluorescence emission spectra in the 400–750 nm region allowed extracting localized information on the nature and amount of chemical species formed upon detemplation at the single particle level. It has been found by means of polarized light dependent UV-Vis absorption measurements that the progressive growth of molecules follows the orientation of the straight channels of ZSM-5 crystals. Oligomerizing template derivatives lead to the subsequent build-up of methyl-substituted benzenium cations and more extended coke-like species, which are thermally stable up to ∼740 K. Complementary confocal fluorescence emission spectra showed nearly equal distribution of these molecules within the entire volume of the thermally treated zeolite crystals. The strongest emission bands were appearing in the orange/red part of the visible spectrum, confirming the presence of large polyaromatic molecules.
Co-reporter:Qingyun Qian, Davide Mores, Jan Kornatowski, Bert M. Weckhuysen
Microporous and Mesoporous Materials 2011 Volume 146(1–3) pp:28-35
Publication Date(Web):December 2011
DOI:10.1016/j.micromeso.2011.05.024
In situ UV–Vis micro-spectroscopy has been employed to reveal physicochemical insight in the template removal process of morpholine-containing micron-sized SAPO-34 crystals at the single particle level. The differences in UV–Vis absorption bands and the related rate of their formation and disappearance demonstrate the generation of different intermediates when the detemplation process is performed in either oxygen or nitrogen. A series of detemplation experiments in oxygen with SAPO-34 crystals ranging between 7.5 and 58 μm indicate the formation of five major Gaussian functions with band maxima at 405, 450, 505, 555 and 608 nm, building up the experimental UV–Vis absorption spectra. These Gaussian functions, assigned to different carbonaceous deposits and their precursors, demonstrate that two separate temperature trends can be distinguished. The first one is related to the formation of small organic compounds, characterized by band maxima at 405 and 450 nm, which lead to a bimodal intensity distribution at the low and high end of the heating ramp for large-sized SAPO-34 crystals, while the more extended poly-aromatic compounds, characterized by band maxima at 555 and 608 nm, generate a mono-modal distribution at intermediate heating temperatures for both small- and large-sized SAPO-34 crystals. These features provide evidence for a crystal size dependent relationship between pore blocking and the formation of small and more extended molecular compounds within SAPO-34 porous networks when performing a thermal detemplation process.Graphical abstractHighlights► The detemplation process in SAPO-34 crystals was studied at the single particle level. ► A faster and more complete removal of morpholine as template occurs in O2 than in N2. ► Small-sized crystals suffer from less severe pore blocking. ► Large-sized crystals experience a slower template burning.
Co-reporter:Bart P. C. Hereijgers
Catalysis Letters 2011 Volume 141( Issue 10) pp:
Publication Date(Web):2011/10/01
DOI:10.1007/s10562-011-0674-3
Co-reporter:Satu T. Korhonen ; Andrew M. Beale ; Mark A. Newton
The Journal of Physical Chemistry C 2011 Volume 115(Issue 4) pp:885-896
Publication Date(Web):May 20, 2010
DOI:10.1021/jp102530y
The performance of silver alumina catalysts and silver aluminate was studied in the selective catalytic reduction (SCR) of NO by propene. The use of boehmite during the impregnation step ensured a strong interaction between the silver species and the alumina surface in the final calcined catalyst. Thus, higher silver loadings (5−7 wt % silver) could be used without significant loss in selectivity to N2 during SCR. The nature of the silver species and the formation of adsorbed surface species during SCR and hydrogen-assisted SCR (H2-SCR) was studied by activity measurements and by combined in situ IR and X-absorption spectroscopic measurements. The combination of these techniques in the same reaction cell allowed simultaneous monitoring of the state of silver and the formation of surface species under realistic reaction conditions. The active silver species on alumina support were concluded to be 2-dimensional oxidic Agnδ+ species. Silver aluminate was ruled out as a possibility for an active phase for the SCR reaction. The oxidic Agnδ+ species were present under both SCR and H2-SCR reaction conditions and even on a prereduced catalyst under the SCR reaction conditions. Hydrogen is proposed to enhance the formation of adsorbed surface species, especially nitrites, but not to change the nature of the active silver sites.
Co-reporter:Dr. Lukasz Karwacki;D. A. Matthijs deWinter;Luis R. Aramburo;Dr. Misjaël N. Lebbink;Dr. Jan A. Post;Dr. Martyn R. Drury;Dr. Bert M. Weckhuysen
Angewandte Chemie International Edition 2011 Volume 50( Issue 6) pp:1294-1298
Publication Date(Web):
DOI:10.1002/anie.201006031
Co-reporter:Emiel deSmit;Matti M. vanSchooneveld;Dr. Fabrizio Cinquini;Dr. Hendrik Bluhm;Dr. Phillippe Sautet;Dr. Frank M. F. deGroot;Dr. Bert M. Weckhuysen
Angewandte Chemie International Edition 2011 Volume 50( Issue 7) pp:1584-1588
Publication Date(Web):
DOI:10.1002/anie.201005282
Co-reporter:Peter J. C. Hausoul;Dr. Andrei N. Parvulescu;Dr. Martin Lutz;Dr. Anthony L. Spek;Dr. Pieter C. A. Bruijnincx;Dr. Robertus J. M. KleinGebbink;Dr. Bert M. Weckhuysen
ChemCatChem 2011 Volume 3( Issue 5) pp:845-852
Publication Date(Web):
DOI:10.1002/cctc.201100048
Abstract
Liquid-chromatography electrospray-ionisation mass spectrometry (LC-ESI-MS) studies on reaction mixtures of the telomerization of 1,3-butadiene with biomass-based polyols revealed that the TOMPP (TOMPP=tris(2-methoxyphenyl)phosphine) ligand is converted towards the corresponding (2,7-octadienyl)phosphonium species during catalysis. The extent of ligand alkylation is substrate dependent and was identified as the primary cause of deactivation for carbohydrate substrates with anomeric hydroxyl groups. Coordination studies of the phosphonium cation with [Pd(dba)2] (dba=dibenzylideneacetone) gave insight into the alkylation mechanism and showed that the formation of the phosphonium cation is fully reversible. The reaction yields the key cationic intermediate [Pd(1,2,3,7,8-η5-octa-2,7-dien-1-yl)(TOMPP)]+, which, in the presence of the iodide anion, results in the formation of [Pd(1,2,3-η3-octa-2,7-dien-1-yl)(I)(TOMPP)]. Both complexes were fully characterized by various techniques including single crystal X-ray crystallography. Based on these results, an extension to the Pd/TOMPP-catalyzed telomerization mechanism was formulated to include the 2,7-octadienylphosphonium cation as a ligand reservoir. Catalytic tests show that the use of [Pd(dba)2] as precatalyst improves the telomerization of glucose and xylose.
Co-reporter:Dr. Lukasz Karwacki;D. A. Matthijs deWinter;Luis R. Aramburo;Dr. Misjaël N. Lebbink;Dr. Jan A. Post;Dr. Martyn R. Drury;Dr. Bert M. Weckhuysen
Angewandte Chemie 2011 Volume 123( Issue 6) pp:1330-1334
Publication Date(Web):
DOI:10.1002/ange.201006031
Co-reporter:Dr. Joseph Zakzeski ;Dr. Bert M. Weckhuysen
ChemSusChem 2011 Volume 4( Issue 3) pp:369-378
Publication Date(Web):
DOI:10.1002/cssc.201000299
Abstract
The solubilization and aqueous phase reforming of lignin, including kraft, soda, and alcell lignin along with sugarcane bagasse, at low temperatures (T≤498 K) and pressures (P≤29 bar) is reported for the first time for the production of aromatic chemicals and hydrogen. Analysis of lignin model compounds and the distribution of products obtained during the lignin aqueous phase reforming revealed that lignin was depolymerized through disruption of the abundant βO4 linkages and, to a lesser extent, the 55’ carbon-carbon linkages to form monomeric aromatic compounds. The alkyl chains contained on these monomeric compounds were readily reformed to produce hydrogen and simple aromatic platform chemicals, particularly guaiacol and syringol, with the distribution of each depending on the lignin source. The methoxy groups present on the aromatic rings were subject to hydrolysis to form methanol, which was also readily reformed to produce hydrogen and carbon dioxide. The composition of the isolated yields of monomeric aromatic compounds and overall lignin conversion based on these isolated yields varied from 10–15 % depending on the lignin sample, with the balance consisting of gaseous products and residual solid material. Furthermore, we introduce the use of a high-pressure autoclave with optical windows and an autoclave with ATR-IR sentinel for on-line in situ spectroscopic monitoring of biomass conversion processes, which provides direct insight into, for example, the solubilization process and aqueous phase reforming reaction of lignin.
Co-reporter:Andrew M. Beale ; Matthew G. O’Brien ; Marta Kasunič ; Amalija Golobič ; Manuel Sanchez-Sanchez ; Alan J. W. Lobo ; Dewi W. Lewis ; David S. Wragg ; Sergey Nikitenko ; Wim Bras
The Journal of Physical Chemistry C 2011 Volume 115(Issue 14) pp:6331-6340
Publication Date(Web):March 7, 2011
DOI:10.1021/jp200043b
The hydrothermal crystallization of ZnAPO-34 (CHA) molecular sieves has been studied for the first time using a combined in situ four technique setup utilizing SAXS/WAXS/XAFS/Raman to follow the various steps that occur during the complex transformation process of an amorphous precursor gel into a crystalline microporous material. These data are also supported by a detailed characterization of both the precursor gel (using Raman, NMR, XAFS, and TEM) and the final crystalline material (NMR, XRD, XAFS, TEM, and energy minimization calculations). Thus, all components during the various stages of reaction have been studied allowing for fundamental insight from the atomic/molecular level up to the bulk scale. On the basis of this multitechnique approach, the following observations are made: (i) The initial formation of a heterogeneous gel containing predominantly separate Al−O−P and Zn−O−P containing species as well as the presence of particles with a broad size distribution were noted. (ii) During sample heating, the SAXS data reveal a second population (14−16 nm) at the onset of crystallization, which were also accompanied by changes in both the ZnO3−O−O3P environment (XAFS) and the template conformer state (Raman). (iii) Before crystallization, Zn2+ species appear heterogeneously distributed throughout the sample, but in the final crystalline CHA phase Zn2+, it is much more homogeneously distributed. One template molecule is found per CHA cage. (iv) Zn2+ is found to promote nano particle growth and that results in the production of increasing amounts of crystalline material. (v) The structure-directing effect of Zn2+ ions leading to CHA formation is most likely initiated via an electrostatic interaction between Zn2+ in a Zn−O−P−O−Al−O−P matrix and the TEA template.
Co-reporter:Bart P. C. Hereijgers ; Tamara M. Eggenhuisen ; Krijn P. de Jong ; Herre Talsma ; Ad M. J. van der Eerden ; Andrew M. Beale
The Journal of Physical Chemistry C 2011 Volume 115(Issue 31) pp:15545-15554
Publication Date(Web):July 7, 2011
DOI:10.1021/jp204919z
Promoting supported gold nanoparticles with lanthanum oxide largely increases the hydrogen selectivity in the partial oxidation of methanol. In this study, the origin of the promotion effect of lanthanum oxide on supported gold catalysts was investigated. The formation of small gold nanoparticles on both the high surface area alumina and the low surface area lanthanum oxide support materials was confirmed by Transmission Electron Microscopy (TEM) and Extended X-ray Absorption Fine Structure (EXAFS). In situ X-ray absorption spectroscopy during partial methanol oxidation revealed the formation of oxidized gold species on the reduced Au/Al2O3 catalyst material, whereas La2O3 was found to facilitate the reduction of initially present oxidic gold species. This was confirmed by a larger measured heat of reaction for the exothermic decomposition of the oxidic gold species supported on lanthanum oxide as was found by differential scanning calorimetry. The catalysts under study did not show significant differences in methanol oxidation activity; however, the Au/Al2O3 catalyst exhibited much higher activity in CO and H2 oxidation. These observed differences in catalytic activity and selectivity of the Al2O3- and La2O3-supported Au catalysts are explained by the differences in redox behavior of the gold nanoparticles. It is proposed that zerovalent gold species limit dissociative H2 adsorption during the partial oxidation of methanol and thus improve the H2 selectivity by reducing CH4 and H2O formation.
Co-reporter:Peter J. C. Hausoul;Dr. Andrei N. Parvulescu;Dr. Martin Lutz;Dr. Anthony L. Spek;Dr. Pieter C. A. Bruijnincx;Dr. Robertus J. M. KleinGebbink;Dr. Bert M. Weckhuysen
ChemCatChem 2011 Volume 3( Issue 5) pp:
Publication Date(Web):
DOI:10.1002/cctc.201190018
Co-reporter:Davide Mores;Dr. Jan Kornatowski ;Dr. Unni Olsbye;Dr.Ir. Bert M. Weckhuysen
Chemistry - A European Journal 2011 Volume 17( Issue 10) pp:2874-2884
Publication Date(Web):
DOI:10.1002/chem.201002624
Abstract
Coke formation during the methanol-to-olefin (MTO) conversion has been studied at the single-particle level with in situ UV/Vis and confocal fluorescence microscopy. For this purpose, large H-ZSM-5 crystals differing in their Si/Al molar ratio have been investigated. During MTO, performed at 623 and 773 K, three major UV/Vis bands assigned to different carbonaceous deposits and their precursors are observed. The absorption at 420 nm, assigned to methyl-substituted aromatic compounds, initiates the buildup of the optically active coke species. With time-on-stream, these carbonaceous compounds expand in size, resulting in the gradual development of a second absorption band at around 500 nm. An additional broad absorption band in the 600 nm region indicates the enhanced formation of extended carbonaceous compounds that form as the reaction temperature is raised. Overall, the rate of coke formation decreases with decreasing aluminum content. Analysis of the reaction kinetics indicates that an increased Brønsted acid site density facilitates the formation of larger coke species and enhances their formation rate. The use of multiple excitation wavelengths in confocal fluorescence microscopy enables the localization of coke compounds with different molecular dimensions in an individual H-ZSM-5 crystal. It demonstrates that small coke species evenly spread throughout the entire H-ZSM-5 crystal, whereas extended coke deposits primarily form near the crystal edges and surfaces. Polarization-dependent UV/Vis spectroscopy measurements illustrate that extended coke species are predominantly formed in the straight channels of H-ZSM-5. In addition, at higher temperatures, fast deactivation leads to the formation of large aromatic compounds within channel intersections and at the external zeolite surface, where the lack of spatial restrictions allows the formation of graphite-like coke.
Co-reporter:Luis R. Aramburo;Dr. Lukasz Karwacki;Dr. Pablo Cubillas;Shunsuke Asahina;D. A. Matthijs deWinter; Martyn R. Drury;Inge L. C. Buurmans;Dr. Eli Stavitski;Davide Mores; Marco Daturi;Philippe Bazin;Dr. Paul Dumas; Fréderic Thibault-Starzyk;Dr. Jan A. Post; Michael W. Anderson; Osamu Terasaki; Bert M. Weckhuysen
Chemistry - A European Journal 2011 Volume 17( Issue 49) pp:13773-13781
Publication Date(Web):
DOI:10.1002/chem.201101361
Abstract
A combination of atomic force microscopy (AFM), high-resolution scanning electron microscopy (HR-SEM), focused-ion-beam scanning electron microscopy (FIB-SEM), X-ray photoelectron spectroscopy (XPS), confocal fluorescence microscopy (CFM), and UV/Vis and synchrotron-based IR microspectroscopy was used to investigate the dealumination processes of zeolite ZSM-5 at the individual crystal level. It was shown that steaming has a significant impact on the porosity, acidity, and reactivity of the zeolite materials. The catalytic performance, tested by the styrene oligomerization and methanol-to-olefin reactions, led to the conclusion that mild steaming conditions resulted in greatly enhanced acidity and reactivity of dealuminated zeolite ZSM-5. Interestingly, only residual surface mesoporosity was generated in the mildly steamed ZSM-5 zeolite, leading to rapid crystal coloration and coking upon catalytic testing and indicating an enhanced deactivation of the zeolites. In contrast, harsh steaming conditions generated 5–50 nm mesopores, extensively improving the accessibility of the zeolites. However, severe dealumination decreased the strength of the Brønsted acid sites, causing a depletion of the overall acidity, which resulted in a major drop in catalytic activity.
Co-reporter:Joseph Zakzeski, Pieter C. A. Bruijnincx, Anna L. Jongerius and Bert M. Weckhuysen
Chemical Reviews 2010 Volume 110(Issue 6) pp:3552
Publication Date(Web):March 10, 2010
DOI:10.1021/cr900354u
Co-reporter:Andrew M. Beale, Simon D. M. Jacques and Bert M. Weckhuysen
Chemical Society Reviews 2010 vol. 39(Issue 12) pp:4656-4672
Publication Date(Web):27 Oct 2010
DOI:10.1039/C0CS00089B
Heterogeneous catalysis is a term normally used to describe a group of catalytic processes, yet it could equally be employed to describe the catalytic solid itself. A better understanding of the chemical and structural variation within such materials is thus a pre-requisite for the rationalising of structure–function relationships and ultimately to the design of new, more sustainable catalytic processes. The past 20 years has witnessed marked improvements in technologies required for analytical measurements at synchrotron sources, including higher photon brightness, nano-focusing, rapid, high resolution data acquisition and in the handling of large volumes of data. It is now possible to image materials using the entire synchrotron radiative profile, thus heralding a new era of in situ/operando measurements of catalytic solids. In this tutorial review we discuss the recent work in this exciting new research area and finally conclude with a future outlook on what will be possible/challenging to measure in the not-too-distant future.
Co-reporter:Eli Stavitski and Bert M. Weckhuysen
Chemical Society Reviews 2010 vol. 39(Issue 12) pp:4615-4625
Publication Date(Web):12 Oct 2010
DOI:10.1039/C0CS00064G
The miniaturization of in situ spectroscopic tools has been recognized as a forefront instrumental development for the characterization of heterogeneous catalysts. With the multitude of micro-spectroscopic methods available fundamental insight into the structure–function relationships of catalytic processes can be obtained. Among these techniques vibrational spectroscopy is one of the most versatile methods and capable to shed insight into the molecular structure of reaction intermediates and products, the chemical state of catalyst materials during reaction as well as the nature of interactions between reactants/intermediates/products and the catalyst surface. In this tutorial review we discuss the recent developments in the field of infrared (IR) and Raman micro-spectroscopy and illustrate their potential. Showcase examples include (1) chemical imaging of spatial heterogeneities during catalyst preparation, (2) high-throughput catalyst screening, (3) transport and adsorption phenomena within catalytic solids and (4) reactivity studies of porous oxides, such as zeolites. Finally, new in situ spectroscopy tools based on vibrational spectroscopy and their potential in the catalysis domain are discussed.
Co-reporter:Leticia Espinosa-Alonso, Andrew M. Beale, and Bert M. Weckhuysen
Accounts of Chemical Research 2010 Volume 43(Issue 9) pp:1279
Publication Date(Web):July 6, 2010
DOI:10.1021/ar100045p
Cylindrical or spherical catalyst bodies with sizes ranging from tens of micrometers to a few millimeters have a wide variety of industrial applications. They are crucial in the oil refining industry and in the manufacture of bulk and fine chemicals. Their stability, activity, and selectivity are largely dependent on their preparation; thus, achieving the optimum catalyst requires a perfect understanding of the physicochemical processes occurring in a catalyst body during its synthesis. The ultimate goal of the catalyst researcher is to visualize these physicochemical processes as the catalyst is being prepared and without interfering with the system. In order to understand this chemistry and improve catalyst design, researchers need better, less invasive tools to observe this chemistry as it occurs, from the first stages in contact with a precursor all the way through its synthesis. In this Account, we provide an overview of the recent advances in the development of space- and time-resolved spectroscopic methods, from invasive techniques to noninvasive ones, to image the physicochemical processes taking place during the preparation of catalyst bodies. Although several preparation methods are available to produce catalyst bodies, the most common method used in industry is the incipient wetness impregnation. It is the most common method used in industry because it is simple and cost-effective. This method consists of three main steps each of which has an important role in the design of a catalytic material: pore volume impregnation, drying, and thermal treatment. During the impregnation step, the interface between the support surface and the precursor of the active phase at the solid−liquid interface is where the critical synthetic chemistry occurs. Gas−solid and solid−solid interfaces are critical during the drying and thermal treatment steps. Because of the length scale of these catalyst bodies, the interfacial chemistry that occurs during preparation is space-dependent. Different processes occurring in the core or in the outer rim of the catalytic solid are enhanced by several factors, such as the impregnation solution pH, the metal ion concentration, the presence of organic additives, and the temperature gradients inside the body. Invasive methods for studying the molecular nature of the metal-ion species during the preparation of catalyst bodies include Raman, UV−vis−NIR, and IR microspectroscopies. Noninvasive techniques include magnetic resonance imaging (MRI). Synchrotron-based techniques such as tomographic energy dispersive diffraction imaging (TEDDI) and X-ray microtomography for noninvasive characterization are also evaluated.
Co-reporter:Andrei N. Parvulescu ; Davide Mores ; Eli Stavitski ; Cristian M. Teodorescu ; Pieter C. A. Bruijnincx ; Robertus J. M. Klein Gebbink
Journal of the American Chemical Society 2010 Volume 132(Issue 30) pp:10429-10439
Publication Date(Web):July 12, 2010
DOI:10.1021/ja102566b
The etherification of biomass-based alcohols with various linear α-olefins under solvent-free conditions was followed in a space- and time-resolved manner on 9 μm large H-Beta zeolite crystals by confocal fluorescence microscopy. This allowed us to visualize the interaction with the substrate and distribution of the coke products into the catalyst at the level of an individual zeolite crystal during the etherification process. The spectroscopic information obtained on the micrometer-scale zeolite was in line with the results obtained with bulk characterization techniques and further confirmed by the catalytic results obtained both for micrometer-scale and nanoscale zeolites. This allowed us to explain the influence of the substrate type (glycerol, glycols, and alkenes) and zeolite properties (Si/Al ratio and particle size) on the etherification activity. The etherification of the biomass-based alcohols takes place mainly on the external surface of the zeolite particles. The gradual blockage of the external surface of the zeolite results in a partial or total loss of etherification activity. The deactivation could be attributed to olefin oligomerization. The high conversions obtained in the etherification of 1,2-propylene glycol with long linear alkenes (up to 80%) and the pronounced deactivation of the zeolite observed in the etherification of glycerol with long linear alkenes (max. 20% conversion) were explained by the spectroscopic measurements and is due to differences in the adsorption, i.e., in the center of the zeolite particle for glycerol and on the external surface in the case of glycols.
Co-reporter:Emiel de Smit ; Fabrizio Cinquini ; Andrew M. Beale ; Olga V. Safonova ; Wouter van Beek ; Philippe Sautet
Journal of the American Chemical Society 2010 Volume 132(Issue 42) pp:14928-14941
Publication Date(Web):October 6, 2010
DOI:10.1021/ja105853q
The stability and reactivity of ϵ, χ, and θ iron carbide phases in Fischer−Tropsch synthesis (FTS) catalysts as a function of relevant reaction conditions was investigated by a synergistic combination of experimental and theoretical methods. Combined in situ X-ray Absorption Fine Structure Spectroscopy/X-ray Diffraction/Raman Spectroscopy was applied to study Fe-based catalysts during pretreatment and, for the first time, at relevant high pressure Fischer−Tropsch synthesis conditions, while Density Functional Theory calculations formed a fundamental basis for understanding the influence of pretreatment and FTS conditions on the formation of bulk iron carbide phases. By combining theory and experiment, it was found that the formation of θ-Fe3C, χ-Fe5C2, and ϵ-carbides can be explained by their relative thermodynamic stability as imposed by gas phase composition and temperature. Furthermore, it was shown that a significant part of the Fe phases was present as amorphous carbide phases during high pressure FTS, sometimes in an equivalent amount to the crystalline iron carbide fraction. A catalyst containing mainly crystalline χ-Fe5C2 was highly susceptible to oxidation during FTS conditions, while a catalyst containing θ-Fe3C and amorphous carbide phases showed a lower activity and selectivity, mainly due to the buildup of carbonaceous deposits on the catalyst surface, suggesting that amorphous phases and the resulting textural properties play an important role in determining final catalyst performance. The findings further uncovered the thermodynamic and kinetic factors inducing the ϵ−χ−θ carbide transformation as a function of the carbon chemical potential μC.
Co-reporter:Joseph Zakzeski, Anna L. Jongerius and Bert M. Weckhuysen
Green Chemistry 2010 vol. 12(Issue 7) pp:1225-1236
Publication Date(Web):24 May 2010
DOI:10.1039/C001389G
Lignin is a component of lignocellulosic biomass from which important aromatic compounds can potentially be obtained. In the present work, Alcell and soda lignin were dissolved in the ionic liquid 1-ethyl-3-methylimidazolium diethylphosphate (EMIM DEP) and subsequently oxidized using several transition metal catalysts and molecular oxygen under mild conditions. CoCl2·6H2O in EMIM DEP proved particularly effective for the oxidation. The catalyst rapidly oxidized benzyl and other alcohol functionalities in lignin, but left phenolic functionality and 5–5′, β-O-4 and phenylcoumaran linkages intact, as determined by analysis of various lignin model compounds and ATR-IR spectroscopy. The catalyst system oxidized the alcohol functionality contained in cinnamyl alcohol to form cinnamaldehyde or cinnamic acid or disrupted the double bond to form benzoic acid or an epoxide. The benzyl functionality in veratryl alcohol, a simple non-phenolic lignin model compound, was selectively oxidized to form veratraldehyde at a maximum turnover frequency of 1440 h−1, compared to 10–15 h−1 reported for earlier systems. Phenolic functional groups contained in guaiacol, syringol, and vanillyl alcohol remained intact, although the benzyl alcohol group in the latter was oxidized to form vanillin. Incorporation of strongly bound tetradentate ligands to the catalyst yielded reduced activity relative to those derived from simple metal salts in EMIM DEP. The influence of reaction conditions, such as temperature, oxygen pressure and NaOH loading, were also investigated. The system represents a potential method in a biorefinery scheme to increase the oxygen functionality in lignin prior to depolymerization or additional functionalization of already depolymerized lignin.
Co-reporter:Inge L. C. Buurmans, Evgeny A. Pidko, Jennifer M. de Groot, Eli Stavitski, Rutger A. van Santen and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 26) pp:7032-7040
Publication Date(Web):14 May 2010
DOI:10.1039/C002442B
A series of H-ZSM-5 crystallites with different framework Si/Al ratios was studied by analyzing the kinetics and reaction mechanism of the oligomerization of 4-fluorostyrene as molecular probe reaction for Brønsted acidity. The formation of carbocationic species was followed by UV-Vis spectroscopy. Three carbocationic products were observed, namely a cyclic dimer, a conjugated linear dimer and a larger, more conjugated carbocation. Rate constants for the formation of all three products show a maximum at a Si/Al ratio of 25. Oligomerization of 4-fluorostyrene within the larger supercages of zeolite H-Y leads solely to cyclic dimers. The experimental observations were rationalized by DFT calculations, which show that the selectivity of the styrene oligomerization is controlled by the steric properties of the intrazeolite micropore voids. Two reaction pathways were considered for the formation of the conjugated linear carbocation. The conventional mechanism involves a hydride transfer between two dimeric hydrocarbons (HCs) in the zeolite pores. We propose an alternative monomolecular path, in which the hydride transfer takes place between a hydrogen atom of a dimeric HC and a zeolitic proton, yielding a conjugated carbocation and molecular H2. Computed free energies indicate that the preference for a particular reaction mechanism is determined by the local shape of the zeolite micropores.
Co-reporter:Emiel de Smit, Frank M. F. de Groot, Raoul Blume, Michael Hävecker, Axel Knop-Gericke and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 3) pp:667-680
Publication Date(Web):19 Nov 2009
DOI:10.1039/B920256K
The effect of Cu on the reduction behavior and surface properties of supported and unsupported Fe-based Fischer–Tropsch synthesis (FTS) catalysts was investigated using in situ X-ray photoelectron spectroscopy (XPS) and in situ X-ray absorption spectroscopy (XAS) in combination with ex situ bulk characterization. During exposure to 0.4 mbar CO–H2 above 180 °C, the reduction of CuO to Cu0 marked the onset of the reduction of Fe3O4 to α-Fe. The promotion effects of Cu are explained by a combination of spillover of H2 and/or CO molecules from metallic Cu0 nuclei to closely associated iron oxide species and textural promotion. XAS showed that in the supported catalyst, Cu+ and Fe2+ species were stabilized by SiO2 and, as a result, Fe species were not reduced significantly beyond Fe3O4 and Fe2+, even after treatment at 350 °C. After the reduction treatment, XPS showed that the concentration of oxygen and carbon surface species was higher in the presence of Cu. Furthermore, it was observed that the unsupported, Cu-containing catalyst showed higher CO2 concentration in the product gas stream during and after reduction and Fe surface species were slightly oxidized after prolonged exposure to CO–H2. These observations suggest that, in addition to facilitating the reduction of the iron oxide phase, Cu also plays a direct role in altering the surface chemistry of Fe-based FTS catalysts.
Co-reporter:L. Espinosa-Alonso, K. P. de Jong and B. M Weckhuysen
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 1) pp:97-107
Publication Date(Web):07 Nov 2009
DOI:10.1039/B915753K
The influence of the Cl−(aq) concentration, solution pH and equilibration time on the PdCl42−(aq) dynamics and molecular structure after impregnation of γ-Al2O3 catalyst bodies has been studied using UV-Vis micro-spectroscopy. To do so, 0.2 wt% Pd catalysts have been prepared from acidic solutions (pH 1 and 5) of the Na2PdCl4 precursor salt with different amounts of NaCl. It was found that egg-shell catalysts are obtained when a less acidic pH (pH 5) is combined with [Cl−(aq)] < 0.6 M and less than 24 h of equilibration time are implemented, while to achieve egg-white catalysts the solution pH should be 1. Moreover, by increasing the equilibration time up to 96 h, the egg-shell profiles vanish to provide a uniform Pd distribution, while the egg-white distribution becomes egg-yolk. Additionally, Pd complexes appeared with different molecular structures depending on the solution pH, equilibration time and macro-distribution achieved. The protocol developed to create different Pd macro-distributions has been applied to prepare two 1 wt% Pd/γ-Al2O3 egg-shell and egg-white catalysts. The Pd dynamics and molecular structure have been followed after impregnation, drying and calcination, demonstrating that the profiles created after impregnation are retained.
Co-reporter:Frank M. F. de Groot Dr.;Emiel de Smit;Matti M. van Schooneveld;Luis R. Aramburo Dr.
ChemPhysChem 2010 Volume 11( Issue 5) pp:951-962
Publication Date(Web):
DOI:10.1002/cphc.200901023
Abstract
The present status of in-situ scanning transmission X-ray microscopy (STXM) is reviewed, with an emphasis on the abilities of the STXM technique in comparison with electron microscopy. The experimental aspects and interpretation of X-ray absorption spectroscopy (XAS) are briefly introduced and the experimental boundary conditions that determine the potential applications for in-situ XAS and in-situ STXM studies are discussed. Nanoscale chemical imaging of catalysts under working conditions is outlined using cobalt and iron Fischer–Tropsch catalysts as showcases. In the discussion, we critically compare STXM-XAS and STEM-EELS (scanning transmission electron microscopy–electron energy loss spectroscopy) measurements and indicate some future directions of in-situ nanoscale imaging of catalytic solids and related nanomaterials.
Co-reporter:Marianne H.F. Kox Dr.;Ana Mijovilovich Dr.;Jesper J.H.B. Sättler;Eli Stavitski Dr. ;BertM. Weckhuysen Dr.
ChemCatChem 2010 Volume 2( Issue 5) pp:564-571
Publication Date(Web):
DOI:10.1002/cctc.200900329
Abstract
X-ray absorption, UV/Vis, and fluorescence microspectroscopy have been used to characterize the catalytic conversion of thiophene derivatives within the micropores of an individual H-ZSM-5 zeolite crystal. Space-resolved information into the Si/Al ratios and sulfur content was provided by X-ray absorption microspectroscopy. X-ray absorption near-edge spectroscopy spectral information and modeling indicated the presence of a sulfur atom, in close proximity of two oxygen atoms, that is, at approximately 2.5 Å. Space-resolved spectroscopic measurements provided experimental evidence for the formation of conjugated carbocationic reaction intermediates by opening of the thiophene ring. The molecular alignment of reaction products within the straight pores of the individual H-ZSM-5 zeolite crystals was observed with polarized light UV/Vis microspectroscopy. In addition, confocal fluorescence microscopy revealed the 3D distribution of different reaction products and demonstrated the influence of the H-ZSM-5 crystal’s intergrowth structure and its related molecular diffusion barriers.
Co-reporter:Marianne H.F. Kox Dr.;Ana Mijovilovich Dr.;Jesper J.H.B. Sättler;Eli Stavitski Dr. ;BertM. Weckhuysen Dr.
ChemCatChem 2010 Volume 2( Issue 5) pp:
Publication Date(Web):
DOI:10.1002/cctc.201090018
Co-reporter:Lukasz Karwacki;HendrikE. vanderBij;Dr. Jan Kornatowski;Dr. Pablo Cubillas;Dr. MartynR. Drury;D.A.Matthijs deWinter;Dr. MichaelW. Anderson;Dr. BertM. Weckhuysen
Angewandte Chemie 2010 Volume 122( Issue 38) pp:6942-6946
Publication Date(Web):
DOI:10.1002/ange.201003273
Co-reporter:Eli Stavitski Dr.;EvgenyA. Pidko Dr.;MarianneH.F. Kox Dr.;EmielJ.M. Hensen Dr.;RutgerA. vanSanten Dr.;BertM. Weckhuysen Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 31) pp:9340-9348
Publication Date(Web):
DOI:10.1002/chem.201000995
Abstract
Large zeolite crystals have been used as model systems for the investigation of diffusion and catalytic reactivity phenomena in microporous host materials for at least two decades. However, their potential in assisting the detection of elusive reactive intermediates appears to have been underestimated. Herein, we show that a complementary use of vibrational and optical spectroscopy in combination with theoretical calculations allows for the unambiguous identification of transient carbocationic species generated upon the acid-catalyzed oligomerization of styrene derivatives within zeolite H-ZSM-5. Thanks to the mediated diffusion of the reactant in the large H-ZSM-5 crystals and minimal external surface the reaction intermediates can be accumulated within zeolite micropores in sufficient concentrations to allow their detection by synchrotron-based IR microspectroscopy. The UV/Vis and IR spectra display strong polarization dependence of on the molecular alignment of the dimeric styrene carbocations imposed by the zeolite channels and cages that can be rationalized in terms of the electronic and vibrational transitions of the intrazeolite carbocations. Based on these findings, a molecular-level picture of the macroscopic arrangement of the reaction intermediates confined within microporous zeolite matrices can be devised.
Co-reporter:Lukasz Karwacki;HendrikE. vanderBij;Dr. Jan Kornatowski;Dr. Pablo Cubillas;Dr. MartynR. Drury;D.A.Matthijs deWinter;Dr. MichaelW. Anderson;Dr. BertM. Weckhuysen
Angewandte Chemie International Edition 2010 Volume 49( Issue 38) pp:6790-6794
Publication Date(Web):
DOI:10.1002/anie.201003273
Co-reporter:Muriel Lepage, Tom Visser, Fouad Soulimani, Ana Iglesias-Juez and Bert M. Weckhuysen
The Journal of Physical Chemistry C 2010 Volume 114(Issue 5) pp:2282-2292
Publication Date(Web):January 12, 2010
DOI:10.1021/jp910371j
Rh nanoparticles supported on a series of zeolite Y samples containing different monovalent (H+, Na+, Rb+, and Cs+) and divalent (Mg2+, Ca2+, Sr2+, and Ba2+) cations have been used as model systems to investigate the effect of promoter elements in the reduction of NO by CO. Infrared (IR) spectroscopy with NO as a probe molecule allowed monitoring of the electronic changes in the local environment of Rh. The IR bands corresponding to linearly adsorbed NO, i.e., linear and dinitrosyl species, were found to shift to lower wavenumbers with increasing ionic radius-to-charge ratio of the cation. Simultaneously, a lower ignition temperature for NO reduction was observed. In addition, the relative intensity of the bridge-bonded NO band as compared to the total absorbance of Rh-bonded NO species decreased with increasing Lewis acidity of the cation, as expressed by the Kamlet−Taft parameter α. The latter observation matches with similar trends, observed in a previous study (Lepage, M., et al., J. Phys. Chem. C 2008, 112, 9394) for the same catalysts using CO as the IR probe molecule, which could be related to the Rh activity for the CO oxidation reaction. The samples of the present study also showed different catalytic activities, although a straightforward correlation between the results obtained with NO IR spectroscopy and the catalytic reduction of NO by CO could not be established.
Co-reporter:Leticia Espinosa-Alonso ; Matthew G. O’Brien ; Simon D. M. Jacques ; Andrew M. Beale ; Krijn P. de Jong ; Paul Barnes
Journal of the American Chemical Society 2009 Volume 131(Issue 46) pp:16932-16938
Publication Date(Web):November 2, 2009
DOI:10.1021/ja907329j
Tomographic energy dispersive diffraction imaging (TEDDI) is a recently developed synchrotron-based characterization technique used to obtain spatially resolved X-ray diffraction and fluorescence information in a noninvasive manner. With the use of a synchrotron beam, three-dimensional (3D) information can be conveniently obtained on the elemental composition and related crystalline phases of the interior of a material. In this work, we show for the first time its application to characterize the structure of a heterogeneous catalyst body in situ during thermal treatment. Ni/γ-Al2O3 hydrogenation catalyst bodies have been chosen as the system of study. As a first example, the heat treatment in N2 of a [Ni(en)3](NO3)2/γ-Al2O3 catalyst body has been studied. In this case, the crystalline [Ni(en)3](NO3)2 precursor was detected in an egg-shell distribution, and its decomposition to form metallic Ni crystallites of around 5 nm was imaged. In the second example, the heat treatment in N2 of a [Ni(en)(H2O)4]Cl2/γ-Al2O3 catalyst body was followed. The initial [Ni(en)(H2O)4]Cl2 precursor was uniformly distributed within the catalyst body as an amorphous material and was decomposed to form metallic Ni crystallites of around 30 nm with a uniform distribution. TEDDI also revealed that the decomposition of [Ni(en)(H2O)4]Cl2 takes place via two intermediate crystalline structures. The first one, which appears at around 180 °C, is related to the restructuring of the Ni precursor on the alumina surface; the second one, assigned to the formation of a limited amount of Ni3C, is observed at 290 °C.
Co-reporter:Leticia Espinosa-Alonso ; Anna A. Lysova ; Peter de Peinder ; Krijn P. de Jong ; Igor V. Koptyug
Journal of the American Chemical Society 2009 Volume 131(Issue 18) pp:6525-6534
Publication Date(Web):April 22, 2009
DOI:10.1021/ja900346k
Magnetic resonance imaging (MRI) was used to study the impregnation step during the preparation of Ni/γ-Al2O3 hydrogenation catalysts with Ni2+ metal ion present in different coordinations. The precursor complexes were [Ni(H2O)6]2+ and [Ni(edtaHx)](2−x)− (where x = 0, 1, 2 and edta = ethylenediaminetetraacetic acid), representing a nonshielded and a shielded paramagnetic complex, respectively. Due to this shielding effect of the ligands, the dynamics of [Ni(H2O)6]2+ or [Ni(edtaHx)](2−x)− were visualized applying T2 or T1 image contrast, respectively. MRI was applied in a quantitative manner to calculate the [Ni(H2O)6]2+ concentration distribution after impregnation when it was present alone in the impregnation solution, or together with the [Ni(edtaHx)](2−x)− species. Moreover, the combination of MRI with UV−vis microspectroscopy allowed the visualization of both species with complementary information on the dynamics and adsorption/desorption phenomena within γ-Al2O3 catalyst bodies. These phenomena yielded nonuniform Ni distributions after impregnation, which are interesting for certain industrial applications.
Co-reporter:Regina Palkovits, Andrei N. Parvulescu, Peter J. C. Hausoul, Cornelis A. Kruithof, Robertus J. M. Klein Gebbink and Bert M. Weckhuysen
Green Chemistry 2009 vol. 11(Issue 8) pp:1155-1160
Publication Date(Web):14 May 2009
DOI:10.1039/B904274A
The telomerization of 1,3-butadiene with various alcohols has been investigated using a catalyst based on a Pd(acac)2 precursor and a phosphine ligand, TOMPP (TOMPP = tris-(o-methoxyphenyl)phosphine). We were able to demonstrate the capability of the catalyst to telomerize 1,3-butadiene with various multifunctional nucleophiles having primary and secondary alcohol functions. High yields of telomer products (>98%) were obtained in very short reaction times (<2 h). The telomerization activity and selectivity of the Pd/TOMPP complex was strongly influenced by the type of alcohol used as substrate. When diols were used, telomerization of 1,3-butadiene with 1,2-propanediol and 1,2-butanediol afforded the highest yield of mono-telomer (over 70%) and for 1,2-butanediol a turnover frequency (TOF) of 300000 h−1 was reached, combined with a turnover number (TON) of 7800.
Co-reporter:BertM. Weckhuysen
ChemCatChem 2009 Volume 1( Issue 1) pp:
Publication Date(Web):
DOI:10.1002/cctc.200900146
Co-reporter:Monica Calatayud Dr.;AgnieszkaM. Ruppert Dr.;BertM. Weckhuysen Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 41) pp:10864-10870
Publication Date(Web):
DOI:10.1002/chem.200900487
Abstract
Alkaline earth metal oxides (MO) are catalytically active in the etherification of glycerol. Density Functional Theory (DFT) calculations have been used to examine the reactivity of glycerol with MO surfaces with M=Mg, Ca, Sr or Ba. More specifically, the optimum glycerol adsorption mode and the strength of glycerol interaction with regular MO (001) surfaces and a stepped CaO surface have been investigated and involves the interaction with acid–base surface sites. The basicity of lattice oxygen atoms is correlated with the adsorption energy: BaO (−3.02 eV) > SrO (−2.85 eV) > CaO (−2.05 eV) > MgO (−1.35 eV). The interactions have an exothermic character, that is, the more basic the alkaline earth metal oxide, the more exothermic is the adsorption process and the higher the dissociation extent. Thus, the dissociation of glycerol increases in the order: MgO (not dissociated) < CaO (partially dissociated < SrO (partially dissociated) < BaO (completely dissociated). The presence of defects is found to play a key role in the mechanism: glycerol interaction with a stepped CaO surface presents the highest adsorption energy (−3.78 eV), and the molecule is found to dissociate at the step. The calculated structural parameters are found to be in good agreement with experimental data on catalyst reactivity. Moreover, the earlier postulated reaction mechanism, which also involves the additional involvement of Lewis acid sites proved to be feasible for CaO and SrO regular surfaces, and for the stepped CaO surface. It was found that for these oxides one of the most favored adsorption modes involves a non-dissociative adsorption of one hydroxyl group of glycerol, which as a result becomes a better leaving group. Therefore, theoretical evidence was found for the possible direct involvement of Lewis acid sites in the catalytic etherification of bio-derived alcohols, such as glycerol, as it is anticipated that these observations can be extended to sugar alcohols as well.
Co-reporter:Bart P.C. Hereijgers ;BertM. Weckhuysen
ChemSusChem 2009 Volume 2( Issue 8) pp:743-748
Publication Date(Web):
DOI:10.1002/cssc.200900108
Abstract
A series of alumina-supported gold catalysts was investigated for the CO-free production of hydrogen by partial oxidation of methanol. The addition of alkaline-earth metal oxide promoters resulted in a significant improvement of the catalytic performance. The methanol conversion was ca. 85 % with all studied catalyst materials, however, the selectivity for hydrogen increased from 15 % to 51 % when going from the unpromoted to a BaO-promoted catalyst. The formation of the undesired byproducts CO, methane, and dimethyl ether was considerably reduced as well. The observed trend in catalyst performance follows the trend in increasing basicity of the studied promoter elements, indicating a chemical effect of the promoter material. Superior catalytic performance, in terms of H2 and CO selectivity, was obtained with a Au/La2O3 catalyst. At 300 °C the hydrogen selectivity reached 80 % with only 2 % CO formation, and the catalyst displayed a stable performance over at least 24 h on-stream. Furthermore, the formation of CO was found to be independent of the oxygen concentration in the feed. The commercial lanthanum oxide used in this study had a low specific surface area, which led to the formation of relative large gold particles. Therefore, the catalytic activity could be enhanced by decreasing the gold particle size through deposition on lanthanum oxide supported on high-surface-area alumina.
Co-reporter:Peter J.C. Hausoul;Pieter C.A. Bruijnincx Dr.;Robertus J.M. KleinGebbink ;BertM. Weckhuysen
ChemSusChem 2009 Volume 2( Issue 9) pp:855-858
Publication Date(Web):
DOI:10.1002/cssc.200900115
Co-reporter:Matthew G. O’Brien, Andrew M. Beale, Simon D. M. Jacques, Thomas Buslaps, Veijo Honkimaki and Bert M. Weckhuysen
The Journal of Physical Chemistry C 2009 Volume 113(Issue 12) pp:4890-4897
Publication Date(Web):2017-2-22
DOI:10.1021/jp810428e
The oxidation of methanol under anaerobic reaction conditions over MoO3 has been studied using an in situ approach, combining ultraviolet−visible (UV−vis), Raman, wide-angle X-ray scattering (WAXS), and online mass spectroscopy (MS) techniques. Comparison of the UV−vis and MS data reveals that during the initial stages of the reaction methanol is chemisorbed onto the oxide’s surface, primarily at defect sites. Reaction then begins, producing formaldehyde, dimethyl ether, and water. At low temperatures, CO and MoO2 are also produced, as reoxidation of the reactive sites cannot occur rapidly enough to avoid additional reduction. After the initial heating, continued reduction of the bulk oxide by Mars−Van Krevelen oxygen transfer to the active surface sites is observed as a change in the total Raman intensity. Most significantly, after 125 min of reaction, bulk MoO2 is observed and here a Rietveld analysis of the MoO3 WAXS data indicates qualitatively that one of the three unique oxygen environments (O1) becomes more active than the others. This is confirmed by changes in the Raman data, indicating that the Mo−O1 bond to this oxygen is broken more quickly. This work has therefore identified the oxygen most likely to be transferred through the bulk of the oxide during the Mars−Van Krevelen oxygen transfer. However, a comparison with previous works and our MS and UV−vis data indicates no particular relationship between the bonding in the bulk oxide and its surface reactivity. Therefore, although O1 is abstracted and transferred through the bulk, it may be replacing other oxygen atoms (i.e., O2 or O3) at the oxide surface. This work then also demonstrates that to fully understand a parent oxide we cannot rely on a bulk view of the entire system, but must obtain separate details about both the surface sites (responsible for selectivity) and the bulk sites (that maintain catalytic activity by oxygen transfer), particularly under oxygen-free conditions.
Co-reporter:BertM. Weckhuysen Dr.
Angewandte Chemie 2009 Volume 121( Issue 27) pp:5008-5043
Publication Date(Web):
DOI:10.1002/ange.200900339
Co-reporter:MarianneH.F. Kox;KatrinF. Domke Dr.;JamesP.R. Day Dr.;Gianluca Rago;Eli Stavitski Dr.;Mischa Bonn Dr.;BertM. Weckhuysen Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 47) pp:
Publication Date(Web):
DOI:10.1002/anie.200905606
Co-reporter:A. Mijovilovich, L. G. M. Pettersson, S. Mangold, M. Janousch, J. Susini, M. Salome, F. M. F. de Groot and B. M. Weckhuysen
The Journal of Physical Chemistry A 2009 Volume 113(Issue 12) pp:2750-2756
Publication Date(Web):March 3, 2009
DOI:10.1021/jp806823c
Sulfur K-edge XANES has been measured for three sulfur model compounds, dibenzothiophene, dibenzothiophene sulfone, and aliphatic sulfur (dl-methionine). The spectra have been simulated with Density Functional Theory (DFT) by using a number of methods, including the half-core-hole approximation. Dipole transition elements were calculated and the transitions were convoluted with linearly increasing Gaussian functions in the first 20 eV of the near-edge region. In the case of dibenzothiophene, relaxation of the first excited states in the presence of the core-hole gave a further improvement. The theoretical results reproduce well the features of the spectra and give insight in the relation between geometric structure and molecular orbitals. Though dl-methionine and dibenzothiophene show a similar sharp rise of the white line, their molecular levels are quite different, pointing out the difficulties in finding useful “fingerprints” in the spectra for specific compounds.
Co-reporter:MarianneH.F. Kox;KatrinF. Domke Dr.;JamesP.R. Day Dr.;Gianluca Rago;Eli Stavitski Dr.;Mischa Bonn Dr.;BertM. Weckhuysen Dr.
Angewandte Chemie 2009 Volume 121( Issue 47) pp:9152-9156
Publication Date(Web):
DOI:10.1002/ange.200904282
Co-reporter:MarianneH.F. Kox;KatrinF. Domke Dr.;JamesP.R. Day Dr.;Gianluca Rago;Eli Stavitski Dr.;Mischa Bonn Dr.;BertM. Weckhuysen Dr.
Angewandte Chemie 2009 Volume 121( Issue 47) pp:
Publication Date(Web):
DOI:10.1002/ange.200905606
Co-reporter:Emiel deSmit;Ingmar Swart Dr.;J.Fredrik Creemer Dr.;Chithra Karunakaran Dr.;Drew Bertwistle;HennyW. Zbergen Dr.;FrankM.F. deGroot Dr.;BertM. Weckhuysen Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 20) pp:3632-3636
Publication Date(Web):
DOI:10.1002/anie.200806003
Co-reporter:Emiel deSmit;Ingmar Swart Dr.;J.Fredrik Creemer Dr.;Chithra Karunakaran Dr.;Drew Bertwistle;HennyW. Zbergen Dr.;FrankM.F. deGroot Dr.;BertM. Weckhuysen Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 20) pp:
Publication Date(Web):
DOI:10.1002/anie.200990101
Co-reporter:BertM. Weckhuysen Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 27) pp:4910-4943
Publication Date(Web):
DOI:10.1002/anie.200900339
Co-reporter:MarianneH.F. Kox;KatrinF. Domke Dr.;JamesP.R. Day Dr.;Gianluca Rago;Eli Stavitski Dr.;Mischa Bonn Dr.;BertM. Weckhuysen Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 47) pp:8990-8994
Publication Date(Web):
DOI:10.1002/anie.200904282
Co-reporter:Matthew G. O’Brien;Andrew M. Beale;Simon D. M. Jacques
Topics in Catalysis 2009 Volume 52( Issue 10) pp:1400-1409
Publication Date(Web):2009 September
DOI:10.1007/s11244-009-9324-z
A setup combining a number of techniques (WAXS, XANES and UV–Vis) has been used to probe the stability of an iron molybdate catalyst during redox cycling. The catalyst was first reduced under anaerobic methanol/helium conditions, producing formaldehyde and then regenerated using air. Although in this test-case the catalyst and conditions differ from that of a commercial catalyst bed we demonstrate how such a setup can reveal new information on catalyst materials. In particular we observe the formation of two phases during reduction; one which we propose to be an oxygen deficient ‘pseudo-molybdate phase’, the other a molybdenum carbide-like phase, both produced as oxygen is removed from the catalyst. Standard in situ techniques could detect such transient phases, however, the information from multiple techniques, allows us to more accurately identify the nature of these materials and to carry out appropriate complementary ex situ measurements to aid in the analysis. This and similar setups therefore offer a way to more quickly and accurately observe reaction pathways within a catalyst, which may for example, result in the deactivation of the material by different routes to those observed previously. Additionally, the specific combination of these techniques with on-line mass spectrometry, allows us to monitor the activity of the catalyst surface and here observe that different catalytic mechanisms may occur during different stages of the redox process. Therefore this setup should allow for the observation of many novel variations in a catalyst’s reactivity, leading to the improvement of current and development of new materials.
Co-reporter:Emiel de Smit and Bert M. Weckhuysen
Chemical Society Reviews 2008 vol. 37(Issue 12) pp:2758-2781
Publication Date(Web):14 Oct 2008
DOI:10.1039/B805427D
Iron-based Fischer–Tropsch catalysts, which are applied in the conversion of CO and H2 into longer hydrocarbon chains, are historically amongst the most intensively studied systems in heterogeneous catalysis. Despite this, fundamental understanding of the complex and dynamic chemistry of the iron–carbon–oxygen system and its implications for the rapid deactivation of the iron-based catalysts is still a developing field. Fischer–Tropsch catalysis is characterized by its multidisciplinary nature and therefore deals with a wide variety of fundamental chemical and physical problems. This critical review will summarize the current state of knowledge of the underlying mechanisms for the activation and eventual deactivation of iron-based Fischer–Tropsch catalysts and suggest systematic approaches for relating chemical identity to performance in next generation iron-based catalyst systems (210 references).
Co-reporter:Eli Stavitski Dr.;MarianneH.F. Kox;Ingmar Swart;FrankM.F. deGroot Dr. ;BertM. Weckhuysen Dr.ir.
Angewandte Chemie 2008 Volume 120( Issue 19) pp:3599-3603
Publication Date(Web):
DOI:10.1002/ange.200705562
Co-reporter:AlwiesW.A.M. vanderHeijden;SimonG. Podkolzin Dr.;MarkE. Jones Dr.;JohannesH. Bitter Dr.;BertM. Weckhuysen Dr.ir.
Angewandte Chemie 2008 Volume 120( Issue 27) pp:5080-5082
Publication Date(Web):
DOI:10.1002/ange.200800270
Co-reporter:Eli Stavitski Dr.;MartynR. Drury Dr.;D.A.Matthijs deWinter;MarianneH.F. Kox;BertM. Weckhuysen Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 30) pp:5637-5640
Publication Date(Web):
DOI:10.1002/anie.200801433
Co-reporter:AlwiesW.A.M. vanderHeijden;SimonG. Podkolzin Dr.;MarkE. Jones Dr.;JohannesH. Bitter Dr.;BertM. Weckhuysen Dr.ir.
Angewandte Chemie International Edition 2008 Volume 47( Issue 27) pp:5002-5004
Publication Date(Web):
DOI:10.1002/anie.200800270
Co-reporter:Regina Palkovits Dr.;Ilenia Nieddu Dr.;RobertusJ.M. KleinGebbink Dr.;BertM. Weckhuysen Dr.
ChemSusChem 2008 Volume 1( Issue 3) pp:193-196
Publication Date(Web):
DOI:10.1002/cssc.200700147
Co-reporter:Regina Palkovits Dr.;Ilenia Nieddu Dr.;RobertusJ.M. KleinGebbink Dr.;BertM. Weckhuysen Dr.
ChemSusChem 2008 Volume 1( Issue 3) pp:
Publication Date(Web):
DOI:10.1002/cssc.200890004
Co-reporter:Muriel Lepage ; Tom Visser ; Fouad Soulimani ; Andrew M. Beale ; Ana Iglesias-Juez ; Ad M. J. van der Eerden
The Journal of Physical Chemistry C 2008 Volume 112(Issue 25) pp:9394-9404
Publication Date(Web):May 29, 2008
DOI:10.1021/jp711743g
Rh particles with an average diameter smaller than 1.5 nm have been supported on a series of zeolite Y samples. These zeolite materials contained different monovalent (H+, Na+, K+, Rb+, and Cs+) and divalent (Mg2+, Ca2+, Sr2+, and Ba2+) cations and were used as model systems to investigate the effect of promoter elements in the oxidation of CO over supported Rh particles in excess of oxygen. Infrared (IR) spectroscopy was carried out to monitor the electronic changes in the local environment of Rh-adsorbed CO. It was found that the bands corresponding to two Rh gem-dicarbonyl species, Rh+(CO)2−(Oz)2 and Rh+(CO)2−(Oz)(H2O), shift to lower wavenumbers with increasing ionic radius/charge ratio of the cation. In addition, the relative intensity of the bridge bonded CO as compared to the total absorbance of Rh-bonded CO species decreases with increasing Lewis acidity, as expressed by the Kamlet−Taft parameter α of the cation. This trend could be directly correlated to the Rh CO oxidation activity, since low temperatures at 50% CO conversion corresponded with catalyst materials with a high contribution of bridge-bonded CO species and hence with small α values. A lower Lewis acidity causes an increased electron density on the framework oxygen atoms and thus an increased electron density on the zeolite-supported Rh particles. Comparable trends have been observed previously on a similar series of cation containing zeolite supported Pt catalyst materials.
Co-reporter:AgnieszkaM. Ruppert Dr.;JohannesD. Meeldijk;BonnyW.M. Kuipers Dr.;BenH. Erné Dr.;BertM. Weckhuysen Dr.
Chemistry - A European Journal 2008 Volume 14( Issue 7) pp:2016-2024
Publication Date(Web):
DOI:10.1002/chem.200701757
Abstract
Glycerol is an attractive renewable building block for the synthesis of di- and triglycerols, which have numerous applications in the cosmetic and pharmaceutical industries. In this work, the selective etherification of glycerol to di- and triglycerol was studied in the presence of alkaline earth metal oxides and the data are compared with those obtained with Na2CO3 as a homogeneous catalyst. It was found that glycerol conversion increased with increasing catalyst basicity; that is, the conversion increases in the order: MgO<CaO<SrO<BaO. The best selectivity values for (di- + tri-) glycerol (>90 % at 60 % conversion) are obtained over CaO, SrO, and BaO. For these catalysts no substantial acrolein formation was observed. Furthermore, at the start of the reaction mainly linear diglycerol was produced, whereas at higher conversion degrees branched diglycerol started to form. In another series of experiments different types of CaO materials were prepared. It was found that these CaO-based materials not only differed in their surface area and number of basic sites, but also in their Lewis acid strength. Within this series the CaO material possessing the strongest Lewis acid sites had the highest catalytic activity, comparable to that of BaO, pointing towards the important role of Lewis acidity for this etherification reaction. Based on these observations a plausible alternative reaction scheme for glycerol etherification is presented, which considers the facilitation of the hydroxyl leaving process. Finally, the stability of the catalytic solids under study was investigated and it was found that colloidal CaO particles of about 50–100 nm can be spontaneously generated during reaction. Catalytic testing of these CaO colloids, after isolation from the reaction medium, revealed a very high etherification activity. Understanding the nature of these Ca-based colloids opens new opportunities for investigating supported colloidal particle catalysts to take advantage of both their hetero- and homogeneous nature.
Co-reporter:JaapA. Bergwerff Dr.;AnnaA. Lysova Dr.;Leticia Espinosa-Alonso;IgorV. Koptyug Dr.;BertM. Weckhuysen Dr.
Chemistry - A European Journal 2008 Volume 14( Issue 8) pp:2363-2374
Publication Date(Web):
DOI:10.1002/chem.200700990
Abstract
An indirect magnetic resonance imaging (MRI) method has been developed to determine in a noninvasive manner the distribution of paramagnetic Co2+ complexes inside Co/Al2O3 catalyst extrudates after impregnation with Co2+/citrate solutions of different pH and citrate concentrations. UV/Vis/NIR microspectroscopic measurements were carried out simultaneously to obtain complementary information on the nature of the Co2+ complexes. In this way, it could be confirmed that the actual distribution of Co2+ inside the extrudates could be derived from the MRI images. By combining these space- and time-resolved techniques, information was obtained on both the strength and the mode of interaction between [Co(H2O)6]2+ and different Co2+ citrate complexes with the Al2O3 support. Complexation of Co2+ by citrate was found to lead to a stronger interaction of Co with the support surface and formation of an eggshell distribution of Co2+ complexes after impregnation. By addition of free citrate and by changing the pH of the impregnation solution, it was possible to obtain the rather uncommon egg-yolk and egg-white distributions of Co2+ inside the extrudates after impregnation. In other words, by carefully altering the chemical composition and pH of the impregnation solution, the macrodistribution of Co2+ complexes inside catalyst extrudates could be fine-tuned from eggshell over egg white and egg yolk to uniform.
Co-reporter:Regina Palkovits;Ilenia Nieddu;CornelisA. Kruithof;RobertusJ.M. KleinGebbink Dr.;BertM. Weckhuysen
Chemistry - A European Journal 2008 Volume 14( Issue 29) pp:8995-9005
Publication Date(Web):
DOI:10.1002/chem.200800792
Abstract
Glycerol is considered a potential renewable building block for the synthesis of existing as well as new chemicals. A promising route is the telomerization of 1,3-butadiene with glycerol leading to C8 chain ethers of glycerol with applications in, for example, surfactant chemistry. Recently, we reported a new set of palladium-based homogeneous catalytic systems for the telomerization of 1,3-butadiene with glycerol and found that palladium complexes bearing methoxy-functionalized triphenylphosphine ligands are highly active catalysts capable of converting crude glycerol without any significant loss of activity. Herein, we present a detailed account of these investigations by reporting on the influence of the butadiene/glycerol ratio, temperature, and reaction time on product selectivity and activity allowing further optimization of catalyst performance. Maximum activity and yield were reached for high 1,3-butadiene/glycerol ratios at a temperature of 90 °C, whereas the selectivity for mono- and diethers of glycerol could be optimized by combining high reaction temperatures and short reaction times with low butadiene/glycerol ratios. Variation of the PdII metal precursors and the metal/ligand ratio showed that palladium precursors with halogen ligands gave unsatisfying results, in contrast to precursors with weakly coordinated ligands such as [Pd(OAc)2] and [Pd(acac)2]. [Pd(dba)2], the only Pd0 precursor tested, gave the best results in terms of activity, which illustrates the importance of the ability to form a Pd0 species in the catalytic cycle. Finally, base addition resulted in a shortening of the reaction time and most likely facilitates the formation of a Pd0 species. Based on these results, we were able to realize the first attempts towards a rational ligand design aimed at a high selectivity for mono- and diether formation.
Co-reporter:Davide Mores;Eli Stavitski Dr.;MarianneH.F. Kox;Jan Kornatowski Dr.;Unni Olsbye ;BertM. Weckhuysen Dr.Ir.
Chemistry - A European Journal 2008 Volume 14( Issue 36) pp:11320-11327
Publication Date(Web):
DOI:10.1002/chem.200801293
Abstract
Formation of coke in large H-ZSM-5 and H-SAPO-34 crystals during the methanol-to-olefin (MTO) reaction has been studied in a space- and time-resolved manner. This has been made possible by applying a high-temperature in-situ cell in combination with micro-spectroscopic techniques. The buildup of optically active carbonaceous species allows detection with UV/Vis microscopy, while a confocal fluorescence microscope in an upright configuration visualises the formation of coke molecules and their precursors inside the catalyst grains. In H-ZSM-5, coke is initially formed at the triangular crystal edges, in which straight channel openings reach directly the external crystal surface. At reaction temperatures ranging from 530 to 745 K, two absorption bands at around 415 and 550 nm were detected due to coke or its precursors. Confocal fluorescence microscopy reveals fluorescent carbonaceous species that initially form in the near-surface area and gradually diffuse inwards the crystal in which internal intergrowth boundaries hinder a facile penetration for the more bulky aromatic compounds. In the case of H-SAPO-34 crystals, an absorption band at around 400 nm arises during the reaction. This band grows in intensity with time and then decreases if the reaction is carried out between 530 and 575 K, whereas at higher temperatures its intensity remains steady with time on stream. Formation of the fluorescent species during the course of the reaction is limited to the near-surface region of the H-SAPO-34 crystals, thereby creating diffusion limitations for the coke front moving towards the middle of the crystal during the MTO reaction. The two applied micro-spectroscopic techniques introduced allow us to distinguish between graphite-like coke deposited on the external crystal surface and aromatic species formed inside the zeolite channels. The use of the methods can be extended to a wide variety of catalytic reactions and materials in which carbonaceous deposits are formed.
Co-reporter:Eli Stavitski Dr.;MarianneH.F. Kox;Ingmar Swart;FrankM.F. deGroot Dr. ;BertM. Weckhuysen Dr.ir.
Angewandte Chemie 2008 Volume 120( Issue 19) pp:
Publication Date(Web):
DOI:10.1002/ange.200890084
Co-reporter:Alwies W. A. M. van der Heijden;Ad J. M. Mens;René Bogerd
Catalysis Letters 2008 Volume 122( Issue 3-4) pp:238-246
Publication Date(Web):2008/05/01
DOI:10.1007/s10562-008-9436-2
Lanthanum oxide-based catalysts are active in the elimination of HCl from C2H5Cl, 1,2-C2H4Cl2 and 1,1,2-C2H3Cl3 leading to the formation of their respective chlorinated ethenes. An oxygen-rich catalytic surface may form CO, CO2 and C2HCl as side products, whereas with chlorine-rich catalytic surfaces a stable product distribution is achieved with 100% selectivity towards the formation of ethenes, such as the valuable C2H3Cl intermediate.
Co-reporter:Eli Stavitski Dr.;MarianneH.F. Kox;Ingmar Swart;FrankM.F. deGroot Dr. ;BertM. Weckhuysen Dr.ir.
Angewandte Chemie International Edition 2008 Volume 47( Issue 19) pp:3543-3547
Publication Date(Web):
DOI:10.1002/anie.200705562
Co-reporter:Eli Stavitski Dr.;MarianneH.F. Kox;Ingmar Swart;FrankM.F. deGroot Dr. ;BertM. Weckhuysen Dr.ir.
Angewandte Chemie International Edition 2008 Volume 47( Issue 19) pp:
Publication Date(Web):
DOI:10.1002/anie.200890084
Co-reporter:Emiel de Smit,
Ingmar Swart,
J. Fredrik Creemer,
Gerard H. Hoveling,
Mary K. Gilles,
Tolek Tyliszczak,
Patricia J. Kooyman,
Henny W. Zandbergen,
Cynthia Morin,
Bert M. Weckhuysen
&
Frank M. F. de Groot
Nature 2008 456(7219) pp:222
Publication Date(Web):2008-11-13
DOI:10.1038/nature07516
The modern chemical industry uses heterogeneous catalysts in almost every production process1. They commonly consist of nanometre-size active components (typically metals or metal oxides) dispersed on a high-surface-area solid support, with performance depending on the catalysts’ nanometre-size features and on interactions involving the active components, the support and the reactant and product molecules. To gain insight into the mechanisms of heterogeneous catalysts, which could guide the design of improved or novel catalysts, it is thus necessary to have a detailed characterization of the physicochemical composition of heterogeneous catalysts in their working state at the nanometre scale1, 2. Scanning probe microscopy methods have been used to study inorganic catalyst phases at subnanometre resolution3, 4, 5, 6, but detailed chemical information of the materials in their working state is often difficult to obtain5, 6, 7. By contrast, optical microspectroscopic approaches offer much flexibility for in situ chemical characterization; however, this comes at the expense of limited spatial resolution8, 9, 10, 11. A recent development promising high spatial resolution and chemical characterization capabilities is scanning transmission X-ray microscopy4, 12, 13, which has been used in a proof-of-principle study to characterize a solid catalyst14. Here we show that when adapting a nanoreactor specially designed for high-resolution electron microscopy7, scanning transmission X-ray microscopy can be used at atmospheric pressure and up to 350 °C to monitor in situ phase changes in a complex iron-based Fisher–Tropsch catalyst and the nature and location of carbon species produced. We expect that our system, which is capable of operating up to 500 °C, will open new opportunities for nanometre-resolution imaging of a range of important chemical processes taking place on solids in gaseous or liquid environments.
Co-reporter:Andrew M. Beale Dr.;Simon D. M. Jacques Dr.;Jaap A. Bergwerff Dr.;Paul Barnes Dr.;Bert M. Weckhuysen Dr.
Angewandte Chemie 2007 Volume 119(Issue 46) pp:
Publication Date(Web):16 NOV 2007
DOI:10.1002/ange.200790233
Tomographische Bildgebung …… durch energiedispersive Beugung wurde als “inneres Auge” zur Bestimmung der Phasen- und Elementverteilung von Metalloxiden in Katalysatorextrudaten genutzt. B. M. Weckhuysen und Mitarbeiter erläutern auf S. 8988 ff. die Funktionsweise dieser Technik am Beispiel von CoMo/Al2O3-Hydrodesulfurierungskatalysatoren sowie ihre Anwendung zur Unterscheidung zwischen kristallinen und nichtkristallinen Phasen. Die Ergebnisse zeigen, dass 1D- und 2D-Analysen meist unzureichend sind, um Zusammenhänge zwischen der Katalysatorleistung und der Präparationsmethode aufzudecken.
Co-reporter:Andrew M. Beale Dr.;Simon D. M. Jacques Dr.;Jaap A. Bergwerff Dr.;Paul Barnes Dr.;Bert M. Weckhuysen Dr.
Angewandte Chemie 2007 Volume 119(Issue 46) pp:
Publication Date(Web):17 OCT 2007
DOI:10.1002/ange.200703673
3D-Karten von Katalysatoren: Die tomographische Bildgebung durch energiedispersive Beugung liefert Informationen über die dreidimensionale Metalloxidverteilung im Inneren eines Katalysatorextrudats während dessen Präparation. Das Bild zeigt eine solche 3D-Karte sowie die Detektorsignale an zwei Stellen im Katalysator.
Co-reporter:Andrew M. Beale Dr.;Simon D. M. Jacques Dr.;Jaap A. Bergwerff Dr.;Paul Barnes Dr.;Bert M. Weckhuysen Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 46) pp:
Publication Date(Web):16 NOV 2007
DOI:10.1002/anie.200790233
Like an 'eye on the inside' …… tomographic energy dispersive diffraction imaging is used to obtain 3D information on the phase and element distribution of metal oxides in catalyst extrudate bodies (the pellets in the cover picture). In the Communication on page 8832 ff., B. M. Weckhuysen and co-workers use Co–Mo/Al2O3 hydrodesulfurization catalysts to showcase the technique and its capacity to discriminate between crystalline and noncrystalline phases. They show that 1D and 2D studies oversimplify the challenge of relating catalyst performance to a preparation method.
Co-reporter:Andrew M. Beale Dr.;Simon D. M. Jacques Dr.;Jaap A. Bergwerff Dr.;Paul Barnes Dr.;Bert M. Weckhuysen Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 46) pp:
Publication Date(Web):17 OCT 2007
DOI:10.1002/anie.200703673
Catalyst extrudate in 3D: Through the use of tomographic energy dispersive diffraction imaging it is possible to obtain detailed three-dimensional insight into the metal oxide distribution inside a catalyst extrudate during its preparation. The picture shows such a 3D map of an extrudate along with examples of the detector signal for two different locations in the catalyst.
Co-reporter:Lukasz Karwacki;Eli Stavitski Dr.;Marianne H. F. Kox;Jan Kornatowski Dr.;Bert M. Weckhuysen Dr. ir.
Angewandte Chemie International Edition 2007 Volume 46(Issue 38) pp:
Publication Date(Web):31 AUG 2007
DOI:10.1002/anie.200702012
Growing together: The template-removal process in a series of zeolite crystals has been mapped by a combination of in situ optical and fluorescence microscopy. Three-dimensional spatial distribution of light-absorbing and -emitting species allowed visualization of the architecture of zeolite intergrowths.
Co-reporter:Eli Stavitski Dr.;Marianne H. F. Kox;Bert M. Weckhuysen Dr. ir.
Chemistry - A European Journal 2007 Volume 13(Issue 25) pp:
Publication Date(Web):15 AUG 2007
DOI:10.1002/chem.200790092
Wanted!…nonbulky and electron-donating species in zeolite channels! In their Full Paper on page 7057 ff., Bert M. Weckhuysen et al. have demonstrated that the use of combined in-situ optical microspectroscopic and fluorescence microscopic techniques under the same reaction conditions provides very valuable insights into the catalytic processes occurring within a zeolite catalyst particle.
Co-reporter:Eli Stavitski Dr.;Marianne H. F. Kox;Bert M. Weckhuysen Dr. ir.
Chemistry - A European Journal 2007 Volume 13(Issue 25) pp:
Publication Date(Web):18 JUL 2007
DOI:10.1002/chem.200700568
A combination of in-situ optical and fluorescence microspectroscopy has been employed to investigate the oligomerization of styrene derivatives occurring in the micropores of coffin-shaped H-ZSM-5 zeolite crystals in a space- and time-resolved manner. The carbocationic intermediates in this reaction act as reporter molecules for catalytic activity, since they exhibit strong optical absorption and fluorescence. In this way, reactant selectivity and restricted transition-state selectivity for 14 substituted styrene molecules can be visualized and quantified. Based on a thorough analysis of the time- and space-resolved UV/Vis spectra, it has been revealed that two main parameters affect the reaction rates, namely, the carbocation stabilization effect and the diffusion hindrance. The stabilization effect was tested by comparison of the reaction rates for 4-methoxystyrene versus 4-methylstyrene and in the series 4-bromo-, 4-chloro and 4-fluorostyrene; in both cases less electronegative substituents were found to accelerate the reaction. As to the steric effect, bulkier chemical groups bring down the reaction rate, as evident from the observation that 4-methoxystyrene is more reactive than 4-ethoxystyrene due to differences in their diffusivity, while heavily substituted styrenes, such as 3,4-dichlorostyrene and 2,3,4,5,6-pentafluorostyrene, cannot enter the zeolite pore system and therefore do not display any reactivity. Furthermore, β-methoxystyrene and trans-β-methylstyrene show limited reactivity as well as restricted reaction-product formation due to steric constraints imposed by the H-ZSM-5 channel system. Finally, polarized-light optical microspectroscopy and fluorescence microscopy demonstrate that dimeric styrene compounds are predominantly formed and aligned within the straight channels at the edges of the crystals, whereas a large fraction of trimeric carbocations along with dimeric compounds are present in the straight channels of the main body of the H-ZSM-5 crystals. Our results reinforce the observation of a non-uniform catalytic behavior within zeolite crystals, with specific parts of the zeolite grains being less accessible and reactive towards reactant molecules. The prospects and potential of this combined in-situ approach for studying large zeolite crystals in the act will be discussed.
Co-reporter:Alwies W. A. M. van der Heijden;Maria Garcia Ramos;Bert M. Weckhuysen Dr. ir.
Chemistry - A European Journal 2007 Volume 13(Issue 34) pp:
Publication Date(Web):11 SEP 2007
DOI:10.1002/chem.200700901
Activity experiments using GC analysis of reactor effluent have been combined with in situ IR spectroscopy to elucidate the reaction steps in the destructive adsorption of CHCl3, CH2Cl2, and CH3Cl over LaOCl. The IR results show that during reaction, LaOCl is covered with carbonate, formate, and methoxy groups. The relative amount of each of these surface intermediates depends on the Cl/H ratio of the reactant. The decomposition of the surface species leads to formation of the reaction products, and is influenced by the temperature and the relative amount of Cl present on the surface. The GC results show that the activity for the destructive adsorption of H-containing chlorinated C1 compounds decreases with increasing hydrogen content of the reactant. The acquired insight into the mechanism of destructive adsorption is crucial to the design of new catalyst materials for the efficient conversion of chlorinated hydrocarbons into nonhazardous products or reusable chemicals.
Co-reporter:Lukasz Karwacki;Eli Stavitski Dr.;Marianne H. F. Kox;Jan Kornatowski Dr.;Bert M. Weckhuysen Dr. ir.
Angewandte Chemie 2007 Volume 119(Issue 38) pp:
Publication Date(Web):31 AUG 2007
DOI:10.1002/ange.200702012
Zusammenwachsen: Der Prozess der Templatentfernung wurde an einer Reihe von Zeolithkristallen in situ mit einer Kombination aus optischer und Fluoreszenzmikroskopie untersucht. Die dreidimensionale räumliche Verteilung lichtabsorbierender und -emittierender Spezies ermöglichte eine Visualisierung der Architektur von Zeolithverwachsungen.
Co-reporter:Luis R. Aramburo, Shewangizaw Teketel, Stian Svelle, Simon R. Bare, Bjørnar Arstad, Henny W. Zandbergen, Unni Olsbye, Frank M.F. de Groot, Bert M. Weckhuysen
Journal of Catalysis (November 2013) Volume 307() pp:185-193
Publication Date(Web):1 November 2013
DOI:10.1016/j.jcat.2013.07.009
•Steaming leads to mesoporosity as well as to a decrease in acid site density and strength.•Deactivation of H-ZSM-5 catalyst during MTH is governed by the formation of coke precursor species.•Upon steaming changes in acidity are more pronounced in the external regions of the catalyst.•STXM reveals a relation between coke precursor species and the distribution of acid sites.H-ZSM-5 catalyst powders before and after a steaming post-treatment have been investigated during the Methanol-To-Hydrocarbons (MTH) process at 350 °C. Bulk and surface characterization techniques have been combined with in situ Scanning Transmission X-ray Microscopy (STXM) at the aluminum and carbon K-edge to study the changes in acidity, porosity, reactivity, and aluminum distribution upon steaming. It was found that steaming post-treatment has a positive impact on the stability of H-ZSM-5 without inducing important changes in the MTH activity and selectivity. The lower MTH stability of non-steamed H-ZSM-5 catalyst powder is related to the formation of poly-aromatic compounds in the outer regions of the catalyst particles, as probed with in situ STXM. In contrast, a limited amount of poly-aromatics was found in the outer rim of steamed H-ZSM-5 catalyst particles. These differences occur as a result of the generation of mesoporosity as well as the reduction in the number and strength of acid sites after steaming, as evidenced by the nanoscale imaging of adsorbed pyridine with STXM.Graphical abstractThe reduced formation of poly-aromatic species in the outer regions of mildly steamed H-ZSM-5 zeolite powders leads to an improved methanol-to-hydrocarbons reaction stability.Download high-res image (63KB)Download full-size image
Co-reporter:Andrew M. Beale, Emma K. Gibson, Matthew G. O’Brien, Simon D.M. Jacques, Robert J. Cernik, Marco Di Michiel, Paul D. Cobden, Özlem Pirgon-Galin, Leon van de Water, Michael J. Watson, Bert M. Weckhuysen
Journal of Catalysis (May 2014) Volume 314() pp:94-100
Publication Date(Web):1 May 2014
DOI:10.1016/j.jcat.2014.04.007
•Effects of Sulfur poisoning on water–gas shift activity of Cu/ZnO catalyst bodies investigated.•XRD, EXAFS, and XRF imaging revealed the presence of sulfur-containing crystalline phases.•Active Cu/ZnO component shrinks with increasing [H2S] reducing water–gas shift activity.•Migration of Cu in the form of CuS and toward the sample periphery also occurs.The effects of sulfur poisoning on the water–gas shift (WGS) activity of industrial Cu/ZnO/Al2O3 catalyst bodies have been studied. The samples were characterized using chemical imaging methods, including XRD-CT, XAFS mapping, and XRF, in order to understand the process by which accelerated sulfur poisoning leads to catalyst deactivation. After ∼90 h on stream, all catalysts exhibited reduced activity; the higher the H2S concentration, the greater the extent of deactivation. Non-invasive XRD-CT measurements performed on intact samples recovered from the reactor revealed the formation of sulfide phases, including sphalerite (β-ZnS) and crystalline CuS, Cu2S, and CuSO4 phases. These sulfide phases were distributed predominantly as a graduated corona around the sample edge reaching ∼1.5 mm thick for experiments performed in the highest concentration of 500 ppm H2S. XAFS mapping, which is particularly sensitive to the local coordination environment around the element being probed, confirmed the presence of mixed Cu/Zn–O/S coordination environments and that the core of the sample remained sulfur-free. A combination of XRD-CT and XRF revealed that CuS appeared to be mobile under reaction conditions resulting in the redistribution of Cu toward the very edge of the samples. A combination of techniques has therefore demonstrated that H2S deactivation of Cu/ZnO/Al2O3 catalyst bodies occurs via phase transformation of the active Cu/ZnO phase into sulfides and redistribution of these components over the sample instead of Cu active site poisoning by Sads species.Graphical abstractDownload high-res image (56KB)Download full-size image
Co-reporter:Wenhao Luo, Upakul Deka, Andrew M. Beale, Ernst R.H. van Eck, Pieter C.A. Bruijnincx, Bert M. Weckhuysen
Journal of Catalysis (May 2013) Volume 301() pp:175-186
Publication Date(Web):1 May 2013
DOI:10.1016/j.jcat.2013.02.003
The catalytic performance of 1 wt% Ru-based catalysts in the hydrogenation of levulinic acid (LA) has been studied at 40 bar H2 and 473 K. This was done by assessing the influence of the support acidity (i.e., Nb2O5, TiO2, H-β, and H-ZSM5) and solvent (i.e., dioxane, 2-ethylhexanoic acid (EHA), and neat LA). The Ru/TiO2 gave excellent selectivity to γ-valerolactone (GVL) (97.5%) at 100% conversion and was remarkably stable even under severe reaction conditions. Ru/H-ZSM5 showed a 45.8% yield of pentanoic acid (PA) and its esters in dioxane, which is the first example of this one-pot conversion directly from LA at 473 K. The gradual deactivation of zeolite-supported catalysts in EHA and neat LA was mainly caused by dealumination, as confirmed by 27Al MAS NMR. Coke buildup originated from angelicalactone and, remarkably, occurred preferentially in the zigzag channels of H-ZSM5 as shown by systematic shifts in the XRD patterns. The GVL ring-opening step is considered to be the rate-determining step on the pathway to PA, necessitating an acidic support.Graphical abstractSelectivity in Ru-catalyzed levulinic acid (LA) hydrogenation depends strongly on support type. Ru/TiO2 is highly selective for γ-valerolactone and very stable even in neat LA, while zeolite-supported catalysts give pentanoic acid in 45.8% yield in a one-pot conversion. Dealumination causes gradual deactivation.Download high-res image (104KB)Download full-size imageHighlights► A direct, one-pot conversion of levulinic acid to pentanoic acid with zeolite-supported Ru catalysts. ► Ru/TiO2 shows excellent selectivity to GVL and remarkable stability. ► Support dealumination is mainly responsible for gradual catalyst deactivation. ► Coke buildup in H-ZSM5 is found to occur preferentially in the zigzag channels.
Co-reporter:Sophie C.C. Wiedemann, Joseph A. Stewart, Fouad Soulimani, Tanja van Bergen-Brenkman, Stephan Langelaar, Bas Wels, Peter de Peinder, Pieter C.A. Bruijnincx, Bert M. Weckhuysen
Journal of Catalysis (July 2014) Volume 316() pp:24-35
Publication Date(Web):1 July 2014
DOI:10.1016/j.jcat.2014.04.018
•Ferrierite catalyses the alkyl branching of oleic acid with high activity and selectivity.•Reuse experiments indicate severe deactivation, despite framework conservation.•Micropore blockage occurs early on in the reaction, suggesting the existence of pore mouth catalysis.•The acid sites are poisoned by the formation of (poly)enylic carbocations.•Long chain alkyl benzenes are found to be the major coke constituents.The formation and nature of coke (precursor) species has been studied during the skeletal isomerisation of oleic acid catalysed by protonated ferrierite, in the presence and absence of a triphenylphosphine promoter. UV–Vis and FT-IR spectroscopic analyses of the spent catalyst materials, complemented by NMR and mass spectrometry of the coke deposits extracted after HF dissolution, provide new insights into the deactivation mechanisms. Initial high catalyst activity and selectivity are quickly lost, despite conservation of the framework integrity, as a result of severe deactivation. Pore blockage is detected very early in the reaction, and only the pore mouth is actively employed. Additionally, polyenylic carbocations formed by hydrogen transfer reactions poison the active sites; they are considered to be the precursors to traces of condensed aromatics detected in the spent catalyst. Dodecyl benzene is the major “coke” constituent, and its precursor probably also competes for the active sites.Download high-res image (98KB)Download full-size image
Co-reporter:Anna L. Jongerius, Robin Jastrzebski, Pieter C.A. Bruijnincx, Bert M. Weckhuysen
Journal of Catalysis (January 2012) Volume 285(Issue 1) pp:315-323
Publication Date(Web):1 January 2012
DOI:10.1016/j.jcat.2011.10.006
In the present work, extensive hydrodeoxygenation (HDO) studies with a commercial sulfided CoMo/Al2O3 catalyst were performed on a library of lignin model compounds at 50 bar hydrogen pressure and 300 °C in dodecane, using a batch autoclave system. The catalyst was activated under hydrogen atmosphere prior to the reaction, and the spent catalyst was analyzed using thermogravimetric analysis. An extended reaction network is proposed, showing that HDO, demethylation, and hydrogenation reactions take place simultaneously. HDO of mono-oxygenated substrates proved to be difficult at the applied conditions. Starting from most positions in the network, phenol, and cresols are therefore the main final products, suggesting the possibility of convergence on a limited number of products from a mixture of substrates. HDO of dimeric model compounds mimicking typical lignin linkages revealed that coumaran alkyl ethers and β-O-4 bonds can be broken, but 5–5′ linkages not.Graphical abstractExtensive hydrodeoxygenation studies with a sulfided CoMo/Al2O3 catalyst were performed on a library of lignin model compounds. An extended reaction network is proposed, showing that HDO, demethylation, and hydrogenation reactions take place simultaneously.Download high-res image (133KB)Download full-size image.Highlights► Extensive study of aromatic oxygenates allowed elucidation of the HDO reaction pathway. ► Hydrodeoxygenation competes with O-demethylation, hydrogenation, and ring methylation. ► CoMo-catalyzed HDO of a library of substrates results nonetheless mainly in phenolics production. ► Abundant lignin–ether linkages are readily cleaved under HDO conditions.
Co-reporter:Wenhao Luo, Pieter C.A. Bruijnincx, Bert M. Weckhuysen
Journal of Catalysis (December 2014) Volume 320() pp:33-41
Publication Date(Web):1 December 2014
DOI:10.1016/j.jcat.2014.09.014
•Wet impregnation gives well-dispersed Ru on H-ZSM5 with well-preserved strong acid sites.•The catalyst gives an excellent yield of 91.3% pentanoic acid directly from levulinic acid.•Any catalyst deactivation proceeds mainly via coke deposition in H-ZSM5.•Activity can be almost fully restored after regeneration by simple coke burn-off.The direct conversion of levulinic acid (LA) to pentanoic acid (PA) has been studied with six 1 wt% Ru/H-ZSM5 catalysts at 40 bar H2 and 473 K in dioxane. The influence of ZSM5 cation form, Si/Al ratio and ruthenium precursor on metal dispersion and acidity has been assessed. A highly active bifunctional 1 wt% Ru/H-ZSM5 catalyst was developed to give a PA yield of 91.3% after 10 h. The PA productivity of 1.157 molPA g−1Ru h−1 is the highest reported to date. The simple preparation method allows for a significant fraction of ruthenium to be located inside the zeolite pores, providing the desired proximity between the hydrogenation function and the strong acid sites, which is key to the conversion of LA into PA. Coke buildup during reaction causes some deactivation, but activity can be almost fully restored after catalyst regeneration by simple coke burn-off.Download high-res image (99KB)Download full-size image
Co-reporter:Bart P.C. Hereijgers, Bert M. Weckhuysen
Journal of Catalysis (22 March 2010) Volume 270(Issue 1) pp:16-25
Publication Date(Web):22 March 2010
DOI:10.1016/j.jcat.2009.12.003
The selective oxidation of cyclohexane to cyclohexanone and cyclohexanol with air over Au/Al2O3, Au/TiO2, and Au/SBA-15 catalysts was investigated and compared with the industrial autoxidation process. In contradiction with the literature results, the Au-based catalysts did not exhibit excellent catalytic performance and the combined selectivity toward cyclohexanone and cyclohexanol decreased to ∼70% with the increasing conversion above 5%. In addition, the observed product evolution and by-product evolution were typical for the autoxidation process, although a significant increase in adipic acid and CO2 formation was observed. Additional oxidation experiments containing a radical scavenger completely inhibited the reaction and provided proof that the oxidation follows a radical-chain mechanism instead of a catalytic mechanism. This explains the low selectivity at increasing conversion. This important deviation from the literature results can be clarified by the complicated but necessary product analysis of both the gas and the liquid phases.Through a detailed analysis of the product distribution during the catalytic oxidation of cyclohexane over Au-based catalysts, a radical-chain mechanism proceeding via peroxo-species was revealed.Download high-res image (58KB)Download full-size image
Co-reporter:Young-Min Chung, Davide Mores, Bert M. Weckhuysen
Applied Catalysis A: General (19 September 2011) Volume 404(Issues 1–2) pp:12-20
Publication Date(Web):19 September 2011
DOI:10.1016/j.apcata.2011.06.030
Co-reporter:Ana Iglesias-Juez, Andrew M. Beale, Karin Maaijen, Tsu Chien Weng, Pieter Glatzel, Bert M. Weckhuysen
Journal of Catalysis (15 December 2010) Volume 276(Issue 2) pp:268-279
Publication Date(Web):15 December 2010
DOI:10.1016/j.jcat.2010.09.018
The catalytic performances of Pt/Al2O3 and Pt–Sn/Al2O3 catalysts for the dehydrogenation of propane through consecutive reaction–regeneration cycles have been studied under realistic reaction conditions. A 10-fold successive dehydrogenation–regeneration cycling study, similar to that employed in an industrial propane dehydrogenation reactor, was performed in order to examine the catalyst activity and stability as well as the propene selectivity. Combined in situ UV–Vis/Raman spectroscopy measurements were taken in order to follow the coke formation processes during propane dehydrogenation. This approach allowed correlating propane conversion and propene formation with the on-line determined Raman D over G band intensity ratio and amount of coke formed. These in situ measurements on coke formation and related catalyst deactivation were supplemented by in situ high-energy resolution fluorescence detected (HERFD) XANES measurements in order to characterize the structural and electronic properties of the supported Pt and PtSn nanoparticles during the successive dehydrogenation–regeneration cycles. This combination of powerful spectroscopic techniques revealed unique information regarding the activity behavior and deactivation mechanism of Pt- and PtSn-based propane dehydrogenation catalysts, enabling us to identify important structure-electronic-performance relationships as well as fundamental insight into the dynamics of PtSn alloy formation processes in Pt nanoparticles at elevated temperatures.XANES and Raman reveal new information on the properties of supported PtSn nanoparticles during propane dehydrogenation–regeneration cycles leading to insight into the dynamics of PtSn alloy formation at elevated temperatures.Download high-res image (96KB)Download full-size image
Co-reporter:Emiel de Smit, Andrew M. Beale, Sergey Nikitenko, Bert M. Weckhuysen
Journal of Catalysis (10 March 2009) Volume 262(Issue 2) pp:244-256
Publication Date(Web):10 March 2009
DOI:10.1016/j.jcat.2008.12.021
The structural properties of three Fe-based Fischer–Tropsch synthesis (FTS) catalysts containing different amounts of Cu, K and SiO2 additives were investigated during pretreatment and FTS in a fixed bed-like reactor using combined in situ X-ray absorption fine structure (XAFS)/wide angle X-ray scattering (WAXS) techniques. This combination enabled acquisition of complementary information regarding the local environment of iron atoms from XAFS and crystalline phases from WAXS during H2 and CO/H2 pretreatment and FTS at 1 bar. The presence of the SiO2 support and promoter elements significantly influenced the structural properties of the catalysts after pretreatment. After FTS, H2 pretreated catalysts mainly consisted of amorphous θ-Fe3C clusters (unsupported catalysts) or amorphous iron (II) silicate (supported catalyst), while the CO/H2 pretreated catalysts all consisted mainly of γ-Fe and χ-Fe5C2. Catalysts activated in CO/H2 showed superior stability and activity, while H2 pretreated unsupported catalysts deactivated rapidly during the first 15 h of FTS.A combined in situ X-ray absorption and diffraction study provided new insights into the local and long range structure of iron-based Fischer–Tropsch catalysts after pretreatment and during synthesis.Download high-res image (182KB)Download full-size image
Co-reporter:Agnieszka M. Ruppert, Andrei N. Parvulescu, Maria Arias, Peter J.C. Hausoul, Pieter C.A. Bruijnincx, Robertus J.M. Klein Gebbink, Bert M. Weckhuysen
Journal of Catalysis (10 December 2009) Volume 268(Issue 2) pp:251-259
Publication Date(Web):10 December 2009
DOI:10.1016/j.jcat.2009.09.023
Heterogeneous etherification of various biomass-based alcohols with 1-octene was investigated as a direct route for the synthesis of long alkyl chain ethers. Several acid catalyst materials including Amberlyst resins and various zeolites were screened as etherification catalysts in a solventless system. It was found that H-Beta zeolites are the most selective catalysts for the etherification of biomass-based alcohols with 1-octene to the corresponding mono-ethers. With H-Beta the conversion of neat glycerol was around 15–20% and increased to 54–89% for glycols such as ethylene glycol and 1,2-propylene glycol, with high selectivities to mono- and di-octyl ethers of 85–97%. Other linear alkenes like 1-dodecene and 1-hexadecene were successfully employed in the direct etherification of glycols as well. Crude glycerol was also etherified, albeit with low conversions. The influence of several reaction parameters on the etherification activity of H-Beta has been investigated together with catalyst recovery and re-use.Direct etherification of biomass-based alcohols with 1-octene can be performed over solid acid catalysts such as zeolite Beta as a new route for synthesis of the valuable long alkyl chain ethers with very high selectivities.Download high-res image (117KB)Download full-size image
Co-reporter:Elena Sacaliuc-Parvulescu, Heiner Friedrich, Regina Palkovits, Bert M. Weckhuysen, T. Alexander Nijhuis
Journal of Catalysis (1 October 2008) Volume 259(Issue 1) pp:43-53
Publication Date(Web):1 October 2008
DOI:10.1016/j.jcat.2008.07.013
Postsynthesis ammonium treatment induces a substantial increase in the catalytic activity of Au/Ti-SBA-15 catalysts for the direct vapor-phase epoxidation of propylene using hydrogen and oxygen. The PO formation rate of a calcined Au/Ti-SBA-15 catalyst prepared by this method increased from 4.3 mgPO h−1 g−1cat to 37.2 mgPO h−1 g−1cat at 200 °C compared with a catalyst prepared in an identical manner without this treatment. The catalysts were characterized by XRD, N2-sorption, UV–vis–NIR DRS, 29Si MAS NMR, FT-IR spectroscopy, and TEM to gain insight into the relationship between ammonia treatment and catalyst activity. 29Si MAS NMR measurements proved that the ammonium nitrate solution caused hydrolysis of SiOSi or TiOSi bonds, resulting in a Ti-SBA-15 surface with a greater amount of surface hydroxyl groups. FT-IR measurements indicated the presence of amine species that favor the homogeneous deposition of Au nanoparticles. This was confirmed by TEM measurements showing greater metal dispersion for NH4NO3-treated Au/Ti-SBA-15 materials.
Co-reporter:Andrew M. Beale, Simon D.M. Jacques, Elena Sacaliuc-Parvalescu, Matthew G. O’Brien, Paul Barnes, Bert M. Weckhuysen
Applied Catalysis A: General (1 July 2009) Volume 363(Issues 1–2) pp:
Publication Date(Web):1 July 2009
DOI:10.1016/j.apcata.2009.05.008
A one-step, low-temperature hydrothermal method has been successfully employed to prepare iron molybdate catalysts with Mo:Fe ratios ranging from 1.5:1 to 3.0:1. The resulting materials were characterized using a number of techniques including: XRD, Raman, N2 adsorption, SEM/EDX, DTA, EDXRD and combined XRD/XAS. The catalytic oxidative dehydrogenation of methanol to formaldehyde has been used as a test reaction. For Mo:Fe ∼ 1.5, phase-pure Fe2(MoO4)3 resulted from syntheses performed at temperatures as low as 100 °C in under 4 h. For samples with a Mo:Fe ∼ 3 detailed analysis of XRD, Raman and EXAFS data revealed the formation of a high surface area possessing, mixed phase material consisting of a poorly crystalline Mo5O14 and an amorphous Fe2(MoO4)3 type precursor. Both phases proved to be thermally unstable above a calcination temperature of 300 °C, going on to form high surface area mixed Fe2(MoO4)3/MoO3. Continued heating of this mixed oxide material resulted in sintering and to a decrease in the surface area. When both mildly (200 °C) and then more severely calcined (300 °C), this mixed phase sample showed a higher selectivity for formaldehyde production than a conventionally prepared (via co-precipitation) iron molybdate catalyst.A one-step, low-temperature hydrothermal method has been successfully employed to prepare high surface area iron molybdate catalysts with Mo:Fe ratios of 1.5:1–3:1. Syntheses performed with a Mo:Fe ∼ 3 resulted in a mixed crystalline Mo5O14/amorphous FeMo oxide phase, with the chemical formula which was unstable above a calcination temperature of 300 °C, forming mixed Fe2(MoO4)3/MoO3. The Mo:Fe ∼ 3 phase after both mild (200 °C) and more severe calcinations (300 °C), showed a higher selectivity for formaldehyde production than a conventionally prepared (via co-precipitation) iron molybdate catalyst.Download full-size image
Co-reporter:Dilek A. Boga, Fang Liu, Pieter C. A. Bruijnincx and Bert M. Weckhuysen
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 1) pp:NaN143-143
Publication Date(Web):2015/09/09
DOI:10.1039/C4CY01711K
The aqueous-phase reforming (APR) of a crude glycerol that originates from an industrial process and the effect of the individual components of crude glycerol on APR activity have been studied over 1 wt% Pt/Mg(Al)O, 1 wt% Pt/Al2O3, 5 wt% Pt/Al2O3 and 5 wt% Pt/C catalysts at 29 bar and 225 °C. The use of a 10 wt% alkaline crude glycerol solution in water, containing 6.85 wt% glycerol, 1.62 wt% soaps, 1.55 wt% methanol, and 0.07 wt% ester, resulted in a dramatic drop in APR activity compared to the corresponding 6.85 wt% solution of pure glycerol in water. Catalytic performance in crude glycerol APR increased in the order: 1 wt% Pt/Al2O3 < 5 wt% Pt/Al2O3 < 1 wt% Pt/Mg(Al)O < 5 wt% Pt/C. A H2 selectivity of only 1% was obtained with crude glycerol over a 1 wt% Pt/Al2O3 catalyst, while the same catalyst material under identical reaction conditions gave 64% H2 with pure glycerol. The cause of deactivation was investigated with synthetic mixtures that mimicked the composition of the crude glycerol and contained glycerol, methanol and varying amounts of NaCl and sodium oleate. The results showed that Na salts of fatty acids have a much more pronounced negative influence on APR activity than NaCl does and greatly inhibit H2 formation. Stearic acid and long chain aliphatics and olefins were shown to be formed and to be involved in the deactivation of the catalyst. The relatively high activity/selectivity of the 1 wt% Pt/Mg(Al)O could be attributed to intercalation of oleate/stearate in the sheets of the layered double hydroxide that is formed under reaction conditions.
Co-reporter:Eli Stavitski and Bert M. Weckhuysen
Chemical Society Reviews 2010 - vol. 39(Issue 12) pp:NaN4625-4625
Publication Date(Web):2010/10/12
DOI:10.1039/C0CS00064G
The miniaturization of in situ spectroscopic tools has been recognized as a forefront instrumental development for the characterization of heterogeneous catalysts. With the multitude of micro-spectroscopic methods available fundamental insight into the structure–function relationships of catalytic processes can be obtained. Among these techniques vibrational spectroscopy is one of the most versatile methods and capable to shed insight into the molecular structure of reaction intermediates and products, the chemical state of catalyst materials during reaction as well as the nature of interactions between reactants/intermediates/products and the catalyst surface. In this tutorial review we discuss the recent developments in the field of infrared (IR) and Raman micro-spectroscopy and illustrate their potential. Showcase examples include (1) chemical imaging of spatial heterogeneities during catalyst preparation, (2) high-throughput catalyst screening, (3) transport and adsorption phenomena within catalytic solids and (4) reactivity studies of porous oxides, such as zeolites. Finally, new in situ spectroscopy tools based on vibrational spectroscopy and their potential in the catalysis domain are discussed.
Co-reporter:Andrew M. Beale, Simon D. M. Jacques and Bert M. Weckhuysen
Chemical Society Reviews 2010 - vol. 39(Issue 12) pp:NaN4672-4672
Publication Date(Web):2010/10/27
DOI:10.1039/C0CS00089B
Heterogeneous catalysis is a term normally used to describe a group of catalytic processes, yet it could equally be employed to describe the catalytic solid itself. A better understanding of the chemical and structural variation within such materials is thus a pre-requisite for the rationalising of structure–function relationships and ultimately to the design of new, more sustainable catalytic processes. The past 20 years has witnessed marked improvements in technologies required for analytical measurements at synchrotron sources, including higher photon brightness, nano-focusing, rapid, high resolution data acquisition and in the handling of large volumes of data. It is now possible to image materials using the entire synchrotron radiative profile, thus heralding a new era of in situ/operando measurements of catalytic solids. In this tutorial review we discuss the recent work in this exciting new research area and finally conclude with a future outlook on what will be possible/challenging to measure in the not-too-distant future.
Co-reporter:Emiel de Smit and Bert M. Weckhuysen
Chemical Society Reviews 2008 - vol. 37(Issue 12) pp:NaN2781-2781
Publication Date(Web):2008/10/14
DOI:10.1039/B805427D
Iron-based Fischer–Tropsch catalysts, which are applied in the conversion of CO and H2 into longer hydrocarbon chains, are historically amongst the most intensively studied systems in heterogeneous catalysis. Despite this, fundamental understanding of the complex and dynamic chemistry of the iron–carbon–oxygen system and its implications for the rapid deactivation of the iron-based catalysts is still a developing field. Fischer–Tropsch catalysis is characterized by its multidisciplinary nature and therefore deals with a wide variety of fundamental chemical and physical problems. This critical review will summarize the current state of knowledge of the underlying mechanisms for the activation and eventual deactivation of iron-based Fischer–Tropsch catalysts and suggest systematic approaches for relating chemical identity to performance in next generation iron-based catalyst systems (210 references).
Co-reporter:D. Cicmil, I. K. van Ravenhorst, J. Meeuwissen, A. Vantomme and B. M. Weckhuysen
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 3) pp:NaN743-743
Publication Date(Web):2015/11/18
DOI:10.1039/C5CY01512J
A study on the influence of H2 and the degree of titanation of a Cr/SiO2 Phillips polymerisation catalyst on the selective oligomerisation of ethylene induced by pre-contacting the catalyst with triethylaluminium (TEAl) is presented. Ethylene oligomerisation reactions were performed at 373 K and 1 bar, inside a quartz reactor of a specially designed operando experimental setup, which allowed examination of the catalysts by UV-vis-NIR diffuse reflectance spectroscopy, while the gas phase was continuously monitored by on-line mass spectroscopy and subsequently analysed by gas chromatography. With this combination of techniques, it was possible to acquire detailed insight into the different distributions of produced oligomers, which were highly dependent on the catalyst structure. The addition of small amounts of Ti significantly changes the electronic environment of Cr oligomerisation sites by the formation of Cr–O–Ti–O–Si linkages, favouring β-H transfer and increasing the selectivity towards butene at the expense of 1-hexene. Moreover, we show that ethylene oligo-/polymerisation reactions follow at least two different pathways, i.e. metallacycle for olefinic species with a broken Schulz-Flory distribution, and Cossee–Arlman for other hydrocarbon species.
Co-reporter:K. H. Cats, J. C. Andrews, O. Stéphan, K. March, C. Karunakaran, F. Meirer, F. M. F. de Groot and B. M. Weckhuysen
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 12) pp:NaN4449-4449
Publication Date(Web):2016/02/16
DOI:10.1039/C5CY01524C
The Fischer–Tropsch synthesis (FTS) reaction is one of the most promising processes to convert alternative energy sources, such as natural gas, coal or biomass, into liquid fuels and other high-value products. Despite its commercial implementation, we still lack fundamental insights into the various deactivation processes taking place during FTS. In this work, a combination of three methods for studying single catalyst particles at different length scales has been developed and applied to study the deactivation of Co/TiO2 Fischer–Tropsch synthesis (FTS) catalysts. By combining transmission X-ray microscopy (TXM), scanning transmission X-ray microscopy (STXM) and scanning transmission electron microscopy-electron energy loss spectroscopy (STEM-EELS) we visualized changes in the structure, aggregate size and distribution of supported Co nanoparticles that occur during FTS. At the microscale, Co nanoparticle aggregates are transported over several μm leading to a more homogeneous Co distribution, while at the nanoscale Co forms a thin layer of ∼1–2 nm around the TiO2 support. The formation of the Co layer is the opposite case to the “classical” strong metal–support interaction (SMSI) in which TiO2 surrounds the Co, and is possibly related to the surface oxidation of Co metal nanoparticles in combination with coke formation. In other words, the observed migration and formation of a thin CoOx layer are similar to a previously discussed reaction-induced spreading of metal oxides across a TiO2 surface.
Co-reporter:Meenakshisundaram Sankar, Qian He, Simon Dawson, Ewa Nowicka, Li Lu, Pieter C. A. Bruijnincx, Andrew M. Beale, Christopher J. Kiely and Bert M. Weckhuysen
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 14) pp:NaN5482-5482
Publication Date(Web):2016/03/23
DOI:10.1039/C6CY00425C
The synthesis and functionalization of imines and amines are key steps in the preparation of many fine chemicals and for pharmaceuticals in particular. Traditionally, metal complexes are used as homogeneous catalysts for these organic transformations. Here we report gold–palladium and ruthenium–palladium nano-alloys supported on TiO2 acting as highly efficient heterogeneous catalysts for the one-pot synthesis of the imine N-benzylideneaniline and the secondary amine N-benzylaniline directly from the easily available and stable nitrobenzene and benzyl alcohol precursors using a hydrogen auto-transfer strategy. These reactions were carried out without any added external hydrogen, sacrificial hydrogen donor or a homogeneous base. The bimetallic catalysts were prepared by the recently developed modified impregnation strategy, giving efficient control of size and nano-alloy composition. Both bimetallic catalysts were found to be far more active than their monometallic analogues due to a synergistic effect. Based on the turnover numbers the catalytic activities follow the order Ru < Pd < Au ≪ Au–Pd < Ru–Pd. Aberration corrected scanning transmission electron microscopy (AC-STEM) and X-ray absorption spectroscopy (XAFS) studies of these catalysts revealed that the reason for the observed synergistic effect is the electronic modification of the metal sites in the case of the Au–Pd system and a size stabilisation effect in the case of the Ru–Pd catalyst.
Co-reporter:Carlo Angelici, Marjolein E. Z. Velthoen, Bert M. Weckhuysen and Pieter C. A. Bruijnincx
Catalysis Science & Technology (2011-Present) 2015 - vol. 5(Issue 5) pp:NaN2879-2879
Publication Date(Web):2015/03/25
DOI:10.1039/C5CY00200A
The Lebedev ethanol-to-butadiene process entails a complex chain of reactions that require catalysts to possess a subtle balance in the number and strength of acidic and basic sites. SiO2–MgO materials can be excellent Lebedev catalysts if properly prepared, as catalyst performance has been found to depend significantly on the synthesis method. To assess the specific requirements for butadiene production in terms of active sites and to link their presence to the specific preparation method applied, five distinct SiO2–MgO catalysts, prepared by wet-kneading and co-precipitation methods, were thoroughly characterized. The amount and strength of the acidic (pyridine-IR and NH3-TPD) and basic (CDCl3-IR and CO2-TPD) sites of the materials as well as the overall acid/base properties in the liquid phase (Hammett indicators) were determined. The number of acidic and strong basic sites could be correlated with the extent of ethylene and diethyl ether by-product formation. The best performing catalysts are those containing a small amount of strong basic sites, combined with an intermediate amount of acidic sites and weak basic ones. These results thus provide further insight into the relation between the amount and strength of acidic/basic sites, preparation method and catalytic performance.
Co-reporter:Luis R. Aramburo, Javier Ruiz-Martínez, Jan P. Hofmann and Bert M. Weckhuysen
Catalysis Science & Technology (2011-Present) 2013 - vol. 3(Issue 5) pp:NaN1214-1214
Publication Date(Web):2012/11/01
DOI:10.1039/C2CY20661G
Confocal fluorescence microscopy has been used in combination with bulky non-reactive dyes (i.e. proflavine, stilbene and nile blue A) and two staining reactions (i.e. fluorescein synthesis and 4-fluorostyrene oligomerisation) to study the effect of steaming on pore accessibility and acidity of large ZSM-5 zeolite crystals. This approach enabled the 3-D visualization of cracks and mesopores connected to the outer zeolite surface as well as mesoporous “cavities” within steamed ZSM-5 zeolite crystals. It has been found that besides the generation of mesoporosity steaming makes the boundaries between the different crystal sub-units accessible for bulky molecules. Additionally, the fluorescein staining reaction reveals prominent formation of structural defects that are connected to the surface of the crystal via the microporous ZSM-5 system and which contain either Brønsted or Lewis acid sites. On the other hand, the 4-fluorostyrene staining reaction shows how mild steaming conditions increase the accessibility towards the Brønsted acid sites, while under severe steaming conditions the Brønsted acidity contained in the internal crystal sub-units is more accessible, although it is preferentially removed close to the surface of the lateral sub-units of ZSM-5 zeolite crystals.
Co-reporter:Peter J. C. Hausoul, Sinedu D. Tefera, Jelle Blekxtoon, Pieter C. A. Bruijnincx, Robertus J. M. Klein Gebbink and Bert M. Weckhuysen
Catalysis Science & Technology (2011-Present) 2013 - vol. 3(Issue 5) pp:NaN1223-1223
Publication Date(Web):2012/11/01
DOI:10.1039/C2CY20522J
The Pd/TOMPP-catalysed (TOMPP = tris(2-methoxyphenyl)phosphine) telomerisation of 1,3-butadiene was studied under solvent- and base-free conditions with phenolic substrates that can be potentially derived from lignin. Large differences in catalytic activity were observed, with reactivity increasing in the order of phenol, p-cresol, guaiacol, creosol and syringol. This reactivity trend can be attributed to the substrates' relative nucleophilicities, as induced by the donating effects of the p-methyl and o-methoxy substituents. The chosen reaction conditions, i.e. temperature, ligand/metal and butadiene/substrate ratios, strongly influenced both the conversion and selectivity of the reaction. Remarkably, the composition of the reaction medium, i.e. the butadiene/substrate ratio, exerted a strong influence on the linear/branched ratio. High conversions and selectivities to the linear products are obtained when excess butadiene is used. The linear telomer products could be readily converted from O-alkylated to C-alkylated phenolics via the thermal Claisen rearrangement. High conversions and selectivities were observed after 2 hours at 200 °C. Branched o-octadienyl phenols were obtained in all cases except for the syringol telomer which gave the linear p-octadienyl product exclusively.
Co-reporter:Clare E. Harvey, Evelien M. van Schrojenstein Lantman, Arjan J. G. Mank and Bert M. Weckhuysen
Chemical Communications 2012 - vol. 48(Issue 12) pp:NaN1744-1744
Publication Date(Web):2012/01/04
DOI:10.1039/C2CC15939B
The integration of Atomic Force Microscopy and Raman spectroscopy is tested for use in heterogeneous catalysis research by a preliminary investigation, the photo-oxidation of rhodamine-6G. Temperature and atmosphere were varied in an in situcell to show compatibility with realistic reaction conditions.
Co-reporter:Matthew G. O'Brien, Simon D. M. Jacques, Marco Di Michiel, Paul Barnes, Bert M. Weckhuysen and Andrew M. Beale
Chemical Science (2010-Present) 2012 - vol. 3(Issue 2) pp:NaN523-523
Publication Date(Web):2011/11/09
DOI:10.1039/C1SC00637A
A combination of synchrotron μ-XRD-CT and μ-absorption-CT (CT = computed tomography) is demonstrated, providing a unique insight into the solid state changes occurring from within crystalline materials. Specifically, we examine here the solid state changes that occur in a millimetre-sized Ni/γ-Al2O3 catalyst body in both 2D and 3D during calcination and CO methanation for the first time. The combination provides a unique insight into the spatial phase distribution of these materials and how these evolve via a series of solid state transformation processes. For example, initially, two Ni-ethylenediamine (en) complexes were observed on the impregnated and dried body; a hydrated and non-hydrated form, which 2D scans reveal possess an egg-shell and egg-yolk distribution, respectively. Furthermore, the μ-XRD data were of sufficient quality so as to be able to reveal that the particles within the ‘egg-shell’ were larger (∼35 nm) than those of the ‘egg-yolk’ (∼19 nm) and that there were more of them. On calcination, both precursors collapsed, yielding metallic fcc Ni particles with a surprisingly uniform average size distribution over the catalyst (∼4 nm). However, a comparison of the scattering at different stages of the experiment suggested that the crystalline structure of some of the Ni remained diffraction ‘silent’. Calcination in oxygen lead to both Ni oxidation and particle sintering, mainly at the exterior, which on pre-reaction reduction (in H2) yielded again fcc Ni particles (∼4 nm interior, ∼6 nm exterior) with a significant reduction in the amorphous Ni component. The catalyst proved active for CO methanation and, during 2 h time on-stream, no change in the structure composition or shape was observed, leading us to conclude that nano-sized fcc Ni particles on γ-Al2O3 are the active component in CO methanation. This work therefore demonstrates both the power of spatially resolved μ-XRD-CT/μ-absorption-CT measurement of catalytic systems and its advantage over more ‘traditional’ single point studies on small sieve fractions.
Co-reporter:Bert M. Weckhuysen and Jihong Yu
Chemical Society Reviews 2015 - vol. 44(Issue 20) pp:NaN7024-7024
Publication Date(Web):2015/09/24
DOI:10.1039/C5CS90100F
A graphical abstract is available for this content
Co-reporter:E. T. C. Vogt and B. M. Weckhuysen
Chemical Society Reviews 2015 - vol. 44(Issue 20) pp:NaN7370-7370
Publication Date(Web):2015/09/18
DOI:10.1039/C5CS00376H
Fluid catalytic cracking (FCC) is one of the major conversion technologies in the oil refinery industry. FCC currently produces the majority of the world's gasoline, as well as an important fraction of propylene for the polymer industry. In this critical review, we give an overview of the latest trends in this field of research. These trends include ways to make it possible to process either very heavy or very light crude oil fractions as well as to co-process biomass-based oxygenates with regular crude oil fractions, and convert these more complex feedstocks in an increasing amount of propylene and diesel-range fuels. After providing some general background of the FCC process, including a short history as well as details on the process, reactor design, chemical reactions involved and catalyst material, we will discuss several trends in FCC catalysis research by focusing on ways to improve the zeolite structure stability, propylene selectivity and the overall catalyst accessibility by (a) the addition of rare earth elements and phosphorus, (b) constructing hierarchical pores systems and (c) the introduction of new zeolite structures. In addition, we present an overview of the state-of-the-art micro-spectroscopy methods for characterizing FCC catalysts at the single particle level. These new characterization tools are able to explain the influence of the harsh FCC processing conditions (e.g. steam) and the presence of various metal poisons (e.g. V, Fe and Ni) in the crude oil feedstocks on the 3-D structure and accessibility of FCC catalyst materials.
Co-reporter:Hendrik E. van der Bij and Bert M. Weckhuysen
Chemical Society Reviews 2015 - vol. 44(Issue 20) pp:NaN7428-7428
Publication Date(Web):2015/06/05
DOI:10.1039/C5CS00109A
Phosphorus and microporous aluminosilicates, better known as zeolites, have a unique but poorly understood relationship. For example, phosphatation of the industrially important zeolite H-ZSM-5 is a well-known, relatively inexpensive and seemingly straightforward post-synthetic modification applied by the chemical industry not only to alter its hydrothermal stability and acidity, but also to increase its selectivity towards light olefins in hydrocarbon catalysis. On the other hand, phosphorus poisoning of zeolite-based catalysts, which are used for removing nitrogen oxides from exhaust fuels, poses a problem for their use in diesel engine catalysts. Despite the wide impact of phosphorus–zeolite chemistry, the exact physicochemical processes that take place require a more profound understanding. This review article provides the reader with a comprehensive and state-of-the-art overview of the academic literature, from the first reports in the late 1970s until the most recent studies. In the first part an in-depth analysis is undertaken, which will reveal universal physicochemical and structural effects of phosphorus–zeolite chemistry on the framework structure, accessibility, and strength of acid sites. The second part discusses the hydrothermal stability of zeolites and clarifies the promotional role that phosphorus plays. The third part of the review paper links the structural and physicochemical effects of phosphorus on zeolite materials with their catalytic performance in a variety of catalytic processes, including alkylation of aromatics, catalytic cracking, methanol-to-hydrocarbon processing, dehydration of bioalcohol, and ammonia selective catalytic reduction (SCR) of NOx. Based on these insights, we discuss potential applications and important directions for further research.
Co-reporter:F. C. Hendriks, D. Valencia, P. C. A. Bruijnincx and B. M. Weckhuysen
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 3) pp:NaN1867-1867
Publication Date(Web):2016/12/21
DOI:10.1039/C6CP07572J
A series of fluorescent probe molecules based on the commercially available trans-4-(4-(diethylamino)styryl)-N-methylpyridinium iodide (DAMPI) scaffold has been developed. The dynamic radii of these DAMPI-type probes covered a range of 5.8 to 10.1 Å and could be changed by varying the alkyl substituents on the amine donor group, with limited effect on the electronic properties. These probe molecules allow for the direct evaluation of the molecular accessibility into confined spaces, more specifically the micropore architecture of zeolite materials. Evaluation of industrially relevant zeolite materials with 8- (CHA), 10- (MFI) and 12-membered ring pores (FAU) showed that steric bulk influences the rate of adsorption, the amount of probe molecule taken up by the zeolite as well as the interaction of the probe molecule with the zeolite material. Furthermore, a positive linear correlation is found between the pore–probe size difference and total probe uptake by the zeolite. The absorption spectra of each probe molecule within the zeolites show that this DAMPI-type compound is chemically bound to the zeolite's acid sites. The new approach shows the general principle of determining size-accessibility relationships in microporous solids with a series of fluorescent probes of systematically tunable size.
Co-reporter:Diego Valencia, Gareth T. Whiting, Rosa E. Bulo and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 3) pp:NaN2086-2086
Publication Date(Web):2015/12/21
DOI:10.1039/C5CP06477E
In an earlier work, protonated thiophene-based oligomers were identified inside ZSM-5 zeolites. The novel compounds exhibited π–π* absorption wavelengths deep within the visible region, earmarking them for possible use as chromophores in a variety of applications. In this computational study, we determine the factors that cause such low-energy transitions, and describe the electronic structure of these remarkable compounds. DFT calculations of conjugated thiophene-based oligomers with up to five monomer units reveal that the main absorption band of each protonated oligomer is strongly red-shifted compared to the unprotonated form. This effect is counterintuitive, since protonation is expected to diminish aromaticity, and thereby increase the HOMO–LUMO gap. We find that upon protonation the π-electrons remain delocalized over the entire π-conjugated molecule, but the positive charge is localized predominantly on the protonated side of the molecule. A possible explanation for this ground-state charge localization is the participation of the C–H bond in the π-system of the protonated ring, locally providing aromatic stabilization for the positive charge. The addition of the proton stabilizes all electronic orbitals, but due to the ground state π-electron distribution away from the added nucleus, the HOMO is stabilized less than the LUMO. The main absorption peak upon protonation corresponds to the charge transfer excitation involving the frontier orbitals, and the small band gap explains the observed red shift. Analogue calculations on thiophene within a ZSM-5 zeolite cluster model confirm the same trends upon protonation as observed in the non-interacting compounds. Understanding the electronic structure of these compounds is very relevant to correlate UV-Vis bands with acidic strength and possibly environment in zeolites and to improve their performance in catalytic and energy related applications.
Co-reporter:Pushkar Singh, Tanja Deckert-Gaudig, Henrik Schneidewind, Konstantin Kirsch, Evelien M. van Schrojenstein Lantman, Bert M. Weckhuysen and Volker Deckert
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 5) pp:NaN2995-2995
Publication Date(Web):2014/12/10
DOI:10.1039/C4CP04850D
Vibrational spectroscopy usually provides structural information averaged over many molecules. We report a larger peak position variation and reproducibly smaller FWHM of TERS spectra compared to SERS spectra indicating that the number of molecules excited in a TERS experiment is extremely low. Thus, orientational averaging effects are suppressed and micro ensembles are investigated. This is shown for a thiophenol molecule adsorbed on Au nanoplates and nanoparticles.
Co-reporter:Gareth T. Whiting, Florian Meirer, Diego Valencia, Machteld M. Mertens, Anton-Jan Bons, Brian M. Weiss, Paul A. Stevens, Emiel de Smit and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 39) pp:NaN21542-21542
Publication Date(Web):2014/08/29
DOI:10.1039/C4CP03649B
Optical absorption and confocal fluorescence micro-spectroscopy were applied to investigate Brønsted acidity in millimetre-sized extrudates of Na(H)-ZSM-5 and SiO2 with varying ZSM-5 content. Partially (residual Na present) and fully proton-exchanged extrudates were employed, using thiophene oligomerization as a probe reaction. Time-resolved in situ optical absorption spectra and time dependent DFT calculations revealed several initial reaction pathways during the oligomerization reaction. In particular, it was found that protonated thiophene monomers reacted by either oligomerization (via reaction with un-reacted thiophene monomers) or ring-opening, depending on the Brønsted acid site density in each sample. Moreover, fully-exchanged extrudates not only have significantly higher reactivity than partially-exchanged samples, but they also favour the formation of ring-opening products, that are not formed on the partially-exchanged samples. Confocal fluorescence microscopy was employed to visualise non-invasively in 3D, the heterogeneity and homogeneity of thiophene oligomers on partially- and fully-exchanged extrudates, respectively. Furthermore, it was observed that extrudates with high binder content produce a higher relative amount of conjugated species, related with a higher quantity of available monomer in the binder, which is able to react further with intermediates adsorbed on active sites. Moreover, these conjugated species appear to form near the external surface of ZSM-5 crystals/agglomerates.
Co-reporter:Jesper J. H. B. Sattler, Andrew M. Beale and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 29) pp:NaN12103-12103
Publication Date(Web):2013/04/11
DOI:10.1039/C3CP50646K
The deactivation of 0.5 wt% Pt/Al2O3 and 0.5 wt% Pt–1.5 wt% Sn/Al2O3 catalysts has been studied by operando Raman spectroscopy during the dehydrogenation of propane and subsequent regeneration in air for 10 successive dehydrogenation–regeneration cycles. Furthermore, the reaction feed was altered by using different propane/propene/hydrogen ratios. It was found that the addition of hydrogen to the feed increases the catalyst performance and decreases the formation of coke deposits, as was revealed by thermogravimetrical analysis. The positive effect of hydrogen on the catalyst performance is comparable to the addition of Sn, a promoter element which increases both the propane conversion and propene selectivity. Operando Raman spectroscopy showed that hydrogen altered the nature of the coke deposits formed during propane dehydrogenation. Due to this approach it was possible to perform a systematic deconvolution procedure on the Raman spectra. By analysing the related intensity, band position and bandwidth of the different Raman features, it was determined that smaller graphite crystallites, which have less defects, are formed when the partial pressure of hydrogen in the feed was increased.
Co-reporter:Luis R. Aramburo, Sue Wirick, Piter S. Miedema, Inge L. C. Buurmans, Frank M. F. de Groot and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 19) pp:NaN6973-6973
Publication Date(Web):2012/01/25
DOI:10.1039/C2CP22848C
The Brønsted acid-catalyzed oligomerization of 4-fluorostyrene has been studied on a series of H-ZSM-5 zeolite powders, steamed under different conditions, with a combination of UV-Vis micro-spectroscopy and Scanning Transmission X-ray Microscopy (STXM). UV-Vis micro-spectroscopy and STXM have been used to monitor the relative formation of cyclic and linear dimeric carbocations as a function of the steaming post-treatment (i.e., parent vs. steaming at 600, 700 and 800 °C). It was found that the UV-Vis band intensity ratios of linear to cyclic dimeric species increase from 0.79 (parent H-ZSM-5) over 1.41 (H-ZSM-5 steamed at 600 °C) and 1.88 (H-ZSM-5 steamed at 700 °C) to 2.33 (H-ZSM-5 steamed at 800 °C). STXM confirms this trend in reaction product selectivity, as the relative intensities of the transitions attributed to the presence of the cyclic dimer in the carbon K-edge spectra decrease with increasing severity of the steaming post-treatment. Furthermore, STXM reveals spatial heterogeneities in reaction product formation within the H-ZSM-5 zeolite powders at the nanoscale. More specifically, a shrinking carbon core–shell distribution was detected within the zeolite aggregates, in which the relative amount of cyclic dimeric species is higher in the core relative to the shell of the zeolite aggregate and the relative amount of cyclic dimeric species in the zeolite core gradually decreases with increasing severity of the steaming post-treatment. These differences are rationalized in terms of spatial differences in Brønsted acidity within H-ZSM-5 zeolite powders as well as by changes in the formation process of linear and dimeric carbocations within H-ZSM-5 micro- and mesopores.
Co-reporter:Lukasz Karwacki and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 9) pp:NaN3685-3685
Publication Date(Web):2010/12/22
DOI:10.1039/C0CP02220A
A combination of in situUV-Vis and confocal fluorescence micro-spectroscopy was used to study the template decomposition process in large zeolite ZSM-5 crystals. Correlation of polarized light dependent UV-Vis absorption spectra with confocal fluorescence emission spectra in the 400–750 nm region allowed extracting localized information on the nature and amount of chemical species formed upon detemplation at the single particle level. It has been found by means of polarized light dependent UV-Vis absorption measurements that the progressive growth of molecules follows the orientation of the straight channels of ZSM-5 crystals. Oligomerizing template derivatives lead to the subsequent build-up of methyl-substituted benzenium cations and more extended coke-like species, which are thermally stable up to ∼740 K. Complementary confocal fluorescence emission spectra showed nearly equal distribution of these molecules within the entire volume of the thermally treated zeolite crystals. The strongest emission bands were appearing in the orange/red part of the visible spectrum, confirming the presence of large polyaromatic molecules.
Co-reporter:Davide Mores, Eli Stavitski, Suzanna P. Verkleij, Antoinette Lombard, Amandine Cabiac, Loïc Rouleau, Joël Patarin, Angélique Simon-Masseron and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 35) pp:NaN15994-15994
Publication Date(Web):2011/08/03
DOI:10.1039/C1CP21324E
A combination of in situUV-Vis and confocal fluorescence micro-spectroscopy is applied to investigate the influence of an external silicalite-1 shell on the Brønsted acidity and coke formation process of individual H-ZSM-5 zeolite crystals. Three probe reactions were used: oligomerization of styrene, methanol-to-olefin (MTO) conversion and aromatization of light naphtha (LNA) derivatives. Oligomerization of styrene leads to the formation of optically active carbocationic oligomers. Different styrene substitutions indicate the conversion ability of the catalyst acid core, a preferred alignment of the oligomers within the straight zeolite channels and a Brønsted acidity gradient throughout the zeolite crystal. Both the MTO conversion and the LNA process lead to limited carbonaceous deposition within the external silicalite-1 layer. This outer shell furthermore prevents the growth of extended coke species at the zeolite external surface. During MTO, the formation of carbonaceous compounds initiates at the center of the H-ZSM-5 zeolite core and expands towards the zeolite exterior. This coke build-up starts with a 420 nm UV-Vis absorption band, assigned to methyl-substituted aromatic carbocations, and a second band around 550 nm, which is indicative of their growth towards larger conjugated systems. Aromatization of linear and branched C5 paraffins causes negligible darkening of the zeolite crystals though it forms fluorescent coke deposits and their precursors within the H-ZSM-5 catalyst. Olefin homologues on the contrary cause pronounced darkening of the zeolite composite. Methyl-branching of these reactants slows down the coke formation rate and produces carbonaceous species that are more restricted in their molecular size.
Co-reporter:Emiel de Smit, Frank M. F. de Groot, Raoul Blume, Michael Hävecker, Axel Knop-Gericke and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 3) pp:NaN680-680
Publication Date(Web):2009/11/19
DOI:10.1039/B920256K
The effect of Cu on the reduction behavior and surface properties of supported and unsupported Fe-based Fischer–Tropsch synthesis (FTS) catalysts was investigated using in situ X-ray photoelectron spectroscopy (XPS) and in situ X-ray absorption spectroscopy (XAS) in combination with ex situ bulk characterization. During exposure to 0.4 mbar CO–H2 above 180 °C, the reduction of CuO to Cu0 marked the onset of the reduction of Fe3O4 to α-Fe. The promotion effects of Cu are explained by a combination of spillover of H2 and/or CO molecules from metallic Cu0 nuclei to closely associated iron oxide species and textural promotion. XAS showed that in the supported catalyst, Cu+ and Fe2+ species were stabilized by SiO2 and, as a result, Fe species were not reduced significantly beyond Fe3O4 and Fe2+, even after treatment at 350 °C. After the reduction treatment, XPS showed that the concentration of oxygen and carbon surface species was higher in the presence of Cu. Furthermore, it was observed that the unsupported, Cu-containing catalyst showed higher CO2 concentration in the product gas stream during and after reduction and Fe surface species were slightly oxidized after prolonged exposure to CO–H2. These observations suggest that, in addition to facilitating the reduction of the iron oxide phase, Cu also plays a direct role in altering the surface chemistry of Fe-based FTS catalysts.
Co-reporter:L. Espinosa-Alonso, K. P. de Jong and B. M Weckhuysen
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 1) pp:NaN107-107
Publication Date(Web):2009/11/07
DOI:10.1039/B915753K
The influence of the Cl−(aq) concentration, solution pH and equilibration time on the PdCl42−(aq) dynamics and molecular structure after impregnation of γ-Al2O3 catalyst bodies has been studied using UV-Vis micro-spectroscopy. To do so, 0.2 wt% Pd catalysts have been prepared from acidic solutions (pH 1 and 5) of the Na2PdCl4 precursor salt with different amounts of NaCl. It was found that egg-shell catalysts are obtained when a less acidic pH (pH 5) is combined with [Cl−(aq)] < 0.6 M and less than 24 h of equilibration time are implemented, while to achieve egg-white catalysts the solution pH should be 1. Moreover, by increasing the equilibration time up to 96 h, the egg-shell profiles vanish to provide a uniform Pd distribution, while the egg-white distribution becomes egg-yolk. Additionally, Pd complexes appeared with different molecular structures depending on the solution pH, equilibration time and macro-distribution achieved. The protocol developed to create different Pd macro-distributions has been applied to prepare two 1 wt% Pd/γ-Al2O3 egg-shell and egg-white catalysts. The Pd dynamics and molecular structure have been followed after impregnation, drying and calcination, demonstrating that the profiles created after impregnation are retained.
Co-reporter:Bart P. C. Hereijgers, Rudy F. Parton and Bert M. Weckhuysen
Catalysis Science & Technology (2011-Present) 2012 - vol. 2(Issue 5) pp:NaN960-960
Publication Date(Web):2012/01/06
DOI:10.1039/C2CY00455K
Olefin epoxidation with cyclohexyl hydroperoxide offers great perspective in increasing the yield from industrial cyclohexane oxidation and the production of epoxides in an apolar medium. Two competing hydroperoxide conversion routes, namely direct epoxidation and thermal decomposition, were identified. The formation of radicals seemed to play a role in both mechanisms. However, olefin epoxidation was found to solely take place at the catalyst. Allylic oxidation of cyclohexene occurs under reaction conditions primarily by molecular oxygen and only constitutes a minor route. The presence of molecular oxygen was found to increase the overall yield of the process by solvent oxidation yielding new cyclohexyl hydroperoxide. Hydrolysis and isomerization of the epoxide were found to be negligible reactions, although the epoxide gets converted at higher concentrations, presumably by the radical initiated polymerization. UV-Vis spectroscopy provided proof for the formation of titanium-hydroperoxide species as the active catalytic site in the direct epoxidation reaction.
Co-reporter:F. Meirer, S. Kalirai, J. Nelson Weker, Y. Liu, J. C. Andrews and B. M. Weckhuysen
Chemical Communications 2015 - vol. 51(Issue 38) pp:NaN8100-8100
Publication Date(Web):2015/04/14
DOI:10.1039/C5CC00401B
Metal accumulation at the catalyst particle surface plays a role in particle agglutination during fluid catalytic cracking.
Co-reporter:Korneel H. Cats, Ines D. Gonzalez-Jimenez, Yijin Liu, Johanna Nelson, Douglas van Campen, Florian Meirer, Ad M. J. van der Eerden, Frank M. F. de Groot, Joy C. Andrews and Bert M. Weckhuysen
Chemical Communications 2013 - vol. 49(Issue 41) pp:NaN4624-4624
Publication Date(Web):2013/04/03
DOI:10.1039/C3CC00160A
Transmission X-ray microscopy has been used to investigate individual Co/TiO2 Fischer–Tropsch (FT) catalyst particles in 2-D and 3-D with 30 nm spatial resolution. Tomographic elemental mapping showed that Co is heterogeneously concentrated in the centre of the catalyst particles. In addition, it was found that Co is mostly metallic during FT at 250 °C and 10 bar. No evidence for Co oxidation was found.
Co-reporter:J. J. H. B. Sattler, I. D. González-Jiménez, A. M. Mens, M. Arias, T. Visser and B. M. Weckhuysen
Chemical Communications 2013 - vol. 49(Issue 15) pp:NaN1520-1520
Publication Date(Web):2013/01/04
DOI:10.1039/C2CC38978A
A novel operando UV-Vis spectroscopic set-up has been constructed and tested for the investigation of catalyst bodies loaded in a pilot-scale reactor under relevant reaction conditions. Spatiotemporal insight into the formation and burning of coke deposits on an industrial CrOx/Al2O3 catalyst during propane dehydrogenation has been obtained.
Co-reporter:I. Lezcano-Gonzalez, U. Deka, B. Arstad, A. Van Yperen-De Deyne, K. Hemelsoet, M. Waroquier, V. Van Speybroeck, B. M. Weckhuysen and A. M. Beale
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 4) pp:NaN1650-1650
Publication Date(Web):2013/11/28
DOI:10.1039/C3CP54132K
Three different types of NH3 species can be simultaneously present on Cu2+-exchanged CHA-type zeolites, commonly used in Ammonia Selective Catalytic Reduction (NH3-SCR) systems. These include ammonium ions (NH4+), formed on the Brønsted acid sites, [Cu(NH3)4]2+ complexes, resulting from NH3 coordination with the Cu2+ Lewis sites, and NH3 adsorbed on extra-framework Al (EFAl) species, in contrast to the only two reacting NH3 species recently reported on Cu-SSZ-13 zeolite. The NH4+ ions react very slowly in comparison to NH3 coordinated to Cu2+ ions and are likely to contribute little to the standard NH3-SCR process, with the Brønsted groups acting primarily as NH3 storage sites. The availability/reactivity of NH4+ ions can be however, notably improved by submitting the zeolite to repeated exchanges with Cu2+, accompanied by a remarkable enhancement in the low temperature activity. Moreover, the presence of EFAl species could also have a positive influence on the reaction rate of the available NH4+ ions. These results have important implications for NH3 storage and availability in Cu-Chabazite-based NH3-SCR systems.
Co-reporter:Hendrik E. van der Bij and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 21) pp:NaN9903-9903
Publication Date(Web):2013/12/18
DOI:10.1039/C3CP54791D
In order to elucidate phosphorus–zeolite H-ZSM-5 interactions, a variety of phosphorus-modified zeolite H-ZSM-5 materials were studied in a multi-spectroscopic manner. By deploying single pulse 27Al, 31P MAS NMR, 2D heteronuclear 27Al–31P correlation (HETCOR), 27Al MQ MAS NMR spectroscopy, TPD of pyridine monitored by FT-IR spectroscopy, and Scanning Transmission X-ray Microscopy (STXM) the interplay and influence of acidity, thermal treatment and phosphorus on the structure and acidity of H-ZSM-5 were established. It was found that while acid treatment did not affect the zeolite structure, thermal treatment leads to the breaking of Si–OH–Al bonds, a decrease in the strong acid site number and the formation of terminal Al–OH groups. No extra-framework aluminium species was observed. Phosphorus precursors interact with the zeolitic framework through hydrogen bonds and physical coordination, as phosphorus species can be simply washed out with hot water. After phosphatation and thermal treatment two effects occur simultaneously, namely (i) phosphorus species transform into water insoluble condensed poly-phosphoric acid and (ii) phosphoric acid binds irreversibly to the terminal Al–OH groups of partially dislodged four-coordinated framework aluminium, forming local silico-aluminophosphate interfaces. These interfaces are potentially the promoters of hydrothermal stability in phosphated zeolite H-ZSM-5.
Co-reporter:Ana Mijovilovich, Sylvain Hamman, Fabrice Thomas, Frank M. F. de Groot and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 13) pp:NaN5604-5604
Publication Date(Web):2011/01/31
DOI:10.1039/C0CP01144D
X-ray Emission Spectroscopy (XES) crossover peaks were shown to be sensitive to the protonation state of solvent molecules in the Zn protein carbonic anhydrase and its model compounds. Here we extend such studies to galactose oxidase models i.e.Cu(II) open d-shell systems, illustrating that XES combined with FEFF8 simulations reflect changes in the protonation state of the phenolate ligand for the copper center.
Co-reporter:Inge L. C. Buurmans, Evgeny A. Pidko, Jennifer M. de Groot, Eli Stavitski, Rutger A. van Santen and Bert M. Weckhuysen
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 26) pp:NaN7040-7040
Publication Date(Web):2010/05/14
DOI:10.1039/C002442B
A series of H-ZSM-5 crystallites with different framework Si/Al ratios was studied by analyzing the kinetics and reaction mechanism of the oligomerization of 4-fluorostyrene as molecular probe reaction for Brønsted acidity. The formation of carbocationic species was followed by UV-Vis spectroscopy. Three carbocationic products were observed, namely a cyclic dimer, a conjugated linear dimer and a larger, more conjugated carbocation. Rate constants for the formation of all three products show a maximum at a Si/Al ratio of 25. Oligomerization of 4-fluorostyrene within the larger supercages of zeolite H-Y leads solely to cyclic dimers. The experimental observations were rationalized by DFT calculations, which show that the selectivity of the styrene oligomerization is controlled by the steric properties of the intrazeolite micropore voids. Two reaction pathways were considered for the formation of the conjugated linear carbocation. The conventional mechanism involves a hydride transfer between two dimeric hydrocarbons (HCs) in the zeolite pores. We propose an alternative monomolecular path, in which the hydride transfer takes place between a hydrogen atom of a dimeric HC and a zeolitic proton, yielding a conjugated carbocation and molecular H2. Computed free energies indicate that the preference for a particular reaction mechanism is determined by the local shape of the zeolite micropores.





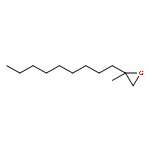
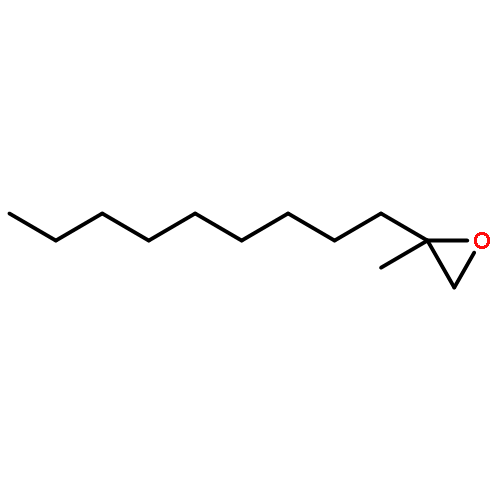
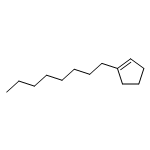


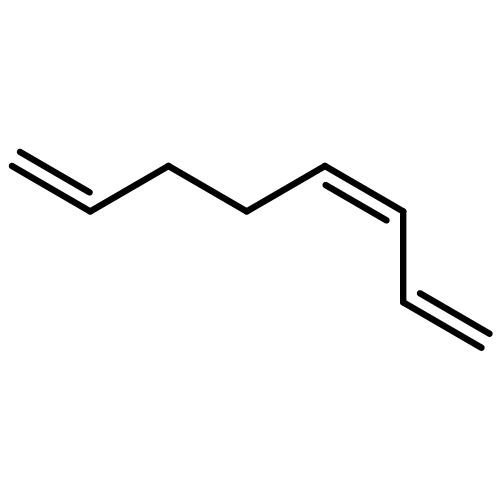














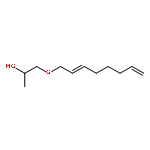
![1,6-OCTADIENE, 8,8'-[1,2-ETHANEDIYLBIS(OXY)]BIS-](http://img.cochemist.com/ccimg/43000/42954-38-1.png)
![1,6-OCTADIENE, 8,8'-[1,2-ETHANEDIYLBIS(OXY)]BIS-](http://img.cochemist.com/ccimg/43000/42954-38-1_b.png)









![13-OXABICYCLO[10.1.0]TRIDECA-4,8-DIENE, (1R*,4E,8Z,12R*)-](http://img.cochemist.com/ccimg/55800/55722-64-0.png)
![13-OXABICYCLO[10.1.0]TRIDECA-4,8-DIENE, (1R*,4E,8Z,12R*)-](http://img.cochemist.com/ccimg/55800/55722-64-0_b.png)


![Benzenemethanol, 3,4-dimethoxy-a-[(2-methoxyphenoxy)methyl]-](http://img.cochemist.com/ccimg/17100/17078-88-5.png)
![Benzenemethanol, 3,4-dimethoxy-a-[(2-methoxyphenoxy)methyl]-](http://img.cochemist.com/ccimg/17100/17078-88-5_b.png)



![(2R,3R,3aR,6R,6aR)-hexahydro-2,6-epoxyfuro[3,2-b]furan-3-ol](http://img.cochemist.com/ccimg/4500/4451-30-3.png)
![(2R,3R,3aR,6R,6aR)-hexahydro-2,6-epoxyfuro[3,2-b]furan-3-ol](http://img.cochemist.com/ccimg/4500/4451-30-3_b.png)
![Benzene, [(1-methyl-2-propenyl)oxy]-](http://img.cochemist.com/ccimg/22600/22509-78-0.png)
![Benzene, [(1-methyl-2-propenyl)oxy]-](http://img.cochemist.com/ccimg/22600/22509-78-0_b.png)
















![3,6-Dioxabicyclo[3.1.0]hexane-2,4-dione(9CI)](http://img.cochemist.com/ccimg/16200/16191-17-6.png)
![3,6-Dioxabicyclo[3.1.0]hexane-2,4-dione(9CI)](http://img.cochemist.com/ccimg/16200/16191-17-6_b.png)
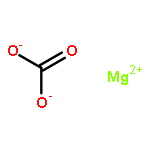











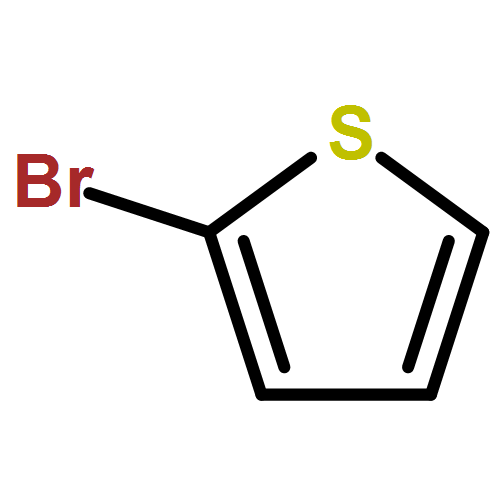





![Benzene, [(1-ethenyl-5-hexenyl)oxy]-](http://img.cochemist.com/ccimg/16000/15972-91-5.png)
![Benzene, [(1-ethenyl-5-hexenyl)oxy]-](http://img.cochemist.com/ccimg/16000/15972-91-5_b.png)
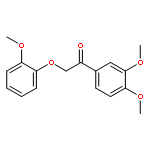
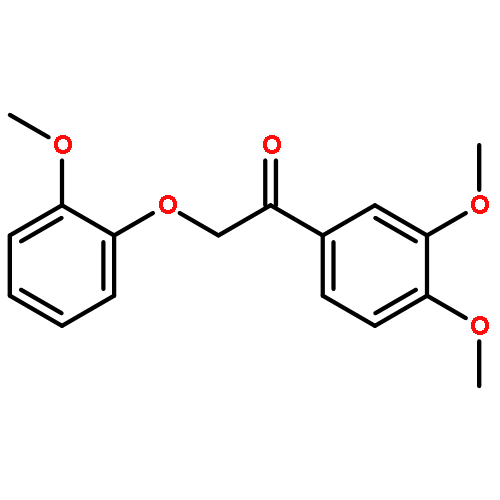










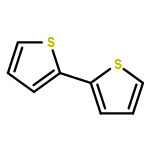
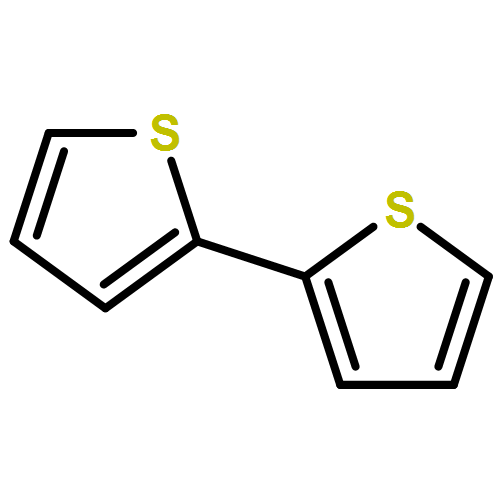























![7-Oxabicyclo[4.1.0]heptan-2-ol](http://img.cochemist.com/ccimg/1200/1192-78-5.png)
![7-Oxabicyclo[4.1.0]heptan-2-ol](http://img.cochemist.com/ccimg/1200/1192-78-5_b.png)
![7-Oxabicyclo[4.1.0]heptane, 1-methyl-4-(1-methylethenyl)-](http://img.cochemist.com/ccimg/1200/1195-92-2.png)
![7-Oxabicyclo[4.1.0]heptane, 1-methyl-4-(1-methylethenyl)-](http://img.cochemist.com/ccimg/1200/1195-92-2_b.png)















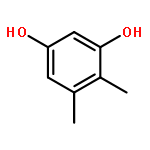
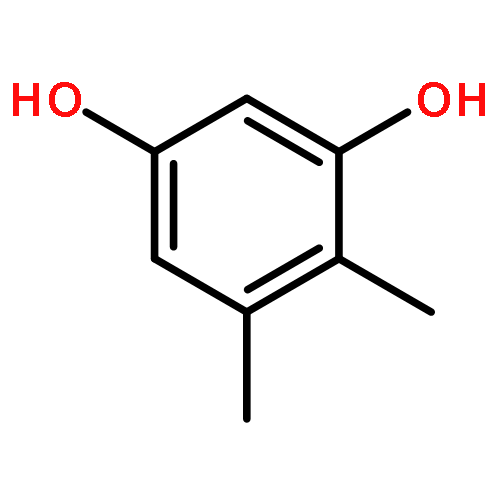




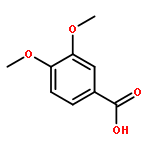
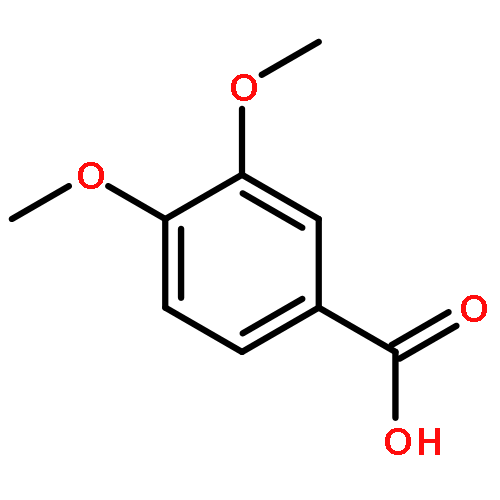


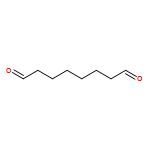
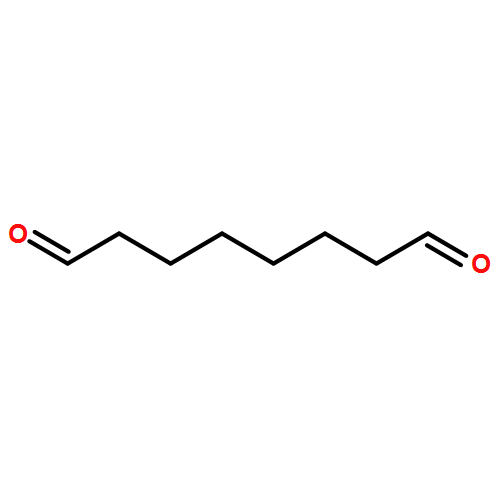
























![1-METHYL-2-[(1-METHYLBENZIMIDAZOL-2-YL)METHYL]BENZIMIDAZOLE](http://img.cochemist.com/ccimg/55600/55514-10-8.png)
![1-METHYL-2-[(1-METHYLBENZIMIDAZOL-2-YL)METHYL]BENZIMIDAZOLE](http://img.cochemist.com/ccimg/55600/55514-10-8_b.png)


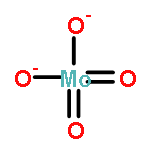
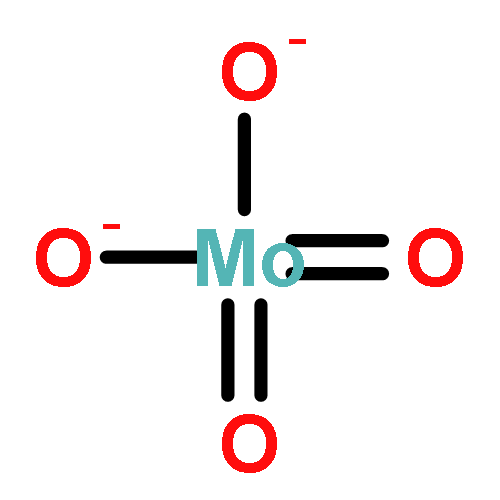




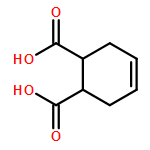
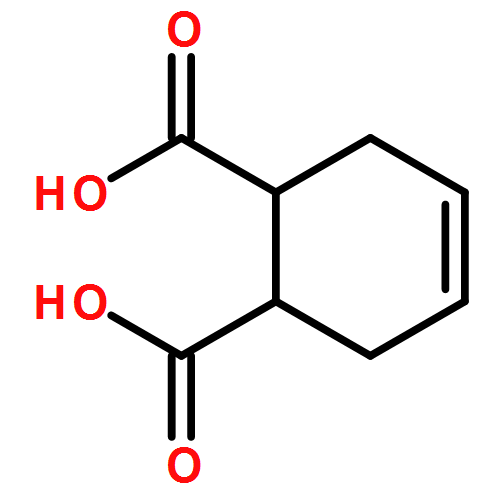




![Ethanol, 2-[(1-methylheptyl)oxy]-](http://img.cochemist.com/ccimg/61200/61192-28-7.png)
![Ethanol, 2-[(1-methylheptyl)oxy]-](http://img.cochemist.com/ccimg/61200/61192-28-7_b.png)

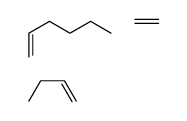


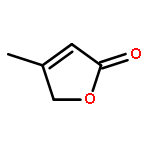


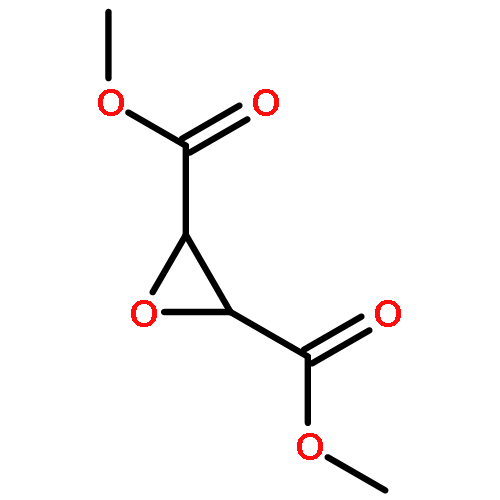
![[1,1'-Biphenyl]-2,2'-diol,3,3'-dimethoxy-5,5'-dimethyl-](http://img.cochemist.com/ccimg/14000/13990-86-8.png)
![[1,1'-Biphenyl]-2,2'-diol,3,3'-dimethoxy-5,5'-dimethyl-](http://img.cochemist.com/ccimg/14000/13990-86-8_b.png)















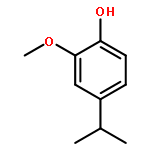

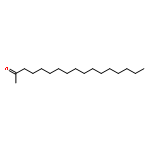
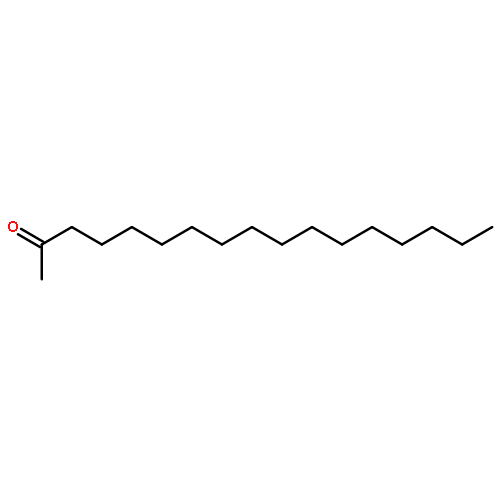
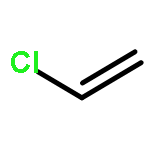
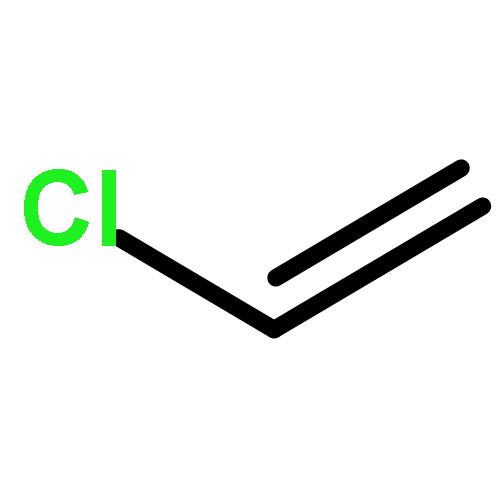






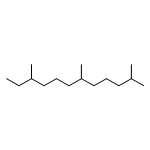
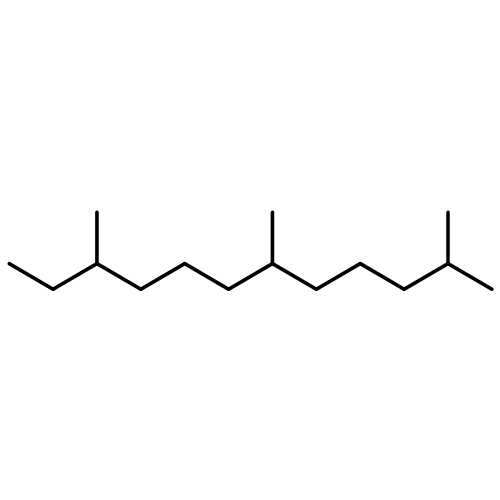












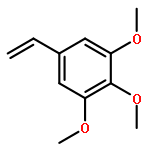

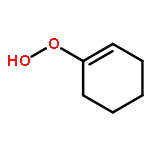
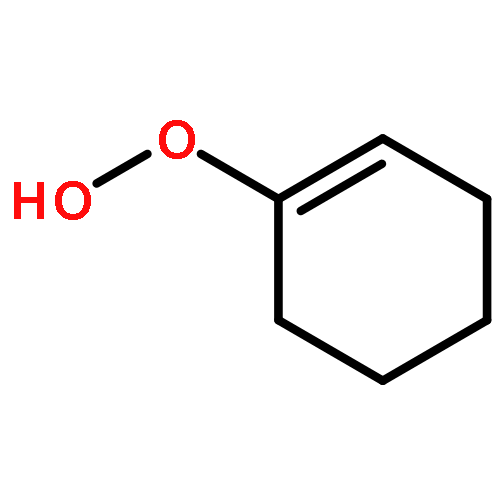

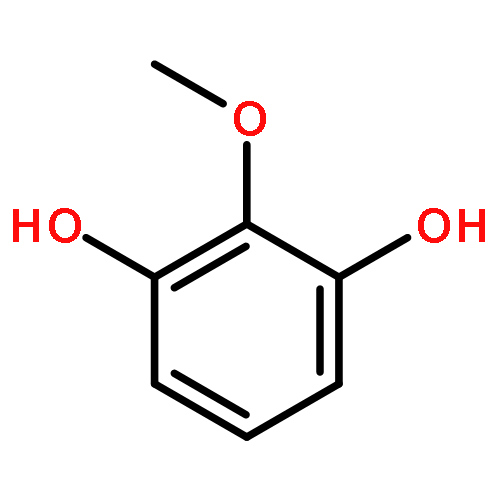
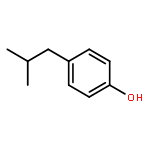












![3,8-Dioxatricyclo[5.1.0.02,4]octane(8CI,9CI)](http://img.cochemist.com/ccimg/300/277-06-5.png)
![3,8-Dioxatricyclo[5.1.0.02,4]octane(8CI,9CI)](http://img.cochemist.com/ccimg/300/277-06-5_b.png)






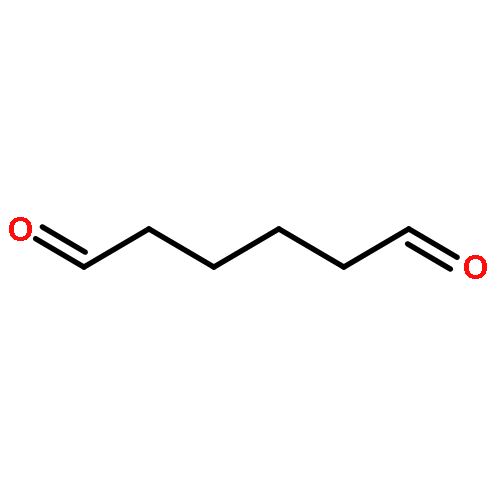


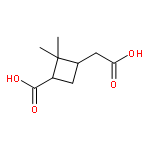

![5,10-Dioxatricyclo[7.1.0.04,6]decane](http://img.cochemist.com/ccimg/300/286-75-9.png)
![5,10-Dioxatricyclo[7.1.0.04,6]decane](http://img.cochemist.com/ccimg/300/286-75-9_b.png)





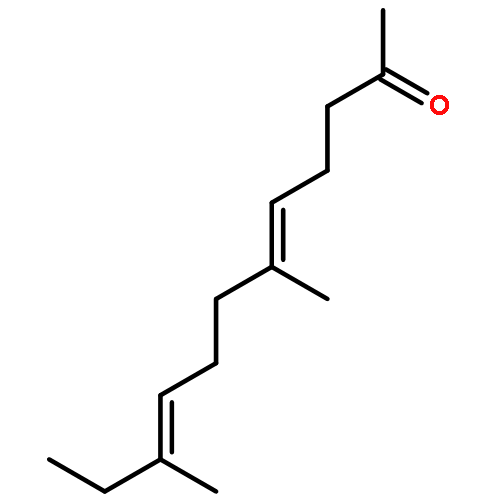
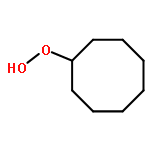
![7-Oxabicyclo[4.1.0]heptane-3,4-dicarboxylic acid](/data/chemimg/69600/49629-93-8.png)
![7-Oxabicyclo[4.1.0]heptane-3,4-dicarboxylic acid](/data/chemimg/69600/49629-93-8_b.png)




























![2-CHLORO-N-[1-(2-FLUOROBENZOYL)PIPERIDIN-4-YL]ACETAMIDE](http://img.cochemist.com/ccimg/56100/56090-54-1.png)
![2-CHLORO-N-[1-(2-FLUOROBENZOYL)PIPERIDIN-4-YL]ACETAMIDE](http://img.cochemist.com/ccimg/56100/56090-54-1_b.png)