Co-reporter:Jie Zhou, Ming Ji, Zhixiang Zhu, Ran Cao, Xiaoguang Chen, Bailing Xu
European Journal of Medicinal Chemistry 2017 Volume 132(Volume 132) pp:
Publication Date(Web):26 May 2017
DOI:10.1016/j.ejmech.2017.03.013
•Novel benzo[d]imidazole derivatives were obtained as highly potent PARP-1 inhibitors.•Compound 10a demonstrated in vivo anti-tumor activity as chemo-sensitizer.•Two co-crystal structures were presented and elicited a unique binding mode.Novel 1H-benzo[d]immidazole-4-carboxamide derivatives bearing five-membered or six-membered N-heterocyclic moieties at the 2-position were designed and synthesized as PARP-1 inhibitors. Structure-activity relationships were conducted and led to a number of potent PARP-1 inhibitors having IC50 values in the single or double digit nanomolar level. Some potent PARP-1 inhibitors also had similar inhibitory activities against PARP-2. Among all the synthesized compounds, compound 10a and 11e displayed strong potentiation effects on temozolomide (TMZ) in MX-1 cells (PF50 = 7.10, PF50 = 4.17). In vivo tumor growth inhibition was investigated using compound 10a in combination with TMZ, and it was demonstrated that compound 10a could strongly potentiate the cytotoxicity of TMZ in MX-1 xenograft tumor model. Two co-crystal structures of compounds 11b and 15e complexed with PARP-1 were achieved and demonstrated a unique binding mode of these benzo-imidazole derivatives.Novel 1H-benzo[d]immidazole-4-carboxamide derivatives bearing five-membered or six-membered N-heterocyclic moieties at the 2-position were synthesized and evaluated as PARP-1 inhibitors.Download high-res image (271KB)Download full-size image
Co-reporter:Hailong Zhao, Ming Ji, Guonan Cui, Jie Zhou, Fangfang Lai, Xiaoguang Chen, Bailing Xu
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 15(Issue 15) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.bmc.2017.05.052
The PARP-2 selective inhibitor is important for clarifying specific roles of PARP-2 in the pathophysiological process and developing desired drugs with reduced off-target side effects. In this work, a series of novel quinazoline-2,4(1H,3H)-dione derivatives was designed and synthesized to explore isoform selective PARP inhibitors. As a result, compound 11a (PARP-1 IC50 = 467 nM, PARP-2 IC50 = 11.5 nM, selectivity PARP-1/PARP-2 = 40.6) was disclosed as the most selective PARP-2 inhibitor with high potency to date. The binding features of compound 11a within PARP-1 and PARP-2 were investigated respectively to provide useful insights for the further construction of new isoform selective inhibitors of PARP-1 and PARP-2 by using CDOCKER program.A series of novel quinazoline-2,4(1H,3H)-dione derivatives was prepared and their inhibitory activities were evaluated against both PARP-1 and PARP-2. Compound 11a was identified as the highly potent and selective PARP-2 inhibitor.Download high-res image (113KB)Download full-size image
Co-reporter:Hailong Zhao, Guonan Cui, Jing Jin, Xiaoguang Chen, Bailing Xu
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 22) pp:5911-5920
Publication Date(Web):15 November 2016
DOI:10.1016/j.bmc.2016.09.049
Pin1 (Protein interacting with NIMA1) is a peptidyl prolyl cis–trans isomerase (PPIase) which specifically catalyze the conformational conversion of the amide bond of pSer/Thr-Pro motifs in its substrate proteins and is a novel promising anticancer target. A series of new thiazole derivatives were designed and synthesized, and their inhibitory activities were measured against human Pin1 using a protease-coupled enzyme assay. Of all the tested compounds, a number of thiazole derivatives bearing an oxalic acid group at 4-position were found to be potent Pin1 inhibitors with IC50 values at low micromolar level. The detailed structure–activity relationships were analyzed and the binding features of compound 10b (IC50 5.38 μM) was predicted using CDOCKER program. The results of this research would provide informative guidance for further optimizing thiazole derivatives as potent Pin1 inhibitors.
Co-reporter:Hai-Long Zhao, Hua-Long Chen, Jie Zhou, Bai-Ling Xu
Chinese Chemical Letters 2016 Volume 27(Issue 2) pp:302-304
Publication Date(Web):February 2016
DOI:10.1016/j.cclet.2015.11.006
Co-reporter:Haiping Yao, Ming Ji, Zhixiang Zhu, Jie Zhou, Ran Cao, Xiaoguang Chen, Bailing Xu
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 4) pp:681-693
Publication Date(Web):15 February 2015
DOI:10.1016/j.bmc.2014.12.071
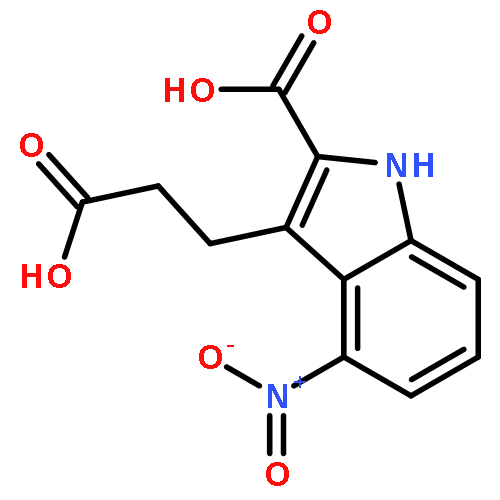
Poly(ADP-ribose)polymerase-1 (PARP-1) has emerged as a promising anticancer drug target due to its key role in the DNA repair process. In this work, a novel series of 1-benzyl-quinazoline-2,4(1H,3H)-dione derivatives were designed and synthesized as human PARP-1 inhibitors, structure–activity relationships were conducted and led to a number of potent PARP-1 inhibitors having IC50 values of single or double digit nanomolar level. Compound 7j was a potent PARP-1 and PARP-2 inhibitor and it could selectively kill the breast cancer cells MX-1 and MDA-MB-468 with mutated BRCA1/2 and PTEN, respectively, in comparison with homologous recombination proficient cell types such as breast cancer cells MDA-MB-231. In addition, compound 7j displayed the strongest potentiation effect on temozolomide in MX-1 cells (PF50 = 3.77) in this series of PARP-1 inhibitors.A series of novel quinazoline-2,4(1H,3H)-dione derivatives were synthesized and evaluated as inhibitors of poly(ADP-ribose)polymerase-1 (PARP-1). X, Y = H or F; R1 = a range of amino acid fragments; IC50: 9.51–5290 nM.
Co-reporter:Jianbo Bie, Shuainan Liu, Zhanmei Li, Yongzhao Mu, Bailing Xu, Zhufang Shen
European Journal of Medicinal Chemistry 2015 90() pp: 394-405
Publication Date(Web):
DOI:10.1016/j.ejmech.2014.11.049
Co-reporter:Jianbo Bie, Shuainan Liu, Jie Zhou, Bailing Xu, Zhufang Shen
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 6) pp:1850-1862
Publication Date(Web):15 March 2014
DOI:10.1016/j.bmc.2014.01.047
Co-reporter:Hai-Long Zhao, Jie Zhou, Hong-Rui Song, Bai-Ling Xu
Chinese Chemical Letters 2014 Volume 25(Issue 3) pp:411-414
Publication Date(Web):March 2014
DOI:10.1016/j.cclet.2013.12.005
A facile and efficient protocol was developed to access 2-alkoxy-4-amino-N-arylthiazole-5-carboxamides through a three-component one-pot reaction, which involved potassium methyl cyanimidodithiocarbonate, 2-halo-N-arylacetamides and alcohols. The easy availability and the broad structural diversity of substrates make the reaction useful for the construction of libraries in drug discovery.A facile and efficient protocol was developed to access 2-alkoxy-4-amino-N-arylthiazole-5-carboxamides through a three-component one-pot reaction, which involved potassium methyl cyanimidodithiocarbonate, 2-halo-N-arylacetamides and alcohols.
Co-reporter:Jie Zhou, Mingyu Ba, Bo Wang, Haibo Zhou, Jianbo Bie, Decai Fu, Yingli Cao, Bailing Xu and Ying Guo
MedChemComm 2014 vol. 5(Issue 4) pp:441-444
Publication Date(Web):11 Dec 2013
DOI:10.1039/C3MD00337J
A series of novel N4-substituted thiophen-3-ylsulfonylquinoxalinone derivatives were designed and synthesized by variations of 2- and 5-position of the thiophene ring. All target molecules were tested for their anti-HIV-1 replication activities and compounds 1b and 1d were found to be the most potent inhibitors with an IC50 value at 10−8 mol L−1 level. The preliminary structure–activity relationships were analyzed on the basis of the binding mode of compound 1b predicted by CDOCKER.
Co-reporter:Jie Zhou, Jing Jin, Yi Zhang, Yuwen Yin, Xiaoguang Chen, Bailing Xu
European Journal of Medicinal Chemistry 2013 Volume 68() pp:222-232
Publication Date(Web):October 2013
DOI:10.1016/j.ejmech.2013.08.006
•Novel benzoimidazole-contained analogs of combretastatin A-4 were synthesized.•The chemical structures of regioisomers 6a–13a and 6b–14b were confirmed.•Four compounds exhibited excellent antiproliferative activities in vitro.•Compound 5 possessed significant in vivo antitumor activity.•Benzoimidazole is a favorable replacement of the B ring on CA-4.A series of novel oxazole-bridged analogs of combretastatin A-4 bearing a benzo[d]-imidazole as B ring were synthesized and evaluated for antiproliferative activities against five human cancer cell lines. Among all the synthesized compounds, the N-unsubstituted benzoimidazole analog 5 and the analogs 6b, 7a and 7b with a small hydrophobic group on nitrogen atom of benzoimidazole ring were identified as the most potent inhibitors of tumor cell growth with IC50 values at nanomolar levels (5, IC50 = 8.4 nM, HT29; 6b, 7a, 7b, IC50 = 9.6 nM, 3.8 nM, 3.0 nM, A549). In a murine H22 tumor xenograft model, compound 5 exhibited significant antitumor activity. The binding mode of compound 5 in the colchicine binding site of tubulin was probed.A series of novel oxazole-bridged analogs of combretastatin A-4 bearing a benzo[d]-imidazole as B ring were synthesized and evaluated for their antiproliferative activities in vitro and in vivo.
Co-reporter:Liang Chen, Zhan Mei Li, Jie Zhou, Hong Rui Song, Bai Ling Xu
Chinese Chemical Letters 2012 Volume 23(Issue 6) pp:695-698
Publication Date(Web):June 2012
DOI:10.1016/j.cclet.2012.04.007
A facile and efficient approach was developed to access 5, 7-disubstitued thiazolo[5,4-d]pyrimidine-4, 6(5H, 7H)-diones through condensation of N-substituted 5-amino-4-carbethoxythiazole with structurally diverse isocyanates in the presence of sodium hydride. The easy availability of substrates and tolerance of structural diversity in this reaction make it attractive to be used for construction of libraries in drug discovery process.
Co-reporter:Chang Liu, Jing Jin, Liang Chen, Jie Zhou, Xiaoguang Chen, Decai Fu, Hongrui Song, Bailing Xu
Bioorganic & Medicinal Chemistry 2012 20(9) pp: 2992-2999
Publication Date(Web):
DOI:10.1016/j.bmc.2012.03.005
Co-reporter:Lina Zhu, Jing Jin, Chang Liu, Chongjing Zhang, Yan Sun, Yanshen Guo, Decai Fu, Xiaoguang Chen, Bailing Xu
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 9) pp:2797-2807
Publication Date(Web):1 May 2011
DOI:10.1016/j.bmc.2011.03.058
A series of novel 2,4-disubstituted quinazoline derivatives were prepared and their inhibitory activities on hPin1 were evaluated. Of all the synthesized compounds, eight compounds displayed inhibitory activities with IC50 value at the level of 10−6 mol/L. Preliminary structure–activity relationships were analyzed in details and the binding mode of the titled compounds was predicted using FlexX algorithm. The design and optimization of novel small molecule Pin1 inhibitors will be guided by the results of this report.A series of novel 2,4-disubstituted quinazoline derivatives were prepared and their inhibitory activities on hPin1 were evaluated. Of all the synthesized compounds, eight compounds displayed inhibitory activities with IC50 value at the level of 10−6 mol/L. Preliminary structure–activity relationships were analyzed in details and the binding mode of the titled compounds was predicted using FlexX algorithm.
Co-reporter:Bailing Xu, Yan Sun, Ying Guo, Yingli Cao, Tao Yu
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 7) pp:2767-2774
Publication Date(Web):1 April 2009
DOI:10.1016/j.bmc.2009.02.039
A series of novel N4-(hetero)arylsulfonylquinoxalinone derivatives were prepared in a straight and efficient way. Of all the synthesized compounds, five compounds exhibited potent anti-HIV-1 replication activities with IC50 value at the level of 10−7 mol/L. Preliminary structure–activity relationships were studied in details and that will shed light on the discovery of more potent non-nucleoside reverse-transcriptase inhibitors.A series of novel N4-(hetero)arylsulfonylquinoxalinone derivatives were prepared in a straight and efficient way. Of all the synthesized compounds, five compounds exhibited potent anti-HIV-1 replication activities with IC50 value at the level of 10−7 mol/L. Preliminary structure–activity relationships were studied in details and that will shed light on the discovery of more potent non-nucleoside reverse-transcriptase inhibitors.
Co-reporter:Bai Ling Xu, Jin Ping Chen, Rong Zhang Qiao, De Cai Fu
Chinese Chemical Letters 2008 Volume 19(Issue 5) pp:537-540
Publication Date(Web):May 2008
DOI:10.1016/j.cclet.2008.03.022
A facile, efficient and novel approach to access 2-substituted-N1-carbethoxy-2,3-dihydro-4(1H)-quinazolinones was developed by condensation of substituted N-carbethoxyanthranilamide with alkyl, aromatic or heteroaromatic aldehydes in the refluxing 2,2,2-trifluoroethanol or hexafluoroisopropanol using p-toluenesulfonic acid as catalyst.


![Propanoic acid, 2-[(2-nitrophenyl)hydrazono]-, ethyl ester](http://img.cochemist.com/ccimg/292900/292853-66-8.png)
![Propanoic acid, 2-[(2-nitrophenyl)hydrazono]-, ethyl ester](http://img.cochemist.com/ccimg/292900/292853-66-8_b.png)
![2,4(1H,3H)-Quinazolinedione, 1-[(3-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/180700/180632-51-3.png)
![2,4(1H,3H)-Quinazolinedione, 1-[(3-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/180700/180632-51-3_b.png)
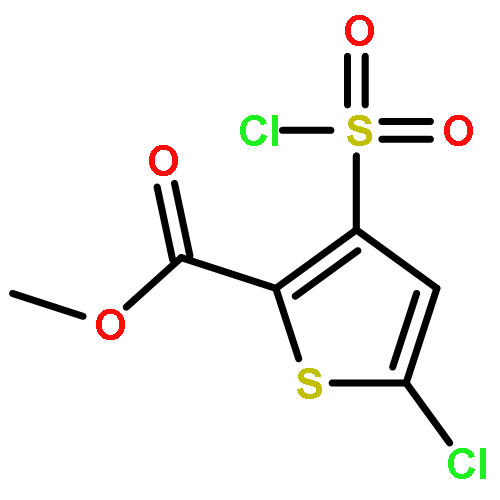
![[2,2'-Bithiophene]-5-carboxylic acid, 4-(chlorosulfonyl)-, methyl ester](http://img.cochemist.com/ccimg/169800/169759-80-2.png)
![[2,2'-Bithiophene]-5-carboxylic acid, 4-(chlorosulfonyl)-, methyl ester](http://img.cochemist.com/ccimg/169800/169759-80-2_b.png)

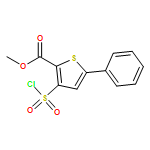
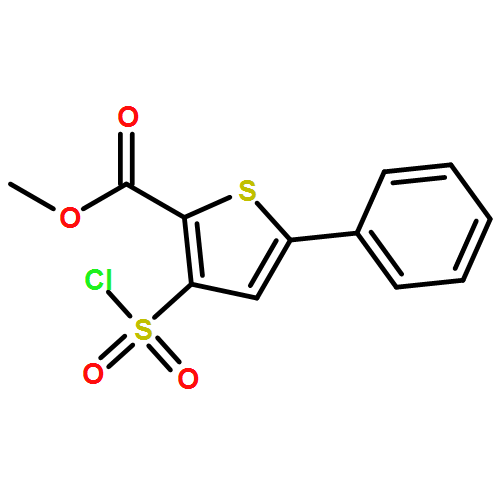

![Quinazoline, 2,4-bis[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/15200/15135-19-0.png)
![Quinazoline, 2,4-bis[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/15200/15135-19-0_b.png)



