Co-reporter:Zhi-Hui Zhang, An-Jun Deng, Lian-Qiu Wu, Lian-Hua Fang, Jin-Qian Yu, Zhi-Hong Li, Tian-Yi Yuan, Wen-Jie Wang, Guan-Hua Du, Hai-Lin Qin
European Journal of Medicinal Chemistry 2014 Volume 86() pp:542-549
Publication Date(Web):30 October 2014
DOI:10.1016/j.ejmech.2014.09.006
•Twenty five 13-substituted quaternary coptisine derivatives were synthesized.•Introduction of alkyls into 13-position led to significant increases of cytotoxicity.•Extending the alkyl side chain increased the cytotoxic activity.•Aromaric methyl and analogs of arylmethyls causes cytotoxicity on IEC-6.•Introducing other arylmethyls into 13-position showed no effect on tested activity.Twenty five 13-substituted quaternary coptisine derivatives were synthesized to test their cytotoxicities against several cancer cell-lines and on intestinal epithelial cell-6 (IEC-6) in vitro to evaluate structure–activity relationship (SAR). Introduction of the alkyl groups into the C-13 position of quaternary coptisine (1) led to significant increase of the cytotoxic activity, while the substitution of arylmethyl groups and others at the same position showed no effect on improving cytotoxicities against the same cancer cell-lines. The cytotoxicities of quaternary 13-alkylcoptisines was significantly reinforced as the length of the aliphatic chain increased, with quaternary 13-n-undecylcoptisine (4l) showing 7, 23, 12, and 9 times, respectively, more active than quaternary coptisine (1) against HCT, A549, Bel7402, and C33A, and being 4, 11, 2, and 3 times, respectively, more active than the positive control, fluorouracil (5-FU), against the same cell-lines, by IC50 values. In comparison to quaternary 13-n-undecylcoptisine (4l) and the above references, quaternary 13-n-dodecylcoptisine (4m) almost showed the same cytotoxicities. In contrast with the n-alkyl chains, the arylmethyl substituents at C-13 displayed low cytotoxicity, except for naphthyl rings or phenyl rings with CF3 or methyl substituents. However, their low cytotoxicity could make them useful as drug candidates for other diseases (bowel, etc).Twenty five 13-substituted quaternary coptisine derivatives were synthesized and tested for their cytotoxicity to evaluate the structure–activity relationships, with explicit influencing factor on improving activity being obtained.
Co-reporter:Yu Yan;Yu-Jie Wu;Tian-Yi Yuan;Li Li;Lian-Hua Fang;Ping Xie;Xiao-Na Xu;Guan-Hua Du;Xiao-Zhen Jiao
Cardiovascular Drugs and Therapy 2014 Volume 28( Issue 5) pp:415-424
Publication Date(Web):2014/10/01
DOI:10.1007/s10557-014-6544-7
In the present study, we investigated the vasodilatory effect of a novel scaffold Rho-kinase inhibitor, DL0805-2, on isolated rat arterial rings including mesenteric, ventral tail, and renal arteries. We also examined the potential mechanisms of its vasodilatory action using mesenteric artery rings.A DMT multiwire myograph system was used to test the tension of isolated small arteries. Several drugs were employed to verify the underlying mechanisms.DL0805-2 (10−7–10−4 M) inhibited KCl (60 mM)-induced vasoconstriction in three types of small artery rings (pEC50: 5.84 ± 0.03, 5.39 ± 0.03, and 5.67 ± 0.02 for mesenteric, renal, and ventral tail artery rings, respectively). Pre-incubation with DL0805-2 (1, 3, or 10 μM) attenuated KCl (10–60 mM) and angiotensin II (AngII; 10−6 M)-induced vasoconstriction in mesenteric artery rings. The relaxant effect on the rat mesenteric artery was partially endothelium-dependent (pEC50: 6.02 ± 0.05 for endothelium-intact and 5.72 ± 0.06 for endothelium-denuded). The influx and release of Ca2+ were inhibited by DL0805-2. In addition, the increased phosphorylation levels of myosin light chain (MLC) and myosin-binding subunit of myosin phosphatase (MYPT1) induced by AngII were blocked by DL0805-2. However, DL0805-2 had little effect on K+ channels.The present results demonstrate that DL0805-2 has a vasorelaxant effect on isolated rat small arteries and may exert its action through the endothelium, Ca2+ channels, and the Rho/ROCK pathway.
Co-reporter:Li-Li Gong, Lian-Hua Fang, Jian-Hao Peng, Ai-Lin Liu, Guan-Hua Du
Journal of Biotechnology (1 February 2010) Volume 145(Issue 3) pp:295-303
Publication Date(Web):1 February 2010
DOI:10.1016/j.jbiotec.2009.12.003
Rho-kinase inhibitors are effective candidates for the treatment of neural and cardiovascular disorders. The present paper reports the discovery of a novel class of ROCK-I inhibitors by integrating virtual screening with high-throughput screening methods. We developed common-feature pharmacophore models based on known representative ROCK inhibitors and employed them to screen a database of 12,280 compounds. We then applied a LigandFit model to reduce the number of hits. A new high-throughput screening model based on Kinase-Glo Luminescent Kinase Assay was established to identify inhibitors observed among the virtual screening models. Ten hits were found to have larger than 70% inhibition at 10 μmol l−1 and were worthy of further investigation.











![Isoquinoline,5-[(2-methyl-1-piperazinyl)sulfonyl]-](http://img.cochemist.com/ccimg/84500/84477-87-2.png)
![Isoquinoline,5-[(2-methyl-1-piperazinyl)sulfonyl]-](http://img.cochemist.com/ccimg/84500/84477-87-2_b.png)
![L-Ornithine,N5-[imino(nitroamino)methyl]-, methyl ester](http://img.cochemist.com/ccimg/51000/50903-99-6.png)
![L-Ornithine,N5-[imino(nitroamino)methyl]-, methyl ester](http://img.cochemist.com/ccimg/51000/50903-99-6_b.png)
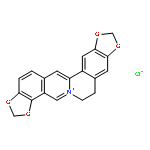
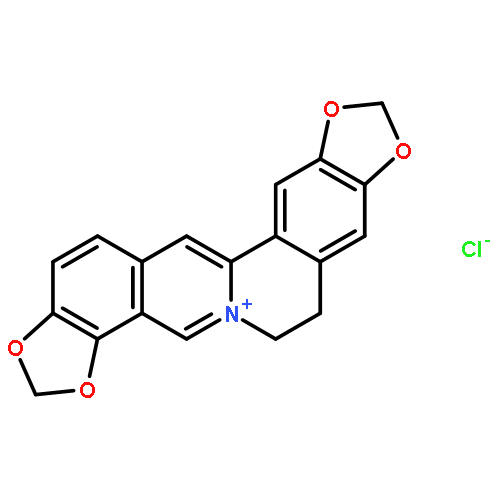


![Bis[1,3]benzodioxolo[5,6-a:4',5'-g]quinolizinium,6,7-dihydro-](http://img.cochemist.com/ccimg/3500/3486-66-6.png)
![Bis[1,3]benzodioxolo[5,6-a:4',5'-g]quinolizinium,6,7-dihydro-](http://img.cochemist.com/ccimg/3500/3486-66-6_b.png)




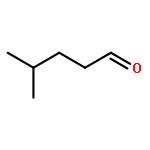
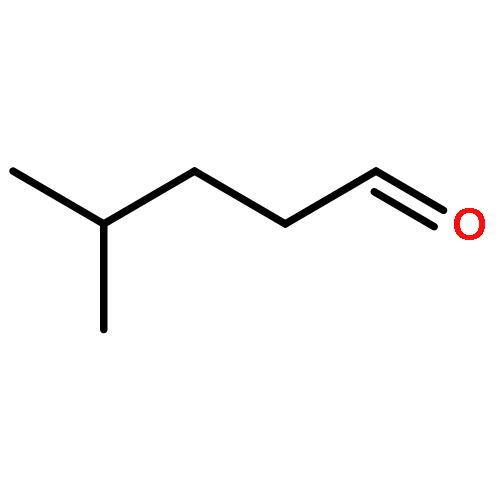
![7,11b-dihydrobenz[b]indeno[1,2-d]pyran-3,6a,9,10(6H)-tetrol](http://img.cochemist.com/ccimg/500/474-07-7.png)
![7,11b-dihydrobenz[b]indeno[1,2-d]pyran-3,6a,9,10(6H)-tetrol](http://img.cochemist.com/ccimg/500/474-07-7_b.png)







