Co-reporter:Bao Liu, Zhou Tian, Ning Zhao, Zhen Liu, and Boping Liu
Industrial & Engineering Chemistry Research May 31, 2017 Volume 56(Issue 21) pp:6164-6164
Publication Date(Web):May 9, 2017
DOI:10.1021/acs.iecr.7b00883
The effects of various polymerization conditions such as cocatalyst concentration, pressure, temperature, and reaction time on ethylene homopolymerization over a SiO2-supported imido-vanadium/silyl-chromate (Cr-iV) bimetallic catalyst were systematically investigated. The monometallic silyl-chromate (S-2) and imido-vanadium (iV) catalyst were also employed for comparison. It was found that the S-2 catalyst produced a polymer with relatively low molecular weight, while the iV catalyst produced ultrahigh-molecular-weight polyethylene (UHMWPE). The bimetallic catalyst was capable of producing reactor blends, with bimodal molecular weight distribution (MWD) and a considerable amount of UHMWPE. Increasing cocatalyst concentration and decreasing polymerization temperature both enhanced the high molecular weight part of the bimodal MWD while the position of the two peaks remained unchanged. The polymerization rates all showed first-order dependences with respect to ethylene pressure for the three catalysts. Ethylene pressure variations caused no changes in the MWD of polymers made by the three catalysts, indicating that transfer to the monomer is the main chain transfer mechanism.
Co-reporter:Wei Zhu;Zhou Tian 田洲;Rui-hua Cheng;Xue-lian He
Chinese Journal of Polymer Science 2017 Volume 35( Issue 12) pp:1474-1487
Publication Date(Web):29 September 2017
DOI:10.1007/s10118-017-1999-1
A series of (SiO2/MgO/ID/MgCl2)·TiClx Ziegler-Natta catalysts for propylene polymerization has been prepared with a new method. These catalysts were synthesized using soluble Mg-compounds as the Mg-source and the preparation progress was relatively simple. The catalyst could copy the spherical shape of the carrier very well. The propylene polymerization results showed that the catalyst revealed the best activity with 9,9-di(methoxymethyl)fluorene (BMMF) as internal donor at 50 °C with the optimal molar ratio Al/Ti = 5, which was much lower than what the industrial polypropylene catalyst used (at least molar ratio Al/Ti = 100), resulting in great cost saving. Additionally, the polymerization kinetics of the catalyst exhibited very stable property after achieving a relatively high value. These catalysts possessed rather high activity and good hydrogen response. The isotactic index (II.) value of the PP products could be higher than 98% in the presence of both internal and external electron donors. Moreover, temperature rising elution fractionation method was used to understand the influence of donors and H2 on the properties of the PP products.
Co-reporter:Ting Fu;Rui-hua Cheng 程瑞华;Xue-lian He;Zhen Liu
Chinese Journal of Polymer Science 2017 Volume 35( Issue 6) pp:739-751
Publication Date(Web):23 April 2017
DOI:10.1007/s10118-017-1939-0
Various (SiO2/MgO/MgCl2)·TiClx Ziegler-Natta catalysts modified by the third metal elements were synthesized by the co-impregnation of water-soluble magnesium and the third metal salts. Several key factors including the electronegativity of the third metal elements, catalyst performances in ethylene homo-polymerization, ethylene/1-hexene copolymerization and hydrogen response were systematically investigated. Both the catalyst performance and the polymer properties are influenced by the introduction of the third metal elements. Compared with the unmodified (SiO2/MgO/MgCl2)·TiClx Ziegler-Natta catalyst, activity and 1-hexene incorporation are enhanced by the introduction of zirconium, vanadium, aluminum and chromium, while deteriorated by the addition of ferrum, nickel, molybdenum and tungsten. Correlations of the catalyst activities and 1-hexene incorporation ability with the electronegativity of the third metal elements are discovered. It is found that the lower electronegativity of the third metal elements leads to the catalyst with higher activity and higher α-olefin co-polymerization ability. The polyethylene produced by a nickel modified catalyst showed broad molecular weight distribution (MWD) and the lowest average molecular weight (MW), while by using a ferrum modified catalyst, the resulting polyethylene had the highest MW, reaching the ultra-high MW area. Vanadium and chromium modified catalysts demonstrated the best hydrogen response.
Co-reporter:Rui Gao;Xuelian He;Yunqi Shao;Yanling Hu;Haiyang Zhang;Zhen Liu
Macromolecular Theory and Simulations 2016 Volume 25( Issue 3) pp:303-311
Publication Date(Web):
DOI:10.1002/mats.201500089
Influences of branch content (BC) and branch length (BL) on isothermal crystallization of precisely branched polyethylene are studied by molecular dynamics simulation. Branch acts as a defect both in nucleation and crystal growth process. BC affects not only crystallization kinetics but also final morphologies. Crystallization rate and crystallinity decrease as BC increases. Morphology Regimes change from lamellae crystal to bundle crystal at critical BC (20/1000 C) because of different folding pattern. 50 CH2 is the critical methyl sequence length to form lamellae crystal. Lamellae thicknesses of final morphologies decrease in gradient corresponding to Morphologies Regimes. BL has no influence on the crystallization kinetics, and only affects the final morphologies when more branches inclusion happens with BL increasing. Trans-rich phenomenon in pre-crystalline state is observed. Crystallization process begins at the end of induction stage when trans state population reaches a critical value, and this value is independent of BC and BL.
Co-reporter:Jingwen Wang;Ruihua Cheng;Xuelian He;Zhen Liu;Ning Zhao
Macromolecular Reaction Engineering 2016 Volume 10( Issue 3) pp:246-260
Publication Date(Web):
DOI:10.1002/mren.201500056
Co-reporter:Qing-Yang Meng;Katie Pepper;Rui-Hua Cheng;Steven M. Howdle;Bo-Ping Liu
Journal of Polymer Science Part A: Polymer Chemistry 2016 Volume 54( Issue 17) pp:2785-2793
Publication Date(Web):
DOI:10.1002/pola.28162
ABSTRACT
The copolymerization of cyclohexene oxide (CHO) and carbon dioxide (CO2) was carried out under supercritical CO2 (scCO2) conditions to afford poly (cyclohexene carbonate) (PCHC) in high yield. The scCO2 provided not only the C1 feedstock but also proved to be a very efficient solvent and processing aid for this copolymerization system. Double metal cyanide (DMC) and salen-Co(III) catalysts were employed, demonstrating excellent CO2/CHO copolymerization with high yield and high selectivity. Surprisingly, our use of scCO2 was found to significantly enhance the copolymerization efficiency and the quality of the final polymer product. Thermally stable and high molecular weight (MW) copolymers were successfully obtained. Optimization led to excellent catalyst yield (656 wt/wt, polymer/catalyst) and selectivity (over 96% toward polycarbonate) that were significantly beyond what could be achieved in conventional solvents. Moreover, detailed thermal analyses demonstrated that the PCHC copolymer produced in scCO2 exhibited higher glass transition temperatures (Tg ∼ 114 °C) compared to polymer formed in dense phase CO2 (Tg ∼ 77 °C), and hence good thermal stability. Additionally, residual catalyst could be removed from the final polymer using scCO2, pointing toward a green method that avoids the use of conventional volatile organic-based solvents for both synthesis and work-up. © 2016 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2016, 54, 2785–2793
Co-reporter:Rui Gao;Xuelian He;Haiyang Zhang;Yunqi Shao;Zhen Liu
Journal of Molecular Modeling 2016 Volume 22( Issue 3) pp:
Publication Date(Web):2016 March
DOI:10.1007/s00894-016-2931-2
A molecular level understanding of the polyethylene (PE) crystallization process was elucidated by molecular dynamics simulation of three states, with varying chain length and temperature. The process can be classified into the following three states: (1) nucleation controlled state, (2) competitive state of crystal growth process and new nuclei formation, and (3) crystal growth controlled state, which could be quantified by the evolution of nuclei number. With increasing chain length, two phenomena occur: the single crystallization mechanism changes from state (1) to (3), and the crystal size increases while the b/a axial ratio in the lateral surface decreases. These changes can be explained from a thermodynamic point of view, in that the van der Waals (vdW) interaction per CH2 unit is strengthened and more nucleation sites are generated for longer chain. Size effect (meaning different surface fractions when the chain collapses into a globule) was an important factor determining vdW energy per unit and the crystallization states of a single PE chain. On the other hand, the crystallization states were independent of chain length for short chains systems with the same size effect. In both conditions, a long chain generates multi-crystal domains, and a short chain prefers a single crystal domain. Our results not only provide molecular level evidence for crystallization states but also clarify the influence of chain length on the crystallization process.
Co-reporter:Minglan Gong, Zhen Liu, Yuanhui Li, Yue Ma, Qiaoqiao Sun, Jialong Zhang, and Boping Liu
Organometallics 2016 Volume 35(Issue 7) pp:972-981
Publication Date(Web):March 29, 2016
DOI:10.1021/acs.organomet.5b01029
The mechanism of selective co-oligomerization of ethylene and 1-hexene by the catalyst [CrCl3(PNPOMe)] (a, PNPOMe = N,N-bis(bis(o-methoxyphenyl)phosphine)methylamine) has been explored in detail using the density functional theory (DFT) method. The full catalytic cycles for the formation of 1-hexene and 1-decenes were calculated on the basis of the metallacyclic mechanism, and the distribution of all decene isomers was explained by locating Gibbs free energy surfaces of various pathways, which is in good agreement with the experimental results. A spin surface crossing through a minimum energy crossing point (MECP) from a sextet to a quartet surface takes place before the formation of metallacyclopentane, which opens up a much lower energy pathway and thus facilitates the following co-oligomerization reactions. It is worth noting that β-hydrogen agostic-assisted hydrogen transfer is of crucial importance for the decomposition of the metallacycle intermediates to give 1-hexene or decenes. Moreover, an analysis of the Cr–O bond distance and NBO charges indicates the important role of a hemiliable methoxy moiety, which acts as a pendant group in the co-oligomerization of ethylene and 1-hexene by CrCl3(PNPOMe) catalyst.
Co-reporter:Jialong Zhang, Pengyuan Qiu, Zhen Liu, Boping Liu, Rami J. Batrice, Mark Botoshansky, and Moris S. Eisen
ACS Catalysis 2015 Volume 5(Issue 6) pp:3562
Publication Date(Web):April 30, 2015
DOI:10.1021/acscatal.5b00240
Our previous experimental report showed a switching behavior from ethylene polymerization to nonselective oligomerization by a novel triphenylsiloxy complex of chromium(II) [(Ph3SiO)Cr·(THF)]2(μ-OSiPh3)2 (1) together with methylaluminoxane (MAO) as a cocatalyst. In this work, combined experimental and computational studies were carried out to shed some light on the nature of the active species and their fascinating switching mechanism. The experimental results and DFT calculations suggested that (i) the chain propagation and chain transfer processes proceed via a Cossee–Arlman mechanism and β-hydrogen transfer to the chromium center, respectively; (ii) the trivalent cationic model [(Ph3SiO)CrIIIMe]+ and [(η6-toluene)CrIIIMe2]+, which could be generated by a disproportionation reaction, are the most plausible active species for ethylene polymerization, and the divalent cationic model [(η6-toluene)CrIIMe]+ might be responsible for ethylene nonselective oligomerization. A switching mechanism from ethylene polymerization to nonselective oligomerization in the 1/MAO catalyst system was proposed on the basis of DFT calculations. These results may have useful implications for studying active species and the mechanism of transition-metal-catalyzed olefin polymerization and oligomerization.Keywords: active species; DFT calculations; ethylene nonselective oligomerization; ethylene polymerization; Phillips catalyst; switching mechanism
Co-reporter:Yu Che, Zhou Tian, Zhen Liu, Rui Zhang, Yuxin Gao, Enguang Zou, Sihan Wang, Boping Liu
Powder Technology 2015 Volume 278() pp:94-110
Publication Date(Web):July 2015
DOI:10.1016/j.powtec.2015.02.022
•Scale-up effect on the hydrodynamics of a pilot-plant FBR is explored.•Remarkable differences in the core–annulus structures and shapes are revealed.•Bed expanding section significantly affects the circular flow patterns.•Operation strategy transition from TPPP to NPPP is analyzed.This work aims to explore the scale-up effect on the hydrodynamic natures in a pilot-plant gas phase ethylene polymerization fluidized bed reactor (FBR) based on two-dimensional (2D) transient Eulerian model integrating the kinetic theory of granular flow (KTGF). Unlike lab scale FBR, significant differences in the core–annulus structures caused by the scale-up effect are revealed. The core region in pilot-plant scale FBR is much broader, and the distribution is nearly flat for two phases. The annulus structure is thin, and exhibits a very small particle velocity fluctuation. The bed expanding section could reduce the solid velocity and improve the flow patterns of gas and polymer particles. Simulations are also performed to assess the effects of polymer particle size and gas velocity on the hydrodynamics for the non-pelletizing polyethylene process. With the increase of polymer particle diameter (446 μm to 1338 μm), the superficial gas velocity should be increased from 0.60 m/s to 0.90 m/s to achieve better steady fluidized state. The results are helpful for understanding how the local mean two phases' velocities and volume fractions vary in pilot-plant reactor for PE production caused by scale-up effect.
Co-reporter:Xing Pan, Zhen Liu, Ruihua Cheng, Xuelian He, Boping Liu
Journal of Organometallic Chemistry 2015 Volume 775() pp:67-75
Publication Date(Web):1 January 2015
DOI:10.1016/j.jorganchem.2014.10.008
•The reaction of CO2 with epoxides over zinc phenoxides studied by DFT.•The activity of catalyst was related to electronic and steric effect of ligands.•Reaction of cyclohexene oxide and CO2 on Zn phenoxides produced alternating copolymer.•Propylene oxide, styrene oxide and epichlorohydrin with CO2 gave cyclic carbonate.•The different behavior of epoxides was related to the steric hindrance of epoxide.The reaction mechanisms between CO2 and different epoxides including cyclohexene oxide (CHO), propylene oxide (PO), styrene oxide (SO) and epichlorohydrin (ECH) over Zn(II) phenoxide catalytic system were investigated using density functional theory (DFT). It was revealed that the reaction between CO2 and CHO over the catalytic system produced alternating copolymer. The higher polymerization activity of the catalyst was usually associated with the higher electron-deficiency and the lower steric hindrance of the zinc center, and the formation of cyclic carbonate from CHO was a two-step elimination reaction process with high activation energy barrier due to the high steric hindrance of CHO. However, the reaction of CO2 with the other three epoxides (PO, SO and ECH) provided only cyclic carbonate via one-step elimination reaction with low activation energy barrier. The catalyst efficiency of zinc phenoxide catalyst was predicted to increase in the following sequence: ECH < PO < SO. In addition, the ring-opening of PO and SO tended to occur at the methine CCH-R–O bond, whereas, the ring-opening of ECH was preferred thermodynamically at the unsubstituted methylene CCH2CCH2–O bond. These theoretical results rationalized well the experimental reports by Darensbourg.The reactivity of three epoxides in the reaction with CO2 over Zn(II) phenoxides were investigated by DFT method. The results showed that the reaction of cyclohexene oxide (CHO) and CO2 produced copolymer, while propylene oxide (PO), styrene oxide (SO) or epichlorohydrin with CO2 provided only cyclic carbonate.
Co-reporter:Jingwen Wang;Ruihua Cheng;Xuelian He;Zhen Liu;Zhou Tian
Macromolecular Chemistry and Physics 2015 Volume 216( Issue 13) pp:1472-1482
Publication Date(Web):
DOI:10.1002/macp.201400599
Co-reporter:Ruihua Cheng;Xin Xue;Weiwei Liu;Ning Zhao;Xuelian He;Zhen Liu
Macromolecular Reaction Engineering 2015 Volume 9( Issue 5) pp:462-472
Publication Date(Web):
DOI:10.1002/mren.201400072
Novel SiO2-supported Cr–V bimetallic oxides catalysts, which were capable of one-pot synthesizing polyethylene with bimodal molecular weight distribution, had been successfully developed. These catalysts were prepared with a simple introduction of vanadium oxide onto the traditional Phillips CrOx/SiO2 catalyst. The polymerization activity over the Cr–V bimetallic catalysts was significantly improved compared with the V-free Phillips CrOx/SiO2 catalyst. Moreover, the bimodal ethylene/1-hexene copolymer prepared by the novel catalyst showed a better short chain branch distribution with more 1-hexene comonomer inserted into the PE chains with high molecular weight, which would be favorable for the long term mechanical properties of the bimodal PE as potential high grade pipe materials.
Co-reporter:Yue Ma, Lisong Wang, Zhen Liu, Ruihua Cheng, Lei Zhong, Yun Yang, Xuelian He, Yuwei Fang, Minoru Terano, Boping Liu
Journal of Molecular Catalysis A: Chemical 2015 401() pp: 1-12
Publication Date(Web):
DOI:10.1016/j.molcata.2015.01.020
Co-reporter:Yu Che, Zhou Tian, Zhen Liu, Rui Zhang, Yuxin Gao, Enguang Zou, Sihan Wang, Boping Liu
Powder Technology 2015 286() pp: 107-123
Publication Date(Web):December 2015
DOI:10.1016/j.powtec.2015.07.049
Co-reporter:Keran Chen, Zhou Tian, Na Luo, and Boping Liu
Industrial & Engineering Chemistry Research 2014 Volume 53(Issue 51) pp:19905-19915
Publication Date(Web):December 2, 2014
DOI:10.1021/ie503456e
This work aims to develop a rigorous model for industrial successive supercritical slurry-phase and gas-phase catalytic polymerization reactors of the Borstar bimodal polyethylene process. The model consists of thermodynamic modeling, multisite Ziegler–Natta polymerization kinetics, and reactor modeling. The perturbed-chain statistical associating fluid theory equation of state (PC-SAFT EOS) with updated parameters allows an accurate description of the phase equilibria of the supercritical slurry-phase and gas-phase system in a wide range of temperatures and pressures. The model predictions of the process variables (i.e., residence time, molar ratio of H2/C2H4 and H2/1-C4H8 of each reactor) and polymer properties (i.e., molecular weight, comonomer content, molecular weight distribution (MWD)) agree well with the industrial plant data under multi-steady-state operation conditions. The model is also capable of simulating the effects on the MWD of bimodal polyethylene resulting from the hydrogen inflow rate in a supercritical slurry-phase loop reactor (SLR) and a gas-phase fluidized bed reactor (FBR), as well as the supercritical solvent propane inflow rate of the SLR.
Co-reporter:Ning Zhao;Ruihua Cheng;Xuelian He;Zhen Liu
Macromolecular Chemistry and Physics 2014 Volume 215( Issue 18) pp:1753-1766
Publication Date(Web):
DOI:10.1002/macp.201400094
Co-reporter:Xing Pan, Zhen Liu, Ruihua Cheng, Dongliang Jin, Xuelian He, Boping Liu
Journal of Organometallic Chemistry 2014 Volume 753() pp:63-71
Publication Date(Web):1 March 2014
DOI:10.1016/j.jorganchem.2013.12.001
•The copolymerization of CO2 with propylene oxide studied by experiment and DFT.•Supported ZnEt2–glycerine–Y(CCl3COO)3 ternary catalyst with SiO2 and Al2O3/SiO2.•Supported catalyst exhibited the highest activity with 3 wt% Al2O3/SiO2 as support.•The activity of the supported catalysts was related to the acidity of the support.•Appropriate electron density of zinc center of the ternary catalyst was crucial.Alternating copolymerization of CO2 and propylene oxide (PO) catalyzed by ZnEt2–glycerine–Y(CCl3COO)3 ternary catalyst was investigated through combined experimental and theoretical approaches. The ternary catalyst showed an increased activity by introducing a support of SiO2. The catalytic activity was further improved when Al2O3 modified SiO2 was used as support. For the supported ternary catalyst using Al2O3 modified SiO2, the catalyst activity increased with increasing of the amount of Al2O3, and the highest activity with 69% of enhancement in catalytic activity compared with the non-supported ternary catalyst was achieved at 3 wt% Al2O3 followed by a further decrease of activity. NH3–TPD measurement confirmed that the surface acidity of the Al2O3 modified SiO2 increased with increasing the amount of Al2O3, which indicated that a proper increment of surface acidity of the SiO2 support was favorable for the improvement of the catalytic activity for the supported ternary catalyst. Moreover, the mechanism for the alternating copolymerization of CO2 and PO over ZnEt2–glycerine binary catalyst and ternary catalyst system has been studied by DFT. The insertion of PO into Zn-carbonate bond was rate-determining and the corresponding activation barrier decreased with increasing of the natural bond order (NBO) charge of the zinc species. The combined experimental and theoretical results suggested that electron deficiency of zinc centers of the ternary catalyst was crucial for the alternating copolymerization of CO2 and PO.The ZnEt2−glycerine−Y(CCl3COO)3 ternary catalyst has been supported on SiO2 and Al2O3/SiO2 to improve catalytic activity. Meantime, copolymerization mechanisms of CO2 and propylene oxide (PO) over five models of ZnEt2--glycerine binary catalyst and ternary catalyst system with different NBO charge on zinc centers are investigated using the DFT method.
Co-reporter:Ning Zhao;Ruihua Cheng;Xuelian He;Zhen Liu;Rui Zhang;Yuxin Gao;Enguang Zou;Sihan Wang
Macromolecular Chemistry and Physics 2014 Volume 215( Issue 15) pp:1434-1445
Publication Date(Web):
DOI:10.1002/macp.201400204
Co-reporter:Yun Yang, Zhen Liu, Ruihua Cheng, Xuelian He, and Boping Liu
Organometallics 2014 Volume 33(Issue 10) pp:2599-2607
Publication Date(Web):May 15, 2014
DOI:10.1021/om500306a
To elucidate fundamental mechanistic aspects of the landmark Chevron–Phillips ethylene trimerization system, a detailed theoretical study has been carried out by DFT methods on an aluminum pyrrolyl chromium catalyst. Reaction pathways for selective ethylene oligomerization have been successfully located on the basis of the metallacycle mechanism. Consistent with experimental results, for the model system ethylene trimerization was proven to be energetically preferred in comparison to ethylene dimerization or further ring expansion toward the formation of higher α-olefins. The Cr(I/III) redox couple was found to be the most likely for the catalytic ethylene trimerization. A careful electronic configuration analysis has been conducted, and the ground state of all active species involved in the catalytic cycle is identified to be S = 3/2 except for the bare active species, which favors a high spin state of S = 5/2. The role of a pendant chlorine functionality has been investigated as well. Variable Cr–Cl bond distance and NBO charge analysis of every intermediate clearly exhibit the hemilabile behavior of the chlorine. This unique hemilability is considered to be a key factor for the selectivity toward 1-hexene formation.
Co-reporter:Zhen Liu, Ruihua Cheng, Xuelian He, and Boping Liu
ACS Catalysis 2013 Volume 3(Issue 6) pp:1172
Publication Date(Web):April 8, 2013
DOI:10.1021/cs400129g
The mechanism of the methylacetylene cyclotrimerization catalyzed by the Phillips Cr/silica catalyst has been studied by DFT investigations based on a Cr(II)/SiO2 cluster model and a silica supported cluster model. Twenty-one kinds of Cr(II)/SiO2·(C3H4)n (n = 1–3) complexes were first optimized successfully. Starting from the most stable chromium(methylacetylene) complex, the following cyclotrimerization of methylacetylene on the quintet surface is prohibited by the spontaneous coupling of the two coordinated methylacetylenes. Instead of overcoming a much higher Gibbs free energy barrier by about 40 kcal/mol on the quintet surface, a spin flipping to the triplet surface at the chromium(methylacetylene) complex only requires 16.9 kcal/mol in Gibbs free energy. After the spin transition, the methyl-chromacyclopropene species was formed immediately on the triplet surface. The triplet dimethyl-chromacyclopentadiene species was generated by insertion of a coordinated methylacetylene into the 3-membered metallacycle ring. The following reaction may follow two pathways: (a) a concerted [4 + 2] cycloaddition or (b) a stepwise pathway (insertion and reductive elimination) via a trimethyl-chromacycloheptatriene species. All the eight [4 + 2] cycloaddition reaction pathways are favored competing with the stepwise pathways. The reactivity of each reaction pathway can be examined in terms of the calculated TOF using the energetic span model. We found that only four [4 + 2] cycloaddition reaction pathways (PES-T1Da, PES-T2Da, PES-T3Da, and PES-T4Da) are responsible for the cyclotrimerization of methylacetylene. The PES-T4Da leads to the production of 1,3,5-trimethylbenzene, while the other three pathways generate 1,2,4-trimethylbenzene. Furthermore, the effects of the silica support and the dispersion correction have been considered for the most plausible reaction pathways PES-T1Da, PES-T2Da, PES-T3Da, and PES-T4Da, respectively. Finally, with a consideration of the effects of the silica support and inclusion of the dispersion correction in the final calculated energies, the ratio of the 1,3,5- to 1,2,4-TMB is 0.32 at 363 K predicting that the 1,2,4-TMB is the dominant product in the cyclotrimerization of methylacetylene catalyzed by the Phillips Cr/silica catalyst.Keywords: DFT; methylacetylene cyclotrimerization; Phillips chromium catalyst; spin flipping; TOF
Co-reporter:Yun Yang, Zhen Liu, Boping Liu, and Robbert Duchateau
ACS Catalysis 2013 Volume 3(Issue 10) pp:2353
Publication Date(Web):September 3, 2013
DOI:10.1021/cs4004968
A series of N,P-based ancillary ligands have been synthesized, and the corresponding catalysts, formed in situ by mixing one of the N,P-ligands, Cr(acac)3 and MAO, have been tested for ethylene oligomerization. Under standard ethylene oligomerization conditions (30 bar ethylene, 60 °C, methylcyclohexane solvent), all of the in situ-formed complexes show catalytic activity, producing oligomers together with varying amounts of polyethylene (PE). Of all these combinations, only the catalyst formed by mixing N-pyrrolyldiphenylphosphine with Cr(acac)3 and MAO is capable of selectively oligomerizing ethylene, producing a mixture of 1-hexene and 1-octene in varying ratios alongside a small amount of PE. Further investigations on this catalyst system revealed that the presence of a low concentration of toluene favors the production of 1-octene. However, in pure toluene as the solvent, the selectivity toward 1-hexene/1-octene is lost and a statistic mixture of α-olefins is produced. Moreover, the choice of the cocatalyst is found to dramatically influence the composition of the liquid products. By careful adjustment of the reaction conditions (temperature, ethylene pressure, catalyst loading, and ligand/Cr ratio), the 1-hexene/1-octene molar ratio can be tuned from 0.3 to 20 and a selectivity for 1-octene formation of up to 74% can be achieved.Keywords: chromium catalyst; DFT calculation; ethylene tetramerization; ligand design; selective ethylene oligomerization; solvent effect
Co-reporter:Rui-hua Cheng;Jun Luo;Zhen Liu;Jing-wen Sun
Chinese Journal of Polymer Science 2013 Volume 31( Issue 4) pp:591-600
Publication Date(Web):2013 April
DOI:10.1007/s10118-013-1252-5
The formations of defective MgCl2 surfaces, and subsequent adsorption of Ti species and electron donor, as well as propylene polymerization over the Ziegler-Natta catalyst have been investigated using density functional theory (DFT) method. Twelve possible support models of regular and defective MgCl2 (110) and (100) surfaces were built. The individual adsorptions of titanium chlorides as mononuclear or dinuclear, and ethyl benzoate (EB) as electron donor, on these models were evaluated. The analysis of energies presented the cases of EB adsorption were generally more stable than titanium chlorides on both surfaces. Thus, EB as internal electron donor mainly prevented TiCl4 from coordinating on the MgCl2 surfaces where mostly non-stereospecific active sites could be formed. Exceptionally, A5 the site model with terminal Cl-vacancy on the MgCl2 support, presented stronger adsorption of TiCl4 than that of EB on (110) surface. Since the TiCl4 and ethyl benzoate (EB) would compete to adsorb on the support surface, it seems reasonable to assume that TiCl4 might predominately occupy this site, which can act as the most plausible active site for propylene polymerization. The first insertion of propylene monomer into the A5 active site model showed that it exhibited good regioselectivity but poor stereospecificity in the absence of electron donor.
Co-reporter:Shiliang Zhang, Qi Dong, Ruihua Cheng, Xuelian He, Quntao Wang, Yan Tang, Yongling Yu, Kan Xie, Jianwen Da, Boping Liu
Journal of Molecular Catalysis A: Chemical 2012 Volume 358() pp:10-22
Publication Date(Web):June 2012
DOI:10.1016/j.molcata.2012.01.021
In this work, a novel SiO2-supported hybrid chromium-based catalyst for ethylene and ethylene/1-hexene polymerization was prepared by using the residual surface hydroxyl groups in Phillips catalyst to further support bis(triphenysilyl) chromate (BC) in order to get the merits from two important chromium-based catalysts namely inorganic Phillips and organic S-2 catalysts. By characterization of the catalysts, investigation of the polymerization behavior and the microstructures of polymers, several vital factors such as cocatalyst, the addition amount of BC, calcination temperature, total chromium loading, polymerization temperature, pressure and hydrogen were systematically investigated. The proper addition weight amount of BC was CrBC ≤ 0.25 wt%. The suitable calcination temperature and total chromium loading were ≤600 °C and ≤0.5 wt%, respectively. With increasing the relative addition amount of CrBC from 0 to 20, 50, 80 and 100 wt% (corresponding from Phillips to HCat-1, HCat-2, HCat-3 and S-2 catalysts), the copolymerization abilities of the above catalysts showed a decreasing tendency. By comparing different chromium catalysts, HCat-2 catalyst showed higher activity than S-2 catalyst and its polymer had higher average molecular weight (MW) than that obtained from Phillips catalyst. HCat-2 catalyst got well-balanced properties mainly including activity, MW of polymers and copolymerization ability.Graphical abstractA novel SiO2-supported inorganic and organic hybrid chromium-based catalyst for ethylene polymerization was prepared by using the residual surface hydroxyl groups in Phillips catalyst to further support bis(triphenysilyl) chromate.Highlights► A novel SiO2-supported chromium-based catalyst for ethylene polymerization. ► Hybridizing inorganic Phillips and organic S-2 catalysts. ► Suitable calcination temperature ≤600 °C and total Cr loading ≤0.5 wt%. ► Proper addition amount of organic bis(triphenysilyl) chromate was CrBC ≤ 0.25 wt%. ► Effects of cocatalyst, temperature, pressure and hydrogen on polymerization.
Co-reporter:Lei Zhong;Zhen Liu;Ruihua Cheng;Siyang Tang;Pengyuan Qiu;Xuelian He;Minoru Terano
ChemCatChem 2012 Volume 4( Issue 6) pp:872-881
Publication Date(Web):
DOI:10.1002/cctc.201100278
Abstract
In spite of the great importance of the Phillips catalyst in commercial polyethylene production and long-term research efforts, the initiation mechanism of polymerization still remains unclear. The effect of formaldehyde desorption on the active site transformation during the induction period of the Phillips catalyst is investigated over cluster models by using DFT. No reaction can be initiated over the cluster model coordinated with two formaldehyde molecules, owing to steric hindrance and electronic donation. The first reaction over cluster models, on which either one or no formaldehyde molecule is adsorbed, follows the metallacyclic mechanism into chromacyclopentane. Subsequent dimerization to 1-butene and metathesis to propylene and ethylene are more favorable over the cluster model adsorbed with one formaldehyde molecule. Only after a complete desorption of formaldehyde does further ring expansion to chromacycloheptane followed by 1-hexene formation become preferential. Spin state crossing from quintet diethylene–CrII complex to triplet chromacyclopentane with a spin acceleration effect is revealed.
Co-reporter:Pengyuan Qiu, Ruihua Cheng, Zhen Liu, Boping Liu, Boris Tumanskii, Moris S. Eisen
Journal of Organometallic Chemistry 2012 699() pp: 48-55
Publication Date(Web):
DOI:10.1016/j.jorganchem.2011.10.033
Co-reporter:Zhen Liu, Ruihua Cheng, Xuelian He, Xiaojun Wu, and Boping Liu
The Journal of Physical Chemistry A 2012 Volume 116(Issue 28) pp:7538-7549
Publication Date(Web):June 14, 2012
DOI:10.1021/jp302529q
In this work, a two-state reaction mechanism for the acetylene cyclotrimerization over a cluster model for the Phillips Cr(II)/silica catalyst were systematically investigated using density functional theory (DFT). Since spin crossover phenomenon was confirmed in the catalytic cycle, an accurate prediction of the energy gap between low- and high-spin states is crucial for the description of a reaction involving a two-state reactivity. Therefore, a massive DFT functional benchmarking test has been conducted on the cluster model by taking a CASPT2 energy gap as a reference. Consequently, B3PW91* with 28% Hartree–Fock exchange energy was selected for the following mechanistic investigation. Each of the possible potential energy surface including singlet, triplet, and quintet surfaces was explored. On the quintet surface, the reaction begins with a coordination of an acetylene on the chromium center to generate a π-coordinated complex. The following oxidative coupling through further coordination with a second acetylene was predicted to be a two-step reaction to generate a chromacyclopentadiene species. This transformation was found to be energetically prohibitive by the presence of the transition state 5TS[C-E] (ΔG‡ = 31.1 kcal/mol). On the triplet surface, however, the coordination of an acetylene generates a chromacyclopropene species without showing any activation barrier. The second acetylene incorporation proceeding via a coordination on the chromium center followed by an insertion into a Cr–C σ-bond of the chromacyclopropene was predicted to be a facile reaction pathway (ΔG‡ = 10.2 kcal/mol). The third acetylene was captured by the cluster model through the formation of a hydrogen bond. The later transformation on the triplet surface was found to be an intermolecular [4 + 2] cycloaddition to finish the cyclization. The lack of the aromaticity of the benzene ring in 3L results in an uncompleted reaction pathway on a single triplet surface. Consequently, a two-state reaction pathway that is connected by two low-lying minimum-energy crossing points (MECPs) of the two surfaces is thus described. It is worthy of note that the third acetylene in the tri(acetylene)chromium complex captured by the cluster model only through the formation of a hydrogen bond rules out the [2 + 2 + 2] concerted one-step reaction pathway proposed by Zecchina et al. [Phys. Chem. Chem. Phys.2003, 5, 4414]. The singlet reaction profile is far higher in energy compared with that proceeded on the triplet and quintet surfaces.
Co-reporter:Shiliang Zhang;Ning Zhao;Yuxiang Wu;Qi Dong;Quntao Wang;Yan Tang;Yongling Yu;Jianwen Da;Xuelian He;Ruihua Cheng
Macromolecular Symposia 2012 Volume 312( Issue 1) pp:63-71
Publication Date(Web):
DOI:10.1002/masy.201100018
Abstract
Summary: Short chain branches distribution (SCBD) is the key factor for high density polyethylene (HDPE) pipe materials to achieve their excellent performance for long term (50 years) applications. However, the precise SCBD characterization of these HDPE materials with relatively low content of comonomer incorporation still remained as a challenge in this field. In this work, two characterization methods, namely temperature rising elution fractionation (TREF) cross step crystallization (SC) (TREF + SC) and TREF cross 13C-NMR (TREF + 13C-NMR), have been respectively used to qualitatively and quantitatively investigate the SCBD for two HDPE pipe materials (PE-1 and PE-2 with different long term performances) with small amount of 1-hexene incorporation prepared from SiO2-supported silyl chromate catalyst system (S-2 catalyst) during UNIPOL gas phase polymerization. The comparison of SCBD between the two samples showed that: although short chain branches of PE-2 with good performance were less than those of PE-1 with bad performance, PE-2 showed less comonomer incorporation on the low crystallinity and low molecular weight (MW) fractions keeping even higher comonomer incorporation on the high crystallinity and high MW parts compared with PE-1. This difference on the SCBD for PE-1 and PE-2 was thought to be the key factor which is responsible for their great difference on environment slow crack resistance (ESCR). Moreover, TREF + SC method further reflected the intra- and inter-molecular heterogeneities of each fraction from the two HDPE samples through the lamella thickness distribution compared with TREF + 13C-NMR.
Co-reporter:Yun Yang, Zhen Liu, Lei Zhong, Pengyuan Qiu, Qi Dong, Ruihua Cheng, Jeffrey Vanderbilt, and Boping Liu
Organometallics 2011 Volume 30(Issue 19) pp:5297-5302
Publication Date(Web):September 7, 2011
DOI:10.1021/om200722r
Experimentally unsolved problems, including the oxidation states of active species and the occurrence of ligand deprotonation in the SNS-Cr ethylene trimerization system, were studied using the density functional theory (DFT) method. The full catalytic cycle was calculated on the basis of the metallacycle mechanism, and Gibbs free energy surfaces of the trimerization reaction were completely located. A detailed spin state analysis revealed that the ground states of intermediates change along the redox cycle and the spin surface crossing occurring at the minimum energy crossing point (MECP) before metallacyclopentane formation was found to open up a much lower energy pathway by spin acceleration. Formation of metallacycloheptane was identified as the rate-determining step in this system. By comparison of the activation energies of the rate-determining step, Cr(I)/Cr(III) active species bearing nondeprotonated ligands were proposed to be most plausibly responsible for ethylene trimerization. Frontier orbitals and natural population analysis were also determined to further elucidate the reason for high 1-hexene selectivity in this system.
Co-reporter:Liuzhong Li;Aiyou Hao;Ruihua Cheng
Frontiers of Chemical Science and Engineering 2011 Volume 5( Issue 1) pp:89-95
Publication Date(Web):2011 March
DOI:10.1007/s11705-010-0558-x
Although an important industrial catalyst for producing high density polyethylene, the SiO2-supported organosilyl chromate UCC S-2 catalyst has not been fully investigated compared with the SiO2-supported oxo chromium Phillips catalyst. In this work, gas phase ethylene polymerization by S-2 catalysts (Cat-1, Cat-2, and Cat-3) was carried out in a high-speed stirredautoclave reactor. The effects of temperature, time, and pressure on kinetics, activity, and product properties were studied. All kinetics were typical built-up types with slow decay. Compared to the simple physical mixtures of Cat-1 and Cat-2, Cat-3 showed higher activity and its product had a broader molecular weight distribution, indicating new active species induced during blending of Cat-1 and Cat-2 in n-hexane. The innovation by a simple catalyst technology created a new application to meet market demands.
Co-reporter:Zhen Liu, Lei Zhong, Yun Yang, Ruihua Cheng, and Boping Liu
The Journal of Physical Chemistry A 2011 Volume 115(Issue 28) pp:8131-8141
Publication Date(Web):June 7, 2011
DOI:10.1021/jp111108p
In this work, the ethylene coordination and dimerization mechanism over Cr(II)OH+ cation were systematically investigated using density functional theory (DFT) and complete active space second-order perturbation theory (CASPT2). It was found that Cr(II)OH+ cation can coordinate with up to four ethylene molecules which gives seven possible stable Cr(II)OH+·(C2H4)n (n = 1–4) π-complexes. We investigated whether ethylene dimerization over Cr(II)OH+ cation proceeds through either a carbene mechanism or a metallacycle mechanism. The potential energy surfaces were characterized using four different functionals (M06L, BLYP, B3LYP, and M06). It was found that the potential energy profiles calculated at the M06 level agreed well with the CASPT2 energy profiles. Since the intermediates involved in the proposed catalytic cycles showed different ground spin states, a reaction pathway involving a spin crossing between two potential energy surfaces was observed. The minimum-energy crossing points (MECPs) that connect the two potential energy surfaces were successfully located. The two-state metallacycle reaction pathway with the formation of chromacyclopentane as the rate-determining step was found to be energetically more favorable than the carbene reaction pathway. 1-Butene was formed from the chromacyclopentane by a two-step reductive elimination pathway through a chromium(IV) hydride intermediate.
Co-reporter:Pengyuan Qiu, Ruihua Cheng, Boping Liu, Boris Tumanskii, Rami J. Batrice, Mark Botoshansky, and Moris S. Eisen
Organometallics 2011 Volume 30(Issue 8) pp:2144-2148
Publication Date(Web):March 18, 2011
DOI:10.1021/om101008j
A novel dinuclear oxygen-bridged chromium(II) complex of [(Ph3SiO)Cr·(THF)]2(μ-OSiPh3)2 (1) was successfully synthesized and structurally characterized. The crystal structure of complex 1 shows that the chromium centers adopt a tetrahedrally distorted square-planar coordination geometry. Complex 1 was tested for ethylene polymerization upon activation with and without aluminum alkyl cocatalysts. No ethylene polymerization activity was observed in the absence of aluminum alkyl cocatalysts; however, in the presence of methylaluminoxane (MAO), complex 1 demonstrates an unexpected transformation from ethylene polymerization to ethylene oligomerization, with the molar ratio of Al to Cr increasing from 50 to 1000. The activation of complex 1 with MAO was investigated by ESR and 29Si NMR spectroscopy. It was found that the Ph3SiO− groups could be transferred from the chromium centers of complex 1 to the aluminum of MAO.
Co-reporter:Xiaochun Cao, Ruihua Cheng, Zhen Liu, Lisong Wang, Qi Dong, Xuelian He, Boping Liu
Journal of Molecular Catalysis A: Chemical 2010 321(1–2) pp: 50-60
Publication Date(Web):
DOI:10.1016/j.molcata.2010.01.018
Co-reporter:Xiaofang Li, Ruihua Cheng, Jun Luo, Qi Dong, Xuelian He, Liuzhong Li, Yongling Yu, Jianwen Da, Boping Liu
Journal of Molecular Catalysis A: Chemical 2010 330(1–2) pp: 56-65
Publication Date(Web):
DOI:10.1016/j.molcata.2010.07.002
Co-reporter:Yuan Qi, Qi Dong, Lei Zhong, Zhen Liu, Pengyuan Qiu, Ruihua Cheng, Xuelian He, Jeffrey Vanderbilt and Boping Liu
Organometallics 2010 Volume 29(Issue 7) pp:1588-1602
Publication Date(Web):March 2, 2010
DOI:10.1021/om900917k
Selective ethylene trimerization into 1-hexene, which is an important comonomer for production of copolymers with high performance, was first commercialized in 2003 using a chromium tris(2-ethylhexanoate) (Cr(EH)3)-based catalyst. One milestone during the development of this catalyst is the addition of 1,2-dimethoxyethane (DME) to Cr(EH)3/partially hydrolyzed tri-isobutyl aluminum (PIBAO) catalyst, resulting in a transformation from ethylene polymerization into trimerization. However, the mechanism of this transformation is unknown. In this work, the role of DME in the switching mechanism from ethylene polymerization to trimerization was investigated by a density functional theory (DFT) method based on the 10 most plausible molecular models for the active species of Cr(EH)3/PIBAO catalyst with or without DME coordination. For neutral models (Cr(I)OR; Cr(I)OR/DME; Cr(I)R; Cr(I)R/DME; R refers to isobutyl ligand), those without DME coordination were found to be dominantly ethylene trimerization sites. After DME coordination, both the weak electron-donating effect and steric effect of DME increased the total energy barriers of metallocycle growth, leading to ethylene trimerization with dimerization as a side reaction. So neutral models mainly presented ethylene trimerization with or without DME coordination and were not consistent with experimental evidence of transformation. For cationic models (Cr(I)+; Cr(I)+/DME; Cr(II)+OR; Cr(II)+OR/DME; Cr(II)+R; Cr(II)+R/DME), metallocycle growth leading to ethylene polymerization occurred on those models without DME. After DME coordination, the strong electron-donating effect of DME was found to facilitate metallocycle expansion, while the dominant role of the steric effect of DME made nine-membered ring formation unfavorable, and thus 1-hexene was liberated from the seven-membered ring. So all the cationic models presented a transformation from ethylene polymerization to trimerization after DME coordination and might be the most plausible active sites for the Cr(EH)3/PIBAO catalyst system. The results provided a much deeper understanding of the highly selective trimerization mechanism and for further development of new catalysts with high performance as well.
Co-reporter:Boping Liu;Ruihua Cheng;Zhen Liu;Pengyuan Qiu;Shiliang Zhang;Toshiaki Taniike;Kunihiko Tashino;Takashi Fujita;Minoru Terano
Macromolecular Symposia 2007 Volume 260(Issue 1) pp:42-48
Publication Date(Web):7 JAN 2008
DOI:10.1002/masy.200751407
In this work, a combination of experimental and computational approaches on the isospecific role of monoester-type internal electron donors (ED) such as phenylpropionate (PhP), ethylheptanoate (EH), methylbenzoate (MB), ethylbenzoate (EB) for TiCl4/ED/MgCl2 Ziegler-Natta catalysts had been performed. The propylene polymerization results revealed that the isospecificity of catalysts increases in the following order: PhP < EH < MB < EB. The subsequent molecular modeling on the electronic properties of the donors and two kinds of cluster model catalysts: TiCl4/ED/MgCl2 and TiCl4/ED/(MgCl2)4 based on density functional theory (DFT) method was carried out. Two kinds of ED coordination on MgCl2 clusters through either O or O within the monoester-type ED had been disclosed. A perfect correlation between the dipole moment of ED, the coordination bond length of O … Mg, the competitive coordination from O with Mg ion and the isospecificity of the catalysts had been established.
Co-reporter:Boping Liu;Ruihua Cheng;Zhen Liu;Pengyuan Qiu;Shiliang Zhang;Toshiaki Taniike;Minoru Terano;Kunihiko Tashino;Takashi Fujita
Macromolecular Symposia 2007 Volume 260(Issue 1) pp:C1-C7
Publication Date(Web):27 FEB 2008
DOI:10.1002/masy.200751429
In this work, a combination of experimental and computational approaches on the isospecific role of monoester-type internal electron donors (ED) such as phenylpropionate (PhP), ethylheptanoate (EH), methylbenzoate (MB), ethylbenzoate (EB) for TiCl4/ED/MgCl2 Ziegler-Natta catalysts had been performed. The propylene polymerization results revealed that the isospecificity of catalysts increases in the following order: PhP < EH < MB < EB. The subsequent molecular modeling on the electronic properties of the donors and two kinds of cluster model catalysts: TiCl4/ED/MgCl2 and TiCl4/ED/(MgCl2)4 based on density functional theory (DFT) method was carried out. Two kinds of ED coordination on MgCl2 clusters through either O or O within the monoester-type ED had been disclosed. A perfect correlation between the dipole moment of ED, the coordination bond length of O … Mg, the competitive coordination from O with Mg ion and the isospecificity of the catalysts had been established.
Co-reporter:Xing Pan, Zhen Liu, Ruihua Cheng, Yun Yang, Lei Zhong, Xuelian He, Boping Liu
Journal of CO2 Utilization (September 2013) Volume 2() pp:39-48
Publication Date(Web):1 September 2013
DOI:10.1016/j.jcou.2013.07.004
•Alternating copolymerization mechanism of CO2 with propylene oxide (PO) studied by DFT.•Six most plausible molecular models of the diethylzinc–water catalyst system.•Condensed species with repeated Zn–O group as the most possible active site.•Polymerization initiated by PO insertion into ZnOR bond followed by CO2 insertion.•Non-condensed species with single Zn–O group leading to cyclic propylene carbonate.The mechanism of alternating copolymerization of CO2 with propylene oxide (PO) catalyzed by the diethylzinc–water system has been studied by means of density functional theory (DFT) calculations. Six most plausible molecular models for the active species of the diethylzinc–water systems (C2H5ZnOH, C2H5(ZnO)2H, C2H5(ZnO)3H, C2H5(ZnO)4H, HOZnOH, C2H5ZnOC2H5) have been constructed. Possible reaction pathways and their corresponding reaction energy barriers have been investigated. It is found that the reaction follows monometallic mechanism and is initiated by PO insertion into the ZnOR bond of the catalyst. The ring-opening of PO is inclined to occur via the methineoxygen bond (CCHO) cleavage. The rate-determining step is the second molecular PO insertion into Zn–carbonate group. CO2 insertion proceeds easily and the reverse reaction of CO2 insertion is also easy to occur. It is likely that the most possible active species is the condensed species with repeated Zn–O group, which shows high Gibbs free energy barrier to the formation of cyclic propylene carbonate. The consecutive PO insertion to give polyether and consecutive CO2 insertion to give dicarbonate linkages have been found to be disfavored. The non-condensed species could be excluded due to its easy formation of cyclic propylene carbonate.The mechanism for alternating copolymerization of CO2 and propylene oxide in diethylzinc–water catalytic system was studied using DFT method by comparing three competing reaction pathways: the formation of polyether, the growth of linear polycarbonate and the formation of cyclic propylene carbonate.Download full-size image
Co-reporter:Qingyang Meng, Ruihua Cheng, Jiajia Li, Tingting Wang, Boping Liu
Journal of CO2 Utilization (December 2016) Volume 16() pp:86-96
Publication Date(Web):1 December 2016
DOI:10.1016/j.jcou.2016.06.011
•Novel ZnGA is achieved with small particle size and increased surface area.•ZnGA/DMC composite catalyst system presents synergistic effects on CO2/PO copolymerization.•ZnGA/DMC composite catalyst shows high catalytic efficiency and selectivity to polycarbonates.•The synthesized copolymer PPC has fairly high molecular weight (Mw = 3.9 × 105 g/mol).The high yield and high molecular weight poly(propylene carbonate) (PPC) was synthesized via copolymerization of propylene oxide (PO) and carbon dioxide (CO2) catalyzed over zinc glutarate/double metal cyanide (ZnGA/DMC) composite catalyst. A fine crystalline ZnGA component was prepared using the crosslinked restrain effect of nonionic surfactant. Combined with a small amount of DMC, the ZnGA/DMC (molar ratio = 10:1) composite catalyst system showed an excellent synergistic effect on CO2/PO copolymerization with higher activity, selectivity, and shorter reaction time than those of the traditional ZnGA catalyst. Under the optimized reaction conditions, the molecular weight of PPC was up to 3.8 × 105 g/mol at the highest yield of 508.0 gpolym/gcat, and the selectivity was over 97.7% towards polycarbonates rather than polyether linkages. The alternating copolymer PPC exhibited good thermostability, high glass transition temperature (42.0 °C) and high decomposition temperature (5% weight loss at 253.4 °C). A synergistic mechanism of ZnGA/DMC composite catalyst was speculated.Download high-res image (155KB)Download full-size image
Co-reporter:Siyang Tang, Zhen Liu, Xiaowei Yan, Ning Li, Ruihua Cheng, Xuelian He, Boping Liu
Applied Catalysis A: General (5 July 2014) Volume 481() pp:39-48
Publication Date(Web):5 July 2014
DOI:10.1016/j.apcata.2014.04.006
Co-reporter:Ruihua Cheng, Chen Xu, Zhen Liu, Qi Dong, Xuelian He, Yuwei Fang, Minoru Terano, Yatao Hu, Thomas J. Pullukat, Boping Liu
Journal of Catalysis (28 July 2010) Volume 273(Issue 2) pp:103-115
Publication Date(Web):28 July 2010
DOI:10.1016/j.jcat.2010.05.002
Ti-modified Phillips catalyst is a most important industrial catalyst widely used in ethylene polymerization, but the mechanism still remains mysterious. In this work, Ti-modified Phillips catalysts were characterized by high-resolution X-ray photoelectron spectroscopy (XPS) and 1H magic-angle-spin solid-state nuclear magnetic resonance (1H MAS solid-state NMR) combined with density functional investigations into the effects of Ti-modification on promotion of polymerization activity and regulation of microstructures of the polymer chains. XPS data revealed Ti-modification caused increase in electron-deficiency and photo-stability of the surface chromate species. 1H NMR provided the first direct evidence of surface residual Ti–OH groups. Modeling results rationalized well the effects of Ti-modification on promotion of polymerization activity, extension of molecular weight distribution (MWD) to lower MW region, improvement of the distribution of inserted co-monomer and enhancement of 2,1-insertion in regioselectivity of Phillips catalysts. It was the first time the Ti-modification on Phillips catalysts were theoretically elucidated.DFT investigations combined with high-resolution XPS and 1H MAS solid-state NMR characterization were carried out to elucidate the mechanism of Ti-modification on Phillips catalyst for ethylene polymerization.Download high-res image (85KB)Download full-size image







![Poly[oxycarbonyloxy(phenyl-1,2-ethanediyl)]](http://img.cochemist.com/ccimg/70700/70694-67-6.png)
![Poly[oxycarbonyloxy(phenyl-1,2-ethanediyl)]](http://img.cochemist.com/ccimg/70700/70694-67-6_b.png)


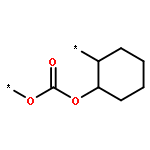











![Poly[oxycarbonyloxy(methyl-1,2-ethanediyl)]](http://img.cochemist.com/ccimg/37300/37248-85-4.png)
![Poly[oxycarbonyloxy(methyl-1,2-ethanediyl)]](http://img.cochemist.com/ccimg/37300/37248-85-4_b.png)