Co-reporter:Andreas Schäfer, Sebastian C. Köhler, Markus Lohe, Michael Wiese, and Martin Hiersemann
The Journal of Organic Chemistry October 6, 2017 Volume 82(Issue 19) pp:10504-10504
Publication Date(Web):September 26, 2017
DOI:10.1021/acs.joc.7b02012
The synthesis of the A–B-cis,B–C-trans-annulated cyclohepta[e]hydrindane core of a gagunin E analogue is reported in detail. The tricarbocyclic scaffold was assembled starting from an easily accessible A ring building block by a (4 + 2)-cycloaddition for annulation of the B ring. A ring-closing metathesis served for construction of the seven-membered C ring. The angular methyl groups were attached by electrophilic cyclopropanation–ring opening. A library based on the most active lead compound was made accessible by esterification of the terpenols with commercially available acids. A transannular etherification reaction gave access to tetracyclic derivatives of the synthetic inhibitors. The members of the compound library of non-natural homoverrucosanoid-derived esters were examined as modulators of the membrane transporter proteins ABCB1 (P-gp), ABCG2 (BCRP), and ABCC1 (MRP1), which are involved in the formation of multidrug resistance (MDR) in cancer chemotherapy.
Co-reporter:Michael K. Krapf, Jennifer Gallus, and Michael Wiese
Journal of Medicinal Chemistry May 25, 2017 Volume 60(Issue 10) pp:4474-4474
Publication Date(Web):May 4, 2017
DOI:10.1021/acs.jmedchem.7b00441
Multidrug resistance (MDR) mediated by ATP-binding cassette (ABC) transport proteins remains a major problem in the chemotherapeutic treatment of cancer and might be overcome by inhibition of the transporter. Because of the lack of understanding, the complex mechanisms involved in the transport process, in particular for breast cancer resistance protein (BCRP/ABCG2), there is a persistent need for studies of inhibitors of ABCG2. In this study, we investigated a systematic series of 4-substituted-2-pyridylquinazolines in terms of their inhibitory potency as well as selectivity toward ABCG2. For comparison, the quinazoline scaffold was reduced to the significantly smaller 4-methylpyrimidine basic structure. Furthermore, the cytotoxicity and the ability to reverse MDR was tested with the chemotherapeutic agents SN-38 and mitoxantrone (MX). Interaction of the compounds with ABCG2 was investigated by a colorimetric ATPase assay. Enzyme kinetic studies were carried out with Hoechst 33342 as fluorescent dye and substrate of ABCG2 to elucidate the compounds binding modes.
Co-reporter:Michael K. Krapf, Jennifer Gallus, Michael Wiese
European Journal of Medicinal Chemistry 2017 Volume 139(Volume 139) pp:
Publication Date(Web):20 October 2017
DOI:10.1016/j.ejmech.2017.08.020
•Quinazolines substituted with a combination of 3,4-dimethoxy and nitro groups are highly potent ABCG2 inhibitors.•Interchangeability between pos. 2 and 4 is proposed for the compounds.•EC50 describing reversal of resistance toward SN-38 and MX is below 20 nM for most potent compounds.•Multiple binding sites are suggested according to interaction with Hoechst 33342 and ATPase results.Expression of ABCG2, a member of the ABC transporter superfamily, has been correlated to the clinical outcome of multiple cancers and is often associated with the occurrence of multidrug resistance (MDR) in chemotherapy. Inhibition of the transport protein by potent and selective inhibitors might be a way to treat cancer more efficiently and improve the therapy of cancer patients.Recently we reported the synthesis of new inhibitors based on a quinazoline scaffold. In the present study more structural variations were explored. Compounds with 3,4-dimethoxy groups and meta or para nitro substituents were found to be highly potent inhibitors of ABCG2. The most potent compound was more than five-fold more potent than Ko143, one of the best inhibitors of ABCG2. To determine the new compounds selectivity toward ABCG2 their inhibitory effects on ABCB1 and ABCC1 were also investigated identifying selective as well as broadspectrum inhibitors. Furthermore, intrinsic cytotoxicity and efficacy regarding the reversal of multidrug resistance toward SN-38 and mitoxantrone were explored. The most potent compounds were able to reverse the resistance toward the cytostatic agents with EC50 values below 20 nM. Additionally, the type of interaction between inhibitors and the ABCG2 substrate Hoechst 33342 was investigated yielding competitive and non-competitive interactions suggesting different modes of binding. Finally the effect of the derivatives on vanadate-sensitive ATPase activity of ABCG2 was determined. According to the different effects on ATPase activity we conclude the existence of different binding sites. This study provides the structural requirements for high potency inhibition and elucidates the interaction with ABCG2 setting the basis for further studies.Download high-res image (264KB)Download full-size image
Co-reporter:Sven Marcel Schmitt; Katja Stefan
Journal of Medicinal Chemistry 2016 Volume 59(Issue 7) pp:3018-3033
Publication Date(Web):March 4, 2016
DOI:10.1021/acs.jmedchem.5b01644
Five series of pyrrolo[3,2-d]pyrimidines were synthesized and evaluated with respect to potency and selectivity toward multidrug resistance-associated protein 1 (MRP1, ABCC1). This transport protein is a major target to overcome multidrug resistance in cancer patients. We investigated differently substituted pyrrolopyrimidines using the doxorubicin selected and MRP1 overexpressing small cell lung cancer cell line H69 AR in a calcein AM and daunorubicin cell accumulation assay. New compounds with high potency and selectivity were identified. Piperazine residues at position 4 bearing large phenylalkyl side chains proved to be beneficial for MRP1 inhibition. Its replacement by an amino group led to decreased activity. Aliphatic and aliphatic–aromatic variations at position 5 and 6 revealed compounds with IC50 values in high nanomolar range. All investigated compounds had low affinity toward P-glycoprotein (P-gp, ABCB1). Pyrrolopyrimidines with small substituents showed moderate inhibition against breast cancer resistance protein (BCRP, ABCG2).
Co-reporter:Michael K. Krapf
Journal of Medicinal Chemistry 2016 Volume 59(Issue 11) pp:5449-5461
Publication Date(Web):May 5, 2016
DOI:10.1021/acs.jmedchem.6b00330
Chemotherapeutic treatment of cancer often fails due to overexpression of the ATP-binding cassette (ABC) transport proteins, like ABCG2, triggering active efflux of various structurally unrelated drugs. This so-called multidrug resistance (MDR) may be reversed by selective, potent, and nontoxic inhibitors of ABCG2. As only a few potent inhibitors are known, new compounds based on a 4-substituted-2-phenylquinazoline scaffold were investigated. Substitution with hydroxy, cyano, nitro, acetamido, and fluoro led to high inhibitory activities toward ABCG2. The ability to reverse MDR of the most active compounds was confirmed in a MTT efficacy assay. Moreover, a negligibly low intrinsic cytotoxicity was found resulting in a high therapeutic ratio. Investigations of the inhibitory activity toward ABCB1 and ABCC1 yielded a high selectivity toward ABCG2 for the quinazoline compounds. Quinoline-based analogues showed lower inhibitory activity and selectivity. The study yielded a variety of promising compounds, some with superior properties compared to those of the standard inhibitor Ko143.
Co-reporter:Anna Spindler; Katja Stefan
Journal of Medicinal Chemistry 2016 Volume 59(Issue 13) pp:6121-6135
Publication Date(Web):June 9, 2016
DOI:10.1021/acs.jmedchem.6b00035
The breast cancer resistance protein (ABCG2) transports chemotherapeutic drugs out of cells, which makes it a major player in mediating multidrug resistance (MDR) of cancer cells. To overcome this mechanism, inhibitors of ABCG2 can be used. Only a few potent and selective ABCG2 inhibitors have been discovered, i.e., fumitremorgin C (FTC), Ko143, and the alkaloid harmine, which contain a tetrahydro-β-carboline or β-carboline backbone, respectively. However, toxicity and or instability prevent their use in vivo. Therefore, there is a need for further potent inhibitors. We synthesized and pharmacologically investigated 37 tetrahydro-β-carboline derivatives. The inhibitory activity of two compounds (51, 52) is comparable to that of Ko143, and they are selective for ABCG2 over ABCB1. Furthermore, they are able to reverse the ABCG2-mediated resistance toward SN-38 and inhibit the ATPase activity. The cytotoxicity data show that their inhibitory effect is substantially higher than their toxicity.
Co-reporter:Veronika F.S. Pape, Szilárd Tóth, András Füredi, Kornélia Szebényi, Anna Lovrics, Pál Szabó, Michael Wiese, Gergely Szakács
European Journal of Medicinal Chemistry 2016 Volume 117() pp:335-354
Publication Date(Web):19 July 2016
DOI:10.1016/j.ejmech.2016.03.078
•A focused library of ONS, NNS and NNN donor chelators was designed and synthesized.•Thiosemicarbazones, arylhydrazones and hydrazinobenzothiazoles were investigated.•In vitro antiproliferative activity was tested in sensitive and MDR cancer cells.•Molecular features influencing the toxicity of anticancer chelators were identified.There is a constant need for new therapies against multidrug resistant (MDR) cancer. An attractive strategy is to develop chelators that display significant antitumor activity in multidrug resistant cancer cell lines overexpressing the drug efflux pump P-glycoprotein. In this study we used a panel of sensitive and MDR cancer cell lines to evaluate the toxicity of picolinylidene and salicylidene thiosemicarbazone, arylhydrazone, as well as picolinylidene and salicylidene hydrazino-benzothiazole derivatives. Our results confirm the collateral sensitivity of MDR cells to isatin-β-thiosemicarbazones, and identify several chelator scaffolds with a potential to overcome multidrug resistance. Analysis of structure-activity-relationships within the investigated compound library indicates that NNS and NNN donor chelators show superior toxicity as compared to ONS derivatives regardless of the resistance status of the cells.
Co-reporter:Sebastian C. Köhler, Katja Silbermann, Michael Wiese
European Journal of Medicinal Chemistry 2016 Volume 124() pp:881-895
Publication Date(Web):29 November 2016
DOI:10.1016/j.ejmech.2016.09.010
•Determination of the influence of substituents on the scaffold of a novel ABCG2 inhibitor class•SAR shows negative impact of electron withdrawing and bulky groups; positive impact of electron donating groups•Compound 32 discovered as highly potent and selective ABCG2 modulator with IC50 values threefold lower than Ko143•Increased activity toward ABCB1 not only by methoxy groups but also by methylthio and hydroxy substitution•Competitive inhibition behavior with Hoechst 33342We recently presented a novel class of ABCG2 modulators based on the third-generation ABCB1 inhibitor tariquidar bearing a 2,5-linked tetrazole instead of an amid linker. We investigated the modulating potential of the compound class by enlarging the substitution pattern on the outer phenyl rings of the scaffold. To identify the structural conditions for achieving a high response, we decided to determine the individual influence of substituents on the scaffold using monosubstituted derivatives. While electron withdrawing groups (with a few exceptions) and bulky moieties decreased the modulating potency, small electron donating groups ensured a high activity level. Interestingly, the unsubstituted derivative 32 reached a similar inhibitory potential as the best derivatives in the previous study. Enzyme kinetic assays indicated that our derivatives have the same binding site as reference inhibitor Ko143. They were found to interact competitively and non-competitively with the substrates Hoechst 33342 and pheophorbide A, respectively.
Co-reporter:Stefanie Kraege, Katja Stefan, Kapil Juvale, Thomas Ross, Thomas Willmes, Michael Wiese
European Journal of Medicinal Chemistry 2016 117() pp: 212-229
Publication Date(Web):19 July 2016
DOI:10.1016/j.ejmech.2016.03.067
•Combination of quinazoline and chalcone scaffolds to heterodimeric inhibitors.•Quinazoline-chalcones are potent inhibitors of ABCG2.•Most potent compound was found to be selective, non-toxic and able to reverse MDR.•Stimulate ATPase activity without being transported.•Three compounds show dual inhibitory behavior.During the last decade it has been found that chalcones and quinazolines are promising inhibitors of ABCG2. The combination of these two scaffolds offers a new class of heterocyclic compounds with potentially high inhibitory activity against ABCG2. For this purpose we investigated 22 different heterodimeric derivatives. In this series only methoxy groups were used as substituents as these had been proven superior for inhibitory activity of chalcones. All compounds were tested for their inhibitory activity, specificity and cytotoxicity. The most potent ABCG2 inhibitor in this series showed an IC50 value of 0.19 μM. It possesses low cytotoxicity (GI50 = 93 μM), the ability to reverse MDR and is nearly selective toward ABCG2. Most compounds containing dimethoxy groups showed slight activity against ABCB1 too. Among these three compounds (17, 19 and 24) showed even higher activity toward ABCB1 than ABCG2. All inhibitors were further screened for their effect on basal ATPase activity. Although the basal ATPase activity was partially stimulated, the compounds were not transported by ABCG2. Thus, quinazoline-chalcones are a new class of effective ABCG2 inhibitors.
Co-reporter:Sebastian C. Köhler
Journal of Medicinal Chemistry 2015 Volume 58(Issue 9) pp:3910-3921
Publication Date(Web):April 9, 2015
DOI:10.1021/acs.jmedchem.5b00188
The breast cancer resistance protein (BCRP, ABCG2) belongs to the superfamily of ATP binding-cassette (ABC) proteins. In addition to other physiological functions, it transports potentially cell-damaging compounds out of the cell using the energy from ATP hydrolysis. Certain tumors overexpressing BCRP were found to become resistant against various anticancer drugs. In previous work, we found that tariquidar analogues lacking the tetrahydroisoquinoline moiety selectively inhibit BCRP. In the present study, we synthesized 21 derivatives of the third-generation P-gp inhibitor HM30181, which is structurally related to tariquidar. The compounds were tested for their inhibitory activities against BCRP and screened against P-glycoprotein (P-gp, ABCB1) and multidrug resistance protein 1 (MRP1, ABCC1) to confirm the selectivity toward BCRP. The most potent compounds are selective toward BCRP and 2-fold more potent than the reference Ko143. Qualitative structure–activity relationship (SAR) analysis revealed that the presence of a methoxy group in the ortho or para position of at least one phenyl ring is beneficial for inhibitory activity. Furthermore, the cytotoxicity and multidrug resistance (MDR)-reversal ability of selected compounds were investigated. It was shown that they have a low cytotoxicity and the ability to reverse the BCRP-mediated SN-38 resistance.
Co-reporter:Federico Marighetti;Dr. Kerstin Steggemann;Maria Karbaum ;Dr. Michael Wiese
ChemMedChem 2015 Volume 10( Issue 4) pp:742-751
Publication Date(Web):
DOI:10.1002/cmdc.201402498
Abstract
We recently reported the synthesis and quantitative structure–activity relationships of a new breast cancer resistance protein (BCRP) inhibitor class. In the study presented herein, we investigated the possibility to better define the scaffold of this compound class by removing or modifying the aromatic ring A with various substituents selected on the basis of their electronic and lipophilic properties. The results show that this aromatic ring is important, but not essential, for activity. Many of the selected substituents led to compounds with low activity, but in some cases activity was retained. Among these, a phenolic hydroxy group proved to impart as much potency to the molecule as a hydroxyethyl side chain, initially considered necessary for activity. This derivative is one of the most active compounds in this class, maintaining an inhibitory activity similar to that of the reference compound; it is also selective for BCRP.
Co-reporter:Kapil Juvale, Jennifer Gallus, Michael Wiese
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 24) pp:7858-7873
Publication Date(Web):15 December 2013
DOI:10.1016/j.bmc.2013.10.007
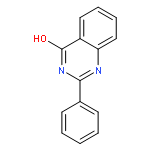
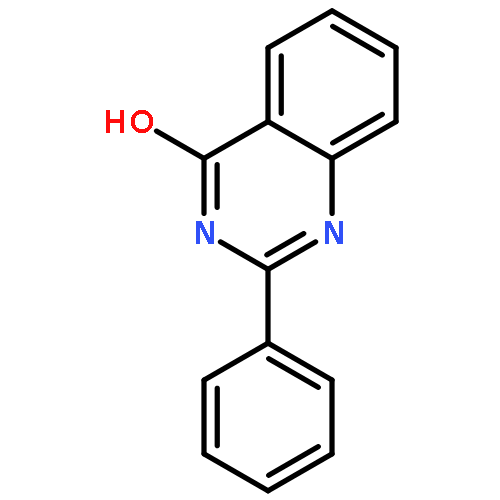
Chemotherapy is one of the major forms of cancer treatment. Unfortunately, tumors are prone to multidrug resistance leading to failure of treatment. Breast cancer resistance protein (BCRP), the second member of ABC transporter subfamily G, has been found to play a major role in drug efflux and hence multidrug resistance. Until now, very few potent and selective BCRP inhibitors like Ko143 have been identified. In the search for more potent and selective BCRP inhibitors, we synthesized and investigated a series of differently substituted quinazoline compounds. Several variations at positions 2, 4, 6 and 7 of the quinazoline scaffold were carried out to develop a structure–activity-relationship analysis for these compounds. It was found that compounds bearing a phenyl substituent at position 2 of the 4-anilinoquinazoline scaffold were most potent. On the aniline ring at position 4 of the quinazoline moiety substituents like NO2, CN, CF3 led to very high BCRP inhibition potencies. The most potent compounds were further investigated for their intrinsic cytotoxicity and their ability to reverse the multidrug resistance. Compound 20, an anilinoquinazoline bearing a phenyl ring at position 2 and meta-nitro substitution on the 4-anilino ring, was found to have the highest therapeutic ratio. The most active compounds from each variation were also investigated for their effect on BCRP expression. It was found that compound 20 has no significant effect on BCRP expression, while compound 31 decreased the surface BCRP expression. The only difference in the two compounds was the presence of a 3,4-dimethoxyphenyl ring in compound 31 instead of phenyl substitution at position 2 of the quinazoline moiety. From the study of all target compounds, compound 20 was the most prominent compound having inhibitory potency even higher than Ko143, the most potent BCRP inhibitor known. Compound 20 was also found to be selective towards BCRP with a very high therapeutic ratio.
Co-reporter:Michael Wiese;Martin Koch
Journal of Cancer Research and Clinical Oncology 2013 Volume 139( Issue 2) pp:259-267
Publication Date(Web):2013/02/01
DOI:10.1007/s00432-012-1317-9
To assign functional properties to gene expression profiles of cervical cancer stages and identify clinically relevant biomarker genes.Microarray samples of 24 normal and 102 cervical cancer biopsies from four publicly available studies were pooled and evaluated. High-quality microarrays were normalized using the CONOR package from the Bioconductor project. Gene expression profiling was performed using variance-component analysis for accessing most reliable probes, which were subsequently processed by gene set enrichment analysis.Of 22.277 probes that were subject to variance-component analysis, eleven probes had low heterogeneity, that is, a W/T ratio between 0.18 and 0.38. Seven of these probes are induced in all cervical cancer stages: they are GINS1, PAK2, DTL, AURKA, PRKDC, NEK2 and CEP55. The other four probes are induced in normal cervix: P11, EMP1, UPK1A and HSPC159. We performed GSEA of 9.873 probes exhibiting less variability, that is, having a W/T ratio of <0.75. Repeatedly, significant gene expression signatures were found that are related to treatment using angiocidin and darapladib. Additionally, expression signatures from immunological disease signatures were found, for example graft versus host disease and acute kidney rejection. Another finding comprises a gene expression signature in stage IB2 that refers to MT1-MMP-dependent migration and invasion. This gene signature is accompanied by gene expression signatures which refer to ECM receptor-mediated interactions.Analysis of cervical cancer patient gene expression data reveals a novel perspective on HPV-mediated transcription processes. This novel point of view contains a better understanding and even might provide improvements to cancer therapy.
Co-reporter:Dr. Ilza K. Pajeva;Dr. Katja Sterz;Matthias Christlieb;Dr. Kerstin Steggemann;Federico Marighetti;Dr. Michael Wiese
ChemMedChem 2013 Volume 8( Issue 10) pp:1701-1713
Publication Date(Web):
DOI:10.1002/cmdc.201300233
Abstract
Tariquidar and elacridar are among the most potent inhibitors of the multidrug resistance transporter P-glycoprotein (P-gp), but how they interact with the protein is yet unknown. In this work, we describe a possible way in which these inhibitors interact with P-gp. We rely on structure–activity relationship analysis of a small group of tariquidar and elacridar analogues that was purposefully selected, designed, and tested. Structural modifications of the compounds relate to the presence or absence of functional groups in the tariquidar and elacridar scaffolds. The activity of the compounds was evaluated by their effects on the accumulation of P-gp substrates rhodamine 123 and Hoechst 33342 in resistant tumor cells. The data allow estimation of the ability of the compounds to interact with the experimentally proposed R- and H-sites to which rhodamine 123 and Hoechst 33342 bind, respectively. Using an inward-facing homology model of human P-gp based on the crystallographic structure of mouse P-gp, we demonstrate that these binding sites may overlap with the binding sites of the QZ59 ligands co-crystallized with mouse P-gp. Based on this SAR analysis, and using flexible alignment and docking, we propose possible binding modes for tariquidar and elacridar. Our results suggest the possibility for the studied compounds to bind to sites that coincide or overlap with the binding sites of rhodamine 123 and Hoechst 33342. These results contribute to further understanding of structure–function relationships of P-gp and can help in the design of selective and potent P-gp inhibitors with potential clinical use.
Co-reporter:Dr. Ilza K. Pajeva;Markus Hanl;Dr. Michael Wiese
ChemMedChem 2013 Volume 8( Issue 5) pp:748-762
Publication Date(Web):
DOI:10.1002/cmdc.201200491
Abstract
The primary aim of this work was to analyze the contacts between residues in the nucleotide binding domains (NBDs) and at the interface between the transmembrane domains (TMDs) and the NBDs in the inward-open homology model of human P-glycoprotein (P-gp). The analysis revealed communication nets through hydrogen bonding in the NBD and at the NBD–TMD interface of each half involving residues from the adenosine triphosphate (ATP) motifs and the coupling helices of the intracellular loops. Similar networks have been identified in P-gp conformations generated by molecular dynamics simulation. Differences have been recorded in the networking between both halves of P-gp. Many of the residue contacts have also been observed in the X-ray crystal structures of other ATP binding cassette (ABC) transporters, which confirms their validity. Next, possible binding pockets involving residues of importance for the TMD–NBD communication were identified. By studying these pockets, binding sites were suggested for rhodamine 123 (R-site) and prazosin (regulatory site) at the NBD–TMD interface that agreed with the experimental data on their location. Additionally, one more R-site in the protein cavity was proposed, in accordance with the available biochemical data. Together with the previously suggested Hoechst 33342 site (H-site), all sites were interpreted with respect to their effects on the protein ATPase activity, in correspondence with the experimental observations. Several residues involved in key contacts in the P-gp NBDs were proposed for further targeted mutagenesis experiments.
Co-reporter:Federico Marighetti;Dr. Kerstin Steggemann;Markus Hanl ;Dr. Michael Wiese
ChemMedChem 2013 Volume 8( Issue 1) pp:125-135
Publication Date(Web):
DOI:10.1002/cmdc.201200377
Abstract
The breast cancer resistance protein (BCRP/ABCG2) is a member of the ABC transporter superfamily. This protein has a number of physiological functions, including protection of the human body from xenobiotics. The overexpression of BCRP in certain tumor cell lines causes cross-resistance against various drugs used in chemotherapeutic treatment. In a previous work we showed that a new class of compounds derived from XR9576 (tariquidar) selectively inhibits BCRP. In this work we synthesized more members of this class, with modification on the second and third aromatic rings. The inhibitory activities against BCRP and P-gp were assayed using a Hoechst 33342 assay for BCRP and a calcein AM assay for P-gp. Finally, quantitative structure–activity relationships for both aromatic rings were established. The results obtained show the importance of the electron density on the third aromatic ring, influenced by substituents, pointing to interactions with aromatic residues of the protein binding site. In the second aromatic ring the activity of compounds is influenced by the steric volume of the substituents.
Co-reporter:Kapil Juvale, Veronika F.S. Pape, Michael Wiese
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 1) pp:346-355
Publication Date(Web):1 January 2012
DOI:10.1016/j.bmc.2011.10.074
Breast cancer resistance protein (BCRP/ABCG2) belongs to the ATP binding cassette family of transport proteins. BCRP has been found to confer multidrug resistance in cancer cells. A strategy to overcome resistance due to BCRP overexpression is the investigation of potent and specific BCRP inhibitors. The aim of the current study was to investigate different multi-substituted chalcones for their BCRP inhibition. We synthesized chalcones and benzochalcones with different substituents (viz. OH, OCH3, Cl) on ring A and B of the chalcone structure. All synthesized compounds were tested by Hoechst 33342 accumulation assay to determine inhibitory activity in MCF-7 MX and MDCK cells expressing BCRP. The compounds were also screened for their P-glycoprotein (P-gp) and Multidrug resistance-associated protein 1 (MRP1) inhibitory activity in the calcein AM accumulation assay and were found to be selective towards inhibition of BCRP. Substituents at position 2′ and 4′ on chalcone ring A were found to be essential for activity; additionally there was a great influence of substituents on ring B. Presence of 3,4-dimethoxy substitution on ring B was found to be optimal, while presence of 2- and 4-chloro substitution also showed a positive effect on BCRP inhibition.
Co-reporter:Kapil Juvale, Michael Wiese
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 21) pp:6766-6769
Publication Date(Web):1 November 2012
DOI:10.1016/j.bmcl.2012.08.024
We investigated several 2-phenylquinazolines with different substitutions at position 4 for their BCRP inhibition. Compounds with phenyl ring attached via an amine-containing linker at position 4 were found to be potent inhibitors of BCRP. In general compounds with meta substitution of phenyl ring at position 4 were found to have higher inhibitory effect, compound 12 being the most potent and selective towards BCRP.
Co-reporter:Dr. Anne Pick ;Dr. Michael Wiese
ChemMedChem 2012 Volume 7( Issue 4) pp:650-662
Publication Date(Web):
DOI:10.1002/cmdc.201100543
Abstract
Multidrug resistance observed in cancer chemotherapy is commonly attributed to overexpression of efflux transporter proteins. These proteins act as ATP-dependent drug efflux pumps, actively extruding chemotherapeutic agents from cells and causing a decrease in intracellular drug accumulation. Besides the well-recognized role of P-glycoprotein (P-gp, ABCB1), the breast cancer resistance protein (BCRP, ABCG2) is becoming increasingly accepted as playing an important role in multidrug resistance. In contrast to P-glycoprotein, only a few inhibitors of ABCG2 are known. According to the literature, tyrosine kinase inhibitors (TKIs) can be considered to be broad-spectrum inhibitors, interacting with ABCB1, ABCC1 and ABCG2. Here, we investigated seven different TKIs, gefitinib, erlotinib, AG1478, PD158780, PD153035, nilotinib and imatinib, for their potential to restore ABCG2 sensitivity to cells. Furthermore, we analyzed the alteration of ABCG2 expression caused by TKIs and demonstrated that EGFR inhibitors such as gefitinib and PD158780 reduced both total and surface expression of ABCG2 in EGRF-positive MDCK BCRP cells by interaction with the PI3K/Akt signaling pathway. The reduced ABCG2 content led to an increased effect of XR9577, a well-known ABCG2 modulator, lowering the concentration required for half maximal inhibition. On the other hand, BCR-ABL inhibitors had no influence on ABCG2 expression and modulator activity. Interestingly, a combination of an EGFR inhibitor with the PI3K/Akt inhibitor LY294002 led to a significant reduction of ABCG2 expression at low concentrations of the drugs. Based on our results, we assume that EGFR exerts a post-transcriptional enhancing effect on ABCG2 expression via the PI3K/Akt signaling pathway, which can be attenuated by EGFR inhibitors. Blocking the key signaling pathway regulating ABCG2 expression with EGFR inhibitors, combined with the inhibition of ABCG2 with potent modulators might be a promising approach to circumvent MDR in cancer cells.
Co-reporter:Anne Pick, Henrik Müller, Ralf Mayer, Britta Haenisch, Ilza K. Pajeva, Mathias Weigt, Heinz Bönisch, Christa E. Müller, Michael Wiese
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 6) pp:2090-2102
Publication Date(Web):15 March 2011
DOI:10.1016/j.bmc.2010.12.043
Flavonoids are an interesting group of natural products ubiquitously present in human diet. Their consumption has been associated with various and differing beneficial health effects. However, several flavonoids have been reported to inhibit the breast cancer resistance protein (BCRP) encoded by the ABCG2 gene. Thus, the consumption of flavonoids with high inhibitory activity could change pharmacokinetics and drug levels of drugs that are BCRP substrates. In cancer patients receiving chemotherapy an increased intake of such flavonoids could lead to adverse effects.We investigated a structurally diverse set of flavonoids, including derivatives with a rare C-methylated structure that were isolated from plants used in traditional medicine. The flavones retusin and ayanin were found to be highly potent inhibitors of BCRP, showing only slightly less potency than Ko143, the most potent ABCG2 inhibitor known so far. The activity data were analyzed by 2D and 3D QSAR analyses and the results revealed the impact of the different substituents at the various positions of the flavonoid core on activity. Additionally, a lateral 2D QSAR analysis of data collected from the literature was performed aiming to derive more general information about the influence of distinct structural features on the inhibitory potency of flavonoids. The comparative QSAR analyses led to a consistent picture of the effects of the different substituents at various positions of the flavone backbone. The following structural features were found to contribute positively to BCRP inhibition: a hydroxyl group in position 5, double bond between position 2 and 3, and a methoxy group in position 3. The exchange of a 3-methoxy group by an OH-group acting also as a hydrogen bond donor, resulted in decrease in activity underlining the potential role of the hydrogen bond acceptor 3-OCH3 for the interaction with BCRP.Summary of structural features influencing the inhibition of BCRP by flavonoids. Plus-circles indicate the positive contribution of structural elements to anti-BCRP activity. Minus-circles illustrate the negative impact on inhibitory potency. Results are based on data from the 3D QSAR approaches of the present study as well as the findings from 2D QSAR analyses of data taken from literature.
Co-reporter:Anna Jacobs;Dana Emmert;Svenja Wieschrath;Christine A. Hrycyna
The Protein Journal 2011 Volume 30( Issue 3) pp:
Publication Date(Web):2011 March
DOI:10.1007/s10930-011-9321-5
Human ABCG2 is an efflux protein belonging to the ATP-binding cassette transporter superfamily. It is expressed in the plasma membrane of different cell types performing various physiological functions. It is the most recently discovered MDR transporter and its structure and function are still not well understood. Thus, expression and functional reconstitution of the protein in different variants and from different sources are important steps for its further investigation. In this work we describe a recombinant synthesis of human ABCG2 R482G from S. cerevisiae. We expressed the human ABCG2 R482G variant in S. cerevisiae and purified the protein from total yeast membranes. Using a panel of sixteen detergents, we analyzed the efficiency of extraction of ABCG2 from membranes by SDS–PAGE and immunoblot analysis. Based on these results, three detergents were selected for further purification studies and two of them, n-octyl-β-D-glucopyranoside and n-dodecyl-β-D-maltopyranoside, yielded functional protein after reconstitution into liposomes. We show here the first example of purified and reconstituted ABCG2 expressed in S. cerevisiae retaining drug-stimulated ATPase activity.
Co-reporter:Anne Pick, Henrik Müller, Michael Wiese
Bioorganic & Medicinal Chemistry Letters 2010 20(1) pp: 180-183
Publication Date(Web):
DOI:10.1016/j.bmcl.2009.11.004
Co-reporter:Anne Pick;Werner Klinkhammer Dr. Dr.
ChemMedChem 2010 Volume 5( Issue 9) pp:1498-1505
Publication Date(Web):
DOI:10.1002/cmdc.201000216
Abstract
A new class of specific breast cancer resistance protein (BCRP) inhibitors was identified, showing no inhibition of the ATP binding cassette (ABC) transporters P-gp and MRP1. Some of these modulators inhibit BCRP with high potency; they are only slightly less potent than Ko143 and could serve as promising lead structures for the design of novel effective BCRP inhibitors. These inhibitors are structurally related to tariquidar (XR9576) and belong to a library of multidrug-resistance modulators synthesized by our research group. The absence of the tetrahydroisoquinoline substructure appears to play a crucial role for specificity; we found that the presence of this substructure is not essential for interaction with BCRP. To determine the type of interaction between pheophorbide A and compounds with and without the tetrahydroisoquinoline substructure, various substrate pheophorbide A concentrations were used in enzyme kinetics assays. The resulting data show that these compounds share a noncompetitive-type interaction with pheophorbide A. Experiments with imatinib and pheophorbide A revealed a mixed-type interaction. The combination of imatinib and compounds with and without the tetrahydroisoquinoline substructure resulted in a positive cooperative effect, indicating that imatinib engages a binding site distinct from that of the new compounds on one side and distinct from that of pheophorbide A on the other side as well. The results of this study suggest that the category of BCRP-specific inhibitors, which includes only fumitremorgin C, Ko143 and analogues, and novobiocin needs to be extended by this new class of inhibitors, which possess three key characteristics: specificity, potency, and low toxicity.
Co-reporter:Hans-Georg Häcker ; Stefan Leyers ; Jeanette Wiendlocha ; Michael Gütschow
Journal of Medicinal Chemistry 2009 Volume 52(Issue 15) pp:4586-4595
Publication Date(Web):July 6, 2009
DOI:10.1021/jm900688v
Four series of aromatic carboxylic acids were prepared with a urea or thiourea moiety at the neighboring position to the carboxyl group and benzene or thiophene as aromatic scaffold. Using a calcein AM assay, these compounds were evaluated as inhibitors of multidrug resistance-associated protein 1 (MRP1) and selected compounds were examined toward P-glycoprotein (P-gp) as well as breast cancer resistance protein (BCRP) to assess selectivity for MRP1. Two 2-thioureidobenzo[b]thiophene-3-carboxylic acids (48, 49) were identified as particularly potent inhibitors of MRP1, with IC50 values of around 1 μM. The structural features of this new family of nontoxic MRP1 inhibitors include a (thio)urea disubstituted with preferentially two alkyl groups at the terminal nitrogen and an additional fused aromatic ring.
Co-reporter:Madhura Vaidya, Mathias Weigt, Michael Wiese
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 10) pp:4070-4082
Publication Date(Web):October 2009
DOI:10.1016/j.ejmech.2009.04.045
Farnesyltransferase is a potential drug target for treating various types of cancers. Three-dimensional quantitative structure–activity relationships (3D-QSAR) for a series of farnesyltransferase inhibitors were investigated using comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) techniques. Pharmacophore search and molecular docking methods were used for construction of the molecular alignments. While the 3D-QSAR models were created for a training set of 33 compounds, their external predictivity was proven using a test set of 12 compounds. The results provided a comprehensive insight into the relationship between the structural features and the activities of farnesyltransferase inhibitors. This investigation will facilitate optimization of the design of new potential farnesyltransferase inhibitors.
Co-reporter:Werner Klinkhammer, Henrik Müller, Christoph Globisch, Ilza K. Pajeva, Michael Wiese
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 6) pp:2524-2535
Publication Date(Web):15 March 2009
DOI:10.1016/j.bmc.2009.01.072
The development of new modulators possessing high efficacy, low toxicity and high selectivity is a pivotal approach to overcoming P-glycoprotein (P-gp) mediated multidrug resistance (MDR) in tumour cells. In this study 39 compounds are presented which have been synthesized and pharmacologically investigated in our laboratory. Similarly to the potent 3rd generation MDR modulator tariquidar (XR9576) the compounds contain a tetrahydroisoquinoline–ethyl-phenylamine substructure that, in contrast to XR9576, is connected to a smaller hydrophobic part, thus leading to molecules of lower molecular weight. The connection between the tetrahydroisoquinoline–ethyl-phenylamine substructure and the hydrophobic part was achieved through four different types of linkers: amide, urea, amide-ether and amide-styryl. A number of structural modifications in the hydrophobic part were created. The calcein AM assay served as test system to determine the P-gp transport inhibitory potencies of the compounds. For the amide linker derivatives a structure–activity relationship analysis was performed outlining which structural modifications contributed to the inhibitory potency. The compounds containing a bicyclic hydrophobic part with a particular substituent in a specific orientation were identified as the most potent amide derivatives. Among the urea derivatives the compounds with highest inhibitory potency possessed an ortho-nitro substituent. The conformational analysis revealed that this position enables the formation of a hydrogen bond to the urea linker thus stabilizing the conformation. Regarding the amide-styryl derivatives the elongation of the amide linker seemed to be most decisive for the observed increase in activity. The most promising candidate in the whole library possess an amide-ether linker and an ortho-nitro substituent in the hydrophobic part. This compound inhibites P-gp slightly less than tariquidar and can serve as a lead structure for new potent P-gp modulators.
Co-reporter:Ilza K. Pajeva
The AAPS Journal 2009 Volume 11( Issue 3) pp:
Publication Date(Web):2009 September
DOI:10.1208/s12248-009-9118-z
The review summarizes the most recent achievements in structure–activity relationship (SAR) studies of tariquidar and its analogs. Tariquidar is one of the most promising representatives of the third generation of multidrug resistance (MDR) modulators created so far. This fact determines the strong interest of different research groups in the development of tariquidar-like structures as selective inhibitors of MDR transporters in resistant human cancer cells. After the discovery of tariquidar, a number of analogs have been synthesized and pharmacologically tested, thus supplying good data for comprehensive analyses of their structure–activity relationships. In the review, the structural and pharmacological data of newly synthesized tariquidar-like compounds are first presented. Next, the main achievements in the SAR studies are described focusing on two main transport proteins: P-glycoprotein and breast cancer resistance protein. The reported results are discussed from the point of view of their significance and importance for future directions in the rational design of effective MDR modulators.
Co-reporter:IlzaK. Pajeva Dr.;Christoph Globisch Dr. Dr.
ChemMedChem 2009 Volume 4( Issue 11) pp:1883-1896
Publication Date(Web):
DOI:10.1002/cmdc.200900282
Abstract
Quinazolinones, indolo- and pyrrolopyrimidines with inhibitory effects toward ABCB1 (P-gp) and ABCC1 (MRP1) transporters were studied by pharmacophore modeling, docking, and 3D QSAR to describe the binding preferences of the proteins. The pharmacophore overlays between dual and/or highly selective inhibitors point to binding sites of different topology and physiochemical properties for MRP1 and P-gp. Docking of selective inhibitors into the P-gp binding cavity by the use of a structural model based on the recently resolved P-gp structure confirms the P-gp pharmacophore features identified, and reveals the interactions of some functional groups and atoms in the structures with particular protein residues. The 3D QSAR analysis of the dual-effect inhibitors allows satisfactory prediction of the selectivity index of the compounds and outlines electrostatics as most important for selectivity. The results from the combined modeling approach complement each other and could improve our understanding of the protein–ligand interactions involved, and could aid in the development of highly selective and potent inhibitors of P-gp and MRP1.
Co-reporter:Katja Sterz;Lars Möllmann;Anna Jacobs;Dieter Baumert Dr.
ChemMedChem 2009 Volume 4( Issue 11) pp:1897-1911
Publication Date(Web):
DOI:10.1002/cmdc.200900283
Abstract
P-glycoprotein (P-gp), a 170 kDa plasma membrane protein, is one of the most relevant ABC transporters involved in the development of multidrug resistance (MDR). Understanding its mechanism of transport as well as its interactions with various substrates are basic requirements for the development of adequate therapeutic approaches to overcome this kind of resistance against a broad spectrum of structurally unrelated cytostatic drugs. P-gp modulators (activators) that exert various effects on the intracellular accumulation of distinct P-gp substrates are useful tools for investigating the interactions between multiple drug binding sites of this transport protein. In this study, a series of 27 different imidazobenzothiazoles and imidazobenzimidazoles structurally related to the known P-gp activators QB102 and QB11 was designed, and their modulating properties were investigated. Most of them were able to stimulate P-gp-mediated efflux of daunorubicin and rhodamine 123 in a concentration-dependent manner, but some compounds also displayed weak inhibitory effects. Additionally, P-gp-mediated efflux of vinblastine and colchicine was inhibited by several compounds. Therefore, we concluded that the novel compounds bind to the H site of P-gp and activate the efflux of specific substrates of the R site in a positive cooperative manner, whereas binding of H-type substrates is inhibited competitively. This hypothesis is confirmed by the observation that the modulators do not influence hydrolysis of ATP or its affinity toward P-gp.
Co-reporter:Henrik Müller, Ilza K. Pajeva, Christoph Globisch, Michael Wiese
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 5) pp:2448-2462
Publication Date(Web):1 March 2008
DOI:10.1016/j.bmc.2007.11.057
Twenty-eight compounds, including 24 structurally related derivatives of tariquidar synthesized in our laboratory, and four XR compounds, reported by Xenova group Ltd, were investigated by the Hoechst 33342 and Calcein AM functional assays for estimation of their inhibitory effects on the transport activity of P-glycoprotein (P-gp). A high correlation between the effects obtained in both assays was observed at the substrate concentrations used. The analyses of kinetics data from experiments at different substrate concentrations revealed non-competitive inhibition in the Calcein AM assay and competitive inhibition in the Hoechst 33342 assay. The 3D structures of the compounds were further aligned on Hoechst 33342 using flexible and pharmacophore alignments. The results suggested that inhibitors could interact with the H-binding site of P-gp and this could potentially be achieved by different ways of binding. The best 3D-QSAR models, generated by CoMFA and CoMSIA, yielded an internal predictive squared correlation coefficient higher than 0.8 and included electrostatic, steric, hydrogen bond acceptor, and hydrophobic fields. Validation of the models on an external test set of 30 XR compounds gave predictive squared correlation coefficients of up to 0.66. An excellent correspondence between the experimental and modeled activities of the test compounds was observed. The models can be used for prediction and rational design of new P-gp inhibitors.
Co-reporter:Christoph Globisch;IlzaK. Pajeva Dr. Dr.
ChemMedChem 2008 Volume 3( Issue 2) pp:280-295
Publication Date(Web):
DOI:10.1002/cmdc.200700249
Abstract
A homology model of P-glycoprotein based on the crystal structure of the multidrug transporter Sav1866 is developed, incorporated into a membrane environment, and optimized. The resulting model is analyzed in relation to the functional state and potential binding sites. The comparison of modeled distances to distances reported in experimental studies between particular residues suggests that the model corresponds most closely to the first ATP hydrolysis step of the protein transport cycle. Comparison to the protein 3D structure confirms this suggestion. Using SiteID and Site Finder programs three membrane related binding regions are identified: a region at the interface between the membrane and cytosol and two regions located in the transmembrane domains. The regions contain binding pockets of different size, orientation, and amino acids. A binding pocket located inside the membrane cavity is also identified. The pockets are analyzed in relation to amino acids shown experimentally to influence the protein function. The results suggest that the protein has multiple binding sites and may bind and/or release substrates in multiple pathways.
Co-reporter:Henrik Müller, Werner Klinkhammer, Christoph Globisch, Matthias U. Kassack, Ilza K. Pajeva, Michael Wiese
Bioorganic & Medicinal Chemistry 2007 Volume 15(Issue 23) pp:7470-7479
Publication Date(Web):1 December 2007
DOI:10.1016/j.bmc.2007.07.024
In this study we describe a simplified, HTS-capable functional assay for the multidrug resistance (MDR) transporter P-glycoprotein (P-gp) based on its substrate Hoechst 33342. The physicochemical properties of Hoechst 33342 and the enormous milieu dependency of its fluorescence intensity allowed performing the assay in a homogeneous manner. This new assay served as an effective tool to estimate the potency of 10 well recognized P-gp substrates and modulators. Further, the potency of these compounds was also estimated in the calcein AM assay. The Hoechst 33342 and calcein AM assays yielded significantly comparable results for all compounds tested. Principal component analysis (PCA) applied to literature data on inhibition of P-gp activity and our results obtained in the Hoechst 33342 and calcein AM assay indicated similarity of compared functional transport assays. However, no correlation could be detected between these functional assays and the ATPase activity assay.
Co-reporter:Christoph Globisch, Ilza K. Pajeva, Michael Wiese
Bioorganic & Medicinal Chemistry 2006 Volume 14(Issue 5) pp:1588-1598
Publication Date(Web):1 March 2006
DOI:10.1016/j.bmc.2005.10.058
Tariquidar (XR9576) analogs, modulators of cancer multidrug resistance (MDR), were subjected to QSAR and 3D-QSAR analyses. The structural features contributing to anti-MDR activity were identified by the Free-Wilson analysis and pharmacophore search using Hoechst 33342 as a template. 3D-QSAR CoMFA and CoMSIA models were derived and tested. The best models yielded an external predictivity of 0.66–0.75 squared correlation coefficient and outlined HB-acceptor, steric, and hydrophobic fields as the most important 3D properties. On the basis of the QSAR and 3D-QSAR analyses it was suggested that the strong inhibitory potency of the compounds studied is related to the presence of a bulky aromatic ring system with a 3rd positioned heteroatom toward the anthranilamide nucleus in the opposite end of the tetrahydroquinoline group. The results can help in directing the rational design of new generations of potent P-glycoprotein MDR modulators.
Co-reporter:Sven Marcel Schmitt, Katja Stefan, Michael Wiese
Biochimica et Biophysica Acta (BBA) - Biomembranes (January 2017) Volume 1859(Issue 1) pp:
Publication Date(Web):January 2017
DOI:10.1016/j.bbamem.2016.10.017
•New classes of selective MRP1 activators•Compounds acting as non-essential activators•Discovery of a triple inhibitor of MRP1, P-gp, BCRPMultidrug resistance (MDR) is the main cause of diminished success in cancer chemotherapy. ABC transport proteins are considered to be one important factor of MDR. Besides P-glycoprotein (P-gp, ABCB1) and Breast Cancer Resistance Protein (BCRP, ABCG2), Multidrug Resistance-associated Protein 1 (MRP1, ABCC1) is associated with non-response to chemotherapy in different cancers. While considerable effort was spent in overcoming MDR during the last two decades, almost nothing is known with respect to activators of transport proteins. In this work we present certain pyrrolo[3,2-d]pyrimidine derivatives with variations at positions 4 and 5 and purine analogs with variations at position 6 as novel activators of MRP1-mediated transport of the MRP1 substrate calcein AM and the anticancer drug daunorubicin in low nanomolar concentration range. Two different MRP1 overexpressing cell lines were used, the doxorubicin-selected human lung cancer cell line H69 AR and the transfected Madin-Darby Canine Kidney cell line MDCK II MRP1. No effect was observed in the sensitive counterparts H69 and MDCK II wild type (wt). Derivatives with higher molecular weight possessed also inhibitory properties at low micromolar concentrations, although most compounds were rather poor MRP1 inhibitors. Purine analogs derived from potent MRP1 inhibitors of the pyrrolopyrimidine class showed equal activating, but no inhibiting effects at all. All tested compounds were non-toxic and had only minor impact on P-gp or BCRP, showing no inhibition or activation.





![4-Quinazolinamine, N-[3-(methylthio)phenyl]-2-phenyl-](http://img.cochemist.com/ccimg/916800/916751-77-4.png)
![4-Quinazolinamine, N-[3-(methylthio)phenyl]-2-phenyl-](http://img.cochemist.com/ccimg/916800/916751-77-4_b.png)












![3,4-Dimethoxy-N-[2-(4-methoxy-phenylcarbamoyl)-phenyl]-benzamide](http://img.cochemist.com/ccimg/827700/827617-24-3.png)
![3,4-Dimethoxy-N-[2-(4-methoxy-phenylcarbamoyl)-phenyl]-benzamide](http://img.cochemist.com/ccimg/827700/827617-24-3_b.png)
![3-[(1S,9AR)-OCTAHYDRO-2H-QUINOLIZIN-1-YL]PROPANOIC ACID](http://img.cochemist.com/ccimg/771600/771573-02-5.png)
![3-[(1S,9AR)-OCTAHYDRO-2H-QUINOLIZIN-1-YL]PROPANOIC ACID](http://img.cochemist.com/ccimg/771600/771573-02-5_b.png)
![ETHANONE, 1-[4-[(6,7-DIMETHOXY-4-QUINAZOLINYL)AMINO]PHENYL]-](http://img.cochemist.com/ccimg/741300/741276-74-4.png)
![ETHANONE, 1-[4-[(6,7-DIMETHOXY-4-QUINAZOLINYL)AMINO]PHENYL]-](http://img.cochemist.com/ccimg/741300/741276-74-4_b.png)


![[3-(1,3-Thiazol-2-yl)phenyl]methylamine](http://img.cochemist.com/ccimg/672400/672324-88-8.png)
![[3-(1,3-Thiazol-2-yl)phenyl]methylamine](http://img.cochemist.com/ccimg/672400/672324-88-8_b.png)

![BENZONITRILE, 3-[(2-PHENYL-4-QUINAZOLINYL)AMINO]-](http://img.cochemist.com/ccimg/474300/474289-70-8.png)
![BENZONITRILE, 3-[(2-PHENYL-4-QUINAZOLINYL)AMINO]-](http://img.cochemist.com/ccimg/474300/474289-70-8_b.png)
![1H-PYRROLO[3,2-D]PYRIMIDINE-7-CARBONITRILE, 4,5-DIHYDRO-4-OXO-5-PHENYL-](http://img.cochemist.com/ccimg/474000/473997-02-3.png)
![1H-PYRROLO[3,2-D]PYRIMIDINE-7-CARBONITRILE, 4,5-DIHYDRO-4-OXO-5-PHENYL-](http://img.cochemist.com/ccimg/474000/473997-02-3_b.png)
![Benzoic acid, 4-[(2-phenyl-4-quinazolinyl)amino]-](http://img.cochemist.com/ccimg/451500/451462-13-8.png)
![Benzoic acid, 4-[(2-phenyl-4-quinazolinyl)amino]-](http://img.cochemist.com/ccimg/451500/451462-13-8_b.png)

![Ethanone, 1-[3-[(2-phenyl-4-quinazolinyl)amino]phenyl]-](http://img.cochemist.com/ccimg/421600/421581-41-1.png)
![Ethanone, 1-[3-[(2-phenyl-4-quinazolinyl)amino]phenyl]-](http://img.cochemist.com/ccimg/421600/421581-41-1_b.png)
![N-(4-methoxyphenyl)-2-[(4-nitrobenzoyl)amino]benzamide](http://img.cochemist.com/ccimg/405000/404901-95-7.png)
![N-(4-methoxyphenyl)-2-[(4-nitrobenzoyl)amino]benzamide](http://img.cochemist.com/ccimg/405000/404901-95-7_b.png)



![1H-Pyrido[3,4-b]indole, 1-(4-bromophenyl)-2,3,4,9-tetrahydro-](http://img.cochemist.com/ccimg/334600/334500-55-9.png)
![1H-Pyrido[3,4-b]indole, 1-(4-bromophenyl)-2,3,4,9-tetrahydro-](http://img.cochemist.com/ccimg/334600/334500-55-9_b.png)
![1H-Pyrido[3,4-b]indole, 1-(3,4-dimethoxyphenyl)-2,3,4,9-tetrahydro-](http://img.cochemist.com/ccimg/310500/310415-88-4.png)
![1H-Pyrido[3,4-b]indole, 1-(3,4-dimethoxyphenyl)-2,3,4,9-tetrahydro-](http://img.cochemist.com/ccimg/310500/310415-88-4_b.png)


![1H-Pyrido[3,4-b]indole, 1-(3,4-difluorophenyl)-2,3,4,9-tetrahydro-](http://img.cochemist.com/ccimg/199700/199678-78-9.png)
![1H-Pyrido[3,4-b]indole, 1-(3,4-difluorophenyl)-2,3,4,9-tetrahydro-](http://img.cochemist.com/ccimg/199700/199678-78-9_b.png)
![Benzamide, 2-amino-N-[4-(aminosulfonyl)phenyl]-](http://img.cochemist.com/ccimg/193900/193896-61-6.png)
![Benzamide, 2-amino-N-[4-(aminosulfonyl)phenyl]-](http://img.cochemist.com/ccimg/193900/193896-61-6_b.png)








![1H-Pyrido[3,4-b]indole, 1-(4-chlorophenyl)-2,3,4,9-tetrahydro-](http://img.cochemist.com/ccimg/139700/139655-03-1.png)
![1H-Pyrido[3,4-b]indole, 1-(4-chlorophenyl)-2,3,4,9-tetrahydro-](http://img.cochemist.com/ccimg/139700/139655-03-1_b.png)
![Pyrimido[4,5-b]indolizine-10-carbonitrile, 4-chloro-6,7,8,9-tetrahydro-](http://img.cochemist.com/ccimg/133000/132994-16-2.png)
![Pyrimido[4,5-b]indolizine-10-carbonitrile, 4-chloro-6,7,8,9-tetrahydro-](http://img.cochemist.com/ccimg/133000/132994-16-2_b.png)
![3,4,6,7,8,9-hexahydro-4-oxo-Pyrimido[4,5-b]indolizine-10-carbonitrile](http://img.cochemist.com/ccimg/133000/132994-09-3.png)
![3,4,6,7,8,9-hexahydro-4-oxo-Pyrimido[4,5-b]indolizine-10-carbonitrile](http://img.cochemist.com/ccimg/133000/132994-09-3_b.png)


![1H-Pyrido[3,4-b]indole, 2,3,4,9-tetrahydro-1-(3-nitrophenyl)-](http://img.cochemist.com/ccimg/121000/120908-68-1.png)
![1H-Pyrido[3,4-b]indole, 2,3,4,9-tetrahydro-1-(3-nitrophenyl)-](http://img.cochemist.com/ccimg/121000/120908-68-1_b.png)
![1H-Pyrido[3,4-b]indole, 1-(2-chlorophenyl)-2,3,4,9-tetrahydro-](http://img.cochemist.com/ccimg/121000/120908-66-9.png)
![1H-Pyrido[3,4-b]indole, 1-(2-chlorophenyl)-2,3,4,9-tetrahydro-](http://img.cochemist.com/ccimg/121000/120908-66-9_b.png)
![1H-Pyrido[3,4-b]indole, 1-(3,4-dichlorophenyl)-2,3,4,9-tetrahydro-](http://img.cochemist.com/ccimg/121000/120908-64-7.png)
![1H-Pyrido[3,4-b]indole, 1-(3,4-dichlorophenyl)-2,3,4,9-tetrahydro-](http://img.cochemist.com/ccimg/121000/120908-64-7_b.png)


![BENZENESULFONIC ACID, [(2-NITROPHENYL)METHYLENE]HYDRAZIDE](http://img.cochemist.com/ccimg/118800/118719-18-9.png)
![BENZENESULFONIC ACID, [(2-NITROPHENYL)METHYLENE]HYDRAZIDE](http://img.cochemist.com/ccimg/118800/118719-18-9_b.png)








![Benzonitrile, 4-[(2-phenyl-4-quinazolinyl)amino]-](http://img.cochemist.com/ccimg/92100/92000-89-0.png)
![Benzonitrile, 4-[(2-phenyl-4-quinazolinyl)amino]-](http://img.cochemist.com/ccimg/92100/92000-89-0_b.png)
![Ethanone, 1-[4-[(2-phenyl-4-quinazolinyl)amino]phenyl]-](http://img.cochemist.com/ccimg/87800/87771-83-3.png)
![Ethanone, 1-[4-[(2-phenyl-4-quinazolinyl)amino]phenyl]-](http://img.cochemist.com/ccimg/87800/87771-83-3_b.png)








![Benzamide, 2-[(4-chlorobenzoyl)amino]-N-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/73400/73343-47-2.png)
![Benzamide, 2-[(4-chlorobenzoyl)amino]-N-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/73400/73343-47-2_b.png)
![1H-Pyrido[3,4-b]indole, 2,3,4,9-tetrahydro-1-phenyl-2-(phenylmethyl)-](http://img.cochemist.com/ccimg/65100/65048-57-9.png)
![1H-Pyrido[3,4-b]indole, 2,3,4,9-tetrahydro-1-phenyl-2-(phenylmethyl)-](http://img.cochemist.com/ccimg/65100/65048-57-9_b.png)
![3,6-diamino-9-[2-(methoxycarbonyl)phenyl]xanthylium chloride](/data/chemimg/622800/62669-70-9.png)
![3,6-diamino-9-[2-(methoxycarbonyl)phenyl]xanthylium chloride](/data/chemimg/622800/62669-70-9_b.png)


![Phenol, 4-[(2-phenyl-4-quinazolinyl)amino]-](http://img.cochemist.com/ccimg/54700/54665-94-0.png)
![Phenol, 4-[(2-phenyl-4-quinazolinyl)amino]-](http://img.cochemist.com/ccimg/54700/54665-94-0_b.png)













![Ethanone, 1-[4-(4-quinazolinylamino)phenyl]-](http://img.cochemist.com/ccimg/19100/19062-70-5.png)
![Ethanone, 1-[4-(4-quinazolinylamino)phenyl]-](http://img.cochemist.com/ccimg/19100/19062-70-5_b.png)










![1H-Pyrido[3,4-b]indole, 2,3,4,9-tetrahydro-1-(3-methoxyphenyl)-](http://img.cochemist.com/ccimg/6700/6649-95-2.png)
![1H-Pyrido[3,4-b]indole, 2,3,4,9-tetrahydro-1-(3-methoxyphenyl)-](http://img.cochemist.com/ccimg/6700/6649-95-2_b.png)






![1-(4-METHOXYPHENYL)-2,3,4,9-TETRAHYDRO-1H-PYRIDO[3,4-B]INDOLE](http://img.cochemist.com/ccimg/3400/3380-73-2.png)
![1-(4-METHOXYPHENYL)-2,3,4,9-TETRAHYDRO-1H-PYRIDO[3,4-B]INDOLE](http://img.cochemist.com/ccimg/3400/3380-73-2_b.png)
![1H-Pyrido[3,4-b]indole, 2,3,4,9-tetrahydro-1-(4-methylphenyl)-](http://img.cochemist.com/ccimg/3400/3380-71-0.png)
![1H-Pyrido[3,4-b]indole, 2,3,4,9-tetrahydro-1-(4-methylphenyl)-](http://img.cochemist.com/ccimg/3400/3380-71-0_b.png)










![[(phenylamino)methylidene]propanedinitrile](http://img.cochemist.com/ccimg/1300/1202-48-8.png)
![[(phenylamino)methylidene]propanedinitrile](http://img.cochemist.com/ccimg/1300/1202-48-8_b.png)