Co-reporter:Donglin Li, Yonggang Yang, Chaozheng Li, Yufang Liu
Journal of Luminescence 2017 Volume 182() pp:15-21
Publication Date(Web):February 2017
DOI:10.1016/j.jlumin.2016.10.002
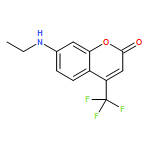
The behavior of three-centered hydrogen bond (THB) in excited state is unveiled firstly. The notion proposed in experiment that the THB configuration can be stable in solution has been verified. And one more stable configuration has been found which has two intramolecular hydrogen bonds compared with THB configuration. The analysis of optimization geometrical configurations shows that the THB weakens upon photoexcitation and is apparent weaker than the two-centered hydrogen bond. The reduced electron density gradient (RDG) maps indicate that the weakening of THB compared with two-centered hydrogen bond is due to the steric repulsion of hydrogen bond donors and acceptors. In addition, fluoresce spectra indicate that the 3-position substituent of coumarin has a large blue-shift on maximum fluorescence wavelength compared with the 7-aminocoumarin C500.
Co-reporter:Gongke Wang, Huimin Hou, Shuangli Wang, Changling Yan, Yufang Liu
Colloids and Surfaces B: Biointerfaces 2017 Volume 157(Volume 157) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.colsurfb.2017.05.071
•Lysozyme binds to silver nanoparticles with a static quenching mechanism.•Hydrophobic interaction plays an essential role in the binding process.•The introduce of silver nanoparticles induce the conformational change of lysozyme.•The multilayer adsorption of lysozyme on silver nanoparticles surface exhibits hysteresis effect.The role of nanoparticle interaction with biomolecules to form a biocorona is the key to nanoparticle behavior and its consequences in the physiological environment. Since the adsorbed biocorona decides the fate of a nanomaterials in vivo, and thus a comprehensive understanding of the dynamic interactions of the proteins with the nanoparticle is imperative. Herein we investigate the interaction of a model protein, lysozyme with silver nanoparticles (AgNPs) using fluorescence, synchronous fluorescence, UV–vis absorption spectrum and circular dichroism (CD) techniques under the physiological conditions. The results indicated that the binding of AgNPs to lysozyme may be a static quenching mechanism. With the analysis of the fluorescence spectral data, the binding constants and the thermodynamic parameters were determined, which suggests that the binding of AgNPs to lysozyme is a spontaneous process. Moreover, it was demonstrated that the main acting forces between AgNPs and lysozyme may be hydrophobic interactions. At the same time, the conformational change of lysozyme induced by AgNPs was investigated with synchronous fluorescence spectroscopy and CD techniques. The results of kinetic studies reveal that the adsorption of lysozyme on AgNPs surface tends to follow pseudo-second-order kinetic characteristic with obvious hysteresis effect.Download high-res image (86KB)Download full-size image
Co-reporter:Chi Ma;Chaozheng Li;Yonggang Yang
RSC Advances (2011-Present) 2017 vol. 7(Issue 22) pp:13561-13569
Publication Date(Web):2017/02/24
DOI:10.1039/C6RA26470K
The newly synthesized blue fluorescence protein (BFP) molecule combined with its derivatives were fully investigated using DFT and TD-DFT methods. The frontier molecular orbitals and NBO charges indicate that the intramolecular benzene ring in the BFP molecule can inhibit the charge redistribution after photo-excitation effectively. The substitution of malonic and malononitrile groups can create the fluorescence quenching phenomenon, which may be caused by their participation in the excited state charge redistribution and act as important electron-donating groups. Also, these substitutions can enhance the configuration stability in the S1 state and hinder the formation of a metastable structure. The malononitrile group substitution can significantly decrease the energy barrier in the S1 state and promote proton transfer.
Co-reporter:Yating Shi;Yarui Shi;Huiling Wei;Hongsheng Zhai
New Journal of Chemistry (1998-Present) 2017 vol. 41(Issue 18) pp:10251-10258
Publication Date(Web):2017/09/11
DOI:10.1039/C7NJ02590D
Two new contorted polycyclic aromatic hydrocarbons (PAHs) 1 and 2 have been synthesized by Perepichka and coworkers (Org. Lett., 2015, 17, 4224). In this work, we aim to clarify the effect of packings of the isomers on the charge-transfer mobility of PAHs by quantum chemistry calculations combined with the Marcus–Hush electron transfer theory. The isomers reveal dissimilar properties with PAH2 having a much smaller energy gap than PAH1. Significantly, the maximum hole mobility of PAH2 is nearly 3.5 times larger than that of PAH1, while the adiabatic ionization potential (IP) value of PAH2 is smaller than that of PAH1. In the solid state, PAH2 packs in a unique two-dimensional herringbone motif with high intrinsic hole mobility and suitable adiabatic ionization potential (IP) values making it a good p-type material.
Co-reporter:Donglin Li;Yonggang Yang;Chaozheng Li
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 24) pp:15849-15855
Publication Date(Web):2017/06/21
DOI:10.1039/C7CP02268A
The fast and accurate detection of amyloid fibrils, which are associated with many neurodegenerative diseases, is important for their early diagnosis. {[50-(p-Hydroxyphenyl)-2,20-bithienyl-5-yl]-methylidene}-propanedinitrile (NIAD-4) is a new promising fluorescent marker for amyloid fibrils, and the photophysical behaviour of NIAD-4 is controversial. Nonadiabatic dynamic simulations, density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations were performed to determine the influence of the environment on NIAD-4 and the photophysical behaviour of NIAD-4. The results indicate that NIAD-4 is in the NIAD-4·3H2O compound form in the ground state in water. The torsion process of NIAD-4 proposed by Hu et al. (Phys. Chem. Chem. Phys. 2016, 18, 28) does not occur in the excited state. In addition, the fluorescence behaviour of NIAD-4 is sensitive to a hydrogen bonding environment, the maximum fluorescence wavelengths of NIAD-4 show considerable red-shifts, and the fluorescence intensity of NIAD-4 increases significantly in a hydrogen bonding environment. Intermolecular hydrogen bonds are vital for the phenomenon observed in the experiment because the fluorescence intensity of NIAD-4 becomes unusually high with increasing solvent polarities. Therefore, the influence of the intermolecular hydrogen bond should be carefully taken into consideration when NIAD-4 is used to probe the amyloid fibrils in hydrogen-bonding surroundings, especially in complex bioenvironments.
Co-reporter:Chaozheng Li;Yonggang Yang;Donglin Li
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 6) pp:4802-4808
Publication Date(Web):2017/02/08
DOI:10.1039/C6CP07716A
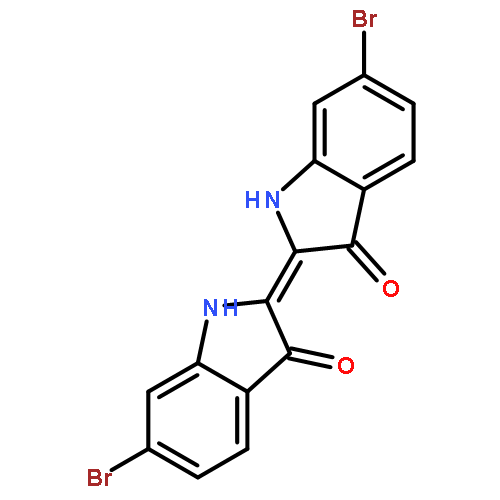
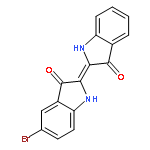
The excited-state double proton transfer (ESDPT) mechanism in a model DNA base pair, 7-azaindole (7AI) dimer, has been debated over the years. Recently, Otero and coworkers concluded that the stepwise mechanism is not possible and the concerted mechanism dominates the dynamics (Chem. Sci., 2015, 6, 5762). In this work, the potential energy surfaces of the 7AI dimer in the ground state (S0) and the lowest energy excited singlet state (S1) were constructed. After vertical excitation to the S1 state, the single proton transfer can occur. The second proton transfer process in the stepwise mechanism is blocked by a high potential barrier (36.4 kcal mol−1), which is consistent with the result proposed by Otero and coworkers. However, the single proton transfer process is compatible with the concerted mechanism and we show that the single proton transfer process rather than the concerted mechanism dominates the dynamics. The concerted process is unfavorable in the S1 state compared with the barrierless single proton transfer process. In addition, the proton transfer process in the S0 state is revealed. The single proton transfer tautomer in the S1 state returns to the S0 state and transfers the second proton via a barrierless process. Finally, the double proton transfer tautomer in the S0 state can recover to the normal dimer through the reverse proton transfer reaction.
Co-reporter:Shi Ya-Rui;Wei Hui-Ling;Shi Ya-Ting;Liu Yu-Fang
CrystEngComm (1999-Present) 2017 vol. 19(Issue 40) pp:6008-6019
Publication Date(Web):2017/10/16
DOI:10.1039/C7CE01359K
In this study, the orientational dependence of the charge transport mechanism and some important factors related to crystal structure are systematically investigated by quantum chemical methods. Organic semiconductors with small reorganization energies and large transfer integrals originating from their π-stacking crystal structures show excellent charge transport properties. The reorganization energy from the geometrical relaxation occurs during the charge transfer process. The transfer integral of a molecular crystal should be attributed to multiple stacking parameters. A semi-classical simulation model to calculate the anisotropic mobility of a crystalline molecule is extended from one to three dimensions. As we predicted, the anisotropic mobility calculated from our model is improved by considering the contribution from every direction in space, rather than just one plane. This theoretical study determines the importance of tuning the molecular geometry and calculation accuracy for high-performance organic semiconductor materials.
Co-reporter:Xueli Jia, Chaozheng Li, Donglin Li, Yufang Liu
Journal of Luminescence 2017 Volume 192(Volume 192) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.jlumin.2017.07.024
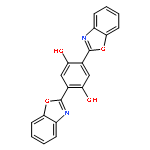
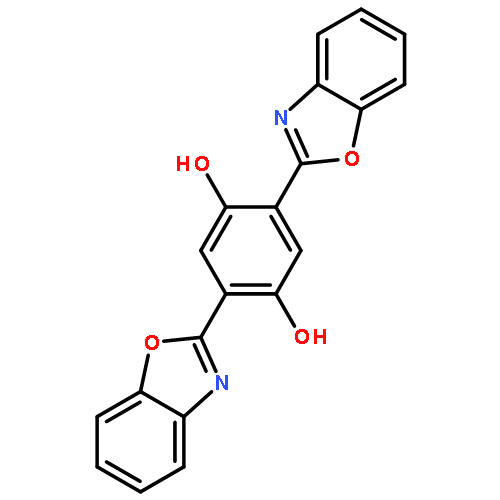
2,5-bis(benzoxazol-2-yl)thiophene-3,4-diol (BBT) is a newly synthesized water-soluble fluorophore dye with two intramolecular hydrogen bonds. It was demonstrated that BBT exhibits the excited-state intramolecular single and double proton transfer emissions in recent experiment. In order to explain the phenomenon, the density functional theory (DFT) and time-dependent density functional theory (TD-DFT) are used to study the ground states and excited states of BBT and its tautomers. The theoretical results confirm that the hydrogen bonds are strengthened upon photoexcitation. To further reveal the proton transfer mechanism, potential energy surfaces of the S0 and S1 states have been constructed. According to the energy potential barrier of 13.69 kcal/mol, it was indicated that the double proton transfer may occur in stepwise rather than simultaneously in the S1 state unless the excited molecule can overcome the barrier.
Co-reporter:Xiaomei Zhang, Hongsheng Zhai, Siyuan Liu, Yufang Liu
Journal of Quantitative Spectroscopy and Radiative Transfer 2017 Volume 196(Volume 196) pp:
Publication Date(Web):1 July 2017
DOI:10.1016/j.jqsrt.2017.04.011
•Total 23 Λ-S states of the PCl+ molecule have been studied with the MRCI+Q method.•The couplings of the states were analyzed via dipolar interaction and SOC effect.•The perturbation that the A2Π state suffered from has been identified.•The SOC effect makes 23 Λ-S states split into 45 Ω states.•The lifetimes of the transitions A2Π1/2-X2Π1/2 and A2Π3/2-X2Π3/2 were determined.The entire 23 Λ-S states of the PCl+ molecule have been studied by using the high-level relativistic MRCI+Q method with full-electron aug-cc-pCVQZ-DK basis set. The potential energy curves(PECs) and wavefunctions of the states have been calculated. From the PECs, the spectroscopic constants of the bound states are also determined, and the good agreements could be found with the experiments. The high density region of states exhibits many PECs' crossings, which lead to complicated interaction of the states. Here, the interactions arising from the dipolar interaction and spin-orbit coupling (SOC) effect have been discussed in detail. Under the influence of the SOC effect, the A2Π state is perturbed by the 14Σ- state. Considering the SOC effect, total 45 Ω states are generated from the original 23 Λ-S states. The transition properties are also predicted, including the transition dipole moments, Franck-Condon factors, and radiative lifetimes. The lifetimes of the transitions A2Π1/2-X2Π1/2 and A2Π3/2-X2Π3/2 are determined to be 478.9 ns and 487.0 ns(v'=0), respectively.The enlarged PECs' crossing in the high density region of electronic statesDownload high-res image (216KB)Download full-size image
Co-reporter:Siyuan Liu, Xiaomei Zhang, Hongsheng Zhai, Yufang Liu
Journal of Quantitative Spectroscopy and Radiative Transfer 2017 Volume 202(Volume 202) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.jqsrt.2017.07.015
•Total 28 Λ-S states of the NTe molecule have been calculated for the first time.•The polarity of the ground state for NTe is determined to be Nδ−Teδ+.•The SOC effects make the lowest 10 Λ-S states split into 30 Ω states.•The Ω-state transitions borrow TDMs from the spin-allowed Λ-S transitions via SOC.The entire 28 Λ-S electronic states correlated to four dissociation limits of the NTe molecule have been reliably characterized for the first time. The potential energy curves (PECs) and wavefunctions of these Λ-S states are calculated using the multi-reference configuration interaction plus Davidson correction (MRCI+Q) approach, with all-electron correlation-consistent basis sets at quadruple-zeta level. The spectroscopic constants of the bound Λ-S states are determined from the PECs, and the excellent agreement with previous work is derived. By analyzing the dipole moment, the polarity of the ground state for NTe is determined to be Nδ−Teδ+, which is contrary to the case of NO. The spin-orbit coupling (SOC) effect is included when the state interaction method is applied. The SOC effect is found to be substantial for the NTe molecule, which makes the lowest 10 Λ-S states split into 30 Ω states. The X2Π states split into two Ω states including X2Π1/2(X11/2) and X2Π3/2(X23/2), and the corresponding SOC splitting is computed to be 1975 cm−1(0.245 eV). Analysis of Λ-S compositions for the Ω-state wavefunction indicates the strong interaction among the Λ-S states. Additionally, the transition properties of the Ω-state transitions 1/2(2)-X11/2, 1/2(4)-X11/2, 3/2(10)-X23/2, and 5/2(1)-X23/2 were predicted. These transitions are all mainly from the spin-forbidden Λ-S transitions, but their transition dipole moments arise from those of the spin-allowed Λ-S transitions via the SOC effect. Accordingly, these transitions have quite long radiative lifetimes that are at the microsecond (ms) level.
Co-reporter:Chaozheng Li, Yonggang Yang, Chi Ma and Yufang Liu
RSC Advances 2016 vol. 6(Issue 6) pp:5134-5140
Publication Date(Web):06 Jan 2016
DOI:10.1039/C5RA23261A
The excited-state intramolecular proton transfer (ESIPT) reactions of 2-(2′-hydroxyphenyl)benzoxazole (HBO), 5-amino-2-(2′-hydroxyphenyl)benzoxazole (5A-HBO) and 6-amino-2-(2′-hydroxyphenyl)benzoxazole (6A-HBO) were investigated with the time-dependent density functional theory (TD-DFT) method at the B3LYP/6-31G(d,p) theoretical level. The primary bond lengths and infrared (IR) vibrational spectra show that the intramolecular hydrogen bond is significantly strengthened in S1 state. The Mulliken's charge distribution and the frontier molecular orbitals (MOs) were analyzed. The result is consistent with the ESIPT mechanism proposed by Han and co-workers. Upon photo-excitation, the intramolecular hydrogen bond of 5A-HBO-enol (1.73 Å) and 6A-HBO-enol (1.74 Å) in the S1 state is weaker than that of HBO-enol (1.69 Å) due to the influence of the amino group in the HBO framework. After vertical excitation to the S1 state, the electronic density redistributes and migrates from the phenol ring to the benzoxazole ring of HBO. While for 5A-HBO and 6A-HBO, it transfers from the amino-benzoxazole moiety to the phenol ring. The analysis of the potential energy curves of HBO, 5A-HBO and 6A-HBO indicates that the ESIPT process of HBO occurs most easily. It is demonstrated that the presence and the position of the amino group in the HBO framework can change the behavior of the intramolecular hydrogen bonds O–H⋯N in the S1 state and thus hinder the ESIPT processes to some extent.
Co-reporter:Chaozheng Li, Donglin Li, Chi Ma, Yufang Liu
Journal of Molecular Liquids 2016 Volume 224(Part A) pp:83-88
Publication Date(Web):December 2016
DOI:10.1016/j.molliq.2016.09.088
•The intramolecular hydrogen bond plays an important role in the ESIPT reaction.•The ESIPT reaction is unlikely occurs in S0 state.•The electron acceptor NO2 and donor NH2 groups influence the ESIPT reactions in different way.The excited-state intramolecular proton transfer (ESIPT) reactions of 2-(2′-hydroxyphenyl)benzimidazole (HBI) derivatives were investigated using time-dependent density functional theory (TD-DFT) method at B3LYP/TZVP theoretical level. The geometric parameters, infrared (IR) vibrational spectra, frontier molecular orbitals (MOs), Mulliken charge distribution analysis, natural bond orbital (NBO) analysis and potential energy curves were calculated to provide the direct information about the effect of electron acceptor and donor groups on the ESIPT reactions. The intramolecular hydrogen bonds are significantly strengthened and the electronic density is redistributed after a vertical excitation to S1 state. The proton transfer (PT) reactions are unlikely occur in S0 state through the analysis of potential energy curves and the Hartree-Fock energy of the stable structures. The presence of the electron acceptor NO2 group can facilitate the ESIPT reaction, while the presence of electron donor NH2 group can hinder the ESIPT reaction. In general, the electron acceptor NO2 group and electron donor NH2 group can influence the ESIPT reactions in a completely different way.
Co-reporter:Chaozheng Li, Chi Ma, Donglin Li, Yufang Liu
Journal of Luminescence 2016 Volume 172() pp:29-33
Publication Date(Web):April 2016
DOI:10.1016/j.jlumin.2015.11.026
Time-dependent density functional theory (TD-DFT) method at B3LYP/6-31G(d,p) theoretical level was employed to investigate the excited-state intramolecular proton transfer (ESIPT) reaction of 6-amino-2-(2′-hydroxyphenyl)benzoxazole in dichloromethane and methanol solvents. Upon photo-excitation, the intramolecular hydrogen bond between the hydroxyl and neighboring N atom is significantly strengthened, which provides a driving force in facilitates the ESIPT reaction. The calculated steady-state absorption and fluorescence spectra agree well with the experimental results. Especially, after the photo-excitation from HOMO (π) to LUMO (π*), the rearrangement of electronic density distribution of frontier molecular orbitals (MOs) is a very important positive factor for the ESIPT process. The potential energy curves confirm that, after photo-excitation, the EXIPT reaction occurs with the H atom of the hydroxyl group remove to the neighboring N atom. By contrast, the potential barrier of the 6A-HBO-MeOH complex in the S1 state falls ca. 3.57 kcal/mol, lower than the isolated 6A-HBO. It is likely that the ESIPT reaction occurs more easily for the 6A HBO-MeOH complex due to the influence of the intermolecular hydrogen bonding.
Co-reporter:Siyuan Liu, Hongsheng Zhai, Yufang Liu
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2016 Volume 162() pp:115-120
Publication Date(Web):5 June 2016
DOI:10.1016/j.saa.2016.03.008
•The PECs of the 15 Λ–S states have been calculated for the first time.•The PECs of the 32 Ω states arising from 15 Λ-S states have been calculated.•The energy degeneracy among B2Σ + (II), C2Δ(I) and 2Σ−(I) states has been analyzed.•The double wells of C2Δ(I) ,2Σ−(I), 2Π(II) and 2Π(III) states have been studied.The internally contracted multi-reference configuration interaction method (MRCI) with Davidson modification and the Douglas–Kroll scalar relativistic correction has been used to calculate the BSe molecule at the level of aug-cc-pV5Z basis set. The calculated electronic states, including 9 doublet and 6 quartet Λ-S states, are correlated to the dissociation limit of B(2Pu) + Se(3Pg) and B(2Pu) + Se(1Dg). The Spin-orbit coupling (SOC) interaction is taken into account via the state interaction approach with the full Breit-Pauli Hamiltonian operator, which causes the entire 15 Λ-S states to split into 32 Ω states. This is the first time that the spin-orbit coupling calculation has been carried out on BSe. The potential energy curves of the Λ-S and Ω electronic states are depicted with the aid of the avoided crossing rule between electronic states of the same symmetry. The spectroscopic constants of the bound Λ-S and Ω states were determined, which are in good agreement with the experimental data. The transition dipole moments (TDMs) and the Franck-Condon factors (FCs) of the transitions from the low-lying bound Ω states A2Π(I)3/2, B2Π(I)1/2 and C2Δ(I)3/2 to the ground state X2Σ+1/2 have also been presented. Based on the previous calculations, the radiative lifetimes of the A2Π(I)3/2, B2Π(I)1/2 and C2Δ(I)3/2 were evaluated.The amplified view of the energy degeneracy of B2Σ+(II), C2Δ(I), 2Σ−(I), 2Δ(II) and 2Σ−(II) states.
Co-reporter:Yarui Shi, Huiling Wei, Yufang Liu
Journal of Molecular Structure 2015 Volume 1083() pp:65-71
Publication Date(Web):5 March 2015
DOI:10.1016/j.molstruc.2014.11.037
•We theoretically calculated the charge-transport and electrochemical data of the TAPP derivatives.•Our calculation is based on the Marcus–Hush electron-transfer theory with DFT calculations.•The core substitution of TAPPs has a drastic influence on the charge-transport mobilities.•Our work makes a profound study on the effect of the different substitution of the TAPPs in the different positions.Tetraazaperopyrenes (TAPPs) derivatives are high-performance n-type organic semiconductor material families with the remarkable long-term stabilities. The charge carrier mobilities in TAPPs derivatives crystals were calculated by the density functional theory (DFT) method combined with the Marcus–Hush electron-transfer theory. The existence of considerable C–H…F–C bonding defines the conformation of the molecular structure and contributes to its stability. We illustrated how it is possible to control the electronic and charge-transport parameters of TAPPs derivatives as a function of the positions, a type of the substituents. It is found that the core substitution of TAPPs has a drastic influence on the charge-transport mobilities. The maximum electron mobility value of the core-brominated 2,9-bis (perfluoroalkyl)-substituted TAPPs is 0.521 cm2 V−1 s−1, which appear in the orientation angle 95° and 275°. The results demonstrate that the TAPPs with bromine substituents in ortho positions exhibit the best charge-transfer efficiency among the four different TAPP derivatives.
Co-reporter:Chaozheng Li, Zhiqiang Fu, Xiaoqian Zhang, Yufang Liu, Yong Wang
Computational and Theoretical Chemistry 2015 Volume 1064() pp:45-50
Publication Date(Web):15 July 2015
DOI:10.1016/j.comptc.2015.04.014
•DFT calculations were carried out to study of the reaction mechanism.•The energy profile and optimized geometries of key intermediates are given.•The solvation effects play a significant role in determining the reaction mechanism.•The structural mimics we got can provides reference for the experiment.In this work, density functional theory (DFT) calculations were carried out to study the role of solvation effects on the reaction of diiron dithiolate complex with CO to form [Fe]-hydrogenase model complex. In the gas phase, the energy barrier of the first transition state TS1 species is ca. 6.1 kcal/mol higher than the second transition state TS2 species. However, when the solvation effects were included, the energy order was reversed, i.e., the energy barrier of TS1 falls ca. 1.2 kcal/mol lower than TS2, indicating that the insertion of the second CO to iron is the rate-determining step in the whole transformation process. The initial insertion of the CO plays an important role in increasing the reaction barrier of the binding of a second CO, which prevented the second step transformation. Thus, the solvation effects play a significant role in determining the reaction mechanism. In addition, the energy of PC species is lower than RC species, demonstrating that this transformation is a significantly exothermic process.
Co-reporter:Chi Ma, Yonggang Yang, Chaozheng Li, and Yufang Liu
The Journal of Physical Chemistry A 2015 Volume 119(Issue 51) pp:12686-12692
Publication Date(Web):November 30, 2015
DOI:10.1021/acs.jpca.5b09430
The 1,3-bis(2-pyridylimino)-4,7-dihydroxyisoindole (BPD) is chosen to investigate the excited-state double proton transfer process (ESDPT). The IR spectra, bond distance, and angle analyses show that the two intramolecular hydrogen bonds in the BPD molecule, formed between hydroxyl group and pyridine-type nitrogen atom, are significantly strengthened in the S1 state. The potential energy surfaces in both S0 and S1 states are scanned with varying O–H bond lengths to visually investigate the double proton transfer mechanism. Compared with previous investigations, the proton transfer process can be interpreted in more detail. The hydrogen bond strengthening promotes the proton transfer in the S1 state effectively. The large Stocks shift observed in the experiment can be explained more comprehensively according to the ESDPT mechanism.
Co-reporter:Hui Li;Yonggang Yang;Dapeng Yang, ;Jinfeng Sun
Journal of Physical Organic Chemistry 2014 Volume 27( Issue 3) pp:170-176
Publication Date(Web):
DOI:10.1002/poc.3255
The time-dependent density functional theory method was performed to investigate the excited-state hydrogen-bonding dynamics of N-(2-hydroxyethyl)-1,8-naphthalimide (2a) and N-(3-hydroxyethyl)-1,8-naphthalimide (3a) in methanol (meoh) solution. The ground and excited-state geometry optimizations, electronic excitation energies, and corresponding oscillation strengths of the low-lying electronically excited states for the complexes 2a + 2meoh and 3a + 2meoh as well as their monomers 2a and 3a were calculated by density functional theory and time-dependent density functional theory methods, respectively. We demonstrated that the three intermolecular hydrogen bonds of 2a + 2meoh and 3a + 2meoh are strengthened after excitation to the S1 state, and thus induce electronic spectral redshift. Moreover, the electronic excitation energies of the hydrogen-bonded complexes in S1 state are correspondingly decreased compared with those of their corresponding monomer 2a and 3a. In addition, the intramolecular charge transfer of the S1 state for complexes 2a + 2meoh and 3a + 2meoh were theoretically investigated by analysis of molecular orbital. Copyright © 2014 John Wiley & Sons, Ltd.
Co-reporter:Hui Li, Yufang Liu, Yonggang Yang, Dapeng Yang, Jinfeng Sun
Journal of Photochemistry and Photobiology A: Chemistry 2014 Volume 291() pp:9-15
Publication Date(Web):1 October 2014
DOI:10.1016/j.jphotochem.2014.06.017
•The intermolecular hydrogen bonding dynamics in the adsorption process has been first investigated in detail.•In the ground state, the trend of hydrogen bonding interactions facilitating adsorption is I > II and III > IV.•In the excited states, the hydrogen bond interactions of complexes I, II and III can facilitate adsorption, while that in complex IV is against adsorption.•The photoexcitation for complex IV should be controlled during the absorption process.The intermolecular hydrogen bonding dynamics between the functionalized activated carbons (AC) and ethyl mercaptan (ETM) were investigated using DFT and TDDFT methods, respectively. The ground-state and excited-state structures, electronic excitation energies and corresponding oscillation strengths of the low-lying electronically excited states for the functionalized ACs and their hydrogen-bonded complexes I, II, III and IV were calculated. It is demonstrated that the stability trend of forming hydrogen-bonded complexes is I > II and III > IV, which coincides with the results from the interaction energies calculation EHB. Moreover, according to Zhao's rule on the excited hydrogen bonding dynamics, it is found that the hydrogen bonds in the complexes I, II and III are significantly strengthened, with the excitation energy of a related excited state being red shifted, the hydrogen bond interactions can facilitate ETM adsorption onto AC-COOCO, AC-CCC and AC-CO, respectively. The intermolecular hydrogen bond of complex IV in the excited state is noteworthily weakened, in which the excitation energy of a related excited state is blue shifted. Therefore, the hydrogen bond interaction in complex IV is against ETM adsorption onto AC-OH in the excited state. The photoexcitation for complex IV should be controlled during the absorption process.
Co-reporter:Yanlei Liu, Hongsheng Zhai, Xiaomei Zhang, Yufang Liu
Chemical Physics 2013 Volume 425() pp:156-161
Publication Date(Web):8 November 2013
DOI:10.1016/j.chemphys.2013.09.002
Highlights
- •
24 Λ–S states are correlated to the dissociation limit of Si(3Pg) + Si+(2Pu) are first reported.
- •
The dissociation energies of the calculated electronic states are predicted in our work.
- •
It is first time that the entire 54 Ω states generated from the 24 Λ–S states have been studied.
- •
PECs of Λ–S and Ω states are depicted with the aid of avoided crossing rule between the same symmetry.
Co-reporter:Xiaomei Zhang, Hongsheng Zhai, Yufang Liu, Jinfeng Sun
Journal of Quantitative Spectroscopy and Radiative Transfer 2013 Volume 119() pp:23-31
Publication Date(Web):April 2013
DOI:10.1016/j.jqsrt.2013.01.020
The entire 12 Λ–S states of CCl+ correlated to ground state atom C+ and Cl are calculated at scalar relativistic MRCI+Q/AV5Z level of theory. Spin–orbit interaction causes the 12 Λ–S states to split into 23 Ω states. The potential energy curves (PECs) of Λ–S and Ω states are depicted with the aid of the avoided crossing rule between the same symmetry. This is the first time that spin–orbit coupling (SOC) calculation has been carried out on CCl+. The spin–orbit coupling effect, leading to many avoided crossings, is found to be substantial for CCl+. The spectroscopic constants of the bound Λ–S and Ω states are determined, where a better agreement with experimental data is found. The predissociations for a3Π and A1Π induced by SOC are analyzed. Moreover, the transition properties, including transition dipole moments and Franck–Condon factors, are derived. Subsequently, the radiative lifetimes of transition a3Π0+–X1Σ+0+ and a3Π1–X1Σ+0+ are calculated.Highlights► Spectroscopic constants for the 1Δ, 1Σ−, 1Σ+(II), 3Σ+(I), 3Δ, 3Σ−, 3П(II) and 1П(II) are firstly reported. ► The dissociation energies of the calculated electronic states are predicted in our work. ► It is first time that the entire 23 Ω states generated from the 12 Λ–S states have been studied. ► The predissociations for a3Пand A1П induced by SOC are firstly analyzed for CCl+. ► The transition properties including the TDMs, FC factors and radiative lifetimes are evaluated.
Co-reporter:Yufang Liu, Xiaomei Zhang, Kun Yu
Computational and Theoretical Chemistry 2012 Volume 991() pp:82-87
Publication Date(Web):1 July 2012
DOI:10.1016/j.comptc.2012.04.001
The internally contracted multi-reference configuration interaction method (MRCI) is used to calculate 22 Λ–S electronic states of BCl radical at the level of aug-cc-pVQZ basis set, where Davidson modification with the Douglas–Kroll scalar relativistic correction is also taken into account. This is the first time multi-reference configuration calculation on the excited state of BCl radical. Spin–orbit coupling effect is introduced by the state interaction approach with the full Breit–Pauli Hamiltonian operator for the two lowest excited and ground Λ–S states. The potential energy curves of the Λ–S and Ω electronic states are depicted according to the avoided crossing rule of the same symmetry. The spectroscopic constants for the bound Λ–S and Ω states are fitted, which are in good agreement with the corresponding observed values. The calculated excitation energy separation of a3Π0−−a3Π0+ is only 0.22 cm−1, while the energy intervals of a3Π0+−a3Π1 and a3Π1−a3Π2 are 47.85 cm−1 and 48.06 cm−1, respectively. In addition, transition dipole moments (TDMs) followed by Franck–Condon factors and radiative lifetimes for the transitions from a3Π0+, a3Π1 and A1Π1 to the ground state X1∑0++ are predicted as well.Graphical abstractIn this paper, 22 Λ–S electronic states of BCl radical are calculated with the internally contracted multi-reference configuration interaction method(MRCI) at the aug-cc-pVQZ level. The calculation is extended to including Davidson modification and the Douglas-Kroll scalar relativistic correction. The spin-orbit coupling effect is introduced by the state interaction approach with the full Breit-Pauli Hamiltonian operator for the two lowest excited and ground Λ–S states.Highlights► This is the first time that the MRCI + Q method has been used and SOC effect has been evaluated for BCl. ► The quintuplet electronic states of BCl radical are reported in our work for the first time. ► Spectroscopic constants for the 3Π(III), 5Π(I), 3Σ−(II) and 3Π(IV) are firstly reported. ► Complete potential energy curves of 22 Λ–S electronic states and 6 Ω states are depicted. ► The dissociation energies of the calculated electronic states are predicted in our work..
Co-reporter:Feng Zhang, Kun Yu, Kaihua Zhang, Yanlei Liu, Kaipin Xu, Yufang Liu
Infrared Physics & Technology (November 2015) Volume 73() pp:275-280
Publication Date(Web):1 November 2015
DOI:10.1016/j.infrared.2015.10.001
•A new emissivity measurement apparatus for near infrared spectrum is reported.•We present an improved method to minimize the measurement error.•The spectral emissivities of TA1, oxidized nickel and 304 austenitic stainless steel are measured and discussed.•The uncertainty of the apparatus is discussed and calculated in detailed.This study develops a new experimental apparatus for infrared spectral emissivity measurements which consists mainly of the following four parts: sample heating system, blackbody furnace, optical system, and data acquisition system. This apparatus focuses on the near-infrared spectral emissivity measurement covering the temperature range from 473 K to 1273 K and the wavelengths between 0.8 μm and 2.2 μm. The apparatus and the measurement method are described in detail, and an improved method is presented to minimize measurement error. The spectral emissivity of pure titanium TA1, oxidized nickel and 304 austenitic stainless steel are measured to validate the reliability and reproducibility of experimental apparatus. The experimental results in this study are in good agreement with those of other literatures. Various uncertainty sources in emissivity measurement are analyzed, and the combined standard uncertainty of this system is less than 3.9%.
Co-reporter:Fang Wang, Ming-Yuan Wang, Feng-Shuo Tian, Yu-Fang Liu, Lei Li, Jing Zhao
Radiation Measurements (June 2015) Volume 77() pp:1-4
Publication Date(Web):1 June 2015
DOI:10.1016/j.radmeas.2015.03.012
•A method to measure the X-ray radiation with low cost and miniaturization.•A general CMOS image sensor is used to detect X-ray.•The system can measure exposure rate and 2D distribution simultaneously.•The Elman algorithm is adopted to improve the precision of the radiation detector.The principle of the X-ray detector which can simultaneously perform the measurement of the exposure rate and 2D (two-dimensional) distribution is described. A commercially available CMOS image sensor has been adopted as the key part to receive X-ray without any scintillators. The correlation between the pixel value (PV) and the absorbed exposure rate of X-ray is studied using the improved Elman neural network. Comparing the optimal adjustment process of the BP (Back Propagation) neural network and the improved Elman neural network, the neural network parameters are selected based on the fitting curve and the error curve. The experiments using the practical production data show that the proposed method achieves high accurate predictions to 10−15, which is consistent with the anticipated value. It is proven that it is possible to detect the exposure rate using the X-ray detector with the improved Elman algorithm for its advantages of fast converges and smooth error curve.
Co-reporter:Donglin Li, Yonggang Yang, Chaozheng Li, Yufang Liu
Environmental Research (April 2017) Volume 154() pp:139-144
Publication Date(Web):1 April 2017
DOI:10.1016/j.envres.2016.12.027
•The hydrogen bond and Van Der Waals interactions play a significant role in MO adsorption by MPW and PANI.•The influence of photoexcitation on adsorption has been studied firstly in our work.•The adsorption of MO on MPW is less favorable in S1 state and the adsorption of MO on PANI is more favorable in S1 state.•The MO adsorption by MPW and PANI are spontaneous and exothermic.The dispersion-corrected density functional theory (DFT-D3) is used to investigate the mechanism of mesoporous pulp waste (MPW) and polyaniline (PANI) adsorptive removal methyl orange (MO) dye from their aqueous solutions. The results are absolutely reliable because of the sufficiently accurate method although such big systems are studied. It is demonstrated that hydrogen bond and Van Der Waals interactions play a significant role in MO adsorption by MPW and PANI. For MO adsorption by MPW, hydrogen bond and Van Der Waals interactions are both weakened in S1 state. In contrast, hydrogen bond and Van Der Waals interactions between PANI and MO are both enhanced in S1 state. The thermodynamic parameters such as enthalpy and free energy change reveal that the MO adsorption by MPW and PANI are spontaneous and exothermic. The adsorption of MO on MPW is less favorable in S1 state and the adsorption of MO on PANI is more favorable in S1 state. Therefore, the photoexcitation should be controlled during the MO adsorption by MPW and applied for MO adsorption by PANI.
Co-reporter:Donglin Li, Yonggang Yang, Chaozheng Li and Yufang Liu
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 24) pp:NaN15855-15855
Publication Date(Web):2017/05/18
DOI:10.1039/C7CP02268A
The fast and accurate detection of amyloid fibrils, which are associated with many neurodegenerative diseases, is important for their early diagnosis. {[50-(p-Hydroxyphenyl)-2,20-bithienyl-5-yl]-methylidene}-propanedinitrile (NIAD-4) is a new promising fluorescent marker for amyloid fibrils, and the photophysical behaviour of NIAD-4 is controversial. Nonadiabatic dynamic simulations, density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations were performed to determine the influence of the environment on NIAD-4 and the photophysical behaviour of NIAD-4. The results indicate that NIAD-4 is in the NIAD-4·3H2O compound form in the ground state in water. The torsion process of NIAD-4 proposed by Hu et al. (Phys. Chem. Chem. Phys. 2016, 18, 28) does not occur in the excited state. In addition, the fluorescence behaviour of NIAD-4 is sensitive to a hydrogen bonding environment, the maximum fluorescence wavelengths of NIAD-4 show considerable red-shifts, and the fluorescence intensity of NIAD-4 increases significantly in a hydrogen bonding environment. Intermolecular hydrogen bonds are vital for the phenomenon observed in the experiment because the fluorescence intensity of NIAD-4 becomes unusually high with increasing solvent polarities. Therefore, the influence of the intermolecular hydrogen bond should be carefully taken into consideration when NIAD-4 is used to probe the amyloid fibrils in hydrogen-bonding surroundings, especially in complex bioenvironments.
Co-reporter:Chaozheng Li, Yonggang Yang, Donglin Li and Yufang Liu
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 6) pp:NaN4808-4808
Publication Date(Web):2017/01/17
DOI:10.1039/C6CP07716A
The excited-state double proton transfer (ESDPT) mechanism in a model DNA base pair, 7-azaindole (7AI) dimer, has been debated over the years. Recently, Otero and coworkers concluded that the stepwise mechanism is not possible and the concerted mechanism dominates the dynamics (Chem. Sci., 2015, 6, 5762). In this work, the potential energy surfaces of the 7AI dimer in the ground state (S0) and the lowest energy excited singlet state (S1) were constructed. After vertical excitation to the S1 state, the single proton transfer can occur. The second proton transfer process in the stepwise mechanism is blocked by a high potential barrier (36.4 kcal mol−1), which is consistent with the result proposed by Otero and coworkers. However, the single proton transfer process is compatible with the concerted mechanism and we show that the single proton transfer process rather than the concerted mechanism dominates the dynamics. The concerted process is unfavorable in the S1 state compared with the barrierless single proton transfer process. In addition, the proton transfer process in the S0 state is revealed. The single proton transfer tautomer in the S1 state returns to the S0 state and transfers the second proton via a barrierless process. Finally, the double proton transfer tautomer in the S0 state can recover to the normal dimer through the reverse proton transfer reaction.








![1H-Benz[de]isoquinoline-1,3(2H)-dione,2-(2-hydroxyethyl)-](/data/chemimg/87900/5450-40-8.png)
![1H-Benz[de]isoquinoline-1,3(2H)-dione,2-(2-hydroxyethyl)-](/data/chemimg/87900/5450-40-8_b.png)