Co-reporter:Peter Comba;Markus Enders;Michael Großhauser;Markus Hiller;Dennis Müller;Hubert Wadepohl
Dalton Transactions 2017 vol. 46(Issue 1) pp:138-149
Publication Date(Web):2016/12/19
DOI:10.1039/C6DT03488H
Reported are the syntheses, structures and magnetic properties, also by NMR spectroscopy in solution, of a series of 13 linear trinuclear 3d–4f compounds with a lanthanide(III) surrounded by two NiII ions, NiII2LnIII, where the central LnIII is hexacoordinate. For three of the crystal structures, an additional H2O molecule is coordinated to the central LnIII ion, leading to a monocapped trigonal prismatic structure. However, NMR spectroscopy indicates that in solution, these complexes also have a hexacoordinate LnIII center. The solution magnetic anisotropies, determined by NMR spectroscopy, indicate that the axial components of the anisotropies are relatively small and that the DyIII derivative might therefore not exhibit single molecule magnetism. The axial anisotropies determined by NMR spectroscopy are in good agreement with the expectations based on the distorted trigonal prismatic ligand field.
Co-reporter:Marija S. Jeremić;Hubert Wadepohl;Vesna V. Kojić;Dimitar S. Jakimov;Ratomir Jelić;Suzana Popović;Zoran D. Matović
RSC Advances (2011-Present) 2017 vol. 7(Issue 9) pp:5282-5296
Publication Date(Web):2017/01/17
DOI:10.1039/C6RA26199J
Two rhodium(III) complexes [Rh(ed3a)(OH2)]·H2O (1) and Na[Rh(ed3a)Cl]·H2O (2) with ethylenediamine-N,N,N′-triacetate (ed3a) have been synthesized and characterized by elemental, spectroscopic and structural analyses. The crystal structure of (1) and (2) and the spectroscopic analysis of the two rhodium(III)–ed3a complexes are discussed in detail. The protonation constants of H3ed3a and the conditional stability constants of its RhIII complexes have been determined in aqueous solution by pH potentiometry and UV-Vis spectrophotometry. Molecular mechanics (MM) and density functional theory (DFT) have been used to model all possible geometric isomers, determine the global energy minimum and compare the computed with the experimentally observed structures. The cytotoxic activity of the new RhIII complexes was evaluated by an MTT assay against four human cancer lines (MCF-7, A549, HT-29 and HeLa) and a normal human cell line (MRC-5). A549, HT-29 and HeLa cells were sensitive to all compounds tested, while the breast carcinoma cell line MCF-7 was only sensitive to the reference compounds (doxorubicin and cisplatin). Western blot (WB) analysis of the effects of the tested compounds indicates that both complexes increase the expression of caspase 3 and consequently the involvement of this enzyme in apoptotic processes of the treated cells. WB also demonstrates proteolytic cleavage of poly-(ADP-ribose)polymerase (PARP) in HeLa cells after treatment with both tested substances. Flow cytometry confirmed apoptotic cell death and showed the induction of cell cycle termination as a possible promoter of apoptosis.
Co-reporter:Peter Comba, Laura Grimm, Chris Orvig, Katharina Rück, and Hubert Wadepohl
Inorganic Chemistry 2016 Volume 55(Issue 24) pp:12531-12543
Publication Date(Web):December 6, 2016
DOI:10.1021/acs.inorgchem.6b01787
The synthesis and CuII, NiII, ZnII, CoII, and GaIII coordination chemistry of the two isomeric hexadentate N5O ligands 6-[[9-hydroxy-1,5-bis(methoxycarbonyl)-7-methyl-6,8-bis(pyridin-2-yl)-3,7-diazabicyclo[3.3.1]nonan-3-yl]methyl]picolinic acid (Hbispa1a) and 6-[[9-hydroxy-1,5-bis(methoxycarbonyl)-7-methyl-2,4-bis(pyridin-2-yl)-3,7-diazabicyclo[3.3.1]nonan-3-yl]methyl]picolinic acid (Hbispa1b), picolinic acid-appended bispidines, are described. The two ligands are highly preorganized for octahedral coordination geometries and are particularly well suited for tetragonal symmetries, i.e., for Jahn–Teller labile ground states. This is confirmed by all data presented: solid-state structures, solution spectroscopy, electrochemistry, and CuII complex stabilities. Differences in the preorganization of the two isomers for the Jahn–Teller labile CuII centers are thoroughly analyzed on the basis of the crystal structures and an angular-overlap-model-based ligand-field analysis.
Co-reporter:Prasenjit Barman, Abayomi S. Faponle, Anil Kumar Vardhaman, Davide Angelone, Anna-Maria Löhr, Wesley R. Browne, Peter Comba, Chivukula V. Sastri, and Sam P. de Visser
Inorganic Chemistry 2016 Volume 55(Issue 20) pp:10170-10181
Publication Date(Web):October 5, 2016
DOI:10.1021/acs.inorgchem.6b01384
Reaction bifurcation processes are often encountered in the oxidation of substrates by enzymes and generally lead to a mixture of products. One particular bifurcation process that is common in biology relates to electron transfer versus oxygen atom transfer by high-valent iron(IV)–oxo complexes, which nature uses for the oxidation of metabolites and drugs. In biomimicry and bioremediation, an important reaction relates to the detoxification of ClOx– in water, which can lead to a mixture of products through bifurcated reactions. Herein we report the first three water-soluble non-heme iron(II) complexes that can generate chlorine dioxide from chlorite at ambient temperature and physiological pH. These complexes are highly active oxygenation oxidants and convert ClO2– into either ClO2 or ClO3¯ via high-valent iron(IV)–oxo intermediates. We characterize the short-lived iron(IV)–oxo species and establish rate constants for the bifurcation mechanism leading to ClO2 and ClO3– products. We show that the ligand architecture of the metal center plays a dominant role by lowering the reduction potential of the metal center. Our experiments are supported by computational modeling, and a predictive valence bond model highlights the various factors relating to the substrate and oxidant that determine the bifurcation pathway and explains the origins of the product distributions. Our combined kinetic, spectroscopic, and computational studies reveal the key components necessary for the future development of efficient chlorite oxidation catalysts.
Co-reporter:Biljana Đ. Glišić, Lidija Senerovic, Peter Comba, Hubert Wadepohl, Aleksandar Veselinovic, Dusan R. Milivojevic, Miloš I. Djuran, Jasmina Nikodinovic-Runic
Journal of Inorganic Biochemistry 2016 Volume 155() pp:115-128
Publication Date(Web):February 2016
DOI:10.1016/j.jinorgbio.2015.11.026
•Synthesis and characterization of silver(I) complexes with phthalazine and quinazoline•Biological activities of the silver(I) complexes•High activity of the silver(I) complexes against Pseudomonas aeruginosa strains•Binding of the silver(I) complexes to genomic DNA from Pseudomonas aeruginosaFive silver(I) complexes with aromatic nitrogen-containing heterocycles, phthalazine (phtz) and quinazoline (qz), were synthesized, characterized and analyzed by single-crystal X-ray diffraction analysis. Although different AgX salts reacted with phtz, only dinuclear silver(I) complexes of the general formula {[Ag(X-O)(phtz-N)]2(μ-phtz-N,N′)2} were formed, X = NO3− (1), CF3SO3− (2) and ClO4− (3). However, reactions of qz with an equimolar amount of AgCF3SO3 and AgBF4 resulted in the formation of polynuclear complexes, {[Ag(CF3SO3-O)(qz-N)]2}n (4) and {[Ag(qz-N)][BF4]}n (5). Complexes 1–5 were evaluated by in vitro antimicrobial studies against a panel of microbial strains that lead to many skin and soft tissue, respiratory, wound and nosocomial infections. The obtained results indicate that all tested silver(I) complexes have good antibacterial activity with MIC (minimum inhibitory concentration) values in the range from 2.9 to 48.0 μM against the investigated strains. Among the investigated strains, these complexes were particularly efficient against pathogenic Pseudomonas aeruginosa (MIC = 2.9–29 μM) and had a marked ability to disrupt clinically relevant biofilms of strains with high inherent resistance to antibiotics. On the other hand, their activity against the fungus Candida albicans was moderate. In order to determine the therapeutic potential of silver(I) complexes 1–5, their antiproliferative effect on the human lung fibroblastic cell line MRC5, has been also evaluated. The binding of complexes 1–5 to the genomic DNA of P. aeruginosa was demonstrated by gel electrophoresis techniques and well supported by molecular docking into the DNA minor groove. All investigated complexes showed an improved cytotoxicity profile in comparison to the clinically used AgNO3.Five new silver(I) complexes with aromatic N-heterocycles phthalazine and quinazoline show potent activity against pathogenic Pseudomonas aeruginosa and can be used to prevent infections due to P. aeruginosa biofilms.
Co-reporter:Simone Bosch, Peter Comba, Lawrence R. Gahan, Gerhard Schenk
Journal of Inorganic Biochemistry 2016 Volume 162() pp:343-355
Publication Date(Web):September 2016
DOI:10.1016/j.jinorgbio.2015.12.028
•The first heterodinuclear model compound that is able to hydrolyze phosphomonoesters•Hydrogen bonding is shown to be of importance for substrate hydrolysis.•Asymmetric dinuclear sites are shown to be of importance for functional modeling.Derivatives of the known dinucleating ligands HL1 (2,6-bis{[bis(pyridin-2-ylmethyl)amino]methyl}-4-methylphenol) and H2L2 (2-{[bis(pyridin-2-ylmethyl)amino]methyl}-6-{[(2-hydroxybenzyl)(pyridine-2-ylmethyl)amino]methyl}-4-methylphenol) with two pivaloylamido hydrogen bond donor substituents, H3L3 and H3L5, have been prepared. The mono-, homo- and heterodinuclear ZnII and GaIII complexes of these ligands have been prepared and characterized. The solution equilibria are discussed on the basis of extensive NMR spectroscopic, mass spectrometric and pH-dependent UV–vis spectroscopic titrations. The phosphoester hydrolysis activity of the complexes has been studied as a function of pH and substrate concentration and analyzed using Michaelis–Menten kinetics. It emerges that the mixed metal (mixed valent) complex of the ligand with an asymmetric disposition of the hydrogen bonding substituents (H3L3) is a functional model for the mixed valent, dinuclear metallohydrolase purple acid phosphatase. This complex combines the essential structural features of the active site of PAP and is the first heterodinuclear model complex mimicking the essential function of PAPs, i.e. the hydrolysis of phosphomonoesters.The GaIIIZnII complex of an asymmetric dinucleating ligand with second coordination sphere hydrogen bond donors is shown to be not only a structural model of purple acid phosphatases but is also able to mimic the essential function of these enzymes, i.e. the hydrolysis of phosphomonoesters.
Co-reporter:Paul V. Bernhardt; Simone Bosch; Peter Comba; Lawrence R. Gahan; Graeme R. Hanson; Valeriu Mereacre; Christopher J. Noble; Annie K. Powell; Gerhard Schenk;Hubert Wadepohl
Inorganic Chemistry 2015 Volume 54(Issue 15) pp:7249-7263
Publication Date(Web):July 21, 2015
DOI:10.1021/acs.inorgchem.5b00628
The active site of mammalian purple acid phosphatases (PAPs) have a dinuclear iron site in two accessible oxidation states (FeIII2 and FeIIIFeII), and the heterovalent is the active form, involved in the regulation of phosphate and phosphorylated metabolite levels in a wide range of organisms. Therefore, two sites with different coordination geometries to stabilize the heterovalent active form and, in addition, with hydrogen bond donors to enable the fixation of the substrate and release of the product, are believed to be required for catalytically competent model systems. Two ligands and their dinuclear iron complexes have been studied in detail. The solid-state structures and properties, studied by X-ray crystallography, magnetism, and Mössbauer spectroscopy, and the solution structural and electronic properties, investigated by mass spectrometry, electronic, nuclear magnetic resonance (NMR), electron paramagnetic resonance (EPR), and Mössbauer spectroscopies and electrochemistry, are discussed in detail in order to understand the structures and relative stabilities in solution. In particular, with one of the ligands, a heterovalent FeIIIFeII species has been produced by chemical oxidation of the FeII2 precursor. The phosphatase reactivities of the complexes, in particular, also of the heterovalent complex, are reported. These studies include pH-dependent as well as substrate concentration dependent studies, leading to pH profiles, catalytic efficiencies and turnover numbers, and indicate that the heterovalent diiron complex discussed here is an accurate PAP model system.
Co-reporter:Peter Comba, Michael Großhauser, Rüdiger Klingeler, Changhyun Koo, Yanhua Lan, Dennis Müller, Jaena Park, Annie Powell, Mark J. Riley, and Hubert Wadepohl
Inorganic Chemistry 2015 Volume 54(Issue 23) pp:11247-11258
Publication Date(Web):November 20, 2015
DOI:10.1021/acs.inorgchem.5b01673
A series of seven isostructural homodinuclear lanthanide complexes are reported. The magnetic properties (ac and dc SQUID measurements) are discussed on the basis of the X-ray structural properties which show that the two lanthanide sites are structurally different. MCD spectroscopy of the dysprosium(III) and neodymium(III) complexes ([DyIII2(L)(OAc)4]+ and [NdIII2(L)(OAc)4]+) allowed us to thoroughly analyze the ligand field, and high-frequency EPR spectroscopy of the gadolinium(III) species ([GdIII2(L)(OAc)4]+) showed the importance of dipolar coupling in these systems. An extensive quantum-chemical analysis of the dysprosium(III) complex ([DyIII2(L)(OAc)4]+), involving an ab initio (CASSCF) wave function, explicit spin–orbit coupling (RASSI-SO), and a ligand field analysis (Lines model and Stevens operators), is in full agreement with all experimental data (SQUID, HF-EPR, MCD) and specifically allowed us to accurately simulate the experimental χT versus T data, which therefore allowed us to establish a qualitative model for all relaxation pathways.
Co-reporter:Mala A. Sainna, Debangsu Sil, Dipankar Sahoo, Bodo Martin, Sankar Prasad Rath, Peter Comba, and Sam P. de Visser
Inorganic Chemistry 2015 Volume 54(Issue 4) pp:1919-1930
Publication Date(Web):January 22, 2015
DOI:10.1021/ic502803b
We report the synthesis, structure, and spectroscopic characterization of 1,2-bis[μ-hydroxo iron(III) 5-(2,3,7,8,12,13,17,18-octaethylporphyrinyl)]ethane with PF6– and SbF6– counteranions. The two iron centers are nonequivalent with admixed intermediate spin state (S = 3/2 with a minor contribution of S = 5/2) on each metal both in the solid and in solution. The molecules are compared with previously known μ-hydroxo complexes with other counterions, such as I3–, BF4–, and ClO4–, which demonstrates that the nature of the counterion can affect the spin-state ordering dramatically. To understand how the spin-state ordering is affected by external perturbations, we also have done a comprehensive computational study. The calculations show that subtle environmental perturbations affect the spin-state ordering and relative energies and are likely to be the root cause of the variation in spin-state ordering observed experimentally.
Co-reporter:Peter Comba, Henning Rudolf and Hubert Wadepohl
Dalton Transactions 2015 vol. 44(Issue 6) pp:2724-2736
Publication Date(Web):14 Nov 2014
DOI:10.1039/C4DT03262D
Reported is the new bispidine-derived hexadentate ligand L (L = 3-(2-methylpyridyl)-7-(bis-2-methylpyridyl)-3,7-diazabicyclo[3.3.1]nonane) with two tertiary amine and four pyridine donor groups. This ligand can form heterodinuclear and mononuclear complexes and, in the mononuclear compounds discussed here, the ligand may coordinate as a pentadentate ligand, with one of the bispyridinemethane-based pyridine groups un- or semi-coordinated, or as a hexadentate ligand, leading to a pentagonal pyramidal coordination geometry or, with an additional monodentate ligand, to a heptacoordinate pentagonal bipyramidal structure. The solution and solid state data presented here indicate that, with the relatively small CuII and high-spin FeII ions the fourth pyridine group is only semi-coordinated for steric reasons and, with the larger high-spin MnII ion genuine heptacoordination is observed but with a relatively large distortion in the pentagonal equatorial plane.
Co-reporter:Dominik Brox, Peter Comba, Dirk-Peter Herten, Esther Kimmle, Michael Morgen, Carmen L. Rühl, Arina Rybina, Holger Stephan, Golo Storch, Hubert Wadepohl
Journal of Inorganic Biochemistry 2015 Volume 148() pp:78-83
Publication Date(Web):July 2015
DOI:10.1016/j.jinorgbio.2015.05.009
•Bispidines are CuII-selective with fast complexation and high stabilities.•Bispidine–dye conjugates with cyanine or rhodamine fluorescence tags are used as on-off CuII sensors.•Quenching of the pseudo-Jahn–Teller effect leads to a decrease of the complex stability.The substitution of tetradentate bispidine ligands with rhodamine and cyanine dye molecules, coupled to an amine donor, forming an amide as potential fifth donor, is described. Bispidines are known to lead to very stable CuII complexes, and the coordination to CuII was expected to efficiently quench the fluorescence of dye molecules. However, at physiological pH the amide is not coordinated, as shown by titration experiments and crystallographic structural data of three possible isomers of these complexes. This may be due to the specific cavity shape of bispidines and the Jahn–Teller lability of the CuII center. While CuII coordination in aqueous solution leads to efficient fluorescence quenching, experiments show that the complex stabilities are not large enough for CuII sensing in biological media, and possibilities are discussed, how this may be achieved by optimized bispidine–dye conjugates.Shown in the pictogram is a highly Cu2 +-selective bispidine ligand, coupled to Cy5.5. Coordination of Cu2 + efficiently quenches the fluorescence of the Cy5.5–bispidine conjugate.
Co-reporter:Dr. Simone Bosch;Dr. Peter Comba; Lawrence R. Gahan;Dr. Graeme R. Hanson;Dr. Christopher Noble;Dr. Gerhard Schenk;Dr. Hubert Wadepohl
Chemistry - A European Journal 2015 Volume 21( Issue 50) pp:18269-18279
Publication Date(Web):
DOI:10.1002/chem.201503348
Abstract
Complexation studies of the dinucleating ligand H3L (H3L=2-{[bis(pyridin-2-ylmethyl)amino]methyl}-6-{[bis(6-pivaloylamidopyridin-2-ylmethyl)amino]methyl}-4-methylphenol), with metal-binding sites A and B, which both provide four donors to a metal ion; a tertiary amine; two pyridines (substituted with amide hydrogen-bond donors in site B), and a bridging phenolate, with ZnII, CuII, and GaIII are reported. The titration of H3L with the three metal ions in solution was monitored by NMR spectroscopy or EPR and UV/Vis/near-IR spectroscopy, as well as by ESI-MS to analyze the selectivity of the two metal-ion sites A and B of this model ligand for metallophosphatases; the spectroscopic assignments are supported by X-ray crystallography results. The first ZnII ion coordinates to site A with unsubstituted pyridine donors and, upon addition of a second equivalent of ZnII, this coordinates to the sterically less accessible site B. From a similar titration with GaIII, it emerges that only a mononuclear complex is obtained, with the GaIII center coordinated to site A. When one equivalent of GaIII is reacted with the mononuclear ZnII complex, ZnII is forced by GaIII to exchange the site; this results in a dinuclear complex with GaIII in site A and ZnII in site B. With CuII, two isomers are observed: one with and the other without a bridging phenolate; these differ significantly in their spectroscopic and magnetic properties.
Co-reporter:Peter Comba, Yong-Min Lee, Wonwoo Nam and Arkadius Waleska
Chemical Communications 2014 vol. 50(Issue 4) pp:412-414
Publication Date(Web):31 Oct 2013
DOI:10.1039/C3CC47013J
Organic substrates (specifically cis-1,2-dimethylcyclohexane, DMCH) are oxidized by O2 in the presence of iron(II)–bispidine complexes. It is shown that this oxidation reaction is not based on O2 activation by the nonheme iron catalysts as in Nature but due to a radical-based initiation, followed by a radical- and ferryl-based catalytic reaction.
Co-reporter:Sam P. de Visser, Matthew G. Quesne, Bodo Martin, Peter Comba and Ulf Ryde
Chemical Communications 2014 vol. 50(Issue 3) pp:262-282
Publication Date(Web):29 Oct 2013
DOI:10.1039/C3CC47148A
With computational resources becoming more efficient and more powerful and at the same time cheaper, computational methods have become more and more popular for studies on biochemical and biomimetic systems. Although large efforts from the scientific community have gone into exploring the possibilities of computational methods for studies on large biochemical systems, such studies are not without pitfalls and often cannot be routinely done but require expert execution. In this review we summarize and highlight advances in computational methodology and its application to enzymatic and biomimetic model complexes. In particular, we emphasize on topical and state-of-the-art methodologies that are able to either reproduce experimental findings, e.g., spectroscopic parameters and rate constants, accurately or make predictions of short-lived intermediates and fast reaction processes in nature. Moreover, we give examples of processes where certain computational methods dramatically fail.
Co-reporter:Peter Comba, Manja Kubeil, Jens Pietzsch, Henning Rudolf, Holger Stephan, and Kristof Zarschler
Inorganic Chemistry 2014 Volume 53(Issue 13) pp:6698-6707
Publication Date(Web):June 6, 2014
DOI:10.1021/ic500476u
The three new dioxo-tetraazamacrocyclic ligands with a fused, very rigid bispidine (3,7-diazabicyclo[3.3.1]nonane) group connecting the two tertiary amine donors, and ethyl, propyl, or benzene groups connecting the two amide donors are highly preorganized and lead to very stable, uncharged CuII complexes. Solution spectroscopy and solid state structures indicate that these are square pyramidal with a solvent molecule occupying the apical position. Cyclic voltammetry defines a reversible CuIII/II couple and a strongly negative irreversible CuII/I couple (ca. −2 V vs Fc/Fc+), indicating that the CuII complexes are very stable in solution. This is supported by superoxide dismutase (SOD) and human serum challenge experiments as well as the biodistribution, which all show that the benzene-based ligand has the highest in vitro and in vivo stability and that this was expected on the basis of the macrocycle ring size and shape and the highest degree of preorganization. This ligand is easy to functionalize for a possible coupling to biological vector molecules and/or fluorescence markers for PET (positron emission tomography) and multimodal imaging (i.e., PET and optical imaging).
Co-reporter:Simone Bosch, Peter Comba, Lawrence R. Gahan, and Gerhard Schenk
Inorganic Chemistry 2014 Volume 53(Issue 17) pp:9036-9051
Publication Date(Web):August 14, 2014
DOI:10.1021/ic5009945
It is becoming increasingly apparent that the secondary coordination sphere can have a crucial role in determining the functional properties of biomimetic metal complexes. We have therefore designed and prepared a variety of ligands as metallo-hydrolase mimics, where hydrogen bonding in the second coordination sphere is able to influence the structure of the primary coordination sphere and the substrate binding. The assessment of a structure–function relationship is based on derivates of 2,6-bis{[bis(pyridin-2-ylmethyl)amino]methyl}-4-methylphenol (HBPMP = HL1) and 2-{[bis(pyridin-2-ylmethyl)amino]methyl}-6-{[(2-hydroxybenzyl)(pyridin-2-ylmethyl)amino]methyl}-4-methylphenol (H2BPBPMP = H2L5), well-known phenolate-based ligands for metallo-hydrolase mimics. The model systems provide similar primary coordination spheres but site-specific modifications in the secondary coordination sphere. Pivaloylamide and amine moieties were chosen to mimic the secondary coordination sphere of the phosphatase models, and the four new ligands H3L2, H3L3, HL4, and H4L6 vary in the type and geometric position of the H-bond donors and acceptors, responsible for the positioning of the substrate and release of the product molecules. Five dinuclear ZnII complexes were prepared and structurally characterized in the solid, and four also in solution. The investigation of the phosphatase activity of four model complexes illustrates the impact of the H-bonding network: the Michaelis–Menten constants (catalyst–substrate binding) for all complexes that support hydrogen bonding are smaller than for the reference complex, and this generally leads to higher catalytic efficiency and higher turnover numbers.
Co-reporter:Peter Comba, Lawrence R. Gahan, Graeme R. Hanson, Marcel Maeder and Michael Westphal
Dalton Transactions 2014 vol. 43(Issue 8) pp:3144-3152
Publication Date(Web):02 Dec 2013
DOI:10.1039/C3DT53135J
The dicopper(II) complexes of six pseudo-octapeptides, synthetic analogues of ascidiacyclamide and the patellamides, found in ascidians of the Pacific and Indian Oceans, are shown to be efficient carbonic anhydrase model complexes with kcat up to 7.3 × 103 s−1 (uncatalyzed: 3.7 × 10−2 s−1; enzyme-catalyzed: 2 × 105–1.4 × 106 s−1) and a turnover number (TON) of at least 1700, limited only by the experimental conditions used. So far, no copper-based natural carbonic anhydrases are known, no faster model systems have been described and the biological role of the patellamide macrocycles is so far unknown. The observed CO2 hydration rates depend on the configuration of the isopropyl side chains of the pseudo-octapeptide scaffold, and the naturally observed R*,S*,R*,S* geometry is shown to lead to more efficient catalysts than the S*,S*,S*,S* isomers. The catalytic efficiency also depends on the heterocyclic donor groups of the pseudo-octapeptides. Interestingly, the dicopper(II) complex of the ligand with four imidazole groups is a more efficient catalyst than that of the close analogue of ascidiacyclamide with two thiazole and two oxazoline rings. The experimental observations indicate that the nucleophilic attack of a CuII-coordinated hydroxide at the CO2 carbon center is rate determining, i.e. formation of the catalyst-CO2 adduct and release of carbonate/bicarbonate are relatively fast processes.
Co-reporter:Peter Comba, Nina Dovalil, Lawrence R. Gahan, Graeme R. Hanson and Michael Westphal
Dalton Transactions 2014 vol. 43(Issue 5) pp:1935-1956
Publication Date(Web):30 Oct 2013
DOI:10.1039/C3DT52664J
Cyclic pseudo-peptides derived from marine metabolites of the genus Lissoclinum bistratum and Lissoclinum patella have attracted scientific interest in the last two decades. Their structural properties and solution dynamics have been analyzed in detail, elaborate synthetic procedures for the natural products and synthetic derivatives developed, the biosynthetic pathways studied and it now is possible to produce them biosynthetically. Initially, these macrocyclic ligands were studied due to their medicinal and pharmaceutical potential – some of the isolated cyclic pseudo-peptides show high cytotoxic and antiviral activity. A major focus in the last decade has been on their CuII coordination chemistry, as a number of studies have indicated that dinuclear CuII complexes of cyclic peptides may be involved in the ascidians’ metabolism, and this is the focus of the present review.
Co-reporter:Dr. Holger Stephan;Dr. Martin Walther;Dr. Silke Fähnemann; Paola Ceroni;Dr. Jennifer K. Molloy;Dr. Giacomo Bergamini;Dr. Fabian Heisig; Christa E. Müller;Werner Kraus; Peter Comba
Chemistry - A European Journal 2014 Volume 20( Issue 51) pp:17011-17018
Publication Date(Web):
DOI:10.1002/chem.201404086
Abstract
The efficient transformation of the hexadentate bispidinol 1 into carbamate derivatives yields functional bispidines enabling convenient functionalization for targeted imaging. The BODIPY-substituted bispidine 3 combines a coordination site for metal ions, such as radioactive 64CuII, with a fluorescent unit. Product 3 was thoroughly characterized by standard analytical methods, single crystal X-ray diffraction, radiolabeling, and photophysical analysis. The luminescence of ligand 3 was found to be strongly dependent on metal ion coordination: CuII quenches the BODIPY fluorescence, whereas NiII and ZnII ions do not affect it. It follows that, in imaging applications with the positron emitter 64CuII, residues of its origin from enriched 64Ni and the decay products 64NiII and 64ZnII, efficiently restore the fluorescence of the ligand. This allows for monitoring of the emitted radiation as well as the fluorescence signal. The stability of the 64CuII3 complex is investigated by transmetalation experiments with ZnII and NiII, using fluorescence and radioactivity detection, and the results confirm the high stability of 64CuII3. In addition, metal complexes of ligand 3 with the lanthanide ions TbIII, EuIII, and NdIII are shown to exhibit emission of the BODIPY ligand and the lanthanide ion, thus enabling dual emission detection.
Co-reporter:Dong Wang, Kallol Ray, Michael J. Collins, Erik R. Farquhar, Jonathan R. Frisch, Laura Gómez, Timothy A. Jackson, Marion Kerscher, Arkadius Waleska, Peter Comba, Miquel Costas and Lawrence Que
Chemical Science 2013 vol. 4(Issue 1) pp:282-291
Publication Date(Web):25 Oct 2012
DOI:10.1039/C2SC21318D
Oxoiron(IV) species have been found to act as the oxidants in the catalytic cycles of several mononuclear nonheme iron enzymes that activate dioxygen. To gain insight into the factors that govern the oxidative reactivity of such complexes, a series of five synthetic S = 1 [FeIV(O)(LN5)]2+ complexes has been characterized with respect to their spectroscopic and electrochemical properties as well as their relative abilities to carry out oxo transfer and hydrogen atom abstraction. The FeO units in these five complexes are supported by neutral pentadentate ligands having a combination of pyridine and tertiary amine donors but with different ligand frameworks. Characterization of the five complexes by X-ray absorption spectroscopy reveals FeO bonds of ca. 1.65 Å in length that give rise to the intense 1s → 3d pre-edge features indicative of iron centers with substantial deviation from centrosymmetry. Resonance Raman studies show that the five complexes exhibit ν(FeO) modes at 825–841 cm−1. Spectropotentiometric experiments in acetonitrile with 0.1 M water reveal that the supporting pentadentate ligands modulate the E1/2(IV/III) redox potentials with values ranging from 0.83 to 1.23 V vs. Fc, providing the first electrochemical determination of the E1/2(IV/III) redox potentials for a series of oxoiron(IV) complexes. The 0.4 V difference in potential may arise from differences in the relative number of pyridine and tertiary amine donors on the LN5 ligand and in the orientations of the pyridine donors relative to the FeO bond that are enforced by the ligand architecture. The rates of oxo-atom transfer (OAT) to thioanisole correlate linearly with the increase in the redox potentials, reflecting the relative electrophilicities of the oxoiron(IV) units. However this linear relationship does not extend to the rates of hydrogen-atom transfer (HAT) from 1,3-cyclohexadiene (CHD), 9,10-dihydroanthracene (DHA), and benzyl alcohol, suggesting that the HAT reactions are not governed by thermodynamics alone. This study represents the first investigation to compare the electrochemical and oxidative properties of a series of S = 1 FeIVO complexes with different ligand frameworks and sheds some light on the complexities of the reactivity of the oxoiron(IV) unit.
Co-reporter:Peter Comba, Sebastian Hunoldt, Michael Morgen, Jens Pietzsch, Holger Stephan, and Hubert Wadepohl
Inorganic Chemistry 2013 Volume 52(Issue 14) pp:8131-8143
Publication Date(Web):July 2, 2013
DOI:10.1021/ic4008685
Pentadentate bispidine ligands (3,7-diazabicyclo[3.3.1]nonanes) are optimized for maximum complex stability and facile functionalization with respect to their coupling to biological vector molecules and/or fluorescence markers for PET (positron emission tomography) and multimodal imaging (i.e., PET and optical imaging). The pentadentate ligand with two tertiary amine donors, two p-methoxy substituted pyridines, and one unsubsituted pyridine group is shown to best fulfill important conditions for PET applications, i.e., fast complexation with CuII and high in vivo stability, and this was predicted from the solution chemistry, in particular the CuII/I redox potentials. Also, solvent partition experiments to model the lipophilicity of the CuII complexes indicate that the bis p-methoxy substituted ligand leads to cationic complexes with an appreciable lipophilicity. This is supported by the biodistribution experiments that show that the complex with the p-methoxy substituted ligand is excreted very quickly and primarily via the renal route and therefore is ideally suited for the development of PET tracers with ligands of this type coupled to biomolecules.
Co-reporter:Peter Comba, Michael Morgen, and Hubert Wadepohl
Inorganic Chemistry 2013 Volume 52(Issue 11) pp:6481-6501
Publication Date(Web):May 10, 2013
DOI:10.1021/ic4004214
Bispidines (3,7-diazabicyclo[3.3.1]nonanes) as very rigid and highly preorganized ligands find broad application in the field of coordination chemistry, and the redox potentials of their transition-metal complexes are of importance in oxidation reactions by high-valent iron complexes, aziridination catalyzed by copper complexes, and imaging by 64Cu positron emission tomography tracers. Here, we show that the redox potentials and stability constants of the copper(II) complexes of 15 tetradentate bispidines can be varied by substitution of the pyridine rings (variation of the redox potential over ca. 450 mV and of the complex stability over approximately 10 log units). It is also shown that these variations are predictable by the pKa values of the pyridine groups as well as by the Hammett parameters of the substituents, and the density functional theory based energy decomposition analysis also allows one to accurately predict the redox potentials and concomitant complex stability. It is shown that the main contribution emerges from the electrostatic interaction energy, and the partial charges of the pyridine donor groups therefore also correlate with the redox potentials.
Co-reporter:Peter Comba, Bodo Martin, Avik Sanyal and Holger Stephan
Dalton Transactions 2013 vol. 42(Issue 31) pp:11066-11073
Publication Date(Web):07 Jun 2013
DOI:10.1039/C3DT51049B
A QSPR scheme for the computation of lipophilicities of 64Cu complexes was developed with a training set of 24 tetraazamacrocylic and bispidine-based CuII compounds and their experimentally available 1-octanol–water distribution coefficients. A minimum number of physically meaningful parameters were used in the scheme, and these are primarily based on data available from molecular mechanics calculations, using an established force field for CuII complexes and a recently developed scheme for the calculation of fluctuating atomic charges. The developed model was also applied to an independent validation set and was found to accurately predict distribution coefficients of potential 64Cu PET (positron emission tomography) systems. A possible next step would be the development of a QSAR-based biodistribution model to track the uptake of imaging agents in different organs and tissues of the body. It is expected that such simple, empirical models of lipophilicity and biodistribution will be very useful in the design and virtual screening of positron emission tomography (PET) imaging agents.
Co-reporter:Peter Comba, Franziska Emmerling, Maik Jakob, Werner Kraus, Manja Kubeil, Michael Morgen, Jens Pietzsch and Holger Stephan
Dalton Transactions 2013 vol. 42(Issue 17) pp:6142-6148
Publication Date(Web):27 Nov 2012
DOI:10.1039/C2DT32356G
The CuII complex of H4TETP (H4TETP = 1,4,8,11-tetraazatetradecane-1,4,8,11-tetrapropionic acid) is five-coordinate with a distorted square-pyramidal structure (τ = 0.45; i.e. the geometry is nearly half-way between square-pyramidal and trigonal-bipyramidal) and a relatively long Cu–N and a short Cu–O bond; the comparison between powder and solution electronic spectroscopy, the frozen solution EPR spectrum and ligand-field-based calculations (angular overlap model, AOM) indicate that the solution and solid state structures are very similar, i.e. the complex has a relatively low “in-plane” and a significant axial ligand field with a dx2−y2 ground state. The ligand-enforced structure is therefore shown to lead to a partially quenched Jahn–Teller distortion and to a relatively low complex stability, lower than with the corresponding acetate-derived ligand H4TETA. This is confirmed by potentiometric titration and by the biodistribution with 64Cu-labeled ligands which show that the uptake in the liver is significantly increased with the H4TETP-based system.
Co-reporter:Peter Comba, Lawrence R. Gahan, Graeme R. Hanson and Michael Westphal
Chemical Communications 2012 vol. 48(Issue 75) pp:9364-9366
Publication Date(Web):27 Jul 2012
DOI:10.1039/C2CC34836E
A possible biological function of cyclic pseudo-octapeptides is presented. The dinuclear copper(II) complex of a synthetic analogue ([Cu2(H2Pat1)(μ-OH)(OH2)2]) of the naturally occurring ascidiacyclamide is known to have a hydroxo-bridged dicopper(II) site which is able to catalytically transform CO2 into CO32−. This complex is shown here to function as a phosphatase mimic, suggesting that the so far unknown biological function of these macrocycles within the ascidians may involve phosphoester hydrolysis.
Co-reporter:Peter Comba ; Christina Haaf ; Stefan Helmle ; Kenneth D. Karlin ; Shanthi Pandian ;Arkadius Waleska
Inorganic Chemistry 2012 Volume 51(Issue 5) pp:2841-2851
Publication Date(Web):February 14, 2012
DOI:10.1021/ic2019296
The reactivity of copper complexes of three different second-generation bispidine-based ligands (bispidine = 3,7-diazabicyclo[3.3.1]nonane; mono- and bis-tetradentate; exclusively tertiary amine donors) with dioxygen [(reversible) binding of dioxygen by copper(I)] is reported. The UV–vis, electrospray ionization mass spectrometry, electron paramagnetic resonance, and vibrational spectra (resonance Raman) of the dioxygen adducts indicate that, depending on the ligand and reaction conditions, several different species (mono- and dinuclear, superoxo, peroxo, and hydroperoxo), partially in equilibrium with each other, are formed. Minor changes in the ligand structure and/or experimental conditions (solvent, temperature, relative concentrations) allow switching between the different forms. With one of the ligands, an end-on peroxodicopper(II) complex and a mononuclear hydroperoxocopper(II) complex could be characterized. With another ligand, reversible dioxygen binding was observed, leading to a metastable superoxocopper(II) complex. The amount of dioxygen involved in the reversible binding to CuI was determined quantitatively. The mechanism of dioxygen binding as well as the preference of each of the three ligands for a particular dioxygen adduct is discussed on the basis of a computational (density functional theory) analysis.
Co-reporter:Mihail Atanasov ; Peter Comba ;Stefan Helmle
Inorganic Chemistry 2012 Volume 51(Issue 17) pp:9357-9368
Publication Date(Web):August 20, 2012
DOI:10.1021/ic301122h
We report here the synthesis and characterization of four dinuclear cyanide-bridged FeIII–CuII complexes, based on a tetra- or a pentadentate bispidine ligand (L1 or L2, respectively; bispidines are 3,7-diazabiyclo[3.3.1]nonane derivatives) coordinated to the CuII center, and a tridentate bipyridineamide (bpca) coordinated to the low-spin FeIII site, with cyanide groups completing the two coordination spheres, one of them bridging between the two metal ions. The four structurally characterized complexes [{Fe(bpca)(CN)3}{Cu(L1·H2O)}]BF4, [{Fe(bpca)(CN)3}{Cu(L2)}][Fe(bpca)(CN)3]·5H2O, [{Fe(bpca)(CN)3}{Cu(L2·MeOH)}]PF6·MeOH·H2O, and [{Fe(bpca)(CN)3}{Cu(L2)}]PF6·2H2O belong to different structural isomers. The most important differences are structurally and electronically enforced (direction of the pseudo-Jahn–Teller mode) strong or weak interactions of the copper(II) center with the cyanide bridge. The related strength of the magnetic coupling of the two centers is analyzed with a combination of experimental magnetic, electron paramagnetic resonance (EPR), electronic spectroscopic data together with a ligand-field theory- and density functional theory (DFT)-based analysis.
Co-reporter:Peter Comba ; Bodo Martin ; Amsaveni Muruganantham ;Johannes Straub
Inorganic Chemistry 2012 Volume 51(Issue 17) pp:9214-9225
Publication Date(Web):August 21, 2012
DOI:10.1021/ic3004917
Oxygen activation by copper(I) complexes with tetra- or pentadentate mono- or dinucleating bispidine ligands is known to lead to unusually stable end-on-[{(bispidine)Cu}2(O2)]2+ complexes (bispidines are methyl-2,4-bis(2-pyridin-yl)-3,7-diazabicyclo-[3.3.1]-nonane-9-diol-1,5-dicarboxylates); catecholase activity of these dinuclear CuII/I systems has been demonstrated experimentally, and the mechanism has been thoroughly analyzed. The present density functional theory (DFT) based study provides an analysis of the electronic structure and catalytic activity of [{(bispidine)Cu}2(O2)]2+. As a result of the unique square pyramidal coordination geometry, the dx2–y2 ground state leads to an unusual σ/π bonding pattern, responsible for the stability of the peroxo complex and the observed catecholase activity with a unique mechanistic pathway. The oxidation of catechol to ortho-quinone (one molecule per catalytic cycle and concomitant formation of one equivalent of H2O2) is shown to occur via an associative, stepwise pathway. The unusual stability of the end-on-peroxo-dicopper(II) complex and isomerization to copper(II) complexes with chelating catecholate ligands, which inhibit the catalytic cycle, are shown to be responsible for an only moderate catalytic activity.
Co-reporter:Mihail Atanasov, Peter Comba, Stefan Helmle, Dennis Müller, and Frank Neese
Inorganic Chemistry 2012 Volume 51(Issue 22) pp:12324-12335
Publication Date(Web):October 26, 2012
DOI:10.1021/ic3016047
The synthesis, single-crystal X-ray structures, electronic absorption spectra, and magnetic properties of six NiII complexes with a tetradentate (L1) and three pentadentate (L2, L3, L4) bispidine ligands (3,7-diazabicyclo[3.3.1]nonane derivatives), Ni(L1·H2O)(OH2)2](PF6)2, [Ni(L1·H2O)(O2NO)]NO3, [Ni(L1·H2O)(OOCCH3)]PF6, [Ni(L2·H2O)NCMe](PF6)2, [Ni(L3·H2O)OH2](PF6)2, and [Ni(L4·H2O)NCMe](PF6)2 are reported. The Ni–donor bonding to pyridine and tertiary amine groups and oxygen- or nitrogen-bound coligands, completing the octahedral coordination sphere of NiII, is analyzed using a combination of ab initio electronic structure calculations (complete active space self-consistent field, CASSCF, followed by N-electron valence perturbation theory, NEVPT2) and angular overlap ligand field analysis. Magnetic properties are rationalized with an analysis of the magnetic anisotropy in terms of zero-field splitting and g-tensor parameters, obtained from first principles, and their correlation with the NiII–donor bonding parameters from the ligand field analysis of the ab initio results. A two-dimensional spectrochemical series of the ligands considered, according to their σ and π bonding to NiII, is also derived.
Co-reporter:Peter Comba, Lawrence R. Gahan, Valeriu Mereacre, Graeme R. Hanson, Annie K. Powell, Gerhard Schenk, and Marta Zajaczkowski-Fischer
Inorganic Chemistry 2012 Volume 51(Issue 22) pp:12195-12209
Publication Date(Web):October 30, 2012
DOI:10.1021/ic301347t
Two new dinucleating ligands (H3L2 and HL3), derivatives of a well-known dinucleating ligand (HL1) with two bis-picolylamine sites connected to a bridging phenolate, with hydrogen-bonding donor groups at two of the pyridine moieties were designed and synthesized. Design of these ligands suggests that they will lead to dinuclear complexes with potential to stabilize phosphoester substrates as monodentate rather than bridging ligands. We report the diferric complexes [FeIII2(H2L2)(OH)]4+ and [FeIII2(L3)(OH)(OH2)2]4+, which have been characterized by spectrophotometric titrations, UV–vis, IR, NMR, EPR, and Mössbauer spectroscopy. The phosphatase activity of the diferric systems, in addition to the partially reduced heterovalent [FeIIIFeII(L3)(OH)(OH2)2]3+ complex, has been investigated, and the complexes are shown to catalytically hydrolyze the activated phosphodiester substrate BDNPP (bis-dinitrophenylphosphate) as well as the corresponding phosphomonoester substrate DNPP (dinitrophenylphosphate). The results indicate that indeed the secondary interactions lead to an increase of the phosphatase activity and to active phosphomonoesterase catalysts. Interestingly, the heterovalent form of the HL3-based complex is more efficient than the diferric complex, and this is also discussed.
Co-reporter:Dr. Peter Comba;Dr. Lawrence R. Gahan;Dr. Graeme R. Hanson;Dr. Valeriu Mereacre;Dr. Christopher J. Noble;Dr. Annie K. Powell;Ion Prisecaru;Dr. Gerhard Schenk;Dr. Marta Zajaczkowski-Fischer
Chemistry - A European Journal 2012 Volume 18( Issue 6) pp:1700-1710
Publication Date(Web):
DOI:10.1002/chem.201100229
Abstract
The synthesis and characterization of a novel dinucleating ligand L (L=4,11-dimethyl-1,8-bis{2-[N-(di-2-pyridylmethyl)amino]ethyl}cyclam) and its μ-oxo-bridged diferric complex [(H2L){FeIII2(O)}(Cl)4]2+ are reported. This diiron(III) complex is the first example of a truly functional purple acid phosphatase (PAP) mimic as it accelerates the hydrolysis of the activated phosphomonoester 2,4-dinitrophenyl phosphate (DNPP). The spectroscopic and kinetic data indicate that only substrates that are monodentately bound to one of the two ferric ions can be attacked by a suitable nucleophile. This is, most probably, a terminal iron(III)-bound hydroxide. DFT calculations support this assumption and also highlight the importance of secondary interactions, exerted by the protonated cyclam platform, for the positioning and activation of the iron(III)-bound substrate. Similar effects are postulated in the native enzyme but addressed in PAP mimics for the first time.
Co-reporter:Dr. Peter Comba;Dr. Nina Dovalil;Dr. Lawrence R. Gahan;Dr. Gebhard Haberhauer;Dr. Graeme R. Hanson;Dr. Christopher J. Noble;Dr. Björn Seibold;Dr. Prabha Vadivelu
Chemistry - A European Journal 2012 Volume 18( Issue 9) pp:2578-2590
Publication Date(Web):
DOI:10.1002/chem.201101975
Abstract
Two synthetic derivatives of the naturally occurring cyclic pseudooctapeptides patellamide A–F and ascidiacyclamide, that is, H4pat2, H4pat3, as well as their CuII complexes are described. These cyclic peptide derivatives differ from the naturally occurring macrocycles by the variation of the incorporated heterocyclic donor groups and the configuration of the amino acids connecting the heterocycles. The exchange of the oxazoline and thiazole groups by dimethylimidazoles or methyloxazoles leads to more rigid macrocycles, and the changes in the configuration of the side chains leads to significant differences in the folding of the cyclic peptides. These variations allow a detailed study of the various possible structural changes on the chemistry of the CuII complexes formed. The coordination of CuII with these macrocyclic species was monitored by high-resolution electrospray mass spectrometry (ESI-MS), spectrophotometric (UV/Vis) and circular dichroic (CD) titrations, and electron paramagnetic resonance (EPR) spectroscopy. Density functional theory (DFT) calculations and molecular mechanics (MM) simulations have been used to model the structures of the CuII complexes and provide a detailed understanding of their geometric preferences and conformational flexibility. This is related to the CuII coordination chemistry and the reactivity of the dinuclear CuII complexes towards CO2 fixation. The variation observed between the natural and various synthetic peptide systems enables conclusions about structure–reactivity correlations, and our results also provide information on why nature might have chosen oxazolines and thiazoles as incorporated heterocycles.
Co-reporter:Peter Comba ; Nina Dovalil ; Graeme R. Hanson ;Gerald Linti
Inorganic Chemistry 2011 Volume 50(Issue 11) pp:5165-5174
Publication Date(Web):May 12, 2011
DOI:10.1021/ic2004694
The synthesis and CuII coordination chemistry of the cyclic pseudo-octapeptide H4pat1, a dimethyl-imidazole analogue of naturally occurring cyclic peptides (patellamide A–F, ascidiacyclamide) is reported. Substitution of the oxazoline and thiazole heterocycles by dimethyl-imidazoles leads to a slightly different structure of the macrocycle in the solid state. The CuII coordination chemistry of H4pat1, monitored with high-resolution electrospray mass spectrometry, spectrophotometric titrations, and EPR spectroscopy, revealed the presence of both mono- and dinuclear CuII complexes. The dimethyl-imidazole analogue shows a high cooperativity in CuII coordination, that is, the preferred formation of dinuclear complexes. The dinuclear unbridged CuII complexes of H4pat1 have unusual EPR features, reminiscent of those of patellamide D: the dipole–dipole interaction of the CuII centers is negligible due to the “magic angle” orientation of the two CuII ions. Density functional theory calculations (DFT) are used to model the structures of the CuII complexes, and the structural assignments from the spectroscopic investigations are supported by the optimized and by X-ray structures of the metal-free macrocycle and dinuclear CuII complexes of H4pat1. The rigidity of the dimethyl-imidazole rings has a significant effect on the structures of the metal-free ligands and CuII complexes and therefore changes the properties of these compounds. This may explain why Nature has chosen the thiazole–oxazoline combination for the patellamides and ascidiacyclamide.
Co-reporter:Mihail Atanasov, Peter Comba, Graeme R. Hanson, Sascha Hausberg, Stefan Helmle, and Hubert Wadepohl
Inorganic Chemistry 2011 Volume 50(Issue 15) pp:6890-6901
Publication Date(Web):June 28, 2011
DOI:10.1021/ic102430a
The synthesis and structural analysis (single crystal X-ray data) of two mononuclear ([Cu(L1)(CN)]BF4 and [Cu(L3)(CN)](BF4)) and three related, cyanide-bridged homodinuclear complexes ([{Cu(L1)}2(CN)](BF4)3·1.35 H2O, [{Cu(L2)}2(CN)](BF4)3 and [{Ni(L3)}2(CN)](BF4)3) with a tetradentate (L1) and two isomeric pentadentate bispidine ligands (L2, L3; bispidines are 3,7-diazabicyclo[3.3.1]nonane derivatives) are reported, together with experimental magnetic, electron paramagnetic resonance (EPR), and electronic spectroscopic data and a ligand-field-theory-based analysis. The temperature dependence of the magnetic susceptibilities and EPR transitions of the dicopper(II) complexes, together with the simulation of the EPR spectra of the mono- and dinuclear complexes leads to an anisotropic set of g- and A-values, zero-field splitting (ZFS) and magnetic exchange parameters (Cu1: gz = 2.055, gx = 2.096, gy = 2.260, Az = 8, Ax = 8, Ay = 195 × 10–4 cm–1, Cu2: g and A as for Cu1 but rotated by the Euler angles α = −6°, β = 100°, Dexc = −0.07 cm–1, Eexc/Dexc = 0.205 for [{Cu(L1)}2(CN)](BF4)3·1.35 H2O; Cu1,2: gz = 2.025, gx = 2.096, gy = 2.240, Az = 8, Ax = 8, Ay = 190 × 10–4cm–1, Dexc = −0.159 cm–1, Eexc/Dexc = 0.080 for [{Cu(L2)}2(CN)](BF4)3). Thorough ligand-field-theory-based analyses, involving all micro states and all relevant interactions (Jahn–Teller and spin–orbit coupling) and DFT calculations of the magnetic exchange leads to good agreement between the experimental observations and theoretical predictions. The direction of the symmetric magnetic anisotropy tensor Dexc in [{Cu(L2)}2(CN)](BF4)3 is close to the Cu···Cu vector (22°), that is, nearly perpendicular to the Jahn–Teller axis of each of the two CuII centers, and this reflects the crystallographically observed geometry. Antisymmetric exchange in [{Cu(L1)}2(CN)](BF4)3·1.35 H2O causes a mixing between the singlet ground state and the triplet excited state, and this also reflects the observed geometry with a rotation of the two CuII sites around the Cu···Cu axis.
Co-reporter:Madhavan Jaccob, Peter Comba, Martin Maurer, Prabha Vadivelu and Ponnambalam Venuvanalingam
Dalton Transactions 2011 vol. 40(Issue 42) pp:11276-11281
Publication Date(Web):29 Sep 2011
DOI:10.1039/C1DT11533B
Iron-bispidine complexes are efficient catalysts for the oxidation of thioanisole to phenylmethylsulfoxide with iodosylbenzene as oxidant. With the tetradentate bispidine ligand L1 (L1 = 2,4-pyridyl-3,7-diazabicyclo[3.3.1]nonane)) the catalytic efficiency is smaller than with the pentadentate bispidine ligand L2 (L2 = 2,4-pyridyl-7-(pyridine-2-ylmethyl)-3,7-diazabicyclo[3.3.1]nonane)). Based on the redox potentials (iron complexes with L1 are stronger oxidants than with L2) and known efficiencies in catalytic olefin oxidation and C–H activation reactions, the expectations were different. A DFT-based analysis is used to explain the apparent contradiction, and this is based on differences in the electronic ground states of the ferryl complexes as well as in the oxygen transfer transition states.
Co-reporter:Peter Comba;Hubert Wadepohl ;Sebastian Wiesner
European Journal of Inorganic Chemistry 2011 Volume 2011( Issue 16) pp:2610-2615
Publication Date(Web):
DOI:10.1002/ejic.201100212
Abstract
High-valent bispidine iron-oxo complexes are among the most efficient nonheme iron oxidation catalysts. Here, we report the synthesis and structural analysis of two derivatives of the known pentadentate bispidine ligand L1 {L1 = 2,4-pyridyl-7-(pyridine-2-ylmethyl)-3,7-diazabicyclo[3.3.1]nonane}: L2 and L3 {L2 = 2,4-pyridyl-7-[1-(pyridine-2-yl)ethyl]-3,7-diazabicyclo[3.3.1]nonane; L3 = 2,4-pyridyl-7-[phenyl(pyridine-2-yl)methyl]-3,7-diazabicyclo[3.3.1]nonane}, and of their FeII complexes. The yield of the catalytic epoxidation of cyclooctene and styrene with iodosylbenzene as oxidant increases from the L1- to the L2- to the L3-based catalyst (e.g., the yield of styrene oxide, with MeCN as solvent, under anaerobic conditions, is 40 %, 90 %, 96 %, respectively), and this is correlated to the FeIII/II reduction potentials {[Fe(L1)(NCMe)]n+ (1.01 V), [Fe(L2)(NCMe)]n+ (1.13 V), and [Fe(L3)(NCMe)]n+ (1.19 V), in MeCN, vs. SCE}. Although this correlation is not unexpected, the interpretation is not entirely trivial, and this is discussed in detail. The rigidity of the bispidine ligands and their preference for relatively large metal ions (low oxidation states, high spin multiplicities) is believed to be responsible for the efficiency of the bispidine-based catalyst systems, and the present results show possible approaches to further improve the performance of these catalysts.
Co-reporter:Peter Comba;Hubert Wadepohl ;Steffen Wunderlich
European Journal of Inorganic Chemistry 2011 Volume 2011( Issue 34) pp:5242-5249
Publication Date(Web):
DOI:10.1002/ejic.201100802
Abstract
The iron–bispidine-catalyzed oxidation and dioxygenation (catechol dioxygenase activity) [i.e., the oxidation of 3,5-di-tert-butylcatecholate (dbc2–) by [FeII(L)X2]n+ (L = 3,7-dimethyl-9-oxo-2,4-bis(2-pyridyl)-3,7-diazabicyclo[3.3.1]nonane-1,5-dicarboxylate methyl ester)] and air (O2) was studied experimentally and supported by the analysis of the X-ray crystal structure of [Fe(L·MeOH)(tcc)][B(Ph)4] with the deactivated tetrachlorocatecholate tcc2– and a DFT-based analysis. The [FeII(L)X2]n+/O2/dbc2– system catalyzes the intradiol cleavage of dbc2– but with a relatively low activity (5 % yield); most of the substrate is oxidized in a two-electron oxidation to the benzoquinone (dbq) product (48 % yield). The crystallographic and DFT-based theoretical analyses indicate that this is due to the high oxidation potential of the FeIII oxidant (fast and efficient electron transfer), that is, the oxidation to the benzoquinone side product is faster, and due to the bonding mode of the catecholate substrate to the FeIII oxidant, with little spin density transferred to the catecholate substrate.
Co-reporter:Peter Comba, Shanthi Pandian, Hubert Wadepohl, Sebastian Wiesner
Inorganica Chimica Acta 2011 Volume 374(Issue 1) pp:422-428
Publication Date(Web):1 August 2011
DOI:10.1016/j.ica.2011.02.047
The structures and spectroscopic properties of various conformations of two diasteromeric pairs of enantiomers of pentacoordinate CuII bispidine complexes with chiral tetradentate ligands are reported. With one of the ligands an interesting type of distortional isomerism was observed experimentally, and this was studied in detail on the basis of the experimental structural and spectroscopic data (UV–Vis–NIR, EPR) and a DFT-, MM- and ligand-field-theory-based analysis.Graphical abstractWith the chiral tetradentate bispidine ligands L, there are two isomeric CuII structures with strikingly different CuII-N distances and N-CuII-N angles. These are thoroughly analyzed by X-ray crystallography, spectroscopy and computational methods.Highlights► One of the most convincing examples of a set of two isomers which are distortional isomers. ► Both are found in the same unit cell and are structurally fully characterized. ► Combination of structural, computational and spectroscopic work. ► Excellent agreement between computed and experimental structures.
Co-reporter:Peter Comba;Gerald Linti
Journal of Inclusion Phenomena and Macrocyclic Chemistry 2011 Volume 71( Issue 3-4) pp:331-337
Publication Date(Web):2011 December
DOI:10.1007/s10847-011-9967-9
In the endo-conformation of the substituted cyclam derivative L, with two trans-disposed di-2-pyridylmethanamine (dipa) coordination sites (endo: both dipa subunits on the same face of cyclam), the bis-dipa-substituted cyclam platform may form hexacoordinate mononuclear complexes with the two dipa subunits coordinated to one metal ion or dinuclear complexes, when the two dipa subunits are coordinated to two metal ions (oligonuclear linear chain complexes with exo-configured ligands L and metal ions coordinated to the cyclam unit have not been observed so far). Here, the structures, relative stabilities and spectroscopic properties of the mononuclear complexes of CuII and ZnII, which are formed in preference to other structural possibilities, are discussed, and the preference for their formation is also evaluated.
Co-reporter:Jordi Benet-Buchholz, Peter Comba, Antoni Llobet, Stephan Roeser, Prabha Vadivelu and Sebastian Wiesner
Dalton Transactions 2010 vol. 39(Issue 13) pp:3315-3320
Publication Date(Web):16 Feb 2010
DOI:10.1039/B924614B
Two RuIVO catalysts with either a pentadentate bispidine ligand L1 or a bidentate pyrazolate L2/terpy L3 combination of ligands have very different efficiencies as oxygen transfer catalysts for the selective oxidation of sulfides to sulfoxides: the [RuII(L1)(solvent)]2+/iodosyl benzene system has an initial TOF of approx. 40 h−1 and quantitative yield, with [RuII(L2)(L3)(solvent)]+ the initial TOF is approx. 12 h−1 with a maximum yield of approx. 60%. By experiment (cyclovoltametry) it is shown that there is S- to O-linkage isomerization of the RuII sulfoxide product complex, and this may partially switch off the catalytic cycle for the L2/L3-based catalyst. It emerges that the reasons for the reduced efficiency in the case of the L2/L3, in comparison with the L1-based catalyst, are a more efficient linkage isomerization, a more stable S-bonded, in comparison with the O-bonded, RuII-based isomer, and inefficient ligand exchange in the product (hydrolysis produces the free sulfoxide and the RuII precatalyst). These interpretations are qualitatively in good agreement with preliminary DFT-based data.
Co-reporter:Peter Comba Dr.;Shunichi Fukuzumi Dr.;Hiroaki Kotani Dr.;Steffen Wunderlich
Angewandte Chemie 2010 Volume 122( Issue 14) pp:2679-2682
Publication Date(Web):
DOI:10.1002/ange.200904427
Co-reporter:Peter Comba;Nina Dovalil
JBIC Journal of Biological Inorganic Chemistry 2010 Volume 15( Issue 7) pp:1129-1135
Publication Date(Web):2010 September
DOI:10.1007/s00775-010-0673-7
The CuII coordination chemistry of three synthetic analogues of westiellamide (H3Lwa) with an [18]azacrown-6 macrocyclic structure and imidazole (H3L1), oxazole (H3L2), or thiazole (H3L3) heterocyclic donors in addition to the peptide groups, is reported. The Nheterocycle–Npeptide–Nheterocycle binding sites are highly preorganized for the coordination to CuII ions. The stability constants of mono- and dinuclear CuII complexes of H3L1, H3L2, and H3L3, obtained by isothermal titration microcalorimetry, are reported. EPR and NMR spectroscopy as well as electrospray ionization mass spectrometry (ESI-MS) were used to characterize the complexes formed in solution. The stabilities of the mononuclear and dinuclear CuII complexes of the three ligands are in the range of 105 M−1, but there are subtle differences; specifically the oxazole-derived ligand has, in contrast to the other two macrocycles, a negative formation entropy for coordination to the first CuII ion and a higher stability for complexation to a second CuII center in comparison with the first CuII center (cooperativity). Differences between the three ligands are also apparent in terms of the formation mechanism. With the oxazole-based ligand H3L2, NMR spectroscopy, EPR spectroscopy, and ESI-MS indicate the formation of a ligand–CuII 2:1 intermediate, and this may explain the differences in the formation entropy as well as the cooperativity.
Co-reporter:Peter Comba Dr. ;Steffen Wunderlich
Chemistry - A European Journal 2010 Volume 16( Issue 24) pp:7293-7299
Publication Date(Web):
DOI:10.1002/chem.201000092
Abstract
When the dichloroiron(II) complex of the tetradentate bispidine ligand L=3,7-dimethyl-9-oxo-2,4-bis(2-pyridyl)-3,7-diazabicyclo[3.3.1]nonane-1,5-dicarboxylate methyl ester is oxidized with H2O2, tBuOOH, or iodosylbenzene, the high-valent FeO complex efficiently oxidizes and halogenates cyclohexane. Kinetic D isotope effects and the preference for the abstraction of tertiary over secondary carbon-bound hydrogen atoms (quantified in the halogenation of adamantane) indicate that CH activation is the rate-determining step. The efficiencies (yields in stoichiometric and turnover numbers in catalytic reactions), product ratios (alcohol vs. bromo- vs. chloroalkane), and kinetic isotope effects depend on the oxidant. These results suggest different pathways with different oxidants, and these may include iron(IV)– and iron(V)–oxo complexes as well as oxygen-based radicals.
Co-reporter:Peter Comba Dr.;Shunichi Fukuzumi Dr.;Hiroaki Kotani Dr.;Steffen Wunderlich
Angewandte Chemie International Edition 2010 Volume 49( Issue 14) pp:2622-2625
Publication Date(Web):
DOI:10.1002/anie.200904427
Co-reporter:Mihail Atanasov, Peter Comba, Sascha Hausberg, Bodo Martin
Coordination Chemistry Reviews 2009 Volume 253(19–20) pp:2306-2314
Publication Date(Web):October 2009
DOI:10.1016/j.ccr.2009.01.033
The anisotropy barrier and blocking temperature of single-molecule magnets (SMMs) depend on the total spin S (and therefore to some extent on the size of the molecule) as well as on the Ising-type axial magnetic anisotropy D. There is a relatively large anisotropy of the magnetic exchange across cyanide bridges and therefore, moderate-sized oligonuclear complexes may lead to appreciably high anisotropy barriers. Also, the geometry of cyanide bridges and the preference of metal ions for a specific linkage isomer allow predicting oligonuclear structures based on relatively simple building blocks of low nuclearity. Theoretical approaches have been developed to understand the nature of the magnetic exchange through the cyanide bridge and to predict magnetic exchange and anisotropy of larger spin clusters on the basis of theoretical approaches which combine ligand field theory with DFT-based methods. These models and consequences for the design and synthesis of SMM materials are discussed in detail.
Co-reporter:Peter Comba, Marion Kerscher
Coordination Chemistry Reviews 2009 Volume 253(5–6) pp:564-574
Publication Date(Web):March 2009
DOI:10.1016/j.ccr.2008.05.019
The computation of electronic structures of transition metal complexes has been developed in recent years to an extent where a large variety of spectroscopic properties and reactivities of mono- and oligonuclear transition metal compounds can be efficiently and reliably computed and interpreted with ab initio and semi-empirical quantum-chemical methods. These computations are often based on known structural data, and the interpretation of the electronic structures usually involves the comparison of computed and experimentally observed spectroscopic data and/or reactivities. The prediction of molecular properties, which eventually may lead to a rational design of novel complexes with given properties, requires as an important additional step a reliable structure prediction. The identification of factors which influence molecular structures of transition metal complexes and the ensuing approaches for a reliable structure optimization are an important basis for electronic structure calculations, and this is discussed in detail.
Co-reporter:Peter Comba, Christina Haaf and Hubert Wadepohl
Inorganic Chemistry 2009 Volume 48(Issue 14) pp:6604-6614
Publication Date(Web):May 20, 2009
DOI:10.1021/ic900571v
Four very rigid second generation bispidine-based ligands (bispidine = 3,7-diazabicyclo[3.3.1]nonane; tetra-, penta- and hexadentate; exclusively tertiary amine donors except for one of the pentadentate ligands, where one of the donors is a pyridyl group) and their CoII, NiII, CuII, and ZnII complexes are reported. The experimentally determined X-ray crystal structures and computational data, based on empirical force field (MM) and approximate density functional theory (DFT) calculations, indicate that these new ligands, which are based on a modular system and therefore allow for a wide range of donor sets and coordination geometries, have rather large cavities (i.e., lead to a preference for +II over +III oxidation states and induce relatively low ligand fields), enforce trigonal geometries (pentacoordinate systems: preference for trigonal bipyramidal, hexacoordinate complexes: preference for trigonal prismatic), and lead, especially for CuII, to very high complex stabilities.
Co-reporter:Peter Comba, Martin Maurer and Prabha Vadivelu
Inorganic Chemistry 2009 Volume 48(Issue 21) pp:10389-10396
Publication Date(Web):October 8, 2009
DOI:10.1021/ic901702s
The iron-bispidine-catalyzed oxidation of cyclohexane with H2O2, where either a tetradentate or a pentadentate bispidine ligand is coordinated to the iron center, yields up to 35% cyclohexanol and cyclohexanone (alcohol/ketone ratio of up to 4). Product distribution (including 18O labeling studies), kinetic isotope effects, and the ratio of tertiary/secondary alcohols with adamantane as a substrate (3°/2°) suggest that (i) H abstraction by a ferryl complex is the rate-determining step and that the emerging cyclohexyl radical is short-lived, (ii) there is a parallel reaction involving oxidation by OH radicals, and (iii) there are considerable differences in the reaction pathways between the tetradentate and pentadentate ligand catalyst. These interpretations are fully supported by a DFT-based computational analysis.
Co-reporter:Stefanie Juran, Martin Walther, Holger Stephan, Ralf Bergmann, Jörg Steinbach, Werner Kraus, Franziska Emmerling and Peter Comba
Bioconjugate Chemistry 2009 Volume 20(Issue 2) pp:347
Publication Date(Web):January 27, 2009
DOI:10.1021/bc800461e
The preparation and use of bispidine derivatives (3,7-diazabicyclo[3.3.1]nonane) as chelate ligands for radioactive copper isotopes for diagnosis (64Cu) or therapy (67Cu) are reported. Starting from the hexadentate bispidine-based bis(amine)tetrakis(pyridine) ligand 1 with a keto and two ester substituents, the corresponding mono-ol 2 and two dicarboxylic acid derivatives 3 and 5 have been synthesized. A range of techniques, including single-crystal X-ray structure analysis, UV/vis spectroscopy, cyclic voltammetry, thin-layer- (TLC), and high-performance liquid chromatography (HPLC), have been used to characterize the structure and stability of the copper(II)-bispidine complexes. A rapid formation (within 1 min) of stable copper(II)-bispidine complexes under mild conditions (ambient temperature, aqueous solution) has been observed. Challenge experiments of these complexes in the presence of a high excess of competing ligands, such as glutathione, cyclam, or superoxide dismutase (SOD), as well as in rat plasma, gave no evidence of demetalation or transchelation. The bifunctional bispidine derivative 5 can be readily functionalized with biologically active molecules at the pendant carboxylate groups. The coupling of a bombesin analogue βhomo-Glu-βAla-βAla-[Cha13,Nle14]BBN(7−14), by condensation of a carboxylate of the bispidine backbone with the N-terminus of the peptide produced the bifunctional ligand 6. The radiocopper(II) complex of this bombesin−bispidine conjugate has a considerable hydrophilicity (log Do/w < −2.4), and this leads to a very fast blood clearance (blood: 0.28 ± 0.02 SUV, 1 h p.i.), low liver tissue accumulation (liver: 1.20 ± 0.27 SUV, 1 h p.i.), and rapid renal-urinary excretion (kidneys: 6.06 ± 2.96 SUV, 1 h p.i.) as shown by biodistribution studies of 64Cu-6 in Wistar rats. Preliminary in vivo studies of 64Cu-6 in NMRI nu/nu mice, bearing the human prostate tumor PC-3 showed an accumulation of the conjugate in the tumor (2.25 ± 0.13 SUV, 12.5 min p.i.; 0.94 ± 0.05 SUV, 55 min p.i.) and allowed a clear visualization of the gastrin-releasing peptide receptor distribution by positron emission tomography (PET).
Co-reporter:Jordi Benet-Buchholz, Peter Comba, Antoni Llobet, Stephan Roeser, Prabha Vadivelu, Hubert Wadepohl and Sebastian Wiesner
Dalton Transactions 2009 (Issue 30) pp:5910-5923
Publication Date(Web):27 May 2009
DOI:10.1039/B902037C
The synthesis and the full characterization of a new ruthenium(II) complex with the pentadentate bispidine ligand L1 is reported and shown to be a very active catalyst for olefin epoxidation. The selectivity in the epoxidation of cis- and trans-β-methylstyrene with the formation of cis and trans products, each, was determined and compared with that of the iron bispidine complex of L1. There is a significant difference in selectivity between the two catalysts in the epoxidation of cis-β-methylstyrene but the epoxidation of trans-β-methylstyrene is highly stereoselective with both catalysts. Based on these results, electrochemical and labeling studies, a radical pathway for the epoxidation and isomerization is proposed, and this is supported by computational data. DFT indicates that, with both catalysts, the epoxidation is based on a stepwise mechanism, which, in the first step leads to a radical-based intermediate. This exists in two configurations, which interconvert with a relatively low energy barrier. The product ratio depends on the relative energies of the two configurations of the radical intermediate and the height of the energy barriers to the cis- and trans-epoxide products. For the Fe-based system there is, as expected, the additional complication of the availability of various spin levels, and multi-state reactivity is observed. The computed structures and energies are in agreement with the observed data.
Co-reporter:Peter Comba ;Christina Haaf;Achim Lienke Dr.;Amsaveni Muruganantham Dr.;Hubert Wadepohl
Chemistry - A European Journal 2009 Volume 15( Issue 41) pp:10880-10887
Publication Date(Web):
DOI:10.1002/chem.200802682
Abstract
The distorted trigonal-bipyramidal CuII complex [Cu(L1)(NCCH3)]2+ of the novel tetradentate bispidine-derived ligand L1 with four tertiary amine donors (L1=1,5-diphenyl-3-methyl-7-(1,4,6-trimethyl-1,4-diazacycloheptane-6-yl)diazabicyclo[3.3.1]nonane-9-one) is a very efficient catalyst for the aziridination of olefins in the presence of a nitrene source. In agreement with the experimental data (in situ spectroscopy, product distribution, and its dependence on the geometry of the substrate and of the nitrene source), a theoretical analysis based on DFT calculations indicates that the active catalyst has the Cu center in its +II oxidation state, that electron transfer is not involved, and that the conversion of the olefin to an aziridine is a stepwise process involving a radical intermediate. The striking change of efficiency and reaction mechanism between classical copper–bispidine complexes and the novel L1-based catalyst is primarily attributed to the structural variation, enforced by the ligand architecture.
Co-reporter:Peter Comba, Sascha Hausberg and Bodo Martin
The Journal of Physical Chemistry A 2009 Volume 113(Issue 24) pp:6751-6755
Publication Date(Web):May 26, 2009
DOI:10.1021/jp900752p
A broken-symmetry method for the calculation of exchange coupling constants from DFT calculations, using the Heisenberg−Dirac−van Vleck spin Hamiltonian, has been validated for a dinuclear copper(II) complex. Hybrid functionals in combination with a large basis set on the metal centers and their first coordination sphere, and a smaller basis set on the ligand backbone are shown to be efficient and acceptable with respect to the computational cost and precision in comparison with experimental data. This method was thoroughly tested with a series of oligonuclear transition metal complexes with CrIII, CuII, FeIII, MnII, MnIII, MnIV, NiII, and VIV as magnetic centers. The computed values of J are within approximately 50 cm−1 of the experimental values for most of the examples; with combined basis sets, there generally is a similar accuracy to that obtained with a large basis set for the entire spin cluster but with significantly reduced computational expense. When the experimentally observed structural data are refined prior to the calculation of the exchange coupling constants, the computed values of J are in most cases in slightly better agreement with the experimental data than those obtained from single point calculations based on the X-ray data.
Co-reporter:Peter Comba;Nina Dovalil;Hyang Hoo Kim
Journal of Inclusion Phenomena and Macrocyclic Chemistry 2009 Volume 65( Issue 1-2) pp:59-64
Publication Date(Web):2009 October
DOI:10.1007/s10847-009-9630-x
Reported are the crystal and solution structures (determined by X-ray crystallography and EPR spectroscopy/simulation of the EPR spectra, respectively) of two dinuclear CuII complexes, coordinated to isomeric dinucleating azetidine-based ligands, whose N3 cavities (pyridine/azetidine/secondary amine) are bridged by para- or meta-substituted phenyl groups. The CuII sites in the two dinuclear systems are similar to each other and as expected from the known structure of the corresponding mononuclear complex. The significant differences between the crystal structures of the mono- and the two dinuclear complexes and between the crystal and the solution structures are due to the elasticity of the CuII coordination sphere, the flexibility of the dinucleating ligands and subtle changes related to weak interactions (crystal lattice, solvation, anion coordination/ion pairing).
Co-reporter:Mihail Atanasov ; Peter Comba ;Claude A. Daul
Inorganic Chemistry 2008 Volume 47(Issue 7) pp:2449-2463
Publication Date(Web):February 27, 2008
DOI:10.1021/ic701702x
Magnetic anisotropy in cyanide-bridged single-molecule magnets (SMMs) with FeIII−CN−MII (M = Cu, Ni) exchange-coupled pairs was analyzed using a density functional theory (DFT)-based ligand field model. A pronounced magnetic anisotropy due to exchange was found for linear FeIII−CN−MII units with fourfold symmetry. This results from spin–orbit coupling of the [FeIII(CN)6]3− unit and was found to be enhanced by a tetragonal field, leading to a 2Eg ground state for FeIII. In contrast, a trigonal field (e.g., due to τ2g Jahn–Teller angular distortions) led to a reduction of the magnetic anisotropy. A large enhancement of the anisotropy was found for the FeIII−CN−NiII exchange pair if anisotropic exchange combined with a negative zero-field splitting energy of the S = 1 ground state of NiII in tetragonally compressed octahedra, while cancellation of the two anisotropic contributions was predicted for tetragonal elongations. A recently developed DFT approach to Jahn–Teller activity in low-spin hexacyanometalates was used to address the influence of dynamic Jahn–Teller coupling on the magnetic anisotropy. Spin Hamiltonian parameters derived for linear Fe−M subunits were combined using a vector-coupling scheme to yield the spin Hamiltonian for the entire spin cluster. The magnetic properties of published oligonuclear transition-metal complexes with ferromagnetic ground states are discussed qualitatively, and predictive concepts for a systematic search of cyanide-based SMM materials are presented.
Co-reporter:Mihail Atanasov ; Christoph Busche ; Peter Comba ; Fadi El Hallak ; Bodo Martin ; Gopalan Rajaraman ; Joris van Slageren ;Hubert Wadepohl
Inorganic Chemistry 2008 Volume 47(Issue 18) pp:8112-8125
Publication Date(Web):August 21, 2008
DOI:10.1021/ic800556c
The reaction of the hexacyanometalates K3[M1(CN)6] (M1 = CrIII, FeIII, CoIII) with the bispidine complexes [M2(L1)(X)]n+ and [M2(L2)(X)]n+ (M2 = MnII, NiII, CuII; L1 = 3-methyl-9-oxo-2,4-di-(2-pyridyl)-7-(2-pyridylmethyl)-3,7-diazabicyclo[3.3.1]nonane-1,5-dicarboxylic acid dimethyl ester; L2 = 3-methyl-9-oxo-7-(2-pyridylmethyl)-2,4-di-(2-quinolyl)-3,7-diazabicyclo[3.3.1]nonane-1,5-dicarboxylic acid dimethyl ester; X = anion or solvent) in water−methanol mixtures affords trinuclear complexes with cis- or trans-arrangement of the bispidine-capped divalent metal centers around the hexacyanometalate. X-ray structural analyses of five members of this family of complexes (cis-Fe[CuL2]2, trans-Fe[CuL1]2, cis-Co[CuL2]2, trans-Cr[MnL1]2, trans-Fe[MnL1]2) and the magnetic data of the entire series are reported. The magnetic data of the cyanide bridged, ferromagnetically coupled cis- and trans-Fe[ML]2 compounds (M = NiII, CuII) with S = 3/2 (CuII) and S = 5/2 (NiII) ground states are analyzed with an extended Heisenberg Hamiltonian which accounts for anisotropy and zero-field splitting, and the data of the CuII systems, for which structures are available, are thoroughly analyzed in terms of an orbital-dependent Heisenberg Hamiltonian, in which both spin−orbit coupling and low-symmetry ligand fields are taken into account. It is shown that the absence of single-molecule magnetic behavior in all spin clusters reported here is due to a large angular distortion of the [Fe(CN)6]3- center and the concomitant quenching of orbital angular momentum of the FeIII (2T2g) ground state.
Co-reporter:Peter Comba Dr.;Marion Kerscher Dr.;GeoffreyA. Lawrance Dr.;Bodo Martin Dr.;Hubert Wadepohl Dr.;Steffen Wunderlich
Angewandte Chemie International Edition 2008 Volume 47( Issue 25) pp:4740-4743
Publication Date(Web):
DOI:10.1002/anie.200800515
Co-reporter:Peter Comba, Martin Maurer and Prabha Vadivelu
The Journal of Physical Chemistry A 2008 Volume 112(Issue 50) pp:13028-13036
Publication Date(Web):September 23, 2008
DOI:10.1021/jp8037895
Experimental data suggest that there are various competing pathways for the catalytic and stoichiometric oxygenation of cyclohexane, assisted by iron-bispidine complexes and using various oxidants (H2O2, O2, PhIO). Density functional theory calculations indicate that both FeIV═O and FeV═O species are accessible and efficiently transfer their oxygen atoms to cyclohexane. The reactivities of the two isomers each and the two possible spin states for the FeIV═O and FeV═O species are sufficiently different to allow an interpretation of the experimental data.
Co-reporter:Peter Comba Dr.;LawrenceR. Gahan Dr.;Gebhard Haberhauer Dr.;GraemeR. Hanson Dr.;ChristopherJ. Noble Dr.;Björn Seibold;AnnaL. vandenBrenk Dr.
Chemistry - A European Journal 2008 Volume 14( Issue 14) pp:4393-4403
Publication Date(Web):
DOI:10.1002/chem.200701778
Abstract
The copper(II) coordination chemistry of westiellamide (H3Lwa), as well as of three synthetic analogues with an [18]azacrown-6 macrocyclic structure but with three imidazole (H3L1), oxazole (H3L2), and thiazole (H3L3) rings instead of oxazoline, is reported. As in the larger patellamide rings, the Nheterocycle-Npeptide-Nheterocycle binding site is highly preorganized for copper(II) coordination. In contrast to earlier reports, the macrocyclic peptides have been found to form stable mono- and dinuclear copper(II) complexes. The coordination of copper(II) has been monitored by high-resolution electrospray mass spectrometry (ESI-MS), spectrophotometric and polarimetric titrations, and EPR and IR spectroscopies, and the structural assignments have been supported by time-dependent studies (UV/Vis/NIR, ESI-MS, and EPR) of the complexation reaction of copper(II) with H3L1. Density functional theory (DFT) calculations have been used to model the structures of the copper(II) complexes on the basis of their spectroscopic data. The copper(II) ion has a distorted square-pyramidal geometry with one or two coordinated solvent molecules (CH3OH) in the mononuclear copper(II) cyclic peptide complexes, but the coordination sphere in [Cu(H2Lwa)(OHCH3)]+ differs from those in the synthetic analogues, [Cu(H2L)(OHCH3)2]+ (L=L1, L2, L3). Dinuclear copper(II) complexes ([CuII2(HL)(μ-X)]+; X=OCH3, OH; L=L1, L2, L3, Lwa) are observed in the mass spectra. While a dipole–dipole coupled EPR spectrum is observed for the dinuclear copper(II) complex of H3L3, the corresponding complexes with H3L (L=L1, L2, Lwa) are EPR-silent. This may be explained in terms of strong antiferromagnetic coupling (H3L1) and/or a low concentration of the dicopper(II) complexes (H3Lwa, H3L2), in agreement with the mass spectrometric observations.
Co-reporter:Peter Comba Dr.;Carolin Lang;Carlos Lopez de Laorden Dr.;Amsaveni Muruganantham;Gopalan Rajaraman Dr.;Hubert Wadepohl Dr. ;Marta Zajaczkowski
Chemistry - A European Journal 2008 Volume 14( Issue 17) pp:5313-5328
Publication Date(Web):
DOI:10.1002/chem.200701910
Abstract
Experimental and DFT-based computational results on the aziridination mechanism and the catalytic activity of (bispidine)copper(I) and -copper(II) complexes are reported and discussed (bispidine=tetra- or pentadentate 3,7-diazabicyclo[3.1.1]nonane derivative with two or three aromatic N donors in addition to the two tertiary amines). There is a correlation between the redox potential of the copper(II/I) couple and the activity of the catalyst. The most active catalyst studied, which has the most positive redox potential among all (bispidine)copper(II) complexes, performs 180 turnovers in 30 min. A detailed hybrid density functional theory (DFT) study provides insight into the structure, spin state, and stability of reactive intermediates and transition states, the oxidation state of the copper center, and the denticity of the nitrene source. Among the possible pathways for the formation of the aziridine product, the stepwise formation of the two NC bonds is shown to be preferred, which also follows from experimental results. Although the triplet state of the catalytically active copper nitrene is lowest in energy, the two possible spin states of the radical intermediate are practically degenerate, and there is a spin crossover at this stage because the triplet energy barrier to the singlet product is exceedingly high.
Co-reporter:Peter Comba Dr.;Marion Kerscher Dr.;GeoffreyA. Lawrance Dr.;Bodo Martin Dr.;Hubert Wadepohl Dr.;Steffen Wunderlich
Angewandte Chemie 2008 Volume 120( Issue 25) pp:4818-4821
Publication Date(Web):
DOI:10.1002/ange.200800515
Co-reporter:Peter Comba;Shigemasa Kuwata;Gerald Linti;Máté Tarnai;Hubert Wadepohl
European Journal of Inorganic Chemistry 2007 Volume 2007(Issue 5) pp:
Publication Date(Web):27 DEC 2006
DOI:10.1002/ejic.200600927
The reaction of 3-methyl-9-oxo-7-picolyl-2,4-di(2-pyridyl)-3,7-diazabicyclo[3.3.1]nonane-1,5-dicarboxylate (L2) with vanadyl sulfate in methanol yields the purple complex[VIV=O(L2′)](ClO4)2, where L2 is hydrated to the corresponding 9-diol L2′. The crystal structure of [VIV=O(L2′)](ClO4)2 has a saturated coordination sphere with a long bond to the tertiary amine N7 trans to the oxido group (2.36 Å). Oxidation of [VIV=O(L2′)](ClO4)2 with an excess of aqueous hydrogen peroxide yields the orange complex [VV=O(O2)(L2′)]ClO4. This can be further oxidized with Ce4+ in MeCN at –40 °Cto the unstable vanadium(V)-oxidosuperoxido complex [VV=O(O2)(L2′)]2+, as shown by EPR spectroscopy. The X-ray crystal structure of [VV=O(O2)(L2′)]ClO4 shows that the N7-pendant pyridine donor is dissociated and the V–N7 bond is elongated (2.56 Å). The O–O distance of the side-on bound peroxide in [VV=O(O2)(L2′)]ClO4 is unexpectedly short (1.28 Å) and is therefore in the range which is typical for a superoxido group. However, the IR spectrum of [VV=O(O2)(L2′)]ClO4 shows 18O isotope shifts, which support the assignment as a peroxido complex, and this is also supported by EPR and UV/Vis spectroscopic analysis. The short O–O bond is attributed to a static disorder rather than to libration of the “non-rigid” peroxido group.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2007)
Co-reporter:Mihail Atanasov, Peter Comba
Journal of Molecular Structure 2007 Volume 838(1–3) pp:157-163
Publication Date(Web):16 July 2007
DOI:10.1016/j.molstruc.2006.12.052
The linear and quadratic Tg ⊗ εg Jahn–Teller effect in the Tg (Tg = 2T2g, 3T1g) ground states of low-spin octahedral cyano complexes of 3d-transition metals (M = TiIII, VIII, MnIII, FeIII, CrII, MnII) has been studied. Vibronic coupling parameters have been derived using density functional theory to calculate energies of Slater determinants, which result from various electron distributions within the t2gn configuration due to the metal based t2g(3d) molecular orbitals (n = 1, 2, 3, 4). Tetragonal elongations are found in the case of TiIII, MnIII and CrII, and compressions for VIII, FeIII and MnII, and these establish the metal–ligand π-back donation as the dominant effect and driving force for the geometry distortions and energy stabilizations towards non-degenerate 2B2g(t2g1,t2g5) and 3A2g(t2g2,t2g4) ground states. The strength of the Jahn–Teller coupling is very weak and found not to follow a monotonic trend across the transition metal series but is shown to be weakest for TiIII, VIII and FeIII, slightly larger for MnIII, increases further to MnII and is found to be the strongest but is still weak for CrII.
Co-reporter:Jochen Bautz Dr. Dr.;Carlos Lopez de Laorden Dr.;Matthias Menzel;Gopalan Rajaraman Dr.
Angewandte Chemie 2007 Volume 119(Issue 42) pp:
Publication Date(Web):17 SEP 2007
DOI:10.1002/ange.200701681
Spurenleser: Der Eisen(II)-Komplex des Bispidinliganden L1 wird mit H2O2 zu [(L1)FeIV(OH)2]2+ (1, siehe Bild) mit mittlerem Spin oxidiert und bildet mit Olefinen cis-Diole 2; der High-Spin-Ferrylkomplex [(L1)FeIVO(OH2)]2+ (3) führt zur Epoxidierung (4). Anhand von Produktverteilungen (Diol/Epoxid) und 18O-Markierungsstudien wird ein Mechanismus entwickelt, der durch DFT-Analysen vollkommen gestützt wird.
Co-reporter:Jochen Bautz Dr. Dr.;Carlos Lopez de Laorden Dr.;Matthias Menzel;Gopalan Rajaraman Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 42) pp:
Publication Date(Web):17 SEP 2007
DOI:10.1002/anie.200701681
Pathfinding: The iron(II) complex of the bispidine ligand L1 is oxidized with H2O2 to intermediate-spin [(L1)FeIV(OH)2]2+ (1, see picture) which forms cis diols 2 from olefins, and the high-spin [(L1)FeIVO(OH2)]2+ ferryl complex (3) results in epoxidation (4). Product distributions (diol/epoxide) and 18O labeling studies are used to propose a mechanism, which is fully supported by DFT analyses.
Co-reporter:Peter Comba Dr.;Stefan Knoppe;Bodo Martin Dr.;Gopalan Rajaraman Dr.;Claudio Rolli;Brett Shapiro ;Timon Stork
Chemistry - A European Journal 2007 Volume 14( Issue 1) pp:344-357
Publication Date(Web):
DOI:10.1002/chem.200700865
Abstract
Mechanistic pathways for the aromatic hydroxylation by [CuII(L1)(TMAO)(O)]− (L1=hippuric acid, TMAO=trimethylamine N-oxide), derived from the ON bond homolysis of its [CuII(L1)(TMAO)2] precursor, were explored by using hybrid density functional theory (B3LYP) and highly correlated ab initio methods (QCISD and CCSD). Published experimental studies suggest that the catalytic reaction is triggered by a terminal copper–oxo species, and a detailed study of electronic structures, bonding, and energetics of the corresponding electromers is presented. Two pathways, a stepwise and a concerted reaction, were considered for the hydroxylation process. The results reveal a clear preference for the concerted pathway, in which the terminal oxygen atom directly attacks the carbon atom of the benzene ring, leading to the ortho-selectively hydroxylated product. Solvent effects were probed by using the PCM and CPCM solvation models, and the PCM model was found to perform better in the present case. Excellent agreement between the experimental and computational results was found, in particular also for changes in reactivity with derivatives of L1.
Co-reporter:Karin Born;André Daubinet
JBIC Journal of Biological Inorganic Chemistry 2007 Volume 12( Issue 1) pp:36-48
Publication Date(Web):2007 January
DOI:10.1007/s00775-006-0161-2
A mechanism for the oxidation of 3,5-di-tert-butylcatechol (dtbc) with dioxygen to the corresponding quinone (dtbq), catalyzed by bispidine-dicopper complexes (bispidines are various mono- and dinucleating derivatives of 3,7-diazabicyclo[3.3.1]nonane with bis-tertiary-amine–bispyridyl or bis-tertiary-amine–trispyridyl donor sets), is proposed on the basis of (1) the stoichiometry of the reaction as well as the stabilities and structures [X-ray, density functional theory (B3LYP, TZV)] of the bispidine-dicopper(II)–3,4,5,6-tetrachlorcatechol intermediates, (2) formation kinetics and structures (molecular mechanics, MOMEC) of the end-on peroxo–dicopper(II) complexes and (3) kinetics of the stoichiometric (anaerobic) and catalytic (aerobic) copper-complex-assisted oxidation of dtbc. This involves (1) the oxidation of the dicopper(I) complexes with dioxygen to the corresponding end-on peroxo–dicopper(II) complexes, (2) coordination of dtbc as a bridging ligand upon liberation of H2O2 and (3) intramolecular electron transfer to produce dtbq, which is liberated, and the dicopper(I) catalyst. Although the bispidine complexes have reactivities comparable to those of recently published catalysts with macrocyclic ligands, which seem to reproduce the enzyme-catalyzed process in various reaction sequences, a strikingly different oxidation mechanism is derived from the bispidine–dicopper-catalyzed reaction.
Co-reporter:Peter Comba, Shigemasa Kuwata, Gerald Linti, Hans Pritzkow, Máté Tarnai and Hubert Wadepohl
Chemical Communications 2006 (Issue 19) pp:2074-2076
Publication Date(Web):11 Apr 2006
DOI:10.1039/B602571D
The reaction of the CoII complex with the rigid bispidine ligand L1 with two tertiary amine and two pyridine donors, [CoII(L1)(OH2)2]2+, with H2O2 and O2 produces [CoII(L2)(OH2)2]3+, where L2 is demethylated at one of the amine donors, and CH2O.
Co-reporter:Peter Comba;Rol Krämer;Andriy Mokhir;Kyaw Naing;Erik Schatz
European Journal of Inorganic Chemistry 2006 Volume 2006(Issue 21) pp:
Publication Date(Web):12 SEP 2006
DOI:10.1002/ejic.200600469
The heteroditopic phenanthroline derivatives 5,6-bis(2-pyridylcarboxamido)-1,10-phenanthroline (H2L1) and 5,6-bis[(4-methoxy-2-pyridyl)carboxamido]-1,10-phenanthroline (H2L2) have been prepared and characterized, together with their luminescent ruthenium(II) complexes [Ru(bpy)2(H2L1,2)](PF6)2 and [Ru(H2L1)3](PF6)2 and the corresponding iron(II) complex [Fe(H2L1)3](PF6)2. In these complexes, the metal ion is coordinated by the bidentate phen site of H2L. The luminescence of the ruthenium complexes (λex = 450 nm, λem ca. 620 nm) is completely quenched by Cu2+ ions in the micromolar concentration range and, to a lesser extent, by other metal ions. At pH 5, the response of the luminescent sensors is highly Cu2+-selective. Heterodinuclear complexes [Ru(bpy)2(LM)](PF6)2, [Ru(LM)3](PF6)2, and [Fe(LM)3](PF6)2 have been isolated for M = Cu2+, Ni2+, Co2+, and Pd2+. It is suggested that M is coordinated to the tetradentate N4 site of L by two deprotonated amide N atoms and two pyridyl groups. This coordination type is confirmed by the EPR spectrum of the compound [RuII(bpy)2(L1CuII)](PF6)2.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Co-reporter:Mihail Atanasov Dr. Dr.;Yaroslaw D. Lampeka Dr.;Gerald Linti Dr.;Thomas Malcherek Dr.;Ronald Miletich Dr.;Alexer I. Prikhod'ko Dr. and;Hans Pritzkow Dr.
Chemistry - A European Journal 2006 Volume 12(Issue 3) pp:
Publication Date(Web):13 OCT 2005
DOI:10.1002/chem.200500365
The reaction of [M(CN)6]3− (M=Cr3+, Mn3+, Fe3+, Co3+) and [M(CN)8]4−/3− (M=Mo4+/5+, W4+/5+) with the trinuclear copper(II) complex of 1,3,5-triazine-2,4,6-triyltris[3-(1,3,5,8,12-pentaazacyclotetradecane)] ([Cu3(L)]6+) leads to partially encapsulated cyanometalates. With hexacyanometalate(III) complexes, [Cu3(L)]6+ forms the isostructural host–guest complexes [{[Cu3(L)(OH2)2][M(CN)6]2}{M(CN)6}]⋅[M(CN)6]⋅30 H2O with one bridging, two partially encapsulated, and one isolated [M(CN)6]3− unit. The octacyanometalates of Mo4+/5+ and W4+/5+ are encapsulated by two tris-macrocyclic host units. Due to the stability of the +IV oxidation state of Mo and W, only assemblies with [M(CN)8]4− were obtained. The Mo4+ and W4+ complexes were crystallized in two different structural forms: [{Cu3(L)(OH2)}2{Mo(CN)8}](NO3)8⋅15 H2O with a structural motif that involves isolated spherical [{Cu3(L)(OH2)}2{M(CN)8}]8+ ions and a “string-of-pearls” type of structure [{[Cu3(L)]2[M(CN)8]}{M(CN)8}](NO3)4⋅ 20 H2O, with [M(CN)8]4− ions that bridge the encapsulated octacyanometalates in a two-dimensional network. The magnetic exchange coupling between the various paramagnetic centers is characterized by temperature-dependent magnetic susceptibility and field-dependent magnetization data. Exchange between the Cu⋅⋅⋅Cu pairs in the [Cu3(L)]6+ “ligand” is weakly antiferromagnetic. Ferromagnetic interactions are observed in the cyanometalate assemblies with Cr3+, exchange coupling of Mn3+ and Fe3+ is very small, and the octacoordinate Mo4+ and W4+ systems have a closed-shell ground state.
Co-reporter:Jochen Bautz Dr.;Michael R. Bukowski Dr.;Marion Kerscher Dr.;Audria Stubna Dr. Dr.;Achim Lienke Dr.;Eckard Münck Dr.;Lawrence Que Jr. Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 34) pp:
Publication Date(Web):21 JUL 2006
DOI:10.1002/anie.200601134
A ferryl cation in water: Iron(II) coordinated by the pentadentate ligand L reacts with H2O2 to form a low-spin FeIVO complex with S=1 (2 in scheme) under conditions similar to those of the classical Fenton reaction. Complex 2 differs from a recently obtained high-spin aquated oxoiron(IV) species and demonstrates the role of the supporting polydentate ligand in controlling spin state and product formation.
Co-reporter:Michael R. Bukowski, Peter Comba, Achim Lienke, Christian Limberg, Carlos Lopez de Laorden, Rubén Mas-Ballesté, Michael Merz,Lawrence Que Jr.
Angewandte Chemie International Edition 2006 45(21) pp:
Publication Date(Web):
DOI:10.1002/anie.200504357
Co-reporter:Jochen Bautz Dr.;Michael R. Bukowski Dr.;Marion Kerscher Dr.;Audria Stubna Dr. Dr.;Achim Lienke Dr.;Eckard Münck Dr.;Lawrence Que Jr. Dr.
Angewandte Chemie 2006 Volume 118(Issue 34) pp:
Publication Date(Web):21 JUL 2006
DOI:10.1002/ange.200601134
Ein Ferryl-Komplex in wässriger Lösung: Eisen(II), koordiniert von einem fünfzähnigen Liganden L, reagiert mit H2O2 unter Bildung eines Low-Spin-FeIVO-Komplexes mit S=1 (2 im Schema) unter Bedingungen, die ähnlich der klassischen Fenton-Reaktion sind. 2 unterscheidet sich deutlich von einer kürzlich erhaltenen wässrigen High-Spin-FeIVO-Spezies und verdeutlicht die Rolle von mehrzähnigen Liganden bei der Kontrolle von Spinzustand und Produktbildung.
Co-reporter:Michael R. Bukowski Dr. Dr.;Achim Lienke Dr.;Christian Limberg Dr.;Carlos Lopez de Laorden Dr.;Rubén Mas-Ballesté Dr.;Michael Merz Dr.;Lawrence Que Jr. Dr.
Angewandte Chemie 2006 Volume 118(Issue 21) pp:
Publication Date(Web):25 APR 2006
DOI:10.1002/ange.200504357
FeII-Komplexe mit zwei isomeren Bispidinliganden katalysieren die Oxidation von Cycloocten mit H2O2 in MeCN unter aeroben Bedingungen mit einer hohen Selektivität für das Epoxid (siehe Schema; L=Bispidinligand). Als Oxidationsmittel wird eine {FeIVO}-Verbindung postuliert, die während der Katalyse durch homolytische Spaltung der O-O-Bindung im {FeIII-OOH}-Intermediat entsteht. Bei Durchführung der Reaktion unter Argon entsteht außer dem Epoxid eine Mischung von cis- und trans-Diolen.
Co-reporter:Peter Comba;Carlos Lopez de Laorden;Hans Pritzkow
Helvetica Chimica Acta 2005 Volume 88(Issue 3) pp:647-664
Publication Date(Web):22 MAR 2005
DOI:10.1002/hlca.200590045
The synthesis of a series of tetra- and pentadentate bispidine-type ligands (bispidine=3,7-diazabicyclo[3.3.1]nonane) – tetradentate ligands are donor-substituted at C(2) and C(4), pentadentate ligands have an additional donor at N(3) or N(7), with pyridine, 2-methylpyridine, or quinoline donor moieties – and of their CuII complexes are reported, together with single-crystal structural analyses and solution studies (electrochemistry, electronic and EPR spectroscopy). Depending on the ligand geometry and on the co-ligands (solvent or counter anion), there are various structural forms (pseudo-Jahn–Teller elongation along all three molecular axes), and the structural data are correlated with the spectroscopic and electrochemical parameters.
Co-reporter:Xian-Ming Zhang, Hai-Shun Wu, Fu-Qiang Zhang, Alexander Prikhod'ko, Shigemasa Kuwata and Peter Comba
Chemical Communications 2004 (Issue 18) pp:2046-2047
Publication Date(Web):13 Aug 2004
DOI:10.1039/B405931J
The novel basket-shaped three-electron-reduced heteropoly blue [H2dmpip]5[K ⊂ P6Mo18O73] was prepared and characterized; it shows reversible one-electron redox properties.
Co-reporter:Peter Comba;Marion Kerscher;Andreas Roodt
European Journal of Inorganic Chemistry 2004 Volume 2004(Issue 23) pp:
Publication Date(Web):20 OCT 2004
DOI:10.1002/ejic.200400518
The electron self-exchange rate of [Cu(L)(OH2)]2+/1+, kexp11 (298.13 K) = 15 ± 11 m−1s−1 {L = dimethyl 3,7-dimethyl-9oxo-2,4-bis(2-pyridyl)-3,7-diazabicyclo[3.3.1]nonane-1,5-dicarboxylate}, was determined by a cross reaction. The analysis, based on classical Marcus theory, indicates that this relatively slow rate is to a large extent due to enthalpic terms (ΔG‡,exp11 = 62.8 ± 3.5 kJ·mol−1, ΔH‡,exp11 = 36.0 ± 2.7 kJ·mol−1 and ΔS‡,exp11 = −92 ± 10 J·mol−1K−1). The activation entropy is significant but not unusually large and the calculated outer-sphere reorganization energy, ΔG*,calcout = 20.5 kJ·mol−1, is at least of the same order of magnitude as the calculated inner-sphere reorganisation energy ΔG*,calcin = 18.6 kJ·mol−1, i.e. the deformation of the solvent sheath is a major reason for the slow electron transfer rate. This is believed to be due to the highly elastic coordination geometry which leads to little strain upon distortion enforced by the electron transfer but to comparably large structural changes and, hence, to a large outer-sphere reorganisation term. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2004)
Co-reporter:Michael R. Bukowski Dr.;Christian Limberg Dr.;Michael Merz Dr.;Lawrence Que Jr. Dr.;Tobias Wistuba Dr.
Angewandte Chemie 2004 Volume 116(Issue 10) pp:
Publication Date(Web):25 FEB 2004
DOI:10.1002/ange.200352523
. ‥und doch so verschieden: Nicht-Häm-Eisenkomplexe können die stereoselektive Oxidation von C-H- und CC-Bindungen katalysieren. Die isomeren FeIII-η1-Hydroperoxo- und FeIII-η2-Peroxokomplexe sind dabei wichtige Zwischenprodukte (siehe Bild). Über die spektroskopischen Eigenschaften der FeIII-Hydroperoxo- und Peroxokomplexe wird berichtet.
Co-reporter:Michael R. Bukowski Prof Dr.;Christian Limberg Dr.;Michael Merz Dr.;Lawrence Que Jr. Dr.;Tobias Wistuba Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 10) pp:
Publication Date(Web):25 FEB 2004
DOI:10.1002/anie.200352523
. ‥and yet so different. Non-heme iron complexes catalyze the stereoselective oxidation of CH and CC bonds. Isomeric FeIII-η1-hydroperoxo and FeIII-η2-peroxo complexes (see picture), important intermediates in these reactions, were studied by EPR, resonance Raman, and UV/Vis spectroscopies.
Co-reporter:Peter Comba Dr.;Andreas Hauser Dr.;Marion Kerscher Dr.;Hans Pritzkow Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 37) pp:
Publication Date(Web):8 SEP 2003
DOI:10.1002/anie.200351900
The long and the short of it: Copper(II) complexes of the pentadentate bispidine ligands exist in two isomeric forms (see structure) with bond-length differences up to 0.5 Å. The stabilization of either isomer may be achieved by a variation of the substituent at N7.
Co-reporter:Peter Comba Dr.;Andreas Hauser Dr.;Marion Kerscher Dr.;Hans Pritzkow Dr.
Angewandte Chemie 2003 Volume 115(Issue 37) pp:
Publication Date(Web):8 SEP 2003
DOI:10.1002/ange.200351900
Spiel mit der Verzerrung: CuII-Komplexe mit fünfzähnigen Bispidinliganden existieren in zwei isomeren Formen, deren Bindungslängen sich um bis zu 0.5 Å unterscheiden (siehe Bild). Die relative Stabilität der beiden Konformere kann durch Variation des Substituenten an N7 gezielt beeinflusst werden.
Co-reporter:Peter Comba, Markus Enders, Michael Großhauser, Markus Hiller, Dennis Müller and Hubert Wadepohl
Dalton Transactions 2017 - vol. 46(Issue 1) pp:NaN149-149
Publication Date(Web):2016/11/30
DOI:10.1039/C6DT03488H
Reported are the syntheses, structures and magnetic properties, also by NMR spectroscopy in solution, of a series of 13 linear trinuclear 3d–4f compounds with a lanthanide(III) surrounded by two NiII ions, NiII2LnIII, where the central LnIII is hexacoordinate. For three of the crystal structures, an additional H2O molecule is coordinated to the central LnIII ion, leading to a monocapped trigonal prismatic structure. However, NMR spectroscopy indicates that in solution, these complexes also have a hexacoordinate LnIII center. The solution magnetic anisotropies, determined by NMR spectroscopy, indicate that the axial components of the anisotropies are relatively small and that the DyIII derivative might therefore not exhibit single molecule magnetism. The axial anisotropies determined by NMR spectroscopy are in good agreement with the expectations based on the distorted trigonal prismatic ligand field.
Co-reporter:Peter Comba, Yong-Min Lee, Wonwoo Nam and Arkadius Waleska
Chemical Communications 2014 - vol. 50(Issue 4) pp:NaN414-414
Publication Date(Web):2013/10/31
DOI:10.1039/C3CC47013J
Organic substrates (specifically cis-1,2-dimethylcyclohexane, DMCH) are oxidized by O2 in the presence of iron(II)–bispidine complexes. It is shown that this oxidation reaction is not based on O2 activation by the nonheme iron catalysts as in Nature but due to a radical-based initiation, followed by a radical- and ferryl-based catalytic reaction.
Co-reporter:Peter Comba, Lawrence R. Gahan, Graeme R. Hanson and Michael Westphal
Chemical Communications 2012 - vol. 48(Issue 75) pp:NaN9366-9366
Publication Date(Web):2012/07/27
DOI:10.1039/C2CC34836E
A possible biological function of cyclic pseudo-octapeptides is presented. The dinuclear copper(II) complex of a synthetic analogue ([Cu2(H2Pat1)(μ-OH)(OH2)2]) of the naturally occurring ascidiacyclamide is known to have a hydroxo-bridged dicopper(II) site which is able to catalytically transform CO2 into CO32−. This complex is shown here to function as a phosphatase mimic, suggesting that the so far unknown biological function of these macrocycles within the ascidians may involve phosphoester hydrolysis.
Co-reporter:Jordi Benet-Buchholz, Peter Comba, Antoni Llobet, Stephan Roeser, Prabha Vadivelu, Hubert Wadepohl and Sebastian Wiesner
Dalton Transactions 2009(Issue 30) pp:NaN5923-5923
Publication Date(Web):2009/05/27
DOI:10.1039/B902037C
The synthesis and the full characterization of a new ruthenium(II) complex with the pentadentate bispidine ligand L1 is reported and shown to be a very active catalyst for olefin epoxidation. The selectivity in the epoxidation of cis- and trans-β-methylstyrene with the formation of cis and trans products, each, was determined and compared with that of the iron bispidine complex of L1. There is a significant difference in selectivity between the two catalysts in the epoxidation of cis-β-methylstyrene but the epoxidation of trans-β-methylstyrene is highly stereoselective with both catalysts. Based on these results, electrochemical and labeling studies, a radical pathway for the epoxidation and isomerization is proposed, and this is supported by computational data. DFT indicates that, with both catalysts, the epoxidation is based on a stepwise mechanism, which, in the first step leads to a radical-based intermediate. This exists in two configurations, which interconvert with a relatively low energy barrier. The product ratio depends on the relative energies of the two configurations of the radical intermediate and the height of the energy barriers to the cis- and trans-epoxide products. For the Fe-based system there is, as expected, the additional complication of the availability of various spin levels, and multi-state reactivity is observed. The computed structures and energies are in agreement with the observed data.
Co-reporter:Jordi Benet-Buchholz, Peter Comba, Antoni Llobet, Stephan Roeser, Prabha Vadivelu and Sebastian Wiesner
Dalton Transactions 2010 - vol. 39(Issue 13) pp:NaN3320-3320
Publication Date(Web):2010/02/16
DOI:10.1039/B924614B
Two RuIVO catalysts with either a pentadentate bispidine ligand L1 or a bidentate pyrazolate L2/terpy L3 combination of ligands have very different efficiencies as oxygen transfer catalysts for the selective oxidation of sulfides to sulfoxides: the [RuII(L1)(solvent)]2+/iodosyl benzene system has an initial TOF of approx. 40 h−1 and quantitative yield, with [RuII(L2)(L3)(solvent)]+ the initial TOF is approx. 12 h−1 with a maximum yield of approx. 60%. By experiment (cyclovoltametry) it is shown that there is S- to O-linkage isomerization of the RuII sulfoxide product complex, and this may partially switch off the catalytic cycle for the L2/L3-based catalyst. It emerges that the reasons for the reduced efficiency in the case of the L2/L3, in comparison with the L1-based catalyst, are a more efficient linkage isomerization, a more stable S-bonded, in comparison with the O-bonded, RuII-based isomer, and inefficient ligand exchange in the product (hydrolysis produces the free sulfoxide and the RuII precatalyst). These interpretations are qualitatively in good agreement with preliminary DFT-based data.
Co-reporter:Madhavan Jaccob, Peter Comba, Martin Maurer, Prabha Vadivelu and Ponnambalam Venuvanalingam
Dalton Transactions 2011 - vol. 40(Issue 42) pp:NaN11281-11281
Publication Date(Web):2011/09/29
DOI:10.1039/C1DT11533B
Iron-bispidine complexes are efficient catalysts for the oxidation of thioanisole to phenylmethylsulfoxide with iodosylbenzene as oxidant. With the tetradentate bispidine ligand L1 (L1 = 2,4-pyridyl-3,7-diazabicyclo[3.3.1]nonane)) the catalytic efficiency is smaller than with the pentadentate bispidine ligand L2 (L2 = 2,4-pyridyl-7-(pyridine-2-ylmethyl)-3,7-diazabicyclo[3.3.1]nonane)). Based on the redox potentials (iron complexes with L1 are stronger oxidants than with L2) and known efficiencies in catalytic olefin oxidation and C–H activation reactions, the expectations were different. A DFT-based analysis is used to explain the apparent contradiction, and this is based on differences in the electronic ground states of the ferryl complexes as well as in the oxygen transfer transition states.
Co-reporter:Peter Comba, Bodo Martin, Avik Sanyal and Holger Stephan
Dalton Transactions 2013 - vol. 42(Issue 31) pp:NaN11073-11073
Publication Date(Web):2013/06/07
DOI:10.1039/C3DT51049B
A QSPR scheme for the computation of lipophilicities of 64Cu complexes was developed with a training set of 24 tetraazamacrocylic and bispidine-based CuII compounds and their experimentally available 1-octanol–water distribution coefficients. A minimum number of physically meaningful parameters were used in the scheme, and these are primarily based on data available from molecular mechanics calculations, using an established force field for CuII complexes and a recently developed scheme for the calculation of fluctuating atomic charges. The developed model was also applied to an independent validation set and was found to accurately predict distribution coefficients of potential 64Cu PET (positron emission tomography) systems. A possible next step would be the development of a QSAR-based biodistribution model to track the uptake of imaging agents in different organs and tissues of the body. It is expected that such simple, empirical models of lipophilicity and biodistribution will be very useful in the design and virtual screening of positron emission tomography (PET) imaging agents.
Co-reporter:Peter Comba, Franziska Emmerling, Maik Jakob, Werner Kraus, Manja Kubeil, Michael Morgen, Jens Pietzsch and Holger Stephan
Dalton Transactions 2013 - vol. 42(Issue 17) pp:NaN6148-6148
Publication Date(Web):2012/11/27
DOI:10.1039/C2DT32356G
The CuII complex of H4TETP (H4TETP = 1,4,8,11-tetraazatetradecane-1,4,8,11-tetrapropionic acid) is five-coordinate with a distorted square-pyramidal structure (τ = 0.45; i.e. the geometry is nearly half-way between square-pyramidal and trigonal-bipyramidal) and a relatively long Cu–N and a short Cu–O bond; the comparison between powder and solution electronic spectroscopy, the frozen solution EPR spectrum and ligand-field-based calculations (angular overlap model, AOM) indicate that the solution and solid state structures are very similar, i.e. the complex has a relatively low “in-plane” and a significant axial ligand field with a dx2−y2 ground state. The ligand-enforced structure is therefore shown to lead to a partially quenched Jahn–Teller distortion and to a relatively low complex stability, lower than with the corresponding acetate-derived ligand H4TETA. This is confirmed by potentiometric titration and by the biodistribution with 64Cu-labeled ligands which show that the uptake in the liver is significantly increased with the H4TETP-based system.
Co-reporter:Peter Comba, Lawrence R. Gahan, Graeme R. Hanson, Marcel Maeder and Michael Westphal
Dalton Transactions 2014 - vol. 43(Issue 8) pp:NaN3152-3152
Publication Date(Web):2013/12/02
DOI:10.1039/C3DT53135J
The dicopper(II) complexes of six pseudo-octapeptides, synthetic analogues of ascidiacyclamide and the patellamides, found in ascidians of the Pacific and Indian Oceans, are shown to be efficient carbonic anhydrase model complexes with kcat up to 7.3 × 103 s−1 (uncatalyzed: 3.7 × 10−2 s−1; enzyme-catalyzed: 2 × 105–1.4 × 106 s−1) and a turnover number (TON) of at least 1700, limited only by the experimental conditions used. So far, no copper-based natural carbonic anhydrases are known, no faster model systems have been described and the biological role of the patellamide macrocycles is so far unknown. The observed CO2 hydration rates depend on the configuration of the isopropyl side chains of the pseudo-octapeptide scaffold, and the naturally observed R*,S*,R*,S* geometry is shown to lead to more efficient catalysts than the S*,S*,S*,S* isomers. The catalytic efficiency also depends on the heterocyclic donor groups of the pseudo-octapeptides. Interestingly, the dicopper(II) complex of the ligand with four imidazole groups is a more efficient catalyst than that of the close analogue of ascidiacyclamide with two thiazole and two oxazoline rings. The experimental observations indicate that the nucleophilic attack of a CuII-coordinated hydroxide at the CO2 carbon center is rate determining, i.e. formation of the catalyst-CO2 adduct and release of carbonate/bicarbonate are relatively fast processes.
Co-reporter:Peter Comba, Nina Dovalil, Lawrence R. Gahan, Graeme R. Hanson and Michael Westphal
Dalton Transactions 2014 - vol. 43(Issue 5) pp:NaN1956-1956
Publication Date(Web):2013/10/30
DOI:10.1039/C3DT52664J
Cyclic pseudo-peptides derived from marine metabolites of the genus Lissoclinum bistratum and Lissoclinum patella have attracted scientific interest in the last two decades. Their structural properties and solution dynamics have been analyzed in detail, elaborate synthetic procedures for the natural products and synthetic derivatives developed, the biosynthetic pathways studied and it now is possible to produce them biosynthetically. Initially, these macrocyclic ligands were studied due to their medicinal and pharmaceutical potential – some of the isolated cyclic pseudo-peptides show high cytotoxic and antiviral activity. A major focus in the last decade has been on their CuII coordination chemistry, as a number of studies have indicated that dinuclear CuII complexes of cyclic peptides may be involved in the ascidians’ metabolism, and this is the focus of the present review.
Co-reporter:Peter Comba, Henning Rudolf and Hubert Wadepohl
Dalton Transactions 2015 - vol. 44(Issue 6) pp:NaN2736-2736
Publication Date(Web):2014/11/14
DOI:10.1039/C4DT03262D
Reported is the new bispidine-derived hexadentate ligand L (L = 3-(2-methylpyridyl)-7-(bis-2-methylpyridyl)-3,7-diazabicyclo[3.3.1]nonane) with two tertiary amine and four pyridine donor groups. This ligand can form heterodinuclear and mononuclear complexes and, in the mononuclear compounds discussed here, the ligand may coordinate as a pentadentate ligand, with one of the bispyridinemethane-based pyridine groups un- or semi-coordinated, or as a hexadentate ligand, leading to a pentagonal pyramidal coordination geometry or, with an additional monodentate ligand, to a heptacoordinate pentagonal bipyramidal structure. The solution and solid state data presented here indicate that, with the relatively small CuII and high-spin FeII ions the fourth pyridine group is only semi-coordinated for steric reasons and, with the larger high-spin MnII ion genuine heptacoordination is observed but with a relatively large distortion in the pentagonal equatorial plane.
Co-reporter:Dong Wang, Kallol Ray, Michael J. Collins, Erik R. Farquhar, Jonathan R. Frisch, Laura Gómez, Timothy A. Jackson, Marion Kerscher, Arkadius Waleska, Peter Comba, Miquel Costas and Lawrence Que
Chemical Science (2010-Present) 2013 - vol. 4(Issue 1) pp:NaN291-291
Publication Date(Web):2012/10/25
DOI:10.1039/C2SC21318D
Oxoiron(IV) species have been found to act as the oxidants in the catalytic cycles of several mononuclear nonheme iron enzymes that activate dioxygen. To gain insight into the factors that govern the oxidative reactivity of such complexes, a series of five synthetic S = 1 [FeIV(O)(LN5)]2+ complexes has been characterized with respect to their spectroscopic and electrochemical properties as well as their relative abilities to carry out oxo transfer and hydrogen atom abstraction. The FeO units in these five complexes are supported by neutral pentadentate ligands having a combination of pyridine and tertiary amine donors but with different ligand frameworks. Characterization of the five complexes by X-ray absorption spectroscopy reveals FeO bonds of ca. 1.65 Å in length that give rise to the intense 1s → 3d pre-edge features indicative of iron centers with substantial deviation from centrosymmetry. Resonance Raman studies show that the five complexes exhibit ν(FeO) modes at 825–841 cm−1. Spectropotentiometric experiments in acetonitrile with 0.1 M water reveal that the supporting pentadentate ligands modulate the E1/2(IV/III) redox potentials with values ranging from 0.83 to 1.23 V vs. Fc, providing the first electrochemical determination of the E1/2(IV/III) redox potentials for a series of oxoiron(IV) complexes. The 0.4 V difference in potential may arise from differences in the relative number of pyridine and tertiary amine donors on the LN5 ligand and in the orientations of the pyridine donors relative to the FeO bond that are enforced by the ligand architecture. The rates of oxo-atom transfer (OAT) to thioanisole correlate linearly with the increase in the redox potentials, reflecting the relative electrophilicities of the oxoiron(IV) units. However this linear relationship does not extend to the rates of hydrogen-atom transfer (HAT) from 1,3-cyclohexadiene (CHD), 9,10-dihydroanthracene (DHA), and benzyl alcohol, suggesting that the HAT reactions are not governed by thermodynamics alone. This study represents the first investigation to compare the electrochemical and oxidative properties of a series of S = 1 FeIVO complexes with different ligand frameworks and sheds some light on the complexities of the reactivity of the oxoiron(IV) unit.
Co-reporter:Sam P. de Visser, Matthew G. Quesne, Bodo Martin, Peter Comba and Ulf Ryde
Chemical Communications 2014 - vol. 50(Issue 3) pp:NaN282-282
Publication Date(Web):2013/10/29
DOI:10.1039/C3CC47148A
With computational resources becoming more efficient and more powerful and at the same time cheaper, computational methods have become more and more popular for studies on biochemical and biomimetic systems. Although large efforts from the scientific community have gone into exploring the possibilities of computational methods for studies on large biochemical systems, such studies are not without pitfalls and often cannot be routinely done but require expert execution. In this review we summarize and highlight advances in computational methodology and its application to enzymatic and biomimetic model complexes. In particular, we emphasize on topical and state-of-the-art methodologies that are able to either reproduce experimental findings, e.g., spectroscopic parameters and rate constants, accurately or make predictions of short-lived intermediates and fast reaction processes in nature. Moreover, we give examples of processes where certain computational methods dramatically fail.
Co-reporter:Peter Comba, Annika Eisenschmidt, Lawrence R. Gahan, Graeme R. Hanson, Nina Mehrkens and Michael Westphal
Dalton Transactions 2016 - vol. 45(Issue 47) pp:NaN18945-18945
Publication Date(Web):2016/11/07
DOI:10.1039/C6DT03787A
The patellamides (cyclic pseudo-octapeptides) are produced by Prochloron, a symbiont of the ascidians, marine invertebrate filter feeders. These pseudo-octapeptides are present in the cytoplasm and a possible natural function of putative metal complexes of these compounds is hydrolase activity, however the true biological role is still unknown. The dinuclear CuII complexes of synthetic patellamide derivatives have been shown in in vitro experiments to be efficient hydrolase model catalysts. Many hydrolase enzymes, specifically phosphatases and carboanhydrases, are ZnII-based enzymes and therefore, we have studied the ZnII and mixed ZnII/CuII solution chemistry of a series of synthetic patellamide derivatives, including solution structural and computational work, with the special focus on model phosphatase chemistry with bis-(2,4-dinitrophenyl)phosphate (BDNPP) as the substrate. The ZnII complexes of a series of ligands are shown to form complexes of similar structure and stability compared to the well-studied CuII analogues and the phosphatase reactivities are also similar. Since the complex stabilities and phosphatase activities are generally a little lower compared to those of CuII and since the concentration of ZnII in Prochloron cells is slightly smaller, we conclude that the CuII complexes of the patellamides are more likely to be of biological importance.





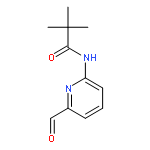
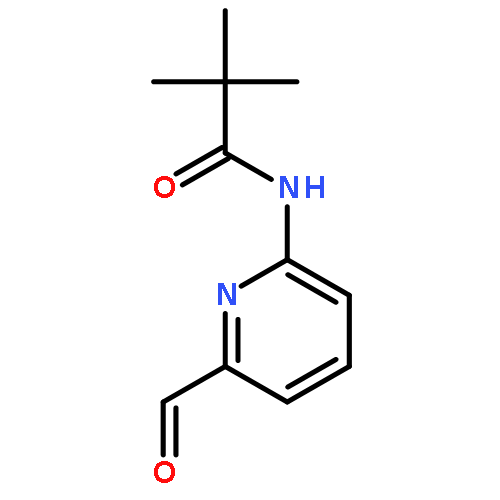


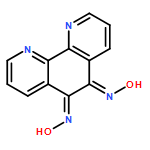
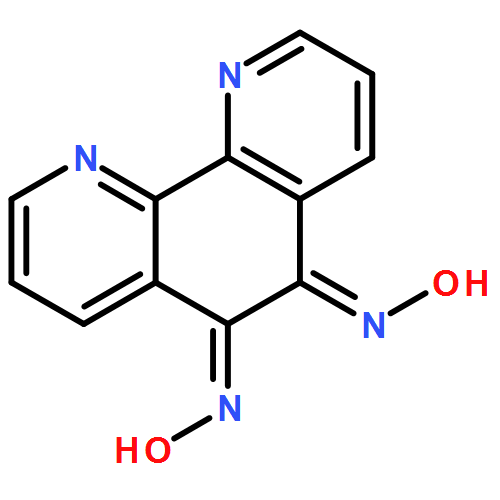

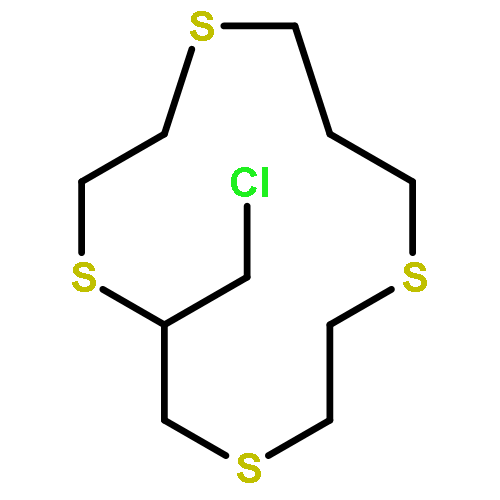
![3-?[[Bis(2-?pyridinylmethyl)?amino]?methyl]?-?2-?hydroxy-?5-?methylbenzaldehyde](/data/chemimg/192819-69-5.png)
![3-?[[Bis(2-?pyridinylmethyl)?amino]?methyl]?-?2-?hydroxy-?5-?methylbenzaldehyde](/data/chemimg/192819-69-5_b.png)
![2-Propanol, 1,1'-[1,2-ethanediylbis(thio)]bis[3-chloro-](http://img.cochemist.com/ccimg/192600/192512-58-6.png)
![2-Propanol, 1,1'-[1,2-ethanediylbis(thio)]bis[3-chloro-](http://img.cochemist.com/ccimg/192600/192512-58-6_b.png)





![1H-Benz[de]isoquinoline-1,3(2H)-dione, 2-(2,2-dimethoxyethyl)-](/data/chemimg/2987300/153721-06-3.png)
![1H-Benz[de]isoquinoline-1,3(2H)-dione, 2-(2,2-dimethoxyethyl)-](/data/chemimg/2987300/153721-06-3_b.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-3-phenyl-, (2R,3S)-rel-](http://img.cochemist.com/ccimg/152000/151909-39-6.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-3-phenyl-, (2R,3S)-rel-](http://img.cochemist.com/ccimg/152000/151909-39-6_b.png)
![6,20-Dioxa-13,27-dithia-3,10,17,24,29,30,31,32-octaazapentacyclo[24.2.1.15,8.112,15.119,22]dotriaconta-5(32),12(31),14,19(30),26(29),28-hexaene-2,9,16,23-tetrone,7,21-dimethyl-4,11-bis(1-methylethyl)-18-(1-methylpropyl)-25-(phenylmethyl)-,(4S,7R,8S,11R,18S,21R,22S,25R)- (9CI)](http://img.cochemist.com/ccimg/140500/140430-46-2.png)
![6,20-Dioxa-13,27-dithia-3,10,17,24,29,30,31,32-octaazapentacyclo[24.2.1.15,8.112,15.119,22]dotriaconta-5(32),12(31),14,19(30),26(29),28-hexaene-2,9,16,23-tetrone,7,21-dimethyl-4,11-bis(1-methylethyl)-18-(1-methylpropyl)-25-(phenylmethyl)-,(4S,7R,8S,11R,18S,21R,22S,25R)- (9CI)](http://img.cochemist.com/ccimg/140500/140430-46-2_b.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-3-phenyl-, trans-](http://img.cochemist.com/ccimg/137600/137595-21-2.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-3-phenyl-, trans-](http://img.cochemist.com/ccimg/137600/137595-21-2_b.png)
![6,20-Dioxa-13,27-dithia-3,10,17,24,29,30,31,32-octaazapentacyclo[24.2.1.15,8.112,15.119,22]dotriaconta-5(32),12(31),14,19(30),26(29),28-hexaene-2,9,16,23-tetrone,7,11,21-trimethyl-4,18-bis[(1S)-1-methylpropyl]-25-(phenylmethyl)-,(4S,7R,8S,11R,18S,21R,22S,25R)-](http://img.cochemist.com/ccimg/120900/120853-15-8.png)
![6,20-Dioxa-13,27-dithia-3,10,17,24,29,30,31,32-octaazapentacyclo[24.2.1.15,8.112,15.119,22]dotriaconta-5(32),12(31),14,19(30),26(29),28-hexaene-2,9,16,23-tetrone,7,11,21-trimethyl-4,18-bis[(1S)-1-methylpropyl]-25-(phenylmethyl)-,(4S,7R,8S,11R,18S,21R,22S,25R)-](http://img.cochemist.com/ccimg/120900/120853-15-8_b.png)


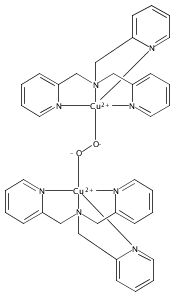
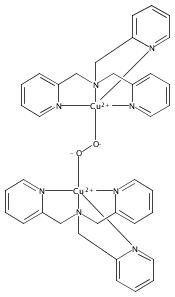
![4-Thiazolecarboxylic acid, 2-[[[(phenylmethoxy)carbonyl]amino]methyl]-, ethyl ester](http://img.cochemist.com/ccimg/112200/112159-27-0.png)
![4-Thiazolecarboxylic acid, 2-[[[(phenylmethoxy)carbonyl]amino]methyl]-, ethyl ester](http://img.cochemist.com/ccimg/112200/112159-27-0_b.png)
![Propanamide, N-[6-(bromomethyl)-2-pyridinyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/111500/111477-43-1.png)
![Propanamide, N-[6-(bromomethyl)-2-pyridinyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/111500/111477-43-1_b.png)




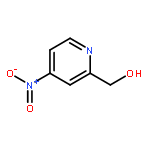
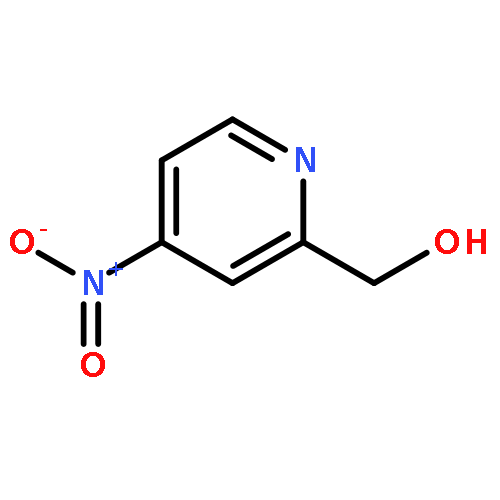
![L-Threonine, N-[(1,1-dimethylethoxy)carbonyl]-L-isoleucyl-](http://img.cochemist.com/ccimg/95400/95364-12-8.png)
![L-Threonine, N-[(1,1-dimethylethoxy)carbonyl]-L-isoleucyl-](http://img.cochemist.com/ccimg/95400/95364-12-8_b.png)
![[12,12'-Bi-1,4,7,10-tetraazacyclotridecane]-11,11',13,13'-tetrone](http://img.cochemist.com/ccimg/92300/92284-95-2.png)
![[12,12'-Bi-1,4,7,10-tetraazacyclotridecane]-11,11',13,13'-tetrone](http://img.cochemist.com/ccimg/92300/92284-95-2_b.png)



![11-benzyl-4-(butan-2-yl)-7,21,25-trimethyl-18-(propan-2-yl)-6,20-dioxa-13,27-dithia-3,10,17,24,29,30,31,32-octaazapentacyclo[24.2.1.1~5,8~.1~12,15~.1~19,22~]dotriaconta-1(28),5(32),12(31),14,19(30),26(29)-hexaene-2,9,16,23-tetrone](http://img.cochemist.com/ccimg/81200/81120-74-3.png)
![11-benzyl-4-(butan-2-yl)-7,21,25-trimethyl-18-(propan-2-yl)-6,20-dioxa-13,27-dithia-3,10,17,24,29,30,31,32-octaazapentacyclo[24.2.1.1~5,8~.1~12,15~.1~19,22~]dotriaconta-1(28),5(32),12(31),14,19(30),26(29)-hexaene-2,9,16,23-tetrone](http://img.cochemist.com/ccimg/81200/81120-74-3_b.png)
![11-benzyl-4-(butan-2-yl)-7,21,25-trimethyl-18-(2-methylpropyl)-6,20-dioxa-13,27-dithia-3,10,17,24,29,30,31,32-octaazapentacyclo[24.2.1.1~5,8~.1~12,15~.1~19,22~]dotriaconta-1(28),5(32),12(31),14,19(30),26(29)-hexaene-2,9,16,23-tetrone](http://img.cochemist.com/ccimg/81100/81098-23-9.png)
![11-benzyl-4-(butan-2-yl)-7,21,25-trimethyl-18-(2-methylpropyl)-6,20-dioxa-13,27-dithia-3,10,17,24,29,30,31,32-octaazapentacyclo[24.2.1.1~5,8~.1~12,15~.1~19,22~]dotriaconta-1(28),5(32),12(31),14,19(30),26(29)-hexaene-2,9,16,23-tetrone](http://img.cochemist.com/ccimg/81100/81098-23-9_b.png)
![1,3-Diazatricyclo[3.3.1.13,7]decan-6-one, 5,7-dimethyl-](http://img.cochemist.com/ccimg/80900/80808-86-2.png)
![1,3-Diazatricyclo[3.3.1.13,7]decan-6-one, 5,7-dimethyl-](http://img.cochemist.com/ccimg/80900/80808-86-2_b.png)
![1,4,8,11-TETRAAZATRICYCLO[9.3.1.1(4,8)]HEXADECANE](http://img.cochemist.com/ccimg/76000/75920-10-4.png)
![1,4,8,11-TETRAAZATRICYCLO[9.3.1.1(4,8)]HEXADECANE](http://img.cochemist.com/ccimg/76000/75920-10-4_b.png)
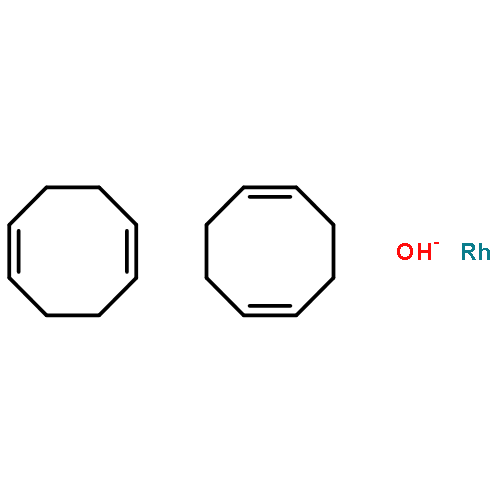
![1-Propanamine, 3,3'-[1,3-propanediylbis(thio)]bis-](http://img.cochemist.com/ccimg/65800/65755-73-9.png)
![1-Propanamine, 3,3'-[1,3-propanediylbis(thio)]bis-](http://img.cochemist.com/ccimg/65800/65755-73-9_b.png)
![Phenol, 2-[[(2-pyridinylmethyl)amino]methyl]-](http://img.cochemist.com/ccimg/63700/63671-68-1.png)
![Phenol, 2-[[(2-pyridinylmethyl)amino]methyl]-](http://img.cochemist.com/ccimg/63700/63671-68-1_b.png)
![Cobalt(2 ), (1,3,6,8,10,13,16,19-octaazabicyclo[6.6.6]eicosane-κN3,κN6,κN10,κN13,κN16,κN19)-, (OC-6-11)-](/data/chemimg/639000/63218-22-4.png)
![Cobalt(2 ), (1,3,6,8,10,13,16,19-octaazabicyclo[6.6.6]eicosane-κN3,κN6,κN10,κN13,κN16,κN19)-, (OC-6-11)-](/data/chemimg/639000/63218-22-4_b.png)
![N-[9-(2-carboxyphenyl)-6-(dimethylamino)-3H-xanthen-3-ylidene]-N-methylmethanaminium perchlorate](http://img.cochemist.com/ccimg/62700/62669-72-1.png)
![N-[9-(2-carboxyphenyl)-6-(dimethylamino)-3H-xanthen-3-ylidene]-N-methylmethanaminium perchlorate](http://img.cochemist.com/ccimg/62700/62669-72-1_b.png)





![Ethanamine, 2,2'-[1,3-propanediylbis(thio)]bis-](http://img.cochemist.com/ccimg/57400/57383-24-1.png)
![Ethanamine, 2,2'-[1,3-propanediylbis(thio)]bis-](http://img.cochemist.com/ccimg/57400/57383-24-1_b.png)
![Benzenamine,2,2'-[1,2-ethanediylbis(thio)]bis-](http://img.cochemist.com/ccimg/52500/52411-33-3.png)
![Benzenamine,2,2'-[1,2-ethanediylbis(thio)]bis-](http://img.cochemist.com/ccimg/52500/52411-33-3_b.png)












































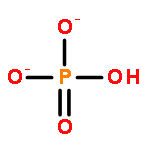
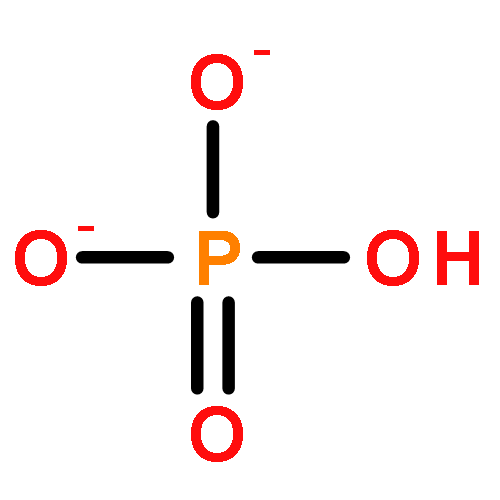



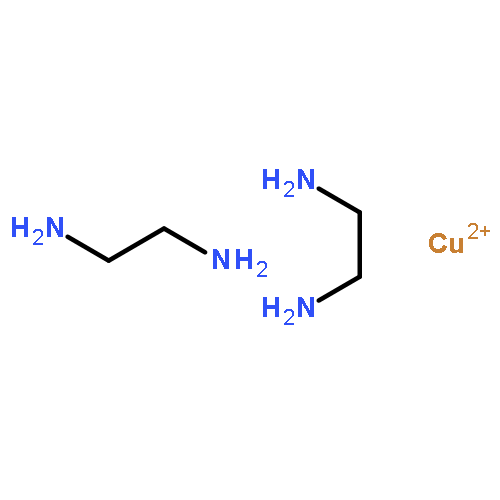








![3-[2-(3-AMINOPROPYLSULFANYL)ETHYLSULFANYL]PROPAN-1-AMINE](http://img.cochemist.com/ccimg/7100/7058-57-3.png)
![3-[2-(3-AMINOPROPYLSULFANYL)ETHYLSULFANYL]PROPAN-1-AMINE](http://img.cochemist.com/ccimg/7100/7058-57-3_b.png)








![9-Oxabicyclo[6.1.0]nonane, (1R,8S)-rel-](http://img.cochemist.com/ccimg/5000/4925-71-7.png)
![9-Oxabicyclo[6.1.0]nonane, (1R,8S)-rel-](http://img.cochemist.com/ccimg/5000/4925-71-7_b.png)


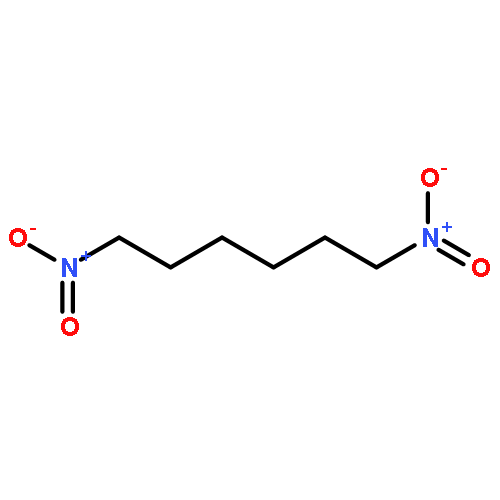


























![6,20-Dioxa-13,27-dithia-3,10,17,24,29,30,31,32-octaazapentacyclo[24.2.1.15,8.112,15.119,22]dotriaconta-5(32),12(31),14,19(30),26(29),28-hexaene-2,9,16,23-tetrone,7-methyl-11,25-bis(1-methylethyl)-4,18-bis[(1S)-1-methylpropyl]-,(4S,7R,8S,11R,18S,22S,25R)-](http://img.cochemist.com/ccimg/81200/81120-73-2.png)
![6,20-Dioxa-13,27-dithia-3,10,17,24,29,30,31,32-octaazapentacyclo[24.2.1.15,8.112,15.119,22]dotriaconta-5(32),12(31),14,19(30),26(29),28-hexaene-2,9,16,23-tetrone,7-methyl-11,25-bis(1-methylethyl)-4,18-bis[(1S)-1-methylpropyl]-,(4S,7R,8S,11R,18S,22S,25R)-](http://img.cochemist.com/ccimg/81200/81120-73-2_b.png)
![Phenol, 2,6-bis[[bis(2-pyridinylmethyl)amino]methyl]-4-methyl-](http://img.cochemist.com/ccimg/80600/80528-41-2.png)
![Phenol, 2,6-bis[[bis(2-pyridinylmethyl)amino]methyl]-4-methyl-](http://img.cochemist.com/ccimg/80600/80528-41-2_b.png)












![Ethanamine,2,2'-[1,2-ethanediylbis(thio)]bis-](http://img.cochemist.com/ccimg/21100/21057-05-6.png)
![Ethanamine,2,2'-[1,2-ethanediylbis(thio)]bis-](http://img.cochemist.com/ccimg/21100/21057-05-6_b.png)
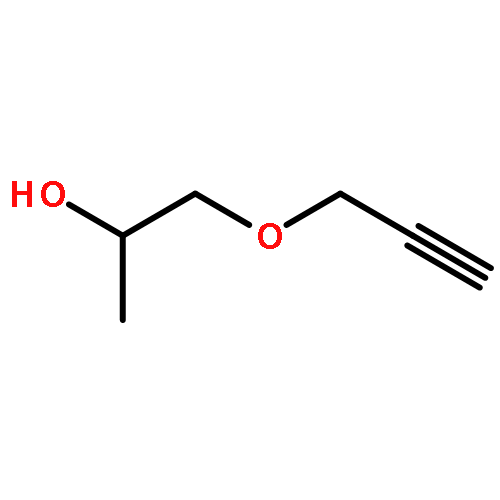
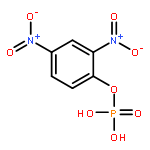

![Aziridine,1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/24400/24395-14-0.png)
![Aziridine,1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/24400/24395-14-0_b.png)
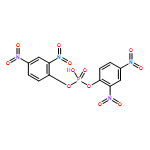
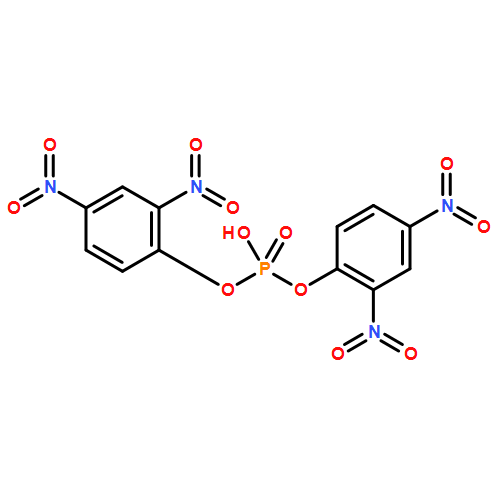
![3,7-Diazabicyclo[3.3.1]nonane](http://img.cochemist.com/ccimg/300/280-74-0.png)
![3,7-Diazabicyclo[3.3.1]nonane](http://img.cochemist.com/ccimg/300/280-74-0_b.png)
![6,13,20-Trioxa-3,10,17,22,23,24-hexaazatetracyclo[17.2.1.15,8.112,15]tetracosa-5(24),12(23),19(22)-triene-2,9,16-trione,7,14,21-trimethyl-4,11,18-tris(1-methylethyl)-, (1S,4S,7R,8S,11S,14R,15S,18S,21R)-](http://img.cochemist.com/ccimg/132000/131998-54-4.png)
![6,13,20-Trioxa-3,10,17,22,23,24-hexaazatetracyclo[17.2.1.15,8.112,15]tetracosa-5(24),12(23),19(22)-triene-2,9,16-trione,7,14,21-trimethyl-4,11,18-tris(1-methylethyl)-, (1S,4S,7R,8S,11S,14R,15S,18S,21R)-](http://img.cochemist.com/ccimg/132000/131998-54-4_b.png)