Co-reporter:Maryam Imani Nejad, Kevin M. Johnson, Nathan E. Price, and Kent S. Gates
Biochemistry December 20, 2016 Volume 55(Issue 50) pp:
Publication Date(Web):November 29, 2016
DOI:10.1021/acs.biochem.6b01080
Nitrogen mustard anticancer drugs generate highly reactive aziridinium ions that alkylate DNA. Monoadducts arising from reaction with position N7 of guanine residues are the major DNA adducts generated by these agents. Interstrand cross-links in which the drug bridges position N7 of two guanine residues are formed in low yields relative to those of the monoadducts but are generally thought to be central to medicinal activity. The N7-alkylguanine residues generated by nitrogen mustards are depurinated to yield abasic (Ap) sites in duplex DNA. Here, we show that Ap sites generated by the nitrogen mustard mechlorethamine lead to interstrand cross-links of a type not previously associated with this drug. Gel electrophoretic data were consistent with early evolution of the expected drug-bridged cross-links, followed by the appearance of Ap-derived cross-links. The evidence is further consistent with a reaction pathway involving alkylation of a guanine residue in a 5′-GT sequence, followed by depurination to generate the Ap site, and cross-link formation via reaction of the Ap aldehyde residue with the opposing adenine residue at this site [Price, N. E., Johnson, K. M., Wang, J., Fekry, M. I., Wang, Y., and Gates, K. S. (2014) J. Am. Chem. Soc. 136, 3483–3490]. The monofunctional DNA-alkylating agents 2-chloro-N,N-diethylethanamine 5, (2-chloroethyl)ethylsulfide 6, and natural product leinamycin similarly were found to induce the formation of Ap-derived cross-links in duplex DNA. This work provides the first characterization of Ap-derived cross-links at sequences in which a cytosine residue is located directly opposing the Ap site. Cross-linking processes of this type could be relevant in medicine and biology because Ap sites with directly opposing cytosine residues occur frequently in genomic DNA via spontaneous or enzymatic depurination of guanine and N7-alkylguanine residues.
Co-reporter:Puminan Punthasee, Adrian R. Laciak, Andrea H. Cummings, Kasi Viswanatharaju Ruddraraju, Sarah M. Lewis, Roman Hillebrand, Harkewal Singh, John J. Tanner, and Kent S. Gates
Biochemistry April 11, 2017 Volume 56(Issue 14) pp:2051-2051
Publication Date(Web):March 27, 2017
DOI:10.1021/acs.biochem.7b00151
Protein tyrosine phosphatase 1B (PTP1B) is a validated drug target, but it has proven difficult to develop medicinally useful, reversible inhibitors of this enzyme. Here we explored covalent strategies for the inactivation of PTP1B using a conjugate composed of an active site-directed 5-aryl-1,2,5-thiadiazolidin-3-one 1,1-dioxide inhibitor connected via a short linker to an electrophilic α-bromoacetamide moiety. Inhibitor–electrophile conjugate 5a caused time-dependent loss of PTP1B activity consistent with a covalent inactivation mechanism. The inactivation occurred with a second-order rate constant of (1.7 ± 0.3) × 102 M–1 min–1. Mass spectrometric analysis of the inactivated enzyme indicated that the primary site of modification was C121, a residue distant from the active site. Previous work provided evidence that covalent modification of the allosteric residue C121 can cause inactivation of PTP1B [Hansen, S. K., Cancilla, M. T., Shiau, T. P., Kung, J., Chen, T., and Erlanson, D. A. (2005) Biochemistry 44, 7704–7712]. Overall, our results are consistent with an unusual enzyme inactivation process in which noncovalent binding of the inhibitor–electrophile conjugate to the active site of PTP1B protects the nucleophilic catalytic C215 residue from covalent modification, thus allowing inactivation of the enzyme via selective modification of allosteric residue C121.
Co-reporter:Kasi Viswanatharaju Ruddraraju, Zachary D. Parsons, Calvin D. Lewis, and Kent S. Gates
The Journal of Organic Chemistry 2017 Volume 82(Issue 1) pp:776-780
Publication Date(Web):December 5, 2016
DOI:10.1021/acs.joc.6b02517
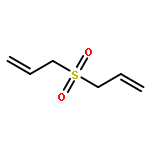
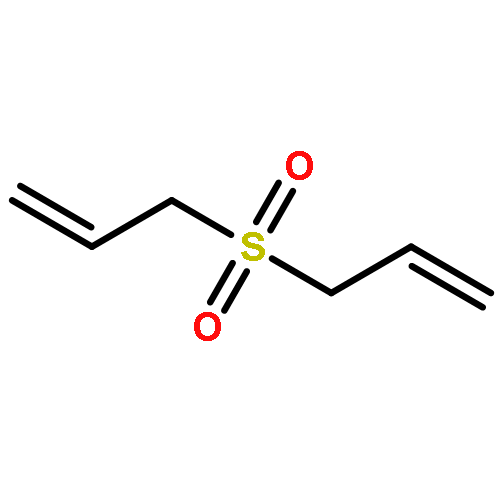
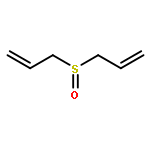
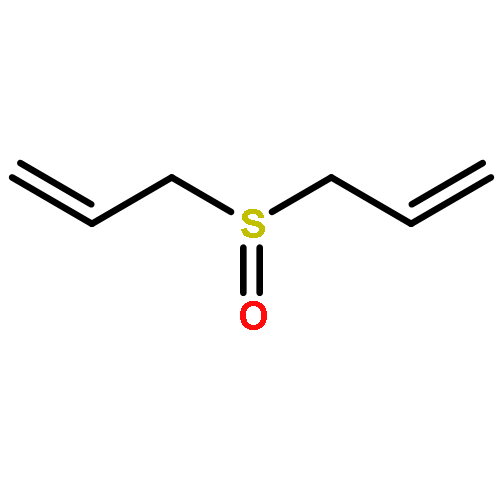
Allyl sulfides are bioactive phytochemicals found in garlic, onion, and other members of the genus Allium. Here we showed that diallyl disulfide and diallyl trisulfide can transfer allyl side chains to low molecular weight thiols. Diallyl monosulfide is inert with respect to this allyl transfer reaction. On the other hand, diallyl sulfone, a known metabolite of diallyl monosulfide, alkylates both amines and thiols under physiologically relevant conditions via isomerization to an electrophilic vinyl sulfone.
Co-reporter:Zachary D. Parsons, Kasi Viswanatharaju Ruddraraju, Nicholas Santo, Kent S. Gates
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 12) pp:2631-2640
Publication Date(Web):15 June 2016
DOI:10.1016/j.bmc.2016.03.054
Redox regulation of protein tyrosine phosphatase 1B (PTP1B) involves oxidative conversion of the active site cysteine thiolate into an electrophilic sulfenyl amide residue. Reduction of the sulfenyl amide by biological thiols regenerates the native cysteine residue. Here we explored fundamental chemical reactions that may enable covalent capture of the sulfenyl amide residue in oxidized PTP1B. Various sulfone-containing carbon acids were found to react readily with a model peptide sulfenyl amide via attack of the sulfonyl carbanion on the electrophilic sulfur center in the sulfenyl amide. Both the products and the rates of these reactions were characterized. The results suggest that capture of a peptide sulfenyl amide residue by sulfone-stabilized carbanions can slow, but not completely prevent, thiol-mediated generation of the corresponding cysteine-containing peptide. Sulfone-containing carbon acids may be useful components in the construction of agents that knock down PTP1B activity in cells via transient covalent capture of the sulfenyl amide oxoform generated during insulin signaling processes.
Co-reporter:Michael J. Catalano, Nathan E. Price, Kent S. Gates
Bioorganic & Medicinal Chemistry Letters 2016 26(11) pp: 2627-2630
Publication Date(Web):1 June 2016
DOI:10.1016/j.bmcl.2016.04.022
Positioning of reactive functional groups within a DNA duplex can enable chemical reactions that otherwise would not occur to an appreciable extent. However, few studies have quantitatively defined the extent to which the enforced proximity of reaction partners in duplex DNA can favor chemical processes. Here, we measured substantial effective molarities (as high as 25 M) afforded by duplex DNA to a reaction involving interstrand cross-link formation between 2′-deoxyadenosine and a 2-deoxyribose abasic (Ap) site.
Co-reporter:Michael J. Catalano; Shuo Liu; Nisana Andersen; Zhiyu Yang; Kevin M. Johnson; Nathan E. Price; Yinsheng Wang
Journal of the American Chemical Society 2015 Volume 137(Issue 11) pp:3933-3945
Publication Date(Web):February 24, 2015
DOI:10.1021/jacs.5b00669
A new type of interstrand cross-link resulting from the reaction of a DNA abasic site with a guanine residue on the opposing strand of the double helix was recently identified, but the chemical connectivity of the cross-link was not rigorously established. The work described here was designed to characterize the chemical structure and properties of dG–AP cross-links generated in duplex DNA. The approach involved characterization of the nucleoside cross-link “remnant” released by enzymatic digestion of DNA duplexes containing the dG–AP cross-link. We first carried out a chemical synthesis and complete spectroscopic structure determination of the putative cross-link remnant 9b composed of a 2-deoxyribose adduct attached to the exocyclic N2-amino group of dG. A reduced analogue of the cross-link remnant was also prepared (11b). Liquid chromatography–tandem mass spectrometric (LC-MS/MS) analysis revealed that the retention times and mass spectral properties of synthetic standards 9b and 11b matched those of the authentic cross-link remnants released by enzymatic digestion of duplexes containing the native and reduced dG–AP cross-link, respectively. These results establish the chemical connectivity of the dG–AP cross-link released from duplex DNA and provide a foundation for detection of this lesion in biological samples. The dG–AP cross-link in duplex DNA was remarkably stable, decomposing with a half-life of 22 days at pH 7 and 23 °C. The intrinsic chemical stability of the dG–AP cross-link suggests that this lesion in duplex DNA may have the power to block DNA-processing enzymes involved in transcription and replication.
Co-reporter:Zhiyu Yang, Nathan E. Price, Kevin M. Johnson, and Kent S. Gates
Biochemistry 2015 Volume 54(Issue 27) pp:4259-4266
Publication Date(Web):June 24, 2015
DOI:10.1021/acs.biochem.5b00482
Interstrand cross-links in cellular DNA are highly deleterious lesions that block transcription and replication. We recently characterized two new structural types of interstrand cross-links derived from the reaction of abasic (Ap) sites with either guanine or adenine residues in duplex DNA. Interestingly, these Ap-derived cross-links are forged by chemically reversible processes, in which the two strands of the duplex are joined by hemiaminal, imine, or aminoglycoside linkages. Therefore, understanding the stability of Ap-derived cross-links may be critical in defining the potential biological consequences of these lesions. Here we employed bacteriophage φ29 DNA polymerase, which can couple DNA synthesis and strand displacement, as a model system to examine whether dA-Ap cross-links can withstand DNA-processing enzymes. We first demonstrated that a chemically stable interstrand cross-link generated by hydride reduction of the dG-Ap cross-link completely blocked primer extension by φ29 DNA polymerase at the last unmodified nucleobase preceding cross-link. We then showed that the nominally reversible dA-Ap cross-link behaved, for all practical purposes, like an irreversible, covalent DNA–DNA cross-link. The dA-Ap cross-link completely blocked progress of the φ29 DNA polymerase at the last unmodified base before the cross-link. This suggests that Ap-derived cross-links have the power to block various DNA-processing enzymes in the cell. In addition, our results reveal φ29 DNA polymerase as a tool for detecting the presence and mapping the location of interstrand cross-links (and possibly other lesions) embedded within regions of duplex DNA.
Co-reporter:Sarmistha Sinha, Xiulong Shen, Fabio Gallazzi, Qian Li, Jaroslaw W. Zmijewski, Jack R. Lancaster Jr., and Kent S. Gates
Chemical Research in Toxicology 2015 Volume 28(Issue 2) pp:175
Publication Date(Web):January 15, 2015
DOI:10.1021/tx500259s
1-Hydroxyphenazine (1-HP) is a virulence factor produced by Pseudomonas aeruginosa. In this study, supercoiled plasmid DNA was employed as an analytical tool for the detection of ROS generation mediated by 1-HP. These assays provided evidence that 1-HP, in conjunction with NADPH alone or NADPH and the enzyme NADPH:cytochrome P450 reductase, mediated the production of superoxide radical under physiological conditions. Experiments with murine macrophage RAW264.7 cells and profluorescent ROS probes dichlorodihydrofluorescein or dihydroethidine provided preliminary evidence that 1-HP mediates the generation of intracellular oxidants. Generation of reactive oxygen species may contribute to the virulence properties of 1-HP in P. aeruginosa infections.
Co-reporter:Sarah M. Lewis, Ya Li, Michael J. Catalano, Adrian R. Laciak, Harkewal Singh, Derrick R. Seiner, Thomas J. Reilly, John J. Tanner, Kent S. Gates
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 20) pp:4549-4552
Publication Date(Web):15 October 2015
DOI:10.1016/j.bmcl.2015.08.065
Isothiocyanates are bioactive dietary phytochemicals that react readily with protein thiol groups. We find that isothiocyanates are time-dependent inactivators of cysteine-dependent protein tyrosine phosphatases (PTPs). Rate constants for the inactivation of PTP1B and SHP-2 by allyl isothiocyanate and sulforaphane range from 2 to 16 M−1 s−1. Results in the context of PTP1B are consistent with a mechanism involving covalent, yet reversible, modification of the enzyme’s active site cysteine residue.
Co-reporter:Xinyue Zhang, Nathan E. Price, Xi Fang, Zhiyu Yang, Li-Qun Gu, and Kent S. Gates
ACS Nano 2015 Volume 9(Issue 12) pp:11812
Publication Date(Web):November 13, 2015
DOI:10.1021/acsnano.5b03923
Nanopore-based sensors have been studied extensively as potential tools for DNA sequencing, characterization of epigenetic modifications such as 5-methylcytosine, and detection of microRNA biomarkers. In the studies described here, the α-hemolysin protein nanopore embedded in a lipid bilayer was used for the detection and characterization of interstrand cross-links in duplex DNA. Interstrand cross-links are important lesions in medicinal chemistry and toxicology because they prevent the strand separation that is required for read-out of genetic information from DNA in cells. In addition, interstrand cross-links are used for the stabilization of duplex DNA in structural biology and materials science. Cross-linked DNA fragments produced unmistakable current signatures in the nanopore experiment. Some cross-linked substrates gave irreversible current blocks of >10 min, while others produced long current blocks (10–100 s) before the double-stranded DNA cross-link translocated through the α-hemolysin channel in a voltage-driven manner. The duration of the current block for the different cross-linked substrates examined here may be dictated by the stability of the duplex region left in the vestibule of the nanopore following partial unzipping of the cross-linked DNA. Construction of calibration curves measuring the frequency of cross-link blocking events (1/τon) as a function of cross-link concentration enabled quantitative determination of the amounts of cross-linked DNA present in samples. The unique current signatures generated by cross-linked DNA in the α-HL nanopore may enable the detection and characterization of DNA cross-links that are important in toxicology, medicine, and materials science.Keywords: abasic site; damaged DNA; interstrand cross-link; nanopore; α-hemolysin;
Co-reporter:Jacqueline GamboaVarela; Kent S. Gates
Angewandte Chemie International Edition 2015 Volume 54( Issue 26) pp:7666-7669
Publication Date(Web):
DOI:10.1002/anie.201502566
Abstract
Interstrand DNA–DNA cross-links are highly toxic to cells because these lesions block the extraction of information from the genetic material. The pathways by which cells repair cross-links are important, but not well understood. The preparation of chemically well-defined cross-linked DNA substrates represents a significant challenge in the study of cross-link repair. Here a simple method is reported that employs “post-synthetic” modifications of commercially available 2′-deoxyoligonucleotides to install a single cross-link in high yield at a specified location within a DNA duplex. The cross-linking process exploits the formation of a hydrazone between a non-natural N4-amino-2′-deoxycytidine nucleobase and the aldehyde residue of an abasic site in duplex DNA. The resulting cross-link is stable under physiological conditions, but can be readily dissociated and re-formed through heating–cooling cycles.
Co-reporter:Jacqueline GamboaVarela; Kent S. Gates
Angewandte Chemie 2015 Volume 127( Issue 26) pp:7776-7779
Publication Date(Web):
DOI:10.1002/ange.201502566
Abstract
Interstrand DNA–DNA cross-links are highly toxic to cells because these lesions block the extraction of information from the genetic material. The pathways by which cells repair cross-links are important, but not well understood. The preparation of chemically well-defined cross-linked DNA substrates represents a significant challenge in the study of cross-link repair. Here a simple method is reported that employs “post-synthetic” modifications of commercially available 2′-deoxyoligonucleotides to install a single cross-link in high yield at a specified location within a DNA duplex. The cross-linking process exploits the formation of a hydrazone between a non-natural N4-amino-2′-deoxycytidine nucleobase and the aldehyde residue of an abasic site in duplex DNA. The resulting cross-link is stable under physiological conditions, but can be readily dissociated and re-formed through heating–cooling cycles.
Co-reporter:Kasi Viswanatharaju Ruddraraju, Zachary D. Parsons, Elizabeth M. Llufrio, Natasha L. Frost, and Kent S. Gates
The Journal of Organic Chemistry 2015 Volume 80(Issue 24) pp:12015-12026
Publication Date(Web):October 30, 2015
DOI:10.1021/acs.joc.5b01949
Protein tyrosine phosphatase 1B (PTP1B) is a validated therapeutic target for the treatment of type 2 diabetes; however, the enzyme has been classified by some as an “undruggable target”. Here we describe studies directed toward the development of agents that covalently capture the sulfenyl amide “oxoform” of PTP1B generated during insulin signaling events. The sulfenyl amide residue found in oxidized PTP1B presents a unique electrophilic sulfur center that may be exploited in drug and probe design. Covalent capture of oxidized PTP1B could permanently disable the intracellular pool of enzyme involved in regulation of insulin signaling. Here, we employed a dipeptide model of oxidized PTP1B to investigate the nucleophilic capture of the sulfenyl amide residue by structurally diverse 1,3-diketones. All of the 1,3-diketones examined here reacted readily with the electrophilic sulfur center in the sulfenyl amide residue to generate stable covalent attachments. Several different types of products were observed, depending upon the substituents present on the 1,3-diketone. The results provide a chemical foundation for the development of agents that covalently capture the oxidized form of PTP1B generated in cells during insulin signaling events.
Co-reporter:Nathan E. Price ; Kevin M. Johnson ; Jin Wang ; Mostafa I. Fekry ; Yinsheng Wang
Journal of the American Chemical Society 2014 Volume 136(Issue 9) pp:3483-3490
Publication Date(Web):February 7, 2014
DOI:10.1021/ja410969x
The loss of a coding nucleobase from the structure of DNA is a common event that generates an abasic (Ap) site (1). Ap sites exist as an equilibrating mixture of a cyclic hemiacetal and a ring-opened aldehyde. Aldehydes are electrophilic functional groups that can form covalent adducts with nucleophilic sites in DNA. Thus, Ap sites present a potentially reactive aldehyde as part of the internal structure of DNA. Here we report evidence that the aldehyde group of Ap sites in duplex DNA can form a covalent adduct with the N6-amino group of adenine residues on the opposing strand. The resulting interstrand DNA–DNA cross-link occurs at 5′-ApT/5′-AA sequences in remarkably high yields (15–70%) under physiologically relevant conditions. This naturally occurring DNA-templated reaction has the potential to generate cross-links in the genetic material of living cells.
Co-reporter:Douglas Melton, Calvin D. Lewis, Nathan E. Price, and Kent S. Gates
Chemical Research in Toxicology 2014 Volume 27(Issue 12) pp:2113
Publication Date(Web):November 6, 2014
DOI:10.1021/tx5003657
Hydralazine (4) is an antihypertensive agent that displays both mutagenic and epigenetic properties. Here, gel electrophoretic, mass spectroscopic, and chemical kinetics methods were used to provide evidence that medicinally relevant concentrations of 4 rapidly form covalent adducts with abasic sites in double- and single-stranded DNA under physiological conditions. These findings raise the intriguing possibility that the genotoxic properties of this clinically used drug arise via reactions with an endogenous DNA lesion rather than with the canonical structure of DNA.
Co-reporter:Xiulong Shen, Anuruddha Rajapakse, Fabio Gallazzi, Venkatraman Junnotula, Tarra Fuchs-Knotts, Rainer Glaser, and Kent S. Gates
Chemical Research in Toxicology 2014 Volume 27(Issue 1) pp:111
Publication Date(Web):December 11, 2013
DOI:10.1021/tx400356y
The 1,2,4-benzotriazine 1,4-dioxides are an important class of potential anticancer drugs that selectively kill the low-oxygen (hypoxic) cells found in solid tumors. These compounds undergo intracellular one-electron enzymatic reduction to yield an oxygen-sensitive drug radical intermediate that partitions forward, under hypoxic conditions, to generate a highly reactive secondary radical that causes cell killing DNA damage. Here, we characterized bioreductively activated, hypoxia-selective DNA-strand cleavage by 1,2,4-benzotriazine 1,4-dioxide. We found that one-electron enzymatic activation of 1,2,4-benzotriazine 1,4-dioxide under hypoxic conditions in the presence of the deuterium atom donor methanol-d4 produced nondeuterated mono-N-oxide metabolites. This and the results of other isotopic labeling studies provided evidence against the generation of atom-abstracting drug radical intermediates and are consistent with a DNA-damage mechanism involving the release of hydroxyl radical from enzymatically activated 1,2,4-benzotriazine 1,4-dioxides.
Co-reporter:Kevin M. Johnson, Zachary D. Parsons, Charles L. Barnes, and Kent S. Gates
The Journal of Organic Chemistry 2014 Volume 79(Issue 16) pp:7520-7531
Publication Date(Web):July 16, 2014
DOI:10.1021/jo501252p
Tirapazamine (3-amino-1,2,4-benzotriazine 1,4-dioxide) is a heterocyclic di-N-oxide that undergoes enzymatic deoxygenation selectively in the oxygen-poor (hypoxic) cells found in solid tumors to generate a mono-N-oxide metabolite. This work explored the idea that the electronic changes resulting from the metabolic deoxygenation of tirapazamine analogues might be exploited to activate a DNA-alkylating species selectively in hypoxic tissue. Toward this end, tirapazamine analogues bearing nitrogen mustard units were prepared. In the case of the tirapazamine analogue 18a bearing a nitrogen mustard unit at the 6-position, it was found that removal of the 4-oxide from the parent di-N-oxide to generate the mono-N-oxide analogue 17a did indeed cause a substantial increase in reactivity of the mustard unit, as measured by hydrolysis rates and DNA-alkylation yields. Hammett sigma values were measured to quantitatively assess the magnitude of the electronic changes induced by metabolic deoxygenation of the 3-amino-1,2,4-benzotriazine 1,4-dioxide heterocycle. The results provide evidence that the 1,2,4-benzotiazine 1,4-dioxide unit can serve as an oxygen-sensing prodrug platform for the selective unmasking of bioactive agents in hypoxic cells.
Co-reporter:Anuruddha Rajapakse, Collette Linder, Ryan D. Morrison, Ujjal Sarkar, Nathan D. Leigh, Charles L. Barnes, J. Scott Daniels, and Kent S. Gates
Chemical Research in Toxicology 2013 Volume 26(Issue 4) pp:555
Publication Date(Web):March 14, 2013
DOI:10.1021/tx300483z
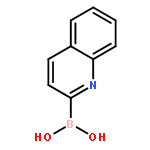
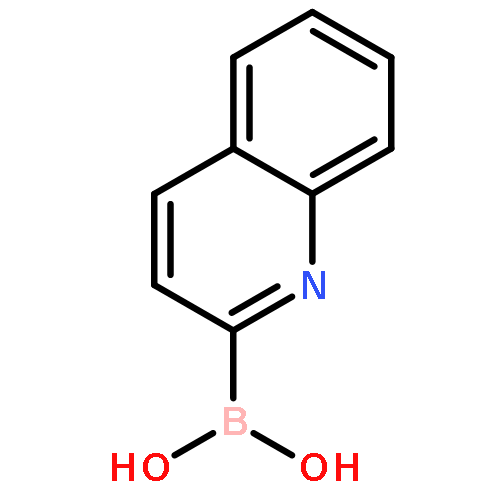
There is substantial interest in small molecules that can be used to detect or kill the hypoxic (low oxygen) cells found in solid tumors. Nitroaryl moieties are useful components in the design of hypoxia-selective imaging agents and prodrugs because one-electron reductases can convert the nitroaryl group to nitroso, hydroxylamino, and amino metabolites selectively under low oxygen conditions. Here, we describe the in vitro, cell free metabolism of a pro-fluorescent substrate, 6-nitroquinoline (1) under both aerobic and hypoxic conditions. Both LC-MS and fluorescence spectroscopic analyses provided evidence that the one-electron reducing enzyme system, xanthine/xanthine oxidase, converted the nonfluorescent parent compound 1 to the known fluorophore 6-aminoquinoline (2) selectively under hypoxic conditions. The presumed intermediate in this reduction process, 6-hydroxylaminoquinoline (6), is fluorescent and can be efficiently converted by xanthine/xanthine oxidase to 2 only under hypoxic conditions. This finding provides evidence for multiple oxygen-sensitive steps in the enzymatic conversion of nitroaryl compounds to the corresponding amino derivatives. In a side reaction that is separate from the bioreductive metabolism of 1, xanthine oxidase converted 1 to 6-nitroquinolin-2(1H)-one (5). These studies may enable the use of 1 as a fluorescent substrate for the detection and profiling of one-electron reductases in cell culture or biopsy samples. In addition, the compound may find use as a fluorogenic probe for the detection of hypoxia in tumor models. The occurrence of side products such as 5 in the enzymatic bioreduction of 1 underscores the importance of metabolite identification in the characterization of hypoxia-selective probes and drugs that employ nitroaryl units as oxygen sensors.
Co-reporter:Zachary D. Parsons and Kent S. Gates
Biochemistry 2013 Volume 52(Issue 37) pp:
Publication Date(Web):August 19, 2013
DOI:10.1021/bi400451m
Protein tyrosine phosphatases (PTPs) play an important role in the regulation of mammalian signal transduction. During some cell signaling processes, the generation of endogenous hydrogen peroxide inactivates selected PTPs via oxidation of the enzyme’s catalytic cysteine thiolate group. Importantly, low-molecular weight and protein thiols in the cell have the potential to regenerate the catalytically active PTPs. Here we examined the recovery of catalytic activity from two oxidatively inactivated PTPs (PTP1B and SHP-2) by various low-molecular weight thiols and the enzyme thioredoxin. All monothiols examined regenerated the catalytic activity of oxidized PTP1B, with apparent rate constants that varied by a factor of approximately 8. In general, molecules bearing low-pKa thiol groups were particularly effective. The biological thiol glutathione repaired oxidized PTP1B with an apparent second-order rate constant of 0.023 ± 0.004 M–1 s–1, while the dithiol dithiothreitol (DTT) displayed an apparent second-order rate constant of 0.325 ± 0.007 M–1 s–1. The enzyme thioredoxin regenerated the catalytic activity of oxidized PTP1B at a substantially faster rate than DTT. Thioredoxin (2 μM) converted oxidized PTP1B to the active form with an observed rate constant of 1.4 × 10–3 s–1. The rates at which these agents regenerated oxidized PTP1B followed the order Trx > DTT > GSHand comparable values observed at 2 μM Trx, 4 mM DTT, and 60 mM GSH. Various disulfides that are byproducts of the reactivation process did not inactivate native PTP1B at concentrations of 1–20 mM. The common biochemical reducing agent tris(2-carboxyethyl)phosphine regenerates enzymatic activity from oxidized PTP1B somewhat faster than the thiol-based reagents, with a rate constant of 1.5 ± 0.5 M–1 s–1. We observed profound kinetic differences between the thiol-dependent regeneration of activity from oxidized PTP1B and SHP-2, highlighting the potential for structural differences in various oxidized PTPs to play a significant role in the rates at which low-molecular weight thiols and thiol-containing enzymes such as thioredoxin and glutaredoxin return catalytic activity to these enzymes during cell signaling events.
Co-reporter:Kripa Keerthi, Anuruddha Rajapakse, Daekyu Sun, Kent S. Gates
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 1) pp:235-241
Publication Date(Web):1 January 2013
DOI:10.1016/j.bmc.2012.10.021
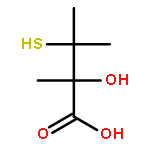
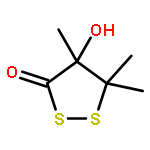
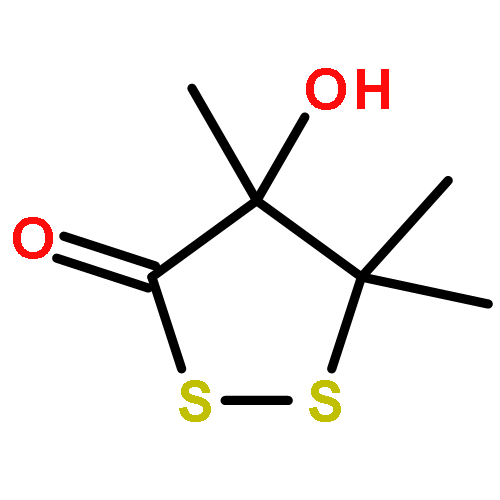
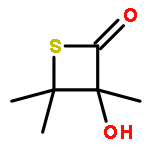
Leinamycin (1) is a Streptomyces-derived natural product that displays nanomolar IC50 values against human cancer cell lines. In the work described here, we report the synthesis and characterization of a small leinamycin analogue 19 that closely resembles the ‘upper-right quadrant’ of the natural product, consisting of an alicyclic 1,2-dithiolan-3-one 1-oxide heterocycle connected to an alkene by a two-carbon linker. The results indicate that this small analogue contains the core set of functional groups required to enable thiol-triggered generation of both redox active polysulfides and an episulfonium ion intermediate via the complex reaction cascade first seen in the natural product leinamycin. The small leinamycin analogue 19 caused thiol-triggered oxidative DNA strand cleavage in a manner similar to the natural product, but did not alkyate duplex DNA effectively. This highlights the central role of the 18-membered macrocycle of leinamycin in driving efficient DNA alkylation by the natural product.A small analogue of the natural product leinamycin was prepared containing the core set of functional groups required to enable thiol-triggered generation of both redox active polysulfides and an episulfonium ion intermediate via the complex reaction cascade first seen in the natural product leinamycin.
Co-reporter:Kevin M. Johnson ; Nathan E. Price ; Jin Wang ; Mostafa I. Fekry ; Sanjay Dutta ; Derrick R. Seiner ; Yinsheng Wang
Journal of the American Chemical Society 2012 Volume 135(Issue 3) pp:1015-1025
Publication Date(Web):December 5, 2012
DOI:10.1021/ja308119q
We recently reported that the aldehyde residue of an abasic (Ap) site in duplex DNA can generate an interstrand cross-link via reaction with a guanine residue on the opposing strand. This finding is intriguing because the highly deleterious nature of interstrand cross-links suggests that even small amounts of Ap-derived cross-links could make a significant contribution to the biological consequences stemming from the generation of Ap sites in cellular DNA. Incubation of 21-bp duplexes containing a central 5′-CAp sequence under conditions of reductive amination (NaCNBH3, pH 5.2) generated much higher yields of cross-linked DNA than reported previously. At pH 7, in the absence of reducing agents, these Ap-containing duplexes also produced cross-linked duplexes that were readily detected on denaturing polyacrylamide gels. Cross-link formation was not highly sensitive to reaction conditions, and the cross-link, once formed, was stable to a variety of workup conditions. Results of multiple experiments including MALDI-TOF mass spectrometry, gel mobility, methoxyamine capping of the Ap aldehyde, inosine-for-guanine replacement, hydroxyl radical footprinting, and LC–MS/MS were consistent with a cross-linking mechanism involving reversible reaction of the Ap aldehyde residue with the N2-amino group of the opposing guanine residue in 5′-CAp sequences to generate hemiaminal, imine, or cyclic hemiaminal cross-links (7–10) that were irreversibly converted under conditions of reductive amination (NaCNBH3/pH 5.2) to a stable amine linkage. Further support for the importance of the exocyclic N2-amino group in this reaction was provided by an experiment showing that installation of a 2-aminopurine-thymine base pair at the cross-linking site produced high yields (15–30%) of a cross-linked duplex at neutral pH, in the absence of NaCNBH3.
Co-reporter:Goutam Chowdhury, Ujjal Sarkar, Susan Pullen, William R. Wilson, Anuruddha Rajapakse, Tarra Fuchs-Knotts, and Kent S. Gates
Chemical Research in Toxicology 2012 Volume 25(Issue 1) pp:197
Publication Date(Web):November 15, 2011
DOI:10.1021/tx2004213
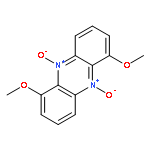
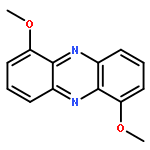
Heterocyclic N-oxides are an interesting class of antitumor agents that selectively kill the hypoxic cells found in solid tumors. The hypoxia-selective activity of the lead compound in this class, tirapazamine, stems from its ability to undergo intracellular one-electron reduction to an oxygen-sensitive drug radical intermediate. In the presence of molecular oxygen, the radical intermediate is back-oxidized to the parent molecule. Under hypoxic conditions, the extended lifetime of the drug radical intermediate enables its conversion to a highly cytotoxic DNA-damaging intermediate via a “deoxygenative” mechanism involving the loss of oxygen from one of its N-oxide groups. The natural product myxin is a phenazine di-N-oxide that displays potent antibiotic activity against a variety of organisms under aerobic conditions. In light of the current view of heterocyclic N-oxides as agents that selectively operate under hypoxic conditions, it is striking that myxin was identified from Sorangium extracts based upon its antibiotic properties under aerobic conditions. Therefore, we set out to examine the molecular mechanisms underlying the biological activity of myxin. We find that myxin causes bioreductively activated, radical-mediated DNA strand cleavage under both aerobic and anaerobic conditions. Our evidence indicates that strand cleavage occurs via a deoxygenative metabolism. We show that myxin displays potent cytotoxicity against the human colorectal cancer cell line HCT-116 under both aerobic and anaerobic conditions that is comparable to the cell-killing properties of tirapazamine under anaerobic conditions. This work sheds light on the processes by which the naturally occurring aromatic N-oxide myxin gains its potent antibiotic properties under aerobic conditions. Furthermore, these studies highlight the general potential for aromatic N-oxides to undergo highly cytotoxic deoxygenative metabolism following enzymatic one-electron reduction under aerobic conditions.
Co-reporter:Jian Yin, Rainer Glaser, and Kent S. Gates
Chemical Research in Toxicology 2012 Volume 25(Issue 3) pp:634
Publication Date(Web):March 5, 2012
DOI:10.1021/tx200546u
The initial steps of the activation of tirapazamine (TPZ, 1, 3-amino-1,2,4-benzotriazine 1,4-N,N-dioxide) under hypoxic conditions consist of the one-electron reduction of 1 to radical anion 2 and the protonation of 2 at O(N4) or O(N1) to form neutral radicals 3 and 4, respectively. There are some questions, however, as to whether radicals 3 and/or 4 will then undergo N–OH homolyses 3 → 5 + ·OH and 4 → 6 + ·OH or, alternatively, whether 3 and/or 4 may react by dehydration and form aminyl radicals via 3 → 11 + H2O and 4 → 12 + H2O or phenyl radicals via 3 → 17 + H2O. These outcomes might depend on the chemistry after the homolysis of 3 and/or 4, that is, dehydration may be the result of a two-step sequence that involves N–OH homolysis and formation of ·OH aggregates of 5 and 6 followed by H-abstraction within the ·OH aggregates to form hydrates of aminyls 11 and 12 or of phenyl 17. We studied these processes with configuration interaction theory, perturbation theory, and density functional theory. All stationary structures of OH aggregates of 5 and 6, of H2O aggregates of 11, 12, and 17, and of the transition state structures for H-abstraction were located and characterized by vibrational analysis and with methods of electron and spin-density analysis. The doublet radical 17 is a normal spin-polarized radical, whereas the doublet radicals 11 and 12 feature quartet instabilities. The computed reaction energies and activation barriers allow for dehydration in principle, but the productivity of all of these channels should be low for kinetic and dynamic reasons. With a view to plausible scenarios for the generation of latent aryl radical species without dehydration, we scanned the potential energy surfaces of 2–4 as a function of the (O)N1–Y (Y = C5a, N2) and (O)N4–Z (Z = C4a, C3) bond lengths. The elongation of any one of these bonds by 0.5 Å requires less than 25 kcal/mol, and this finding strongly suggests the possibility of bimolecular reactions of the spin-trap molecules with 2–4 concomitant with triazene ring-opening.
Co-reporter:Jian Yin, Rainer Glaser, and Kent S. Gates
Chemical Research in Toxicology 2012 Volume 25(Issue 3) pp:620
Publication Date(Web):March 5, 2012
DOI:10.1021/tx2005458
Tirapazamine (TPZ, 1, 3-amino-1,2,4-benzotriazine 1,4-N,N-dioxide), the radical anion 2 formed by one-electron reduction of 1, and neutral radicals 3 and 4 formed by protonation of 2 at O(N4) or O(N1), respectively, and their N–OH homolyses 3 → 5 + ·OH and 4 → 6 + ·OH have been studied with configuration interaction theory, perturbation theory, and density functional theory. A comprehensive comparative analysis is presented of structures and electronic structures and with focus on the development of an understanding of the spin-density distributions of the radical species. The skeletons of radicals 3 and 4 are distinctly nonplanar, several stereoisomeric structures are discussed, and there exists an intrinsic preference for 3 over 4. The N-oxides 1, 5, and 6 have closed-shell singlet ground states and low-lying, singlet biradical (SP-1, SP-6) or biradicaloid (SP-5) excited states. The doublet radicals 2, 3, and 4 are heavily spin-polarized. Most of the spin density of the doublet radicals 2, 3, and 4 is located in one (N,O)-region, and in particular, 3 and 4 are not C3-centered radicals. Significant amounts of spin density occur in both rings in the singlet biradical(oid) excited states of 1, 5, and 6. The dipole moment of the N2–C3(X) bond is large, and the nature of X provides a powerful handle to modulate the N2–C3 bond polarity with opposite effects on the two NO regions. Our studies show very low proton affinities of radical anion 2 and suggest that the pKa of radical [2+H] might be lower than 6. Implications are discussed regarding the formation of hydroxyl from 3 and/or 4, regarding the ability of 5 and 6 to react with carbon-centered radicals in a manner that ultimately leads to oxygen transfer, and regarding the interpretation of the EPR spectra of reduced TPZ species and of their spin-trap adducts.
Co-reporter:Marjorie Hoffman, Anuruddha Rajapakse, Xiulong Shen, and Kent S. Gates
Chemical Research in Toxicology 2012 Volume 25(Issue 8) pp:1609
Publication Date(Web):May 23, 2012
DOI:10.1021/tx300066z
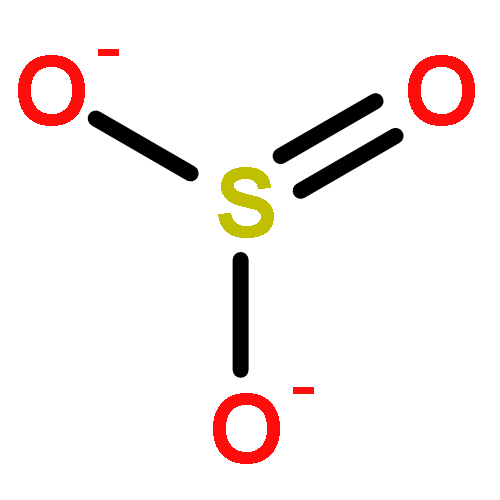
Hydrogen sulfide (H2S) has long been known for its toxic properties; however, in recent years, evidence has emerged that this small, gaseous molecule may serve as an endogenous cell-signaling agent. Though perhaps surprising in light of its potential role as an endogenous signaling agent, a number of studies have provided evidence that H2S is a DNA-damaging mutagen. In the work reported here, the chemical mechanisms of DNA damage by H2S were examined. Using a plasmid-based DNA strand cleavage assay, we found that micromolar concentrations of H2S generated single-strand DNA cleavage. Mechanistic studies indicate that this process involved autoxidation of H2S to generate superoxide, hydrogen peroxide, and, ultimately, the well-known DNA-damaging agent hydroxyl radical via a trace metal-mediated Fenton-type reaction. Strand cleavage by H2S proceeded in the presence of physiological thiol concentrations, and the known byproducts of H2S oxidation such as thiosulfate, sulfite, and sulfate do not contribute to the strand cleavage process. However, initially generated oxidation products such as persulfide (S22–) likely undergo rapid autoxidation reactions that contribute to the generation of superoxide. The potential relevance of autoxidation processes to the genotoxic and cell signaling properties of H2S is discussed.
Co-reporter:Santhosh Sivaramakrishnan, Leonid Breydo, Daekyu Sun, Kent S. Gates
Bioorganic & Medicinal Chemistry Letters 2012 22(11) pp: 3791-3794
Publication Date(Web):
DOI:10.1016/j.bmcl.2012.04.003
Co-reporter:Anuruddha Rajapakse and Kent S. Gates
The Journal of Organic Chemistry 2012 Volume 77(Issue 7) pp:3531-3537
Publication Date(Web):March 14, 2012
DOI:10.1021/jo3004748
Regions of low oxygen concentration (hypoxia) occur in both normal human physiology and under pathophysiological conditions. Fluorescent probes for the direct imaging of cellular hypoxia could be useful tools that complement radiochemical imaging and immunohistochemical staining methods. In this work, we set out to characterize the hypoxia-selective enzymatic metabolism of a simple nitroaryl probe, 6-nitroquinoline (1). We envisioned that this compound might undergo hypoxia-selective, bioreductive conversion to the fluorescent product, 6-aminoquinoline (2). The probe 1 was, indeed, converted to a fluorescent product selectively under hypoxic conditions by the one-electron reducing enzyme NADPH:cytochrome P450 reductase. However, inspection of the fluorescence spectrum and LC–MS analysis of the reaction mixture revealed that the expected product 2 was not formed. Rather, the 63-fold increase in fluorescence emission at 445 nm resulting from the hypoxic metabolism of 1 was due to formation of the azoxy-helicene product, pyrido[3,2-f]quinolino[6,5-c]cinnoline 3-oxide (4). The generation of 4 involves an unusual biaryl bond formation under reductive conditions. The mechanism of this process remains uncertain but could proceed via combination of a nitroaryl radical anion with a neutral nitrosoaryl radical, followed by tautomerization and intramolecular condensation between the resulting hydroxylamine and nitroso functional groups. Bioreductive metabolism of nitroaryl compounds represents a promising strategy for the selective delivery of cytotoxic agents and fluorescent markers to hypoxic tissue, but the results described here provide an important glimpse of the chemical complexity that can be associated with the enzymatic one-electron reduction of nitroaryl compounds.
Co-reporter:Haiying Zhou ; Harkewal Singh ; Zachary D. Parsons ; Sarah M. Lewis ; Sanjib Bhattacharya ; Derrick R. Seiner ; Jason N. LaButti ; Thomas J. Reilly ; John J. Tanner
Journal of the American Chemical Society 2011 Volume 133(Issue 40) pp:15803-15805
Publication Date(Web):September 13, 2011
DOI:10.1021/ja2077137
Hydrogen peroxide is a cell signaling agent that inactivates protein tyrosine phosphatases (PTPs) via oxidation of their catalytic cysteine residue. PTPs are inactivated rapidly during H2O2-mediated cellular signal transduction processes, but, paradoxically, hydrogen peroxide is a rather sluggish PTP inactivator in vitro. Here we present evidence that the biological buffer bicarbonate/CO2 potentiates the ability of H2O2 to inactivate PTPs. The results of biochemical experiments and high-resolution crystallographic analysis are consistent with a mechanism involving oxidation of the catalytic cysteine residue by peroxymonocarbonate generated via the reaction of H2O2 with HCO3–/CO2.
Co-reporter:Mostafa I. Fekry ; Jozsef Szekely ; Sanjay Dutta ; Leonid Breydo ; Hong Zang
Journal of the American Chemical Society 2011 Volume 133(Issue 44) pp:17641-17651
Publication Date(Web):September 28, 2011
DOI:10.1021/ja2046149
Molecular recognition and chemical modification of DNA are important in medicinal chemistry, toxicology, and biotechnology. Historically, natural products have revealed many interesting and unexpected mechanisms for noncovalent DNA binding and covalent DNA modification. The studies reported here characterize the molecular mechanisms underlying the efficient alkylation of duplex DNA by the Streptomyces-derived natural product leinamycin. Previous studies suggested that alkylation of duplex DNA by activated leinamycin (2) is driven by noncovalent association of the natural product with the double helix. This is striking because leinamycin does not contain a classical noncovalent DNA-binding motif, such as an intercalating unit, a groove binder, or a polycation. The experiments described here provide evidence that leinamycin is an atypical DNA-intercalating agent. A competition binding assay involving daunomycin-mediated inhibition of DNA alkylation by leinamycin provided evidence that activated leinamycin binds to duplex DNA with an apparent binding constant of approximately 4.3 ± 0.4 × 103 M–1. Activated leinamycin caused duplex unwinding and hydrodynamic changes in DNA-containing solutions that are indicative of DNA intercalation. Characterization of the reaction of activated leinamycin with palindromic duplexes containing 5′-CG and 5′-GC target sites, bulge-containing duplexes, and 5-methylcytosine-containing duplexes provided evidence regarding the orientation of leinamycin with respect to target guanine residues. The data allow construction of a model for the leinamycin–DNA complex suggesting how a modest DNA-binding constant combines with proper positioning of the natural product to drive efficient alkylation of guanine residues in the major groove of duplex DNA.
Co-reporter:Mostafa I. Fekry, Peter A. Tipton, and Kent S. Gates
ACS Chemical Biology 2011 Volume 6(Issue 2) pp:127
Publication Date(Web):January 18, 2011
DOI:10.1021/cb2000023
It was claimed in a recent publication that a strain of Halomonadacea bacteria (GFAJ-1) isolated from the arsenic-rich waters of Mono Lake, California is able to substitute arsenic for phosphorus in its macromolecules and small molecule metabolites. In this short Perspective, we consider chemical and biochemical issues surrounding the central claim that Halomonadacea GFAJ-1 is able to survive while incorporating kinetically labile arsenodiester linkages into the backbone of its DNA. Chemical precedents suggest that arsenodiester linkages in the putative arsenic-containing DNA of GFAJ-1 would undergo very rapid hydrolytic cleavage in water at 25 °C with an estimated half-life of 0.06 s. In contrast, the phosphodiester linkages of native DNA undergo spontaneous hydrolysis with a half-life of approximately 30,000,000 y at 25 °C. Overcoming such dramatic kinetic instability in its genetic material would present serious challenges to Halomonadacea GFAJ-1.
Co-reporter:Mostafa I. Fekry, Nathan E. Price, Hong Zang, Chaofeng Huang, Michael Harmata, Paul Brown, J. Scott Daniels, and Kent S. Gates
Chemical Research in Toxicology 2011 Volume 24(Issue 2) pp:217
Publication Date(Web):January 20, 2011
DOI:10.1021/tx100282b
Some biologically active chemicals are relatively stable in the extracellular environment but, upon entering the cell, undergo biotransformation into reactive intermediates that covalently modify DNA. The diverse chemical reactions involved in the bioactivation of DNA-damaging agents are both fundamentally interesting and of practical importance in medicinal chemistry and toxicology. The work described here examines the bioactivation of α-haloacrolyl-containing molecules. The α-haloacrolyl moiety is found in a variety of cytotoxic natural products including clionastatin B, bromovulone III, discorahabdins A, B, and C, and trichodenone C, in mutagens such as 2-bromoacrolein and 3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone (MX), and in the anticancer drug candidates brostallicin and PNU-151807. Using α-bromo-2-cyclopentenone (1) as a model compound, the activation of α-haloacrolyl-containing molecules by biological thiols was explored. The results indicate that both low molecular weight and peptide thiols readily undergo conjugate addition to 1. The resulting products are consistent with a mechanism in which initial addition of thiols to 1 is followed by intramolecular displacement of bromide to yield a DNA-alkylating episulfonium ion intermediate. The reaction of thiol-activated 1 with DNA produces labile lesions at deoxyguanosine residues. The sequence specificity and salt dependence of this process is consistent with involvement of an episulfonium ion intermediate. The alkylated guanine residue resulting from the thiol-triggered reaction of 1 with duplex DNA was characterized using mass spectrometry. The results provide new insight regarding the mechanisms by which thiols can bioactivate small molecules and offer a more complete understanding of the molecular mechanisms underlying the biological activity of cytotoxic, mutagenic, and medicinal compounds containing the α-haloacrolyl group.
Co-reporter:Anuruddha Rajapakse;Charles L. Barnes
Journal of Chemical Crystallography 2011 Volume 41( Issue 11) pp:
Publication Date(Web):2011 November
DOI:10.1007/s10870-011-0162-z
The helicene, pyrido[3,2-f]quinolino[6,5-c]cinnoline 5-oxide, was prepared by treatment of 6-hydroxylaminoquinoline with xanthine oxidase or treatment of 6-nitroquinoline with glucose in 30% NaOH and the product characterized using NMR, high resolution mass spectrometry, and X-ray crystallography. The hydrogens on carbons 7 and 12 of the terminal aromatic rings are separated by 2.495 Å creating an angle of 25.0° between the planes of the two quinoline ring systems. In the crystal, water molecules serve to link the helicenes into a one dimensional chain structure forming a hydrogen bonded bridge between N2 of one molecule and N4 of another. The molecule (C18H10N4O·H2O) crystallized in the monoclinic P21/n space group. Unit cell parameters for pyrido[3,2-f]quinolino[6,5-c]cinnoline 5-oxide monohydrate: a = 7.0829(12), b = 18.559(3), c = 11.0985(19) Å, β = 107.736(2)°, and Ζ = 4.
Co-reporter:Sanjib Bhattacharyya, Haiying Zhou, Derrick R. Seiner, Kent S. Gates
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 16) pp:5945-5949
Publication Date(Web):15 August 2010
DOI:10.1016/j.bmc.2010.06.084
Dithiolethiones upregulate the expression of cancer-preventive proteins via modification of thiol residues in the Keap1–Nrf2 transcription factor complex. In addition to Keap1–Nrf2, dithiolethiones have the potential to modify a variety of cysteine-containing proteins in the cell. Such ‘off target’ reactions could contribute to either side effects or cancer-preventive efficacy. Evidence is presented here that cancer chemopreventive dithiolethiones inactivate protein tyrosine phosphatases via covalent, but thiol-labile, modification of active site residues. This observation may explain a number of previously reported cellular responses to dithiolethiones.
Co-reporter:Venkatraman Junnotula, Anuruddha Rajapakse, Leire Arbillaga, Adela López de Cerain, Beatriz Solano, Raquel Villar, Antonio Monge, Kent S. Gates
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 9) pp:3125-3132
Publication Date(Web):1 May 2010
DOI:10.1016/j.bmc.2010.03.042
The heterocyclic N-oxide, 3-amino-1,2,4-benzotriazine 1,4-dioxide (tirapazamine, 1), shows promising antitumor activity in preclinical studies, but there is a continuing need to explore new compounds in this general structural category. In the work described here, we examined the properties of 7-chloro-2-thienylcarbonyl-3-trifluoromethylquinoxaline 1,4-dioxide (9h). We find that 9h causes redox-activated, hypoxia-selective DNA cleavage that mirrors the lead compound, tirapazamine, in both mechanism and potency. Furthermore, we find that 9h displays hypoxia-selective cytotoxicity against human cancer cell lines.
Co-reporter:Santhosh Sivaramakrishnan, Andrea H. Cummings, Kent S. Gates
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 2) pp:444-447
Publication Date(Web):15 January 2010
DOI:10.1016/j.bmcl.2009.12.001
Model reactions offer a chemical mechanism by which formation of a sulfenyl amide residue at the active site of the redox-regulated protein tyrosine phosphatase PTP1B protects the cysteine redox switch in this enzyme against irreversible oxidative destruction. The results suggest that ‘overoxidation’ of the sulfenyl amide redox switch to the sulfinyl amide in proteins is a chemically reversible event, because the sulfinyl amide can be easily returned to the native cysteine thiol residue via reactions with cellular thiols.Model reactions offer a chemical mechanism by which formation of a sulfenyl amide residue at the active site of the redox-regulated protein tyrosine phosphatase PTP1B protects the cysteine redox switch in this enzyme against irreversible oxidative destruction. The results suggest that ‘overoxidation’ of the sulfenyl amide redox switch to the sulfinyl amide in proteins is a chemically reversible event, because the sulfinyl amide can be easily returned to the native cysteine thiol residue via reactions with cellular thiols.
Co-reporter:Ujjal Sarkar;Rainer Glaser;Zack D. Parsons
Journal of Chemical Crystallography 2010 Volume 40( Issue 7) pp:624-629
Publication Date(Web):2010 July
DOI:10.1007/s10870-010-9707-9
1,2,4-Benzotriazine 1,4-di-N-oxides are potent antitumor drug candidates that undergo in vivo bioreduction leading to selective DNA damage in the low oxygen (hypoxic) cells found in tumors. Tirapazamine (TPZ) is the lead compound in this family. Here we report on the synthesis, crystal structure, and conformational analysis of a new analog, 3-cyclopropyl-1,2,4-benzotriazine 1,4-di-N-oxide (3). Compound 3 (C10H10N3O2) crystallized in the monoclinic space group C2/c. Unit cell parameters for 3: a = 16.6306 (12), b = 7.799 (5), c = 16.0113 (11) Å, α = 90, β = 119.0440 (10), γ = 90, and z = 8.
Co-reporter:Kent S. Gates
Chemical Research in Toxicology 2009 Volume 22(Issue 11) pp:1747
Publication Date(Web):September 16, 2009
DOI:10.1021/tx900242k
The sequence of heterocyclic bases on the interior of the DNA double helix constitutes the genetic code that drives the operation of all living organisms. With this said, it is not surprising that chemical modification of cellular DNA can have profound biological consequences. Therefore, the organic chemistry of DNA damage is fundamentally important to diverse fields including medicinal chemistry, toxicology, and biotechnology. This review is designed to provide a brief overview of the common types of chemical reactions that lead to DNA damage under physiological conditions.
Co-reporter:Jason N. LaButti, Kent S. Gates
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 1) pp:218-221
Publication Date(Web):1 January 2009
DOI:10.1016/j.bmcl.2008.10.133
It has been suggested that peroxymonophosphate could serve as an endogenous hydrogen peroxide-derived regulator of cellular protein tyrosine phosphatase activity under physiological or pathophysiological conditions. To facilitate further consideration of the potential role of peroxymonophosphate in biological systems we present studies related to the preparation, characterization, stability, and fluorometric detection of this agent.
Co-reporter:Mostafa I Fekry;Kent S Gates
Nature Chemical Biology 2009 5(10) pp:710-711
Publication Date(Web):2009-10-01
DOI:10.1038/nchembio.224
Hydrolysis of the phosphodiester linkages in DNA is a notoriously difficult reaction. Deoxyribozymes that use Zn2+ and Mn2+ ions to accelerate this reaction by a factor of one hundred million (or more) have been identified and characterized.Hydrolysis of the DNA backbone is an important reaction in biology and in the laboratory manipulation of genetic material. While many enzymes catalyze the sequence-selective hydrolysis of DNA, it has been difficult to design nonprotein catalysts for this purpose.
Co-reporter:Kripa Keerthi, Santhosh Sivaramakrishnan and Kent S. Gates
Chemical Research in Toxicology 2008 Volume 21(Issue 7) pp:1368
Publication Date(Web):May 23, 2008
DOI:10.1021/tx8000187
Sulfenic acids (RSOH) are among the most common sulfur-centered reactive intermediates generated in biological systems. Given the biological occurrence of sulfenic acids, it is important to explore the reactivity of these intermediates under physiological conditions. The Morin rearrangement is a synthetic process developed for the conversion of penicillin derivatives into cephalosporins that proceeds via nucleophilic attack of an alkene on a sulfenic acid intermediate. In its classic form, the Morin reaction involves initial elimination of a sulfenic acid from a cyclic sulfoxide, followed by intramolecular cyclization of the resulting alkene and sulfenic acid groups to generate an episulfonium ion intermediate that undergoes further reaction to yield ring-expanded products. On the basis of the existing literature, it is difficult to assess whether the reaction between an alkene and a sulfenic group can occur under mild conditions because the conditions required to generate the sulfenic acid from the sulfoxide precursor in the Morin reaction typically involve high temperatures and strong acid. In the work described here, β-sulfinylketone precursors were used to generate a “Morin type” sulfenic acid intermediate under mild conditions. This approach made it possible to demonstrate that the intramolecular cyclization of an alkene with a phenylsulfenic acid to generate an episulfonium ion intermediate can occur in neutral aqueous solution at room temperature.
Co-reporter:Rainer Glaser, Yongqiang Sui, Ujjal Sarkar and Kent S. Gates
The Journal of Physical Chemistry A 2008 Volume 112(Issue 21) pp:4800-4814
Publication Date(Web):May 22, 2008
DOI:10.1021/jp8011987
Radicals resulting from one-electron reduction of (N-methylpyridinium-4-yl) methyl esters have been reported to yield (N-methylpyridinium-4-yl) methyl radical, or N-methyl-γ-picoliniumyl for short, by heterolytic cleavage of carboxylate. This new reaction could provide the foundation for a new structural class of bioreductively activated, hypoxia-selective antitumor agents. N-methyl-γ-picoliniumyl radicals are likely to damage DNA by way of H-abstraction and it is of paramount significance to assess their H-abstraction capabilities. In this context, the benzylic C−H homolyses were studied of toluene (T), γ-picoline (P, 4-methylpyridine), and N-methyl-γ-picolinium (1c, 1,4-dimethylpyridinium). With a view to providing capacity for DNA intercalation the properties also were examined of the annulated derivatives 2c (1,4-dimethylquinolinium), 3c (9,10-dimethylacridinium), and 4c (1,4-dimethylbenzo[g]quinolinium). The benzylic C−H homolyses were studied with density functional theory (DFT), perturbation theory (up to MP4SDTQ), and configuration interaction methods (QCISD(T), CCSD(T)). Although there are many similarities between the results obtained here with DFT and CI theory, a number of significant differences occur and these are shown to be caused by methodological differences in the spin density distributions of the radicals. The quality of the wave functions is established by demonstration of internal consistencies and with reference to a number of observable quantities. The analysis of spin polarization emphasizes the need for a clear distinction between “electron delocalization” and “spin delocalization” in annulated radicals. Aside from their relevance for the rational design of new antitumor drugs, the conceptional insights presented here also will inform the understanding of ferromagnetic materials, of spin-based signaling processes, and of spin topologies in metalloenzymes.
Co-reporter:Kripa Keerthi and Kent S. Gates
Organic & Biomolecular Chemistry 2007 vol. 5(Issue 10) pp:1595-1600
Publication Date(Web):2007/04/13
DOI:10.1039/B701179B
Attack of cellular thiols on the antitumor natural product leinamycin is believed to generate a sulfenate intermediate that undergoes subsequent rearrangement to a DNA-alkylating episulfonium ion. Here, 2-(trimethylsilyl)ethyl sulfoxides were employed in a fluoride-triggered generation of sulfenate anions related to the putative leinamycin-sulfenate. The resulting sulfenates enter smoothly into a leinamycin-type rearrangement reaction to afford an episulfonium ion alkylating agent. The results provide evidence that the sulfenate ion is, indeed, a competent intermediate in the leinamycin rearrangement. Further, the molecules examined here may provide a foundation for the design of functional leinamycin analogues that bypass the unstable and synthetically challenging 1,2-dithiolan-3-one 1-oxide moiety found in the natural product.
Co-reporter:Derrick R. Seiner, Jason N. LaButti and Kent S. Gates
Chemical Research in Toxicology 2007 Volume 20(Issue 9) pp:1315
Publication Date(Web):July 27, 2007
DOI:10.1021/tx700213s
Human cells are exposed to the electrophilic α,β-unsaturated aldehyde acrolein from a variety of sources. The reaction of acrolein with functionally critical protein thiol residues can yield important biological consequences. Protein tyrosine phosphatases (PTPs) are an important class of cysteine-dependent enzymes whose reactivity with acrolein previously has not been well-characterized. These enzymes catalyze the dephosphorylation of phosphotyrosine residues on proteins via a phosphocysteine intermediate. PTPs work in tandem with protein tyrosine kinases to regulate a number of critically important mammalian signal transduction pathways. We find that acrolein is a potent time-dependent inactivator of the enzyme PTP1B (kinact = 0.02 ± 0.005 s−1 and KI = 2.3 ± 0.6 × 10−4 M). The enzyme activity does not return upon gel filtration of the inactivated enzyme, and addition of the competitive phosphatase inhibitor vanadate slows inactivation of PTP1B by acrolein. Together, these observations suggest that acrolein covalently modifies the active site of PTP1B. Mass spectrometric analysis reveals that acrolein modifies the catalytic cysteine residue at the active site of the enzyme. Aliphatic aldehydes such as glyoxal, acetaldehyde, and propanal are relatively weak inactivators of PTP1B under the conditions employed here. Similarly, unsaturated aldehydes such as crotonaldehyde and 3-methyl-2-butenal bearing substitution at the alkene terminus are poor inactivators of the enzyme. Overall, the data suggest that enzyme inactivation occurs via conjugate addition of the catalytic cysteine residue to the carbon–carbon double bond of acrolein. The results indicate that inactivation of PTPs should be considered as a possible contributor to the diverse biological activities of acrolein and structurally related α,β-unsaturated aldehydes.
Co-reporter:Delshanee Kotandeniya, Brian Ganley, Kent S. Gates
Bioorganic & Medicinal Chemistry Letters 2002 Volume 12(Issue 17) pp:2325-2329
Publication Date(Web):2 September 2002
DOI:10.1016/S0960-894X(02)00468-7
Tirapazamine is a bioreductively activated DNA-damaging agent that selectively kills the hypoxic cells found in solid tumors. In this work, base excision repair enzymes were used to provide evidence that tirapazamine causes significant amounts of damage to both purine and pyrimidine residues in double-stranded DNA.In this work, base excision repair enzymes that remove oxidatively damaged DNA bases, leaving behind easily detected strand breaks, were used to provide evidence that tirapazamine causes significant amount of damage to both purine and pyrimidine residues in double-stranded DNA.
Co-reporter:Zhiyu Yang, Maryam Imani Nejad, Jacqueline Gamboa Varela, Nathan E. Price, Yinsheng Wang, Kent S. Gates
DNA Repair (April 2017) Volume 52() pp:1-11
Publication Date(Web):1 April 2017
DOI:10.1016/j.dnarep.2017.02.011
•Current models for replication-dependent cross-link repair involve activation of the Fanconi anemia pathway is activated and cross-links are “unhooked” by the action of structure-specific endonucleases such as XPF-ERCC1 that make incisions flanking the cross-link.•Recent work provides evidence for a new, incision-independent unhooking mechanism involving the intrusion of a base excision repair enzyme, NEIL3, into the world of cross-link repair.•The evidence suggests that the glycosylase action of NEIL3 unhooks interstrand cross-links derived from an abasic site or the psoralen derivative trioxsalen.Interstrand DNA–DNA cross-links are highly toxic lesions that are important in medicinal chemistry, toxicology, and endogenous biology. In current models of replication-dependent repair, stalling of a replication fork activates the Fanconi anemia pathway and cross-links are “unhooked” by the action of structure-specific endonucleases such as XPF-ERCC1 that make incisions flanking the cross-link. This process generates a double-strand break, which must be subsequently repaired by homologous recombination. Recent work provided evidence for a new, incision-independent unhooking mechanism involving intrusion of a base excision repair (BER) enzyme, NEIL3, into the world of cross-link repair. The evidence suggests that the glycosylase action of NEIL3 unhooks interstrand cross-links derived from an abasic site or the psoralen derivative trioxsalen. If the incision-independent NEIL3 pathway is blocked, repair reverts to the incision-dependent route. In light of the new model invoking participation of NEIL3 in cross-link repair, we consider the possibility that various BER glycosylases or other DNA-processing enzymes might participate in the unhooking of chemically diverse interstrand DNA cross-links.Download high-res image (65KB)Download full-size image
Co-reporter:Kripa Keerthi and Kent S. Gates
Organic & Biomolecular Chemistry 2007 - vol. 5(Issue 10) pp:NaN1600-1600
Publication Date(Web):2007/04/13
DOI:10.1039/B701179B
Attack of cellular thiols on the antitumor natural product leinamycin is believed to generate a sulfenate intermediate that undergoes subsequent rearrangement to a DNA-alkylating episulfonium ion. Here, 2-(trimethylsilyl)ethyl sulfoxides were employed in a fluoride-triggered generation of sulfenate anions related to the putative leinamycin-sulfenate. The resulting sulfenates enter smoothly into a leinamycin-type rearrangement reaction to afford an episulfonium ion alkylating agent. The results provide evidence that the sulfenate ion is, indeed, a competent intermediate in the leinamycin rearrangement. Further, the molecules examined here may provide a foundation for the design of functional leinamycin analogues that bypass the unstable and synthetically challenging 1,2-dithiolan-3-one 1-oxide moiety found in the natural product.

![N-Boc-2-{2-[2-(2-amino-ethoxy)-ethoxy]-ethoxy} ethylamine](http://img.cochemist.com/ccimg/101200/101187-40-0.png)
![N-Boc-2-{2-[2-(2-amino-ethoxy)-ethoxy]-ethoxy} ethylamine](http://img.cochemist.com/ccimg/101200/101187-40-0_b.png)










![GLYCINE, N-[2-[(2-HYDROXYETHYL)DITHIO]BENZOYL]-, ETHYL ESTER](http://img.cochemist.com/ccimg/863200/863194-16-5.png)
![GLYCINE, N-[2-[(2-HYDROXYETHYL)DITHIO]BENZOYL]-, ETHYL ESTER](http://img.cochemist.com/ccimg/863200/863194-16-5_b.png)
![3-Oxa-12,21-dithia-15,22-diazatetracyclo[16.2.1.110,13.02,4]docosa-6,8,10,13(22)-tetraene-18-aceticacid, a-hydroxy-a,1,14-trimethyl-5,16-dioxo-,[1S-[1R*,2S*,4S*,6E,8Z,14S*,18S*(R*)]]- (9CI)](http://img.cochemist.com/ccimg/179000/178943-66-3.png)
![3-Oxa-12,21-dithia-15,22-diazatetracyclo[16.2.1.110,13.02,4]docosa-6,8,10,13(22)-tetraene-18-aceticacid, a-hydroxy-a,1,14-trimethyl-5,16-dioxo-,[1S-[1R*,2S*,4S*,6E,8Z,14S*,18S*(R*)]]- (9CI)](http://img.cochemist.com/ccimg/179000/178943-66-3_b.png)
![7,20-Dithia-4,21-diazatricyclo[14.3.1.16,9]heneicosa-6(21),8,10,12-tetraene-1-aceticacid, a,15,17-trihydroxy-a,5,17-trimethyl-3,14-dioxo-, (aS,1R,5R,10Z,12E,15S,16S,17R)- (9CI)](http://img.cochemist.com/ccimg/179000/178943-64-1.png)
![7,20-Dithia-4,21-diazatricyclo[14.3.1.16,9]heneicosa-6(21),8,10,12-tetraene-1-aceticacid, a,15,17-trihydroxy-a,5,17-trimethyl-3,14-dioxo-, (aS,1R,5R,10Z,12E,15S,16S,17R)- (9CI)](http://img.cochemist.com/ccimg/179000/178943-64-1_b.png)
![L-Valine, N-[N-[(1,1-dimethylethoxy)carbonyl]-L-cysteinyl]-, methyl ester](http://img.cochemist.com/ccimg/139200/139167-66-1.png)
![L-Valine, N-[N-[(1,1-dimethylethoxy)carbonyl]-L-cysteinyl]-, methyl ester](http://img.cochemist.com/ccimg/139200/139167-66-1_b.png)








































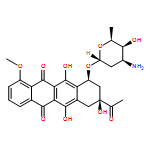
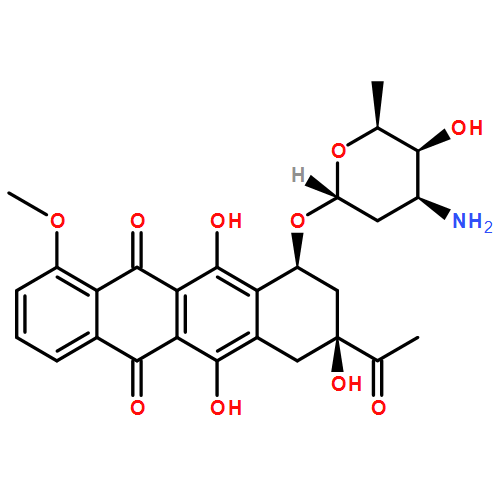
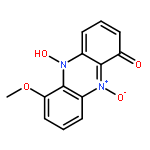







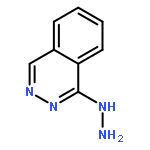







![Spiro[1,2-dithiolane-3,6'-[19]thia[3,20]diazabicyclo[15.2.1]eicosa[1(20),9,13,15,17]pentaene]-4',5,12'-trione,4,11'-dihydroxy-2',4,9'-trimethyl-, 2-oxide, (2S,2'R,3R,4R,9'E,11'R,13'E,15'Z)-](http://img.cochemist.com/ccimg/120600/120500-15-4.png)
![Spiro[1,2-dithiolane-3,6'-[19]thia[3,20]diazabicyclo[15.2.1]eicosa[1(20),9,13,15,17]pentaene]-4',5,12'-trione,4,11'-dihydroxy-2',4,9'-trimethyl-, 2-oxide, (2S,2'R,3R,4R,9'E,11'R,13'E,15'Z)-](http://img.cochemist.com/ccimg/120600/120500-15-4_b.png)


![3-Aminobenzo[e][1,2,4]triazine 1,4-dioxide](http://img.cochemist.com/ccimg/27400/27314-97-2.png)
![3-Aminobenzo[e][1,2,4]triazine 1,4-dioxide](http://img.cochemist.com/ccimg/27400/27314-97-2_b.png)