Co-reporter:David A. Russell;Julien J. Freudenreich;Joe J. Ciardiello;Hannah F. Sore
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 7) pp:1593-1596
Publication Date(Web):2017/02/15
DOI:10.1039/C6OB02659A
We describe stereocontrolled semi-syntheses of deguelin and tephrosin, anti-cancer rotenoids isolated from Tephrosia vogelii. Firstly, we present a new two-step transformation of rotenone into rot-2′-enonic acid via a zinc-mediated ring opening of rotenone hydrobromide. Secondly, following conversion of rot-2′-enonic acid into deguelin, a chromium-mediated hydroxylation provides tephrosin as a single diastereoisomer. An Étard-like reaction mechanism is proposed to account for the stereochemical outcome. Our syntheses of deguelin and tephrosin are operationally simple, scalable and high yielding, offering considerable advantages over previous methods.
Co-reporter:Y. R. Baker;J. T. Hodgkinson;B. I. Florea;E. Alza;W. R. J. D. Galloway;L. Grimm;S. M. Geddis;H. S. Overkleeft;M. Welch;D. R. Spring
Chemical Science (2010-Present) 2017 vol. 8(Issue 11) pp:7403-7411
Publication Date(Web):2017/10/23
DOI:10.1039/C7SC01270E
Many bacterial species, including the human pathogen Pseudomonas aeruginosa, employ a mechanism of intercellular communication known as quorum sensing (QS), which is mediated by signalling molecules termed autoinducers. The Pseudomonas Quinolone Signal (PQS) and 2-Heptyl-3H-4-Quinolone (HHQ) are autoinducers in P. aeruginosa, and they are considered important factors in the progress of infections by this clinically relevant organism. Herein, we report the development of HHQ and PQS photoaffinity-based probes for chemical proteomic studies. Application of these probes led to the identification of previously unsuspected putative HHQ and PQS binders, thereby providing new insights into QS at a proteomic level and revealing potential new small molecule targets for virulence attenuation strategies. Notably, we found evidence that PQS binds RhlR, the cognate receptor in the Rhl QS sub-system of P. aeruginosa. This is the first indication of interaction between the Rhl and PQS systems at the protein/ligand level, which suggests that RhlR should be considered a highly attractive target for antivirulence strategies.
Co-reporter:Claudia De Fusco, Paul Brear, Jessica Iegre, Kathy Hadje Georgiou, Hannah F. Sore, Marko Hyvönen, David R. Spring
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 13(Issue 13) pp:
Publication Date(Web):1 July 2017
DOI:10.1016/j.bmc.2017.04.037
Recently we reported the discovery of a potent and selective CK2α inhibitor CAM4066. This compound inhibits CK2 activity by exploiting a pocket located outside the ATP binding site (αD pocket). Here we describe in detail the journey that led to the discovery of CAM4066 using the challenging fragment linking strategy. Specifically, we aimed to develop inhibitors by linking a high-affinity fragment anchored in the αD site to a weakly binding warhead fragment occupying the ATP site. Moreover, we describe the remarkable impact that molecular modelling had on the development of this novel chemical tool. The work described herein shows potential for the development of a novel class of CK2 inhibitors.Download high-res image (71KB)Download full-size image
Co-reporter:J.J. Ciardiello, H.L. Stewart, H.F. Sore, W.R.J.D. Galloway, D.R. Spring
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 11(Issue 11) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.bmc.2017.02.060
Recent years have witnessed a global decline in the productivity and advancement of the pharmaceutical industry. A major contributing factor to this is the downturn in drug discovery successes. This can be attributed to the lack of structural (particularly scaffold) diversity and structural complexity exhibited by current small molecule screening collections.Macrocycles have been shown to exhibit a diverse range of biological properties, with over 100 natural product-derived examples currently marketed as FDA-approved drugs. Despite this, synthetic macrocycles are widely considered to be a poorly explored structural class within drug discovery, which can be attributed to their synthetic intractability.Herein we describe a novel complexity-to-diversity strategy for the diversity-oriented synthesis of novel, structurally complex and diverse macrocyclic scaffolds from natural product starting materials. This approach exploits the inherent structural (including functional) and stereochemical complexity of natural products in order to rapidly generate diversity and complexity. Readily-accessible natural product-derived intermediates serve as structural templates which can be divergently functionalized with different building blocks to generate a diverse range of acyclic precursors. Subsequent macrocyclisation then furnishes compounds that are each based around a distinct molecular scaffold. Thus, high levels of library scaffold diversity can be rapidly achieved. In this proof-of-concept study, the natural product quinine was used as the foundation for library synthesis, and six novel structurally diverse, highly complex and functionalized macrocycles were generated.Download high-res image (104KB)Download full-size image
Co-reporter:Terence T.-L. Kwan;Omar Boutureira;Elizabeth C. Frye;Stephen J. Walsh;Moni K. Gupta;Stephen Wallace;Yuteng Wu;Fengzhi Zhang;Hannah F. Sore;Warren R. J. D. Galloway;Jason W. Chin;Martin Welch;Gonçalo J. L. Bernardes
Chemical Science (2010-Present) 2017 vol. 8(Issue 5) pp:3871-3878
Publication Date(Web):2017/05/03
DOI:10.1039/C6SC05313K
Transition metal catalysis has emerged as a powerful strategy to expand synthetic flexibility of protein modification. Herein, we report a cationic Ru(II) system that enables the first example of alkyne hydrosilylation between dimethylarylsilanes and O-propargyl-functionalized proteins using a substoichiometric amount or low-loading of Ru(II) catalyst to achieve the first C–Si bond formation on full-length substrates. The reaction proceeds under physiological conditions at a rate comparable to other widely used bioorthogonal reactions. Moreover, the resultant gem-disubstituted vinylsilane linkage can be further elaborated through thiol–ene coupling or fluoride-induced protodesilylation, demonstrating its utility in further rounds of targeted modifications.
Co-reporter:Tze Han Sum;Tze Jing Sum;Súil Collins;Warren R. J. D. Galloway;David G. Twigg;Florian Hollfelder
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 21) pp:4554-4570
Publication Date(Web):2017/05/31
DOI:10.1039/C7OB00804J
Biflavonoids are associated with a variety of biologically useful properties. However, synthetic biflavonoids are poorly explored within drug discovery. There is considerable structural diversity possible within this compound class and large regions of potentially biologically relevant biflavonoid chemical space remain untapped or underexplored. Herein, we report the development of a modular and divergent strategy towards biflavonoid derivatives which enabled the step-economical preparation of a structurally diverse collection of novel unnatural biflavonoids. Preliminary studies established that the strategy could also be successfully extended to the preparation of very rare triflavonoids, which are also expected to be useful tools for biological intervention. Prompted by previous inhibitory studies with flavonoid libraries, amyloid anti-aggregation screening was performed, which led to the identification of several structurally novel inhibitors of the aggregation of the amyloid β peptide (Aβ42). Aggregated Aβ42 is a pathological hallmark of Alzheimer's disease and the use of small molecules to inhibit the aggregation process has been identified as a potentially valuable therapeutic strategy for disease treatment. Methylated biaurones were associated with highest levels of potency (the most active compound had an IC50 value of 16 μM), establishing this scaffold as a starting point for inhibitor development.
Co-reporter:Dr. Yuteng Wu;Dr. Fabrizio Villa;Dr. Joseph Maman;Dr. Yu Heng Lau;Lina Dobnikar;Dr. Aline C. Simon; Karim Labib; David R. Spring; Luca Pellegrini
Angewandte Chemie 2017 Volume 129(Issue 42) pp:13046-13052
Publication Date(Web):2017/10/09
DOI:10.1002/ange.201705611
AbstractThe exploitation of synthetic lethality by small-molecule targeting of pathways that maintain genomic stability is an attractive chemotherapeutic approach. The Ctf4/AND-1 protein hub, which links DNA replication, repair, and chromosome segregation, represents a novel target for the synthetic lethality approach. Herein, we report the design, optimization, and validation of double-click stapled peptides encoding the Ctf4-interacting peptide (CIP) of the replicative helicase subunit Sld5. By screening stapling positions in the Sld5 CIP, we identified an unorthodox i,i+6 stapled peptide with improved, submicromolar binding to Ctf4. The mode of interaction with Ctf4 was confirmed by a crystal structure of the stapled Sld5 peptide bound to Ctf4. The stapled Sld5 peptide was able to displace the Ctf4 partner DNA polymerase α from the replisome in yeast extracts. Our study provides proof-of-principle evidence for the development of small-molecule inhibitors of the human CTF4 orthologue AND-1.
Co-reporter:Dr. Yuteng Wu;Dr. Fabrizio Villa;Dr. Joseph Maman;Dr. Yu Heng Lau;Lina Dobnikar;Dr. Aline C. Simon; Karim Labib; David R. Spring; Luca Pellegrini
Angewandte Chemie International Edition 2017 Volume 56(Issue 42) pp:12866-12872
Publication Date(Web):2017/10/09
DOI:10.1002/anie.201705611
AbstractThe exploitation of synthetic lethality by small-molecule targeting of pathways that maintain genomic stability is an attractive chemotherapeutic approach. The Ctf4/AND-1 protein hub, which links DNA replication, repair, and chromosome segregation, represents a novel target for the synthetic lethality approach. Herein, we report the design, optimization, and validation of double-click stapled peptides encoding the Ctf4-interacting peptide (CIP) of the replicative helicase subunit Sld5. By screening stapling positions in the Sld5 CIP, we identified an unorthodox i,i+6 stapled peptide with improved, submicromolar binding to Ctf4. The mode of interaction with Ctf4 was confirmed by a crystal structure of the stapled Sld5 peptide bound to Ctf4. The stapled Sld5 peptide was able to displace the Ctf4 partner DNA polymerase α from the replisome in yeast extracts. Our study provides proof-of-principle evidence for the development of small-molecule inhibitors of the human CTF4 orthologue AND-1.
Co-reporter:T. A. Alanine, W. R. J. D. Galloway, S. Bartlett, J. J. Ciardiello, T. M. McGuire and D. R. Spring
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 3) pp:1031-1038
Publication Date(Web):24 Nov 2015
DOI:10.1039/C5OB01784J
Pyrido[1,2-a]pyrimidin-2-ones represent a pharmaceutically interesting class of heterocycles. The structurally related pyrido[1,2-a]pyrimidin-4-ones are associated with a broad range of useful biological properties. Furthermore, quinolizinone-type scaffolds of these sorts with a bridgehead nitrogen are expected to display interesting physico–chemical properties. However, pyrido[1,2-a]pyrimidin-2-ones are largely under-represented in current small molecule screening libraries and the physical and biological properties of the pyrido[1,2-a]pyrimidin-2-one scaffold have been poorly explored (indeed, the same can be said for unsaturated bicyclic compounds with a bridgehead nitrogen in general). Herein, we report the development of a new strategy for the concise synthesis of substituted pyrido[1,2-a]pyrimidin-2-ones from readily available starting materials. The synthetic route involved the acylation of the lithium amide bases of 2-aminopyridines with alkynoate esters to form alkynamides, which were then cyclised under thermal conditions. The use of lithium amide anions ensured excellent regioselectivity for the 2-oxo-isomer over the undesired 4-oxo-isomer, which offers a distinct advantage over some existing methods for the synthesis of pyrido[1,2-a]pyrimidin-2-ones. Notably, different aminoazines could also be employed in this approach, which enabled access to several very unusual bicyclic systems with higher nitrogen contents. This methodology thus represents an important contribution towards the synthesis of pyrido[1,2-a]pyrimidin-2-ones and other rare azabicycles with a ring-junction nitrogen. These heterocycles represent attractive structural templates for drug discovery.
Co-reporter:Flavia Salvaggio;James T. Hodgkinson;Laura Carro;Stephen M. Geddis;Warren R. J. D. Galloway;Martin Welch
European Journal of Organic Chemistry 2016 Volume 2016( Issue 3) pp:434-437
Publication Date(Web):
DOI:10.1002/ejoc.201501400
Abstract
The synthesis of four quinolone natural products from the actinomycete Pseudonocardia sp. is reported. The key step involved a sp2–sp3 Suzuki–Miyaura reaction between a common boronic ester lateral chain and various functionalised quinolone cores. The quinolones slowed growth of E. coli and S. aureus by inducing extended lag phases.
Co-reporter:Joe J. Ciardiello, Warren R.J.D. Galloway, Cornelius J. O'Connor, Hannah F. Sore, Jamie E. Stokes, Yuteng Wu, David R. Spring
Tetrahedron 2016 Volume 72(Issue 25) pp:3567-3578
Publication Date(Web):23 June 2016
DOI:10.1016/j.tet.2015.10.061
Naturally-derived macrocyclic compounds are associated with a diverse range of biological activities, including antibacterial effects, and there are over 100 marketed macrocycle drugs derived from natural products. However, synthetic macrocycles are widely considered to be poorly explored in antibiotic development (indeed, within drug discovery in general). This has been attributed to challenges associated with the generation of such compounds. Whilst there are synthetic methods that can produce large collections of structurally similar macrocycles (i.e., compounds with varying appendages based around similar core macrocyclic ring architectures) there is a relative dearth of strategies for the efficient generation of more structurally diverse macrocycle collections in which there is greater variation in the nature of macrocyclic scaffolds present. Such macrocycle collections should contain compounds with a broad range of biological activities (including antibacterial activities) and the requisite robust synthetic methodology useful for analogue synthesis and lead optimization once an active compound has been identified in a biological screen. Herein, we describe a new and expedient diversity-oriented synthesis (DOS) strategy for the generation of a library of novel structurally diverse macrocyclic compounds with a high level of scaffold diversity. The strategy is concise, proceeds from readily-available starting materials, is modular in nature and features a variety of macrocyclisation techniques. In this proof-of-concept study, the synthesis of several previously unreported macrocyclic compounds was achieved. Each of these macrocycles was based around a distinct molecular scaffold and contained natural product-like structural features (e.g., three-dimensionality and multiple hydrogen bond donors and acceptors) as well as synthetic handles for potential further elaboration. The successful generation of these macrocycles demonstrates the feasibility of the new DOS strategy as a synthetic platform for library generation.Figure optionsDownload full-size imageDownload high-quality image (111 K)Download as PowerPoint slide
Co-reporter:Yuteng Wu;Lasse B. Olsen;Dr. Yu Heng Lau;Claus Hatt Jensen;Dr. Maxim Rossmann;Dr. Ysobel R. Baker;Dr. Hannah F. Sore;Súil Collins; David R. Spring
ChemBioChem 2016 Volume 17( Issue 8) pp:689-692
Publication Date(Web):
DOI:10.1002/cbic.201500648
Abstract
Photoaffinity labelling is a useful method for studying how proteins interact with ligands and biomolecules, and can help identify and characterise new targets for the development of new therapeutics. We present the design and synthesis of a novel multifunctional benzophenone linker that serves as both a photo-crosslinking motif and a peptide stapling reagent. Using double-click stapling, we attached the benzophenone to the peptide via the staple linker, rather than by modifying the peptide sequence with a photo-crosslinking amino acid. When applied to a p53-derived peptide, the resulting photoreactive stapled peptide was able to preferentially crosslink with MDM2 in the presence of competing protein. This multifunctional linker also features an extra alkyne handle for downstream applications such as pull-down assays, and can be used to investigate the target selectivity of stapled peptides.
Co-reporter:Yu Heng Lau, Peterson de Andrade, Yuteng Wu and David R. Spring
Chemical Society Reviews 2015 vol. 44(Issue 1) pp:91-102
Publication Date(Web):08 Sep 2014
DOI:10.1039/C4CS00246F
Peptide stapling is a strategy for constraining short peptides typically in an alpha-helical conformation. Stapling is carried out by covalently linking the side-chains of two amino acids, thereby forming a peptide macrocycle. There is an expanding repertoire of stapling techniques based on different macrocyclisation chemistries. In this tutorial review, we categorise and analyse key examples of peptide stapling in terms of their synthesis and applicability to biological systems.
Co-reporter:Esther Alza, Luca Laraia, Brett M. Ibbeson, Súil Collins, Warren R. J. D. Galloway, Jamie E. Stokes, Ashok R. Venkitaraman and David R. Spring
Chemical Science 2015 vol. 6(Issue 1) pp:390-396
Publication Date(Web):09 Sep 2014
DOI:10.1039/C4SC02547D
The synthesis of a previously undescribed sp3-rich 6-5-5-6 tetracyclic ring scaffold using a palladium catalysed domino Heck–Suzuki reaction is reported. This reaction is high-yielding, selective for the domino process over the direct Suzuki reaction and tolerant towards a variety of boronic acids. The novel scaffold can also be accessed via domino Heck–Stille and radical cyclisations. Compounds based around this scaffold were found to be effective antimitotic agents in a human cancer cell line. Detailed phenotypic profiling showed that the compounds affected the congression of chromosomes to give mitotic arrest and apoptotic cell death. Thus, a novel structural class of antimitotic agents that does not disrupt the tubulin network has been identified.
Co-reporter:Robert J. H. Scanes, Oleg Grossmann, André Grossmann, and David R. Spring
Organic Letters 2015 Volume 17(Issue 10) pp:2462-2465
Publication Date(Web):April 27, 2015
DOI:10.1021/acs.orglett.5b00971
The enantioselective intramolecular Rauhut–Currier reaction has been developed using a bifunctional dipeptidic phosphane catalyst, providing a direct access to biologically active α-methylene-δ-valerolactones in high yields and enantiomeric excesses. The novel catalyst is accessible in only four steps from commercial sources and exhibits unusual binding selectivities for a small molecule, suggesting the possibility for long-range interactions between the catalyst and the substrate.
Co-reporter:Albert Isidro-Llobet, Kathy Hadje Georgiou, Warren R. J. D. Galloway, Elisa Giacomini, Mette R. Hansen, Gabriela Méndez-Abt, Yaw Sing Tan, Laura Carro, Hannah F. Sore and David R. Spring
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 15) pp:4570-4580
Publication Date(Web):11 Mar 2015
DOI:10.1039/C5OB00371G
Macrocyclic peptidomimetics are associated with a broad range of biological activities. However, despite such potentially valuable properties, the macrocyclic peptidomimetic structural class is generally considered as being poorly explored within drug discovery. This has been attributed to the lack of general methods for producing collections of macrocyclic peptidomimetics with high levels of structural, and thus shape, diversity. In particular, there is a lack of scaffold diversity in current macrocyclic peptidomimetic libraries; indeed, the efficient construction of diverse molecular scaffolds presents a formidable general challenge to the synthetic chemist. Herein we describe a new, advanced strategy for the diversity-oriented synthesis (DOS) of macrocyclic peptidomimetics that enables the combinatorial variation of molecular scaffolds (core macrocyclic ring architectures). The generality and robustness of this DOS strategy is demonstrated by the step-efficient synthesis of a structurally diverse library of over 200 macrocyclic peptidomimetic compounds, each based around a distinct molecular scaffold and isolated in milligram quantities, from readily available building-blocks. To the best of our knowledge this represents an unprecedented level of scaffold diversity in a synthetically derived library of macrocyclic peptidomimetics. Cheminformatic analysis indicated that the library compounds access regions of chemical space that are distinct from those addressed by top-selling brand-name drugs and macrocyclic natural products, illustrating the value of our DOS approach to sample regions of chemical space underexploited in current drug discovery efforts. An analysis of three-dimensional molecular shapes illustrated that the DOS library has a relatively high level of shape diversity.
Co-reporter:Michelle S. Frei, Matthew K. Bilyard, Thomas A. Alanine, Warren R.J.D. Galloway, Jamie E. Stokes, David R. Spring
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 11) pp:2666-2679
Publication Date(Web):1 June 2015
DOI:10.1016/j.bmc.2014.11.037
Herein, we report on work towards the development of a new strategy for the synthesis of rare and biologically interesting indolizin-5(3H)-ones, which is based around the use of ring-closing metathesis to construct the carbocyclic ring system. This study has provided insights into the general stability of indolizin-5(3H)-ones and their tendency to exist as the tautomeric indolizin-5-ols. Furthermore, this approach has allowed access to other novel structurally related compounds based around unusual 6,5-azabicyclic scaffolds, which are also difficult to generate using typical methods. The azabicyclic compounds synthesized in this study reside in attractive regions of heterocyclic chemical space that are underexploited in current drug and agrochemical discovery efforts.
Co-reporter:Tze Han Sum, Tze Jing Sum, Jamie E. Stokes, Warren R.J.D. Galloway, David R. Spring
Tetrahedron 2015 Volume 71(26–27) pp:4557-4564
Publication Date(Web):1 July 2015
DOI:10.1016/j.tet.2015.02.017
Dihydrochalcones and 5-deoxyflavones are types of compounds possessing various biologically interesting properties. Herein, we report the concise and divergent total syntheses of several naturally occurring dihydrochalcones and 5-deoxyflavones from readily available starting materials. The divergent strategy is based around manipulation of a common chalcone scaffold and features application of Algar–Flynn–Oyamada oxidation and benzoquinone C–H activation methodologies. These are the first reported total syntheses of these biologically interesting compounds and the concise and flexible route should be readily amenable to future analogue generation. Furthermore, this work provides an illustration of the utility of divergent synthesis for the expedient and step-economical preparation of natural product libraries.
Co-reporter:Dr. Yu Heng Lau;Yuteng Wu;Dr. Maxim Rossmann;Dr. Ban Xiong Tan;Dr. Peterson deAndrade;Dr. Yaw Sing Tan;Dr. Chra Verma;Dr. Grahame J. McKenzie; Ashok R. Venkitaraman;Dr. Marko Hyvönen; David R. Spring
Angewandte Chemie International Edition 2015 Volume 54( Issue 51) pp:15410-15413
Publication Date(Web):
DOI:10.1002/anie.201508416
Abstract
Peptide stapling is a method for designing macrocyclic alpha-helical inhibitors of protein–protein interactions. However, obtaining a cell-active inhibitor can require significant optimization. We report a novel stapling technique based on a double strain-promoted azide–alkyne reaction, and exploit its biocompatibility to accelerate the discovery of cell-active stapled peptides. As a proof of concept, MDM2-binding peptides were stapled in parallel, directly in cell culture medium in 96-well plates, and simultaneously evaluated in a p53 reporter assay. This in situ stapling/screening process gave an optimal candidate that showed improved proteolytic stability and nanomolar binding to MDM2 in subsequent biophysical assays. α-Helicity was confirmed by a crystal structure of the MDM2-peptide complex. This work introduces in situ stapling as a versatile biocompatible technique with many other potential high-throughput biological applications.
Co-reporter:Dr. Yu Heng Lau;Yuteng Wu;Dr. Maxim Rossmann;Dr. Ban Xiong Tan;Dr. Peterson deAndrade;Dr. Yaw Sing Tan;Dr. Chra Verma;Dr. Grahame J. McKenzie; Ashok R. Venkitaraman;Dr. Marko Hyvönen; David R. Spring
Angewandte Chemie 2015 Volume 127( Issue 51) pp:15630-15633
Publication Date(Web):
DOI:10.1002/ange.201508416
Abstract
Peptide stapling is a method for designing macrocyclic alpha-helical inhibitors of protein–protein interactions. However, obtaining a cell-active inhibitor can require significant optimization. We report a novel stapling technique based on a double strain-promoted azide–alkyne reaction, and exploit its biocompatibility to accelerate the discovery of cell-active stapled peptides. As a proof of concept, MDM2-binding peptides were stapled in parallel, directly in cell culture medium in 96-well plates, and simultaneously evaluated in a p53 reporter assay. This in situ stapling/screening process gave an optimal candidate that showed improved proteolytic stability and nanomolar binding to MDM2 in subsequent biophysical assays. α-Helicity was confirmed by a crystal structure of the MDM2-peptide complex. This work introduces in situ stapling as a versatile biocompatible technique with many other potential high-throughput biological applications.
Co-reporter:Fengzhi Zhang and David R. Spring
Chemical Society Reviews 2014 vol. 43(Issue 20) pp:6906-6919
Publication Date(Web):01 Jul 2014
DOI:10.1039/C4CS00137K
The use of coordinating moieties as directing groups for the functionalisation of aromatic carbon–hydrogen (C–H) bonds has become an efficient strategy for the selective construction of new carbon–carbon (C–C) and carbon–heteroatom (C–X) bonds in arenes. However many directing groups cannot be easily removed/modified from the products after C–H functionalisation, thus limiting the structural diversity of the products. This limitation can be overcome by employing removable/modifiable or traceless directing groups which can be easily attached to the starting materials and detached from the products. In this tutorial review, we give an overview of recent advances in this emerging field which have dramatically increased the synthetic applicability of C–H functionalisation processes.
Co-reporter:Yu Heng Lau, Peterson de Andrade, Soo-Tng Quah, Maxim Rossmann, Luca Laraia, Niklas Sköld, Tze Jing Sum, Pamela J. E. Rowling, Thomas L. Joseph, Chandra Verma, Marko Hyvönen, Laura S. Itzhaki, Ashok R. Venkitaraman, Christopher J. Brown, David P. Lane and David R. Spring
Chemical Science 2014 vol. 5(Issue 5) pp:1804-1809
Publication Date(Web):10 Mar 2014
DOI:10.1039/C4SC00045E
Stapled peptides are a promising class of alpha-helix mimetic inhibitors for protein–protein interactions. We report the divergent synthesis of “functionalised” stapled peptides via an efficient two-component strategy. Starting from a single unprotected diazido peptide, dialkynyl staple linkers bearing different unprotected functional motifs are introduced to create different alpha-helical peptides in one step, functionalised on the staple linkage itself. Applying this concept to the p53/MDM2 interaction, we improve the cell permeability and p53 activating capability of an otherwise impermeable p53 stapled peptide by introducing cationic groups on the staple linkage, rather than modifying the peptide sequence.
Co-reporter:Yu Heng Lau, Peterson de Andrade, Niklas Sköld, Grahame J. McKenzie, Ashok R. Venkitaraman, Chandra Verma, David P. Lane and David R. Spring
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 24) pp:4074-4077
Publication Date(Web):02 May 2014
DOI:10.1039/C4OB00742E
Stapling peptides for inhibiting the p53/MDM2 interaction is a promising strategy for developing anti-cancer therapeutic leads. We evaluate double-click stapled peptides formed from p53-based diazidopeptides with different staple positions and azido amino acid side-chain lengths, determining the impact of these variations on MDM2 binding and cellular activity. We also demonstrate a K24R mutation, necessary for cellular activity in hydrocarbon-stapled p53 peptides, is not required for analogous ‘double-click’ peptides.
Co-reporter:Thomas A. Alanine;Warren R. J. D. Galloway;Thomas M. McGuire
European Journal of Organic Chemistry 2014 Volume 2014( Issue 26) pp:5767-5776
Publication Date(Web):
DOI:10.1002/ejoc.201402648
Abstract
The 4H-quinolizin-4-one scaffold is of significant pharmaceutical interest. This heterocyclic structure is predicted to have attractive physico-chemical properties and is present in a variety of biologically active molecules. Despite these interesting characteristics, 4H-quinolizin-4-ones are largely under-represented in current small molecule screening libraries, and, therefore, this scaffold has been poorly investigated. Herein, a new strategy is reported for the syntheses of these rare and biologically interesting 4H-quinolizin-4-ones. This modular route involves the regioselective N-alkylation of 6-halo-2-pyridones followed by a Stille cross-coupling, ring-closing metathesis, and palladium-catalyzed dehydrogenation reaction sequence. This method furnishes the target compounds in good yields and allows for access to unusual substitution patterns that are difficult to achieve by using other synthetic strategies.
Co-reporter:Luca Laraia, Jamie Stokes, Amy Emery, Grahame J. McKenzie, Ashok R. Venkitaraman, and David R. Spring
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 5) pp:598-603
Publication Date(Web):February 24, 2014
DOI:10.1021/ml5000564
Tubulin modulating agents such as the taxanes are among the most effective antimitotic cancer drugs, although resistance and toxicity present significant problems in their clinical use. However, most tubulin modulators are derived from complex natural products, which can make modification of their structure to address these problems difficult. Here, we report the discovery of new antimitotic compounds with simple structures that can be rapidly synthesized, through the phenotypic screening of a diverse compound library for the induction of mitotic arrest. We first identified a compound, which induced mitotic arrest in human cells at submicromolar concentrations. Its simple structure enabled rapid exploration of activity, defining a biphenylacetamide moiety required for activity, A family of analogues was synthesized, yielding optimized compounds that caused mitotic arrest and cell death in the low nanomolar range, comparable to clinically used antimitotic agents. These compounds can be synthesized in 1–3 steps and good yields. We show that one such compound targets tubulin, partially inhibiting colchicine but not vinblastine binding, suggesting that it acts allosterically to the known colchicine-binding site. Thus, our results exemplify the use of phenotypic screening to identify novel antimitotic compounds from diverse chemical libraries and characterize a family of biphenylacetamides (biphenabulins) that show promise for further development.Keywords: allosteric; antimitotics; colchicine; Phenotypic screening; tubulin;
Co-reporter:Dr. André Grossmann;Sean Bartlett;Matej Janecek;Dr. James T. Hodgkinson ; David R. Spring
Angewandte Chemie International Edition 2014 Volume 53( Issue 48) pp:13093-13097
Publication Date(Web):
DOI:10.1002/anie.201406865
Abstract
Small-molecule modulators of biological targets play a crucial role in biology and medicine. In this context, diversity-oriented synthesis (DOS) provides strategies toward generating small molecules with a broad range of unique scaffolds, and hence three-dimensionality, to target a broad area of biological space. In this study, an organocatalysis-derived DOS library of macrocycles was synthesized by exploiting the pluripotency of aldehydes. The orthogonal combination of multiple diversity-generating organocatalytic steps with alkene metathesis enabled the synthesis of 51 distinct macrocyclic structures bearing 48 unique scaffolds in only two to four steps without the need for protecting groups. Furthermore, merging organocatalysis and alkene metathesis in a one-pot protocol facilitated the synthesis of drug-like macrocycles with natural-product-like levels of shape diversity in a single step.
Co-reporter:Yu Heng Lau;Dr. Peterson de Andrade;Dr. Grahame J. McKenzie; Ashok R. Venkitaraman; David R. Spring
ChemBioChem 2014 Volume 15( Issue 18) pp:2680-2683
Publication Date(Web):
DOI:10.1002/cbic.201402374
Abstract
We investigated linear aliphatic dialkynes as a new structural class of i,i+7 linkers for the double-click stapling of p53-based peptides. The optimal combination of azido amino acids and dialkynyl linker length for MDM2 binding was determined. In a direct comparison between aliphatic and aromatic staple scaffolds, the aliphatic staples resulted in superior binding to MDM2 in vitro and superior p53-activating capability in cells when using a diazidopeptide derived from phage display. This work demonstrates that the nature of the staple scaffold is an important factor that can affect peptide bioactivity in cells.
Co-reporter:Dr. André Grossmann;Sean Bartlett;Matej Janecek;Dr. James T. Hodgkinson ; David R. Spring
Angewandte Chemie 2014 Volume 126( Issue 48) pp:13309-13313
Publication Date(Web):
DOI:10.1002/ange.201406865
Abstract
Small-molecule modulators of biological targets play a crucial role in biology and medicine. In this context, diversity-oriented synthesis (DOS) provides strategies toward generating small molecules with a broad range of unique scaffolds, and hence three-dimensionality, to target a broad area of biological space. In this study, an organocatalysis-derived DOS library of macrocycles was synthesized by exploiting the pluripotency of aldehydes. The orthogonal combination of multiple diversity-generating organocatalytic steps with alkene metathesis enabled the synthesis of 51 distinct macrocyclic structures bearing 48 unique scaffolds in only two to four steps without the need for protecting groups. Furthermore, merging organocatalysis and alkene metathesis in a one-pot protocol facilitated the synthesis of drug-like macrocycles with natural-product-like levels of shape diversity in a single step.
Co-reporter:Tianyu Liu, Zhaochao Xu, David R. Spring, and Jingnan Cui
Organic Letters 2013 Volume 15(Issue 9) pp:2310-2313
Publication Date(Web):April 24, 2013
DOI:10.1021/ol400973v
In this work, a 1,8-naphthalimide-derived fluorescent probe for H2S based on the thiolysis of dinitrophenyl ether is reported. This probe exhibits turn-on fluorescence detection of H2S in bovine serum and lysosome-targetable fluorescent imaging of H2S with excellent selectivity.
Co-reporter:Paola Mestichelli, Matthew J. Scott, Warren R. J. D. Galloway, Jamie Selwyn, Jeremy S. Parker, and David R. Spring
Organic Letters 2013 Volume 15(Issue 21) pp:5448-5451
Publication Date(Web):October 17, 2013
DOI:10.1021/ol4025259
A new method for the synthesis of tricyclic biaryl ether-linked ring systems incorporating seven-, eight-, and nine-membered ring amines is presented. In the presence of catalytic quantities of copper(I), readily accessible acyclic precursors undergo an intramolecular carbon–oxygen bond-forming reaction facilitated by a “templating” chelating nitrogen atom. The methodology displays a broad substrate scope, is practical, and generates rare and biologically interesting tricyclic heteroaromatic products that are difficult to access by other means.
Co-reporter:Hannah F. Sore, Warren R. J. D. Galloway and David R. Spring
Chemical Society Reviews 2012 vol. 41(Issue 5) pp:1845-1866
Publication Date(Web):02 Nov 2011
DOI:10.1039/C1CS15181A
The palladium catalysed cross-coupling of organosilicon reagents with organo halides and pseudo-halides has developed over the past 30 years into an efficient and attractive carbon–carbon bond forming strategy. Extensive research within this field to expand and diversify on the scope of the organosilicon coupling reaction will continue to promote its use in the synthesis of biologically and pharmaceutically important organic molecules. The recent advances made within this area are explored in this critical review (199 references).
Co-reporter:Cornelius J. O' Connor, Henning S. G. Beckmann and David R. Spring
Chemical Society Reviews 2012 vol. 41(Issue 12) pp:4444-4456
Publication Date(Web):10 Apr 2012
DOI:10.1039/C2CS35023H
Small molecule modulators of biological function can be discovered by the screening of compound libraries. However, it became apparent that some human disease related targets could not be addressed by the libraries commonly used which typically are comprised of large numbers of structurally similar compounds. The last decade has seen a paradigm shift in library construction, with particular emphasis now being placed on increasing a library's structural, and thus functional diversity, rather than only its size. Diversity-oriented synthesis (DOS) aims to generate such structural diversity efficiently. This tutorial review has been written to introduce the subject to a broad audience and recent achievements in both the preparation and the screening of structurally diverse compound collections against so-called ‘undruggable’ targets are highlighted.
Co-reporter:Zhaochao Xu, Xin Liu, Jie Pan and David R. Spring
Chemical Communications 2012 vol. 48(Issue 39) pp:4764-4766
Publication Date(Web):05 Mar 2012
DOI:10.1039/C2CC30963G
We report a coumarin-derived fluorescent sensor for Zn2+ termed CTS. CTS shows excellent binding selectivity for Zn2+ over competing metal ions due to the transformable ability of CTS, that is the displacement of other metal ions by Zn2+, which induces transformation of chelation from an amide to an imidic acid tautomeric form.
Co-reporter:James T. Hodgkinson, Warren R. J. D. Galloway, Megan Wright, Ioulia K. Mati, Rebecca L. Nicholson, Martin Welch and David R. Spring
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 30) pp:6032-6044
Publication Date(Web):09 Mar 2012
DOI:10.1039/C2OB25198A
Many species of bacteria employ a mechanism of intercellular communication known as quorum sensing which is mediated by small diffusible signalling molecules termed autoinducers. The most common class of autoinducer used by Gram-negative bacteria are N-acylated-L-homoserine lactones (AHLs). Pseudomonas aeruginosa is a clinically important bacterium which is known to use AHL-mediated quorum sensing systems to regulate a variety of processes associated with virulence. Thus the selective disruption of AHL-based quorum sensing represents a strategy to attenuate the pathogenicity of this bacterium. Herein we describe the design, synthesis and biological evaluation of a collection of structurally novel AHL mimics. A number of new compounds capable of modulating the LasR-dependent quorum sensing system of P. aeruginosa were identified, which could have value as molecular tools to study and manipulate this signalling pathway. Worthy of particular note, this research has delivered novel potent quorum sensing antagonists, which strongly inhibit the production of virulence factors in a wild type strain of this pathogenic bacterium.
Co-reporter:Shaojun Zheng, Luca Laraia, Cornelius J. O' Connor, David Sorrell, Yaw Sing Tan, Zhaochao Xu, Ashok R. Venkitaraman, Wenjun Wu and David R. Spring
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 13) pp:2590-2593
Publication Date(Web):30 Jan 2012
DOI:10.1039/C2OB25065A
A novel synthesis of the ellagitannin natural product tellimagrandin I and a series of medium ring analogues is described. These compounds were all subsequently screened for redox activity, ability to precipitate protein and cellular phenotype in HeLa cells. From this we have shown that all properties can be modulated independently by varying ring size and by moving the ester out of conjugation with the biaryl ring system. Increasing ring size increased redox activity and cytotoxicity, leading to the identification of a compound (10) which was significantly more cytotoxic. In addition compounds identified with a redox active scaffold and low cytotoxicity may be employed as a new class of redox probes.
Co-reporter:Kieron M. G. O'Connell, Henning S. G. Beckmann, Luca Laraia, Helen T. Horsley, Andreas Bender, Ashok R. Venkitaraman and David R. Spring
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 37) pp:7545-7551
Publication Date(Web):03 Aug 2012
DOI:10.1039/C2OB26272J
Macrocyclic compounds represent a structural class with exceptional potential for biological activity; however, they have historically been underrepresented in screening collections and synthetic libraries. In this article we report the development of a highly step-efficient strategy for the diversity-oriented synthesis of complex macrocyclic architectures, using a modular approach based on the two-directional synthesis of bifunctional linear precursors and their subsequent combination in a two-directional macrocyclisation process. In this proof of principle study, the synthesis of 14 such compounds was achieved. Cheminformatic analysis of the compounds produced suggests that they reside in biologically relevant regions of chemical space and the compounds were screened for activity against two cancer cell lines.
Co-reporter:Elizabeth C. Frye;Dr. Cornelius J. O' Connor;David G. Twigg;Bryony Elbert;Luca Laraia;Dr. David G. Hulcoop; Ashok R. Venkitaraman;Dr. David R. Spring
Chemistry - A European Journal 2012 Volume 18( Issue 28) pp:8774-8779
Publication Date(Web):
DOI:10.1002/chem.201200431
Abstract
The Hiyama cross-coupling reaction is a powerful method for carbon–carbon bond formation. To date, the substrate scope of this reaction has predominantly been limited to sp2–sp2 coupling reactions. Herein, the palladium-catalysed Hiyama type cross-coupling of vinyldisiloxanes with benzylic and allylic bromides, chlorides, tosylates and mesylates is reported. A wide variety of functional groups were tolerated, and the synthetic utility of the methodology was exemplified through the efficient total synthesis of the cytotoxic natural product bussealin A. In addition, the antiproliferative ability of bussealin A was evaluated in two cancer-cell lines.
Co-reporter:Shaojun Zheng;Dr. Sarah J. Aves;Luca Laraia;Dr. Warren R. J. D. Galloway;Dr. Kurt G. Pike; Wenjun Wu;Dr. David R. Spring
Chemistry - A European Journal 2012 Volume 18( Issue 11) pp:3193-3198
Publication Date(Web):
DOI:10.1002/chem.201103530
Abstract
The natural product deoxyschizandrin has been shown to have a wide range of biological activities. In recent years the therapeutic potential of this compound against cancers has attracted significant interest. Herein we describe a concise de novo total synthesis of deoxyschizandrin based around a double organocuprate oxidation strategy. In addition, we present the results of biological studies exploring the ability of deoxyschizandrin and synthetic precursors lacking the medium ring biaryl unit to inhibit the proliferation of a human cancer cell line. These studies led to the identification of a structurally novel agent with in vitro anticancer activity.
Co-reporter:Warren R. J. D. Galloway, James T. Hodgkinson, Steven D. Bowden, Martin Welch, and David R. Spring
Chemical Reviews 2011 Volume 111(Issue 1) pp:28
Publication Date(Web):December 23, 2010
DOI:10.1021/cr100109t
Co-reporter:Cornelius J. O' Connor, Luca Laraia and David R. Spring
Chemical Society Reviews 2011 vol. 40(Issue 8) pp:4332-4345
Publication Date(Web):12 May 2011
DOI:10.1039/C1CS15053G
Chemical genetics can be defined as the study of biological systems using small molecule tools. Cell permeable and selective small molecules modulate gene product function rapidly, reversibly and can be administered conditionally in either a cellular or organismal context. The small molecule approach provides exacting temporal and quantitative control and is therefore an extremely powerful tool for dissecting biological processes. This tutorial review has been written to introduce the subject to a broad audience and highlights recent developments within the field in four key areas of biology: modulating protein–protein interactions, malaria research, hepatitis C virus research, and disrupting RNA interference pathways.
Co-reporter:Warren R.J.D. Galloway, David R. Spring
Chemistry & Biology 2011 Volume 18(Issue 10) pp:1209-1210
Publication Date(Web):28 October 2011
DOI:10.1016/j.chembiol.2011.10.002
The development of a method for the amplification of PNA tags (Svensen et al., in this issue of Chemistry & Biology) should expand the range of biological targets amenable to screening using PNA-encoded combinatorial libraries and thus facilitate the discovery of new biologically useful agents.
Co-reporter:David J. Huggins, Ashok R. Venkitaraman, and David R. Spring
ACS Chemical Biology 2011 Volume 6(Issue 3) pp:208
Publication Date(Web):January 24, 2011
DOI:10.1021/cb100420r
Traditionally a pursuit of large pharmaceutical companies, high-throughput screening assays are becoming increasingly common within academic and government laboratories. This shift has been instrumental in enabling projects that have not been commercially viable, such as chemical probe discovery and screening against high-risk targets. Once an assay has been prepared and validated, it must be fed with screening compounds. Crafting a successful collection of small molecules for screening poses a significant challenge. An optimized collection will minimize false positives while maximizing hit rates of compounds that are amenable to lead generation and optimization. Without due consideration of the relevant protein targets and the downstream screening assays, compound filtering and selection can fail to explore the great extent of chemical diversity and eschew valuable novelty. Herein, we discuss the different factors to be considered and methods that may be employed when assembling a structurally diverse compound collection for screening. Rational methods for selecting diverse chemical libraries are essential for their effective use in high-throughput screens.
Co-reporter:James T. Hodgkinson, Warren R. J. D. Galloway, Shreya Saraf, Ian R. Baxendale, Steven V. Ley, Mark Ladlow, Martin Welch and David R. Spring
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 1) pp:57-61
Publication Date(Web):22 Oct 2010
DOI:10.1039/C0OB00652A
Expedient syntheses of Pseudomonas quinolone signal (PQS) and related structural analogues using microwave and flow methods are reported.
Co-reporter:James T. Hodgkinson, Warren R.J.D. Galloway, Mariangela Casoli, Harriet Keane, Xianbin Su, George P.C. Salmond, Martin Welch, David R. Spring
Tetrahedron Letters 2011 Volume 52(Issue 26) pp:3291-3294
Publication Date(Web):29 June 2011
DOI:10.1016/j.tetlet.2011.04.059
The ready availability of native quorum sensing molecules and related structural analogues is of significant biological interest in the development of methods to manipulate bacterial quorum sensing systems in a useful fashion. In this Letter we report robust routes for the synthesis of a range of N-acylated-l-homoserine lactone (AHL) quorum sensing molecules. Crucially, we have analysed the enantiopurity of the final AHLs and in all cases, excellent levels were observed.
Co-reporter:Dr. Christine M. Boehner;Elizabeth C. Frye;Kieron M. G. O'Connell;Dr. Warren R. J. D. Galloway;Dr. Hannah F. Sore;Dr. Patricia Garcia Dominguez;Dr. David Norton;Dr. David G. Hulcoop;Dr. Martin Owen;Dr. Gillian Turner;Dr. Claire Crawford;Dr. Helen Horsley;Dr. David R. Spring
Chemistry - A European Journal 2011 Volume 17( Issue 47) pp:13230-13239
Publication Date(Web):
DOI:10.1002/chem.201102285
Abstract
The prevalence of the biaryl structural motif in biologically interesting and synthetically important molecules has inspired considerable interest in the development of methods for aryl–aryl bond formation. Herein we describe a novel strategy for this process involving the fluoride-free, palladium-catalysed cross-coupling of readily accessible aryldisiloxanes and aryl bromides. Using a statistical-based optimisation process, preparatively useful reaction conditions were formulated to allow the cross-coupling of a wide range of different substrates. This methodology represents an attractive, cost-efficient, flexible and robust alternative to the traditional transition-metal-catalysed routes typically used to generate molecules containing the privileged biaryl scaffold.
Co-reporter:Dr. Jayne L. Kenwright;Dr. Warren R. J. D. Galloway;Dr. David T. Blackwell;Dr. Albert Isidro-Llobet;James Hodgkinson;Dr. Lars Wortmann;Dr. Steven D. Bowden;Dr. Martin Welch;Dr. David R. Spring
Chemistry - A European Journal 2011 Volume 17( Issue 10) pp:2981-2986
Publication Date(Web):
DOI:10.1002/chem.201002093
Abstract
Herein, a new copper-catalysed strategy for the synthesis of rare nitrogen-linked seven-, eight- and nine-membered biaryl ring systems is described. It is proposed that the reaction proceeds through a highly activated intramolecularly co-ordinated copper catalyst. The process is technically simple, proceeds under relatively mild conditions, displays a broad substrate scope and forms biologically valuable products that are difficult to synthesise by other methods. We envisage that this methodology will prove useful in a wide synthetic context, with possible applications in both target-oriented and diversity-oriented synthesis.
Co-reporter:Dr. Zhaochao Xu;Dr. David R. Spring; Juyoung Yoon
Chemistry – An Asian Journal 2011 Volume 6( Issue 8) pp:2114-2122
Publication Date(Web):
DOI:10.1002/asia.201100120
Abstract
It is still a challenging task to discriminate adenosine-5′-triphosphate (ATP) from various nucleoside triphosphates, such as GTP, CTP, UTP, and TTP. The ability to distinguish ATP from adenosine diphosphate (ADP) by fluorescent signals is also urgently desired. Herein, we report two pyrene-based zinc complexes as nucleoside polyphosphate receptors with high selectivity for ATP and ADP based on fluorescence and NMR studies. A unique pyrene–adenine–pyrene sandwich assembly was observed in the case of compound 1 with ATP or ADP, resulting in the increase of monomer fluorescence intensity; whereas the other bases of nucleoside triphosphates, such as GTP, CTP, UTP, and TTP were not sandwiched, resulting in a switch in the monomer–excimer fluorescence of pyrene. The different binding patterns of various nucleobases with a pyrene–pyrene assembly make 1 a highly selective fluorescent sensor for ANP (N=di, tri). In the case of compound 2, the first 0.5 equivalents of ATP induced a strong excimer emission, whilst ADP induced a large enhancement in the monomeric fluorescent peak. This fluorescence change makes 2 an efficient sensor to discriminate ATP from ADP.
Co-reporter:Dr. Zhaochao Xu;Dr. N. Jiten Singh;Dr. Sook Kyung Kim;Dr. David R. Spring; Kwang S. Kim; Juyoung Yoon
Chemistry - A European Journal 2011 Volume 17( Issue 4) pp:1163-1170
Publication Date(Web):
DOI:10.1002/chem.201002105
Abstract
Intermolecular interactions that involve aromatic rings are key processes in both chemical and biological recognition. It is common knowledge that the existence of anion–π interactions between anions and electron-deficient (π-acidic) aromatics indicates that electron-rich (π-basic) aromatics are expected to be repulsive to anions due to their electron-donating character. Here we report the first concrete theoretical and experimental evidence of the anion–π interaction between electron-rich alkylbenzene rings and a fluoride ion in CH3CN. The cyclophane cavity bridged with three naphthoimidazolium groups selectively complexes a fluoride ion by means of a combination of anion–π interactions and (CH)+⋅⋅⋅F−-type ionic hydrogen bonds. 1H NMR, 19F NMR, and fluorescence spectra of 1 and 2 with fluoride ions are examined to show that only 2 can host a fluoride ion in the cavity between two alkylbenzene rings to form a sandwich complex. In addition, the cage compounds can serve as highly selective and ratiometric fluorescent sensors for a fluoride ion. With the addition of 1 equiv of F−, a strongly increased fluorescence emission centered at 385 nm appears at the expense of the fluorescence emission of 2 centered at 474 nm. Finally, isothermal titration calorimetry (ITC) experiments were performed to obtain the binding constants of the compounds 1 and 2 with F− as well as Gibbs free energy. The 2-F− complex is more stable than the 1-F− complex by 1.87 kcal mol−1, which is attributable to the stronger anion–π interaction between F− and triethylbenzene.
Co-reporter:Warren R. J. D. Galloway;James T. Hodgkinson;Paula Bello;Agostino Cilibrizzi;Tiffanie Murillo;Albert Isidro-Llobet;Martin Welch;Andreas Bender
PNAS 2011 Volume 108 (Issue 17 ) pp:6793-6798
Publication Date(Web):2011-04-26
DOI:10.1073/pnas.1015267108
Structurally diverse libraries of novel small molecules represent important sources of biologically active agents. In this
paper we report the development of a diversity-oriented synthesis strategy for the generation of diverse small molecules based
around a common macrocyclic peptidomimetic framework, containing structural motifs present in many naturally occurring bioactive
compounds. Macrocyclic peptidomimetics are largely underrepresented in current small-molecule screening collections owing
primarily to synthetic intractability; thus novel molecules based around these structures represent targets of significant
interest, both from a biological and a synthetic perspective. In a proof-of-concept study, the synthesis of a library of 14
such compounds was achieved. Analysis of chemical space coverage confirmed that the compound structures indeed occupy underrepresented
areas of chemistry in screening collections. Crucial to the success of this approach was the development of novel methodologies
for the macrocyclic ring closure of chiral α-azido acids and for the synthesis of diketopiperazines using solid-supported
N methylmorpholine. Owing to their robust and flexible natures, it is envisaged that both new methodologies will prove to be
valuable in a wider synthetic context.
Co-reporter:Zhaochao Xu, Juyoung Yoon and David R. Spring
Chemical Society Reviews 2010 vol. 39(Issue 6) pp:1996-2006
Publication Date(Web):28 Apr 2010
DOI:10.1039/B916287A
In the past decade, fluorescent chemosensors for zinc ion (Zn2+) have attracted great attention because of the biological significance of zinc combined with the simplicity and high sensitivity of fluorescence assays. Chemosensors can be divided into a fluorophore, a spacer and a receptor unit; the receptor is the central processing unit (CPU) of a chemosensor. This tutorial review will classify zinc chemosensors based on receptor types.
Co-reporter:Hannah F. Sore, David T. Blackwell, Simon J. F. MacDonald and David R. Spring
Organic Letters 2010 Volume 12(Issue 12) pp:2806-2809
Publication Date(Web):May 21, 2010
DOI:10.1021/ol100895d
The regio- and stereoselective synthesis and subsequent Hiyama cross coupling of pentafluorophenyldimethylvinylsilanes has been developed, thus providing a convenient and robust method for the diversity-oriented synthesis of (E)-, (Z)- and α-disubstituted alkenes from terminal alkynes. Pentafluorophenyldimethylvinylsilanes undergo cross-coupling reactions with excellent selectivity and in good yields, offering an attractive alternative to existing masked silanols.
Co-reporter:Zhaochao Xu, Juyoung Yoon and David R. Spring
Chemical Communications 2010 vol. 46(Issue 15) pp:2563-2565
Publication Date(Web):24 Feb 2010
DOI:10.1039/C000441C
A fluorescent probe was designed and shown to detect Cu2+ ratiometrically and selectively in aqueous solutions based on naphthalimide excimer–monomer switching.
Co-reporter:Zhaochao Xu, Su Jung Han, Chongmok Lee, Juyoung Yoon and David R. Spring
Chemical Communications 2010 vol. 46(Issue 10) pp:1679-1681
Publication Date(Web):12 Jan 2010
DOI:10.1039/B924503K
A carbonyl group was positioned between 1,8-naphthalimide and di-2-picolylamine (DPA) and played a key role of displaying fluorescence enhancements with heavy and transition metal (HTM) ions through increasing the oxidation potential of the fluorophore, blocking HTM ions from sterically interacting with the naphthalimide fluorophore, and by acting as a sacrificial donor.
Co-reporter:Mónica Díaz-Gavilán, Warren R. J. D. Galloway, Kieron M. G. O’Connell, James T. Hodkingson and David R. Spring
Chemical Communications 2010 vol. 46(Issue 5) pp:776-778
Publication Date(Web):14 Dec 2009
DOI:10.1039/B917965H
A diversity-oriented synthesis involving a cascade sequence, taking linear aminoalkenes to polycyclic scaffolds reminiscent of natural alkaloids, is presented.
Co-reporter:Zhaochao Xu, Shaojun Zheng, Juyoung Yoon and David R. Spring
Analyst 2010 vol. 135(Issue 10) pp:2554-2559
Publication Date(Web):06 Sep 2010
DOI:10.1039/C0AN00405G
An ideal fluorescent probe should show the strongest affinity with the relevant target (binding-selectivity) by means of a selective fluorescence change (signal-selectivity). [15]aneNO2S2 (1,4-dioxa-7,13-dithia-10-azacyclopentadecane) based probes usually show high binding selectivity for Ag+ but signal selectivity for Hg2+, because Ag+ can quench or silence the fluorescence. To amplify the Ag+ binding to the greatest extent, a carbonyl group was positioned between 1,8-naphthalimide and [15]aneNO2S2 which played a key role of displaying selective fluorescence enhancements with Ag+ through increasing the oxidation potential of the fluorophore, blocking Ag+ from sterically interacting with the naphthalimide fluorophore, and by acting as a sacrificial donor. Probe 2 can detect Ag+ with a selective fluorescence enhancement (∼14 fold) and high affinity (Ka = 1.64 × 105 M−1).
Co-reporter:Christine M. Boehner, David M. Marsden, Hannah F. Sore, David Norton, David R. Spring
Tetrahedron Letters 2010 Volume 51(Issue 45) pp:5930-5932
Publication Date(Web):10 November 2010
DOI:10.1016/j.tetlet.2010.09.024
A tetrafluorophenol acrylamide monomer unit was synthesised, co-polymerised and grafted onto a glass slide to form individual gel spots. As a proof of principle study, a small library of amides was rapidly synthesised within these gel spots using ‘catch-and-release’ chemistry, including the biologically interesting quorum sensing acyl-homoserine lactones. The tetrafluorophenol acrylamide gel provides an efficient platform to synthesise and screen small molecules for biological activity.The development of tetrafluorophenol acrylamide 3D gels as an effective platform for the synthesis of small molecules has been achieved. Furthermore, this offers the potential to synthesise and screen compounds for biological activity on the same slide.
Co-reporter:Warren R.J.D. Galloway, James T. Hodgkinson, Martin Welch, David R. Spring
Chemistry & Biology 2009 Volume 16(Issue 9) pp:913-914
Publication Date(Web):25 September 2009
DOI:10.1016/j.chembiol.2009.09.006
The publication of the crystal structures of a bacterial quorum sensing receptor complexed with a variety of ligands (Zou and Nair, 2009) provides a much-needed molecular rationale for the modulation of this intercellular signaling process, which may facilitate the development of new therapeutic agents.
Co-reporter:Warren R.J.D. Galloway, James T. Hodgkinson, Steven Bowden, Martin Welch, David R. Spring
Trends in Microbiology (September 2012) Volume 20(Issue 9) pp:449-458
Publication Date(Web):1 September 2012
DOI:10.1016/j.tim.2012.06.003
Quorum sensing is a form of intercellular communication used by many species of bacteria that facilitates concerted interactions between the cells comprising a population. The phenotypes regulated by quorum sensing are extremely diverse, with many having a significant impact upon healthcare, agriculture, and the environment. Consequently there has been significant interest in developing methods to manipulate this signalling process and recent years have witnessed significant theoretical and practical developments. A wide range of small molecule modulators of quorum sensing systems has been discovered, providing an expansive chemical toolbox for the study and modulation of this signalling mechanism. In this review, a selection of recent case studies which illustrate the value of both activators and inhibitors of quorum sensing in Gram-negative bacteria are discussed.
Co-reporter:Kieron M. G. O'Connell, Henning S. G. Beckmann, Luca Laraia, Helen T. Horsley, Andreas Bender, Ashok R. Venkitaraman and David R. Spring
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 37) pp:NaN7551-7551
Publication Date(Web):2012/08/03
DOI:10.1039/C2OB26272J
Macrocyclic compounds represent a structural class with exceptional potential for biological activity; however, they have historically been underrepresented in screening collections and synthetic libraries. In this article we report the development of a highly step-efficient strategy for the diversity-oriented synthesis of complex macrocyclic architectures, using a modular approach based on the two-directional synthesis of bifunctional linear precursors and their subsequent combination in a two-directional macrocyclisation process. In this proof of principle study, the synthesis of 14 such compounds was achieved. Cheminformatic analysis of the compounds produced suggests that they reside in biologically relevant regions of chemical space and the compounds were screened for activity against two cancer cell lines.
Co-reporter:James T. Hodgkinson, Warren R. J. D. Galloway, Shreya Saraf, Ian R. Baxendale, Steven V. Ley, Mark Ladlow, Martin Welch and David R. Spring
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 1) pp:NaN61-61
Publication Date(Web):2010/10/22
DOI:10.1039/C0OB00652A
Expedient syntheses of Pseudomonas quinolone signal (PQS) and related structural analogues using microwave and flow methods are reported.
Co-reporter:Tze Han Sum, Tze Jing Sum, Súil Collins, Warren R. J. D. Galloway, David G. Twigg, Florian Hollfelder and David R. Spring
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 21) pp:NaN4570-4570
Publication Date(Web):2017/05/12
DOI:10.1039/C7OB00804J
Biflavonoids are associated with a variety of biologically useful properties. However, synthetic biflavonoids are poorly explored within drug discovery. There is considerable structural diversity possible within this compound class and large regions of potentially biologically relevant biflavonoid chemical space remain untapped or underexplored. Herein, we report the development of a modular and divergent strategy towards biflavonoid derivatives which enabled the step-economical preparation of a structurally diverse collection of novel unnatural biflavonoids. Preliminary studies established that the strategy could also be successfully extended to the preparation of very rare triflavonoids, which are also expected to be useful tools for biological intervention. Prompted by previous inhibitory studies with flavonoid libraries, amyloid anti-aggregation screening was performed, which led to the identification of several structurally novel inhibitors of the aggregation of the amyloid β peptide (Aβ42). Aggregated Aβ42 is a pathological hallmark of Alzheimer's disease and the use of small molecules to inhibit the aggregation process has been identified as a potentially valuable therapeutic strategy for disease treatment. Methylated biaurones were associated with highest levels of potency (the most active compound had an IC50 value of 16 μM), establishing this scaffold as a starting point for inhibitor development.
Co-reporter:Cornelius J. O' Connor, Henning S. G. Beckmann and David R. Spring
Chemical Society Reviews 2012 - vol. 41(Issue 12) pp:NaN4456-4456
Publication Date(Web):2012/04/10
DOI:10.1039/C2CS35023H
Small molecule modulators of biological function can be discovered by the screening of compound libraries. However, it became apparent that some human disease related targets could not be addressed by the libraries commonly used which typically are comprised of large numbers of structurally similar compounds. The last decade has seen a paradigm shift in library construction, with particular emphasis now being placed on increasing a library's structural, and thus functional diversity, rather than only its size. Diversity-oriented synthesis (DOS) aims to generate such structural diversity efficiently. This tutorial review has been written to introduce the subject to a broad audience and recent achievements in both the preparation and the screening of structurally diverse compound collections against so-called ‘undruggable’ targets are highlighted.
Co-reporter:Mónica Díaz-Gavilán, Warren R. J. D. Galloway, Kieron M. G. O’Connell, James T. Hodkingson and David R. Spring
Chemical Communications 2010 - vol. 46(Issue 5) pp:NaN778-778
Publication Date(Web):2009/12/14
DOI:10.1039/B917965H
A diversity-oriented synthesis involving a cascade sequence, taking linear aminoalkenes to polycyclic scaffolds reminiscent of natural alkaloids, is presented.
Co-reporter:Hannah F. Sore, Warren R. J. D. Galloway and David R. Spring
Chemical Society Reviews 2012 - vol. 41(Issue 5) pp:NaN1866-1866
Publication Date(Web):2011/11/02
DOI:10.1039/C1CS15181A
The palladium catalysed cross-coupling of organosilicon reagents with organo halides and pseudo-halides has developed over the past 30 years into an efficient and attractive carbon–carbon bond forming strategy. Extensive research within this field to expand and diversify on the scope of the organosilicon coupling reaction will continue to promote its use in the synthesis of biologically and pharmaceutically important organic molecules. The recent advances made within this area are explored in this critical review (199 references).
Co-reporter:Zhaochao Xu, Juyoung Yoon and David R. Spring
Chemical Society Reviews 2010 - vol. 39(Issue 6) pp:NaN2006-2006
Publication Date(Web):2010/04/28
DOI:10.1039/B916287A
In the past decade, fluorescent chemosensors for zinc ion (Zn2+) have attracted great attention because of the biological significance of zinc combined with the simplicity and high sensitivity of fluorescence assays. Chemosensors can be divided into a fluorophore, a spacer and a receptor unit; the receptor is the central processing unit (CPU) of a chemosensor. This tutorial review will classify zinc chemosensors based on receptor types.
Co-reporter:Albert Isidro-Llobet, Kathy Hadje Georgiou, Warren R. J. D. Galloway, Elisa Giacomini, Mette R. Hansen, Gabriela Méndez-Abt, Yaw Sing Tan, Laura Carro, Hannah F. Sore and David R. Spring
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 15) pp:NaN4580-4580
Publication Date(Web):2015/03/11
DOI:10.1039/C5OB00371G
Macrocyclic peptidomimetics are associated with a broad range of biological activities. However, despite such potentially valuable properties, the macrocyclic peptidomimetic structural class is generally considered as being poorly explored within drug discovery. This has been attributed to the lack of general methods for producing collections of macrocyclic peptidomimetics with high levels of structural, and thus shape, diversity. In particular, there is a lack of scaffold diversity in current macrocyclic peptidomimetic libraries; indeed, the efficient construction of diverse molecular scaffolds presents a formidable general challenge to the synthetic chemist. Herein we describe a new, advanced strategy for the diversity-oriented synthesis (DOS) of macrocyclic peptidomimetics that enables the combinatorial variation of molecular scaffolds (core macrocyclic ring architectures). The generality and robustness of this DOS strategy is demonstrated by the step-efficient synthesis of a structurally diverse library of over 200 macrocyclic peptidomimetic compounds, each based around a distinct molecular scaffold and isolated in milligram quantities, from readily available building-blocks. To the best of our knowledge this represents an unprecedented level of scaffold diversity in a synthetically derived library of macrocyclic peptidomimetics. Cheminformatic analysis indicated that the library compounds access regions of chemical space that are distinct from those addressed by top-selling brand-name drugs and macrocyclic natural products, illustrating the value of our DOS approach to sample regions of chemical space underexploited in current drug discovery efforts. An analysis of three-dimensional molecular shapes illustrated that the DOS library has a relatively high level of shape diversity.
Co-reporter:Shaojun Zheng, Luca Laraia, Cornelius J. O' Connor, David Sorrell, Yaw Sing Tan, Zhaochao Xu, Ashok R. Venkitaraman, Wenjun Wu and David R. Spring
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 13) pp:NaN2593-2593
Publication Date(Web):2012/01/30
DOI:10.1039/C2OB25065A
A novel synthesis of the ellagitannin natural product tellimagrandin I and a series of medium ring analogues is described. These compounds were all subsequently screened for redox activity, ability to precipitate protein and cellular phenotype in HeLa cells. From this we have shown that all properties can be modulated independently by varying ring size and by moving the ester out of conjugation with the biaryl ring system. Increasing ring size increased redox activity and cytotoxicity, leading to the identification of a compound (10) which was significantly more cytotoxic. In addition compounds identified with a redox active scaffold and low cytotoxicity may be employed as a new class of redox probes.
Co-reporter:Zhaochao Xu, Su Jung Han, Chongmok Lee, Juyoung Yoon and David R. Spring
Chemical Communications 2010 - vol. 46(Issue 10) pp:NaN1681-1681
Publication Date(Web):2010/01/12
DOI:10.1039/B924503K
A carbonyl group was positioned between 1,8-naphthalimide and di-2-picolylamine (DPA) and played a key role of displaying fluorescence enhancements with heavy and transition metal (HTM) ions through increasing the oxidation potential of the fluorophore, blocking HTM ions from sterically interacting with the naphthalimide fluorophore, and by acting as a sacrificial donor.
Co-reporter:Yu Heng Lau, Peterson de Andrade, Yuteng Wu and David R. Spring
Chemical Society Reviews 2015 - vol. 44(Issue 1) pp:NaN102-102
Publication Date(Web):2014/09/08
DOI:10.1039/C4CS00246F
Peptide stapling is a strategy for constraining short peptides typically in an alpha-helical conformation. Stapling is carried out by covalently linking the side-chains of two amino acids, thereby forming a peptide macrocycle. There is an expanding repertoire of stapling techniques based on different macrocyclisation chemistries. In this tutorial review, we categorise and analyse key examples of peptide stapling in terms of their synthesis and applicability to biological systems.
Co-reporter:T. A. Alanine, W. R. J. D. Galloway, S. Bartlett, J. J. Ciardiello, T. M. McGuire and D. R. Spring
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 3) pp:NaN1038-1038
Publication Date(Web):2015/11/24
DOI:10.1039/C5OB01784J
Pyrido[1,2-a]pyrimidin-2-ones represent a pharmaceutically interesting class of heterocycles. The structurally related pyrido[1,2-a]pyrimidin-4-ones are associated with a broad range of useful biological properties. Furthermore, quinolizinone-type scaffolds of these sorts with a bridgehead nitrogen are expected to display interesting physico–chemical properties. However, pyrido[1,2-a]pyrimidin-2-ones are largely under-represented in current small molecule screening libraries and the physical and biological properties of the pyrido[1,2-a]pyrimidin-2-one scaffold have been poorly explored (indeed, the same can be said for unsaturated bicyclic compounds with a bridgehead nitrogen in general). Herein, we report the development of a new strategy for the concise synthesis of substituted pyrido[1,2-a]pyrimidin-2-ones from readily available starting materials. The synthetic route involved the acylation of the lithium amide bases of 2-aminopyridines with alkynoate esters to form alkynamides, which were then cyclised under thermal conditions. The use of lithium amide anions ensured excellent regioselectivity for the 2-oxo-isomer over the undesired 4-oxo-isomer, which offers a distinct advantage over some existing methods for the synthesis of pyrido[1,2-a]pyrimidin-2-ones. Notably, different aminoazines could also be employed in this approach, which enabled access to several very unusual bicyclic systems with higher nitrogen contents. This methodology thus represents an important contribution towards the synthesis of pyrido[1,2-a]pyrimidin-2-ones and other rare azabicycles with a ring-junction nitrogen. These heterocycles represent attractive structural templates for drug discovery.
Co-reporter:David A. Russell, Julien J. Freudenreich, Joe J. Ciardiello, Hannah F. Sore and David R. Spring
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 7) pp:NaN1596-1596
Publication Date(Web):2017/01/24
DOI:10.1039/C6OB02659A
We describe stereocontrolled semi-syntheses of deguelin and tephrosin, anti-cancer rotenoids isolated from Tephrosia vogelii. Firstly, we present a new two-step transformation of rotenone into rot-2′-enonic acid via a zinc-mediated ring opening of rotenone hydrobromide. Secondly, following conversion of rot-2′-enonic acid into deguelin, a chromium-mediated hydroxylation provides tephrosin as a single diastereoisomer. An Étard-like reaction mechanism is proposed to account for the stereochemical outcome. Our syntheses of deguelin and tephrosin are operationally simple, scalable and high yielding, offering considerable advantages over previous methods.
Co-reporter:Terence T.-L. Kwan, Omar Boutureira, Elizabeth C. Frye, Stephen J. Walsh, Moni K. Gupta, Stephen Wallace, Yuteng Wu, Fengzhi Zhang, Hannah F. Sore, Warren R. J. D. Galloway, Jason W. Chin, Martin Welch, Gonçalo J. L. Bernardes and David R. Spring
Chemical Science (2010-Present) 2017 - vol. 8(Issue 5) pp:NaN3878-3878
Publication Date(Web):2017/03/14
DOI:10.1039/C6SC05313K
Transition metal catalysis has emerged as a powerful strategy to expand synthetic flexibility of protein modification. Herein, we report a cationic Ru(II) system that enables the first example of alkyne hydrosilylation between dimethylarylsilanes and O-propargyl-functionalized proteins using a substoichiometric amount or low-loading of Ru(II) catalyst to achieve the first C–Si bond formation on full-length substrates. The reaction proceeds under physiological conditions at a rate comparable to other widely used bioorthogonal reactions. Moreover, the resultant gem-disubstituted vinylsilane linkage can be further elaborated through thiol–ene coupling or fluoride-induced protodesilylation, demonstrating its utility in further rounds of targeted modifications.
Co-reporter:James T. Hodgkinson, Warren R. J. D. Galloway, Megan Wright, Ioulia K. Mati, Rebecca L. Nicholson, Martin Welch and David R. Spring
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 30) pp:NaN6044-6044
Publication Date(Web):2012/03/09
DOI:10.1039/C2OB25198A
Many species of bacteria employ a mechanism of intercellular communication known as quorum sensing which is mediated by small diffusible signalling molecules termed autoinducers. The most common class of autoinducer used by Gram-negative bacteria are N-acylated-L-homoserine lactones (AHLs). Pseudomonas aeruginosa is a clinically important bacterium which is known to use AHL-mediated quorum sensing systems to regulate a variety of processes associated with virulence. Thus the selective disruption of AHL-based quorum sensing represents a strategy to attenuate the pathogenicity of this bacterium. Herein we describe the design, synthesis and biological evaluation of a collection of structurally novel AHL mimics. A number of new compounds capable of modulating the LasR-dependent quorum sensing system of P. aeruginosa were identified, which could have value as molecular tools to study and manipulate this signalling pathway. Worthy of particular note, this research has delivered novel potent quorum sensing antagonists, which strongly inhibit the production of virulence factors in a wild type strain of this pathogenic bacterium.
Co-reporter:Cornelius J. O' Connor, Luca Laraia and David R. Spring
Chemical Society Reviews 2011 - vol. 40(Issue 8) pp:NaN4345-4345
Publication Date(Web):2011/05/12
DOI:10.1039/C1CS15053G
Chemical genetics can be defined as the study of biological systems using small molecule tools. Cell permeable and selective small molecules modulate gene product function rapidly, reversibly and can be administered conditionally in either a cellular or organismal context. The small molecule approach provides exacting temporal and quantitative control and is therefore an extremely powerful tool for dissecting biological processes. This tutorial review has been written to introduce the subject to a broad audience and highlights recent developments within the field in four key areas of biology: modulating protein–protein interactions, malaria research, hepatitis C virus research, and disrupting RNA interference pathways.
Co-reporter:Esther Alza, Luca Laraia, Brett M. Ibbeson, Súil Collins, Warren R. J. D. Galloway, Jamie E. Stokes, Ashok R. Venkitaraman and David R. Spring
Chemical Science (2010-Present) 2015 - vol. 6(Issue 1) pp:NaN396-396
Publication Date(Web):2014/09/09
DOI:10.1039/C4SC02547D
The synthesis of a previously undescribed sp3-rich 6-5-5-6 tetracyclic ring scaffold using a palladium catalysed domino Heck–Suzuki reaction is reported. This reaction is high-yielding, selective for the domino process over the direct Suzuki reaction and tolerant towards a variety of boronic acids. The novel scaffold can also be accessed via domino Heck–Stille and radical cyclisations. Compounds based around this scaffold were found to be effective antimitotic agents in a human cancer cell line. Detailed phenotypic profiling showed that the compounds affected the congression of chromosomes to give mitotic arrest and apoptotic cell death. Thus, a novel structural class of antimitotic agents that does not disrupt the tubulin network has been identified.
Co-reporter:Yu Heng Lau, Peterson de Andrade, Soo-Tng Quah, Maxim Rossmann, Luca Laraia, Niklas Sköld, Tze Jing Sum, Pamela J. E. Rowling, Thomas L. Joseph, Chandra Verma, Marko Hyvönen, Laura S. Itzhaki, Ashok R. Venkitaraman, Christopher J. Brown, David P. Lane and David R. Spring
Chemical Science (2010-Present) 2014 - vol. 5(Issue 5) pp:NaN1809-1809
Publication Date(Web):2014/03/10
DOI:10.1039/C4SC00045E
Stapled peptides are a promising class of alpha-helix mimetic inhibitors for protein–protein interactions. We report the divergent synthesis of “functionalised” stapled peptides via an efficient two-component strategy. Starting from a single unprotected diazido peptide, dialkynyl staple linkers bearing different unprotected functional motifs are introduced to create different alpha-helical peptides in one step, functionalised on the staple linkage itself. Applying this concept to the p53/MDM2 interaction, we improve the cell permeability and p53 activating capability of an otherwise impermeable p53 stapled peptide by introducing cationic groups on the staple linkage, rather than modifying the peptide sequence.
Co-reporter:Zhaochao Xu, Xin Liu, Jie Pan and David R. Spring
Chemical Communications 2012 - vol. 48(Issue 39) pp:NaN4766-4766
Publication Date(Web):2012/03/05
DOI:10.1039/C2CC30963G
We report a coumarin-derived fluorescent sensor for Zn2+ termed CTS. CTS shows excellent binding selectivity for Zn2+ over competing metal ions due to the transformable ability of CTS, that is the displacement of other metal ions by Zn2+, which induces transformation of chelation from an amide to an imidic acid tautomeric form.
Co-reporter:Zhaochao Xu, Juyoung Yoon and David R. Spring
Chemical Communications 2010 - vol. 46(Issue 15) pp:NaN2565-2565
Publication Date(Web):2010/02/24
DOI:10.1039/C000441C
A fluorescent probe was designed and shown to detect Cu2+ ratiometrically and selectively in aqueous solutions based on naphthalimide excimer–monomer switching.
Co-reporter:Yu Heng Lau, Peterson de Andrade, Niklas Sköld, Grahame J. McKenzie, Ashok R. Venkitaraman, Chandra Verma, David P. Lane and David R. Spring
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 24) pp:NaN4077-4077
Publication Date(Web):2014/05/02
DOI:10.1039/C4OB00742E
Stapling peptides for inhibiting the p53/MDM2 interaction is a promising strategy for developing anti-cancer therapeutic leads. We evaluate double-click stapled peptides formed from p53-based diazidopeptides with different staple positions and azido amino acid side-chain lengths, determining the impact of these variations on MDM2 binding and cellular activity. We also demonstrate a K24R mutation, necessary for cellular activity in hydrocarbon-stapled p53 peptides, is not required for analogous ‘double-click’ peptides.

![[1,1'-Biphenyl]-3,3'-dicarboxylic acid, 5,5'-diiodo-, diethyl ester](http://img.cochemist.com/ccimg/852100/852050-91-0.png)
![[1,1'-Biphenyl]-3,3'-dicarboxylic acid, 5,5'-diiodo-, diethyl ester](http://img.cochemist.com/ccimg/852100/852050-91-0_b.png)
![1H-Indole-3-butanamide, N-[(3S)-tetrahydro-2-oxo-3-furanyl]-](http://img.cochemist.com/ccimg/868100/868054-78-8.png)
![1H-Indole-3-butanamide, N-[(3S)-tetrahydro-2-oxo-3-furanyl]-](http://img.cochemist.com/ccimg/868100/868054-78-8_b.png)
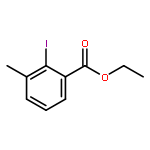
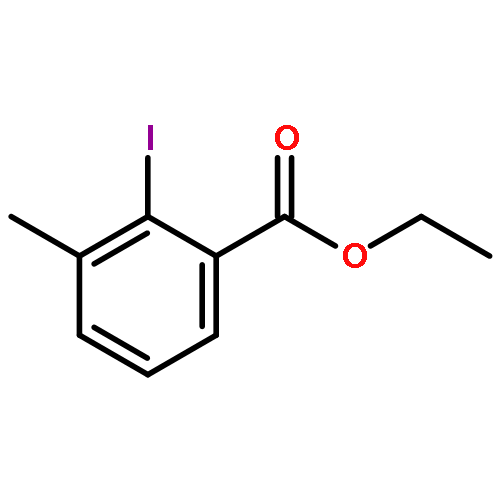


![Benzoic acid, 2,5-diiodo-, 2-[[(2-iodobenzoyl)oxy]methyl]phenyl ester](http://img.cochemist.com/ccimg/852100/852050-88-5.png)
![Benzoic acid, 2,5-diiodo-, 2-[[(2-iodobenzoyl)oxy]methyl]phenyl ester](http://img.cochemist.com/ccimg/852100/852050-88-5_b.png)




![5H-Dibenzo[b,e][1,4]diazepine, 10,11-dihydro-](http://img.cochemist.com/ccimg/4900/4810-47-3.png)
![5H-Dibenzo[b,e][1,4]diazepine, 10,11-dihydro-](http://img.cochemist.com/ccimg/4900/4810-47-3_b.png)


![Dibenzo[b,d][1,6]dioxecin, 6,7,8,9-tetrahydro-](http://img.cochemist.com/ccimg/34200/34131-06-1.png)
![Dibenzo[b,d][1,6]dioxecin, 6,7,8,9-tetrahydro-](http://img.cochemist.com/ccimg/34200/34131-06-1_b.png)


![Benzene, 1-[(1E)-2-phenylethenyl]-3-(trifluoromethyl)-](http://img.cochemist.com/ccimg/900/891-70-3.png)
![Benzene, 1-[(1E)-2-phenylethenyl]-3-(trifluoromethyl)-](http://img.cochemist.com/ccimg/900/891-70-3_b.png)





![Dibenzo[f,h][1,4]dioxecin-5,10-dione, 7,8-dihydro-](http://img.cochemist.com/ccimg/153000/152936-28-2.png)
![Dibenzo[f,h][1,4]dioxecin-5,10-dione, 7,8-dihydro-](http://img.cochemist.com/ccimg/153000/152936-28-2_b.png)
![PYRIDINE, 3-[(1E)-2-PHENYLETHENYL]-](http://img.cochemist.com/ccimg/5100/5097-91-6.png)
![PYRIDINE, 3-[(1E)-2-PHENYLETHENYL]-](http://img.cochemist.com/ccimg/5100/5097-91-6_b.png)




![BENZENEACETAMIDE, 4-BROMO-N-[(3S)-TETRAHYDRO-2-OXO-3-FURANYL]-](http://img.cochemist.com/ccimg/868100/868054-85-7.png)
![BENZENEACETAMIDE, 4-BROMO-N-[(3S)-TETRAHYDRO-2-OXO-3-FURANYL]-](http://img.cochemist.com/ccimg/868100/868054-85-7_b.png)
![4(1H)-Quinolinone, 2-[(2E)-3,7-dimethyl-2,6-octadienyl]-1,3-dimethyl-](http://img.cochemist.com/ccimg/189400/189372-43-8.png)
![4(1H)-Quinolinone, 2-[(2E)-3,7-dimethyl-2,6-octadienyl]-1,3-dimethyl-](http://img.cochemist.com/ccimg/189400/189372-43-8_b.png)

![[1,1'-BIPHENYL]-2,2'-DICARBOXYLIC ACID, 6,6'-DIMETHYL-, DIETHYL ESTER](http://img.cochemist.com/ccimg/71900/71871-12-0.png)
![[1,1'-BIPHENYL]-2,2'-DICARBOXYLIC ACID, 6,6'-DIMETHYL-, DIETHYL ESTER](http://img.cochemist.com/ccimg/71900/71871-12-0_b.png)
![BENZAMIDE, N-[2-(1-PIPERIDINYL)ETHYL]-](http://img.cochemist.com/ccimg/4400/4380-82-9.png)
![BENZAMIDE, N-[2-(1-PIPERIDINYL)ETHYL]-](http://img.cochemist.com/ccimg/4400/4380-82-9_b.png)


![[1,1'-BIPHENYL]-2,2'-DICARBOXYLIC ACID, 4,4'-DIIODO-, DIETHYL ESTER](http://img.cochemist.com/ccimg/852100/852050-90-9.png)
![[1,1'-BIPHENYL]-2,2'-DICARBOXYLIC ACID, 4,4'-DIIODO-, DIETHYL ESTER](http://img.cochemist.com/ccimg/852100/852050-90-9_b.png)



![Ethanone, 1-[4-[(1E)-2-phenylethenyl]phenyl]-](/data/chemimg/1785200/20488-42-0.png)
![Ethanone, 1-[4-[(1E)-2-phenylethenyl]phenyl]-](/data/chemimg/1785200/20488-42-0_b.png)
![[1,1'-Biphenyl]-4-amine, 4'-methoxy-N,N-dimethyl-](/data/chemimg/1470800/18158-44-6.png)
![[1,1'-Biphenyl]-4-amine, 4'-methoxy-N,N-dimethyl-](/data/chemimg/1470800/18158-44-6_b.png)
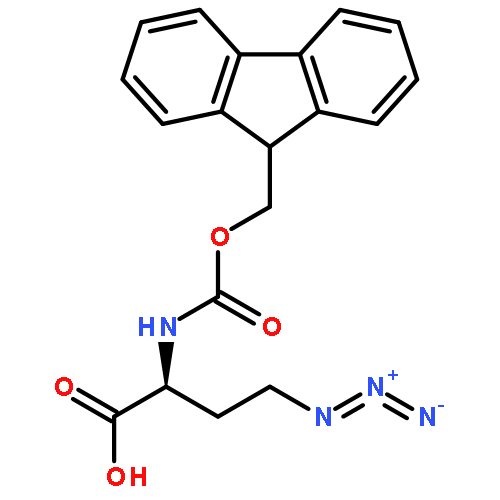

![Dodecanamide, 2,2-difluoro-3-oxo-N-[(3S)-tetrahydro-2-oxo-3-furanyl]-](http://img.cochemist.com/ccimg/218200/218150-36-8.png)
![Dodecanamide, 2,2-difluoro-3-oxo-N-[(3S)-tetrahydro-2-oxo-3-furanyl]-](http://img.cochemist.com/ccimg/218200/218150-36-8_b.png)

![(5Z)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one oxime](http://img.cochemist.com/ccimg/18000/17910-25-7.png)
![(5Z)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one oxime](http://img.cochemist.com/ccimg/18000/17910-25-7_b.png)

![Ethanone, 1-[4-[(1Z)-2-phenylethenyl]phenyl]-](http://img.cochemist.com/ccimg/20500/20488-43-1.png)
![Ethanone, 1-[4-[(1Z)-2-phenylethenyl]phenyl]-](http://img.cochemist.com/ccimg/20500/20488-43-1_b.png)



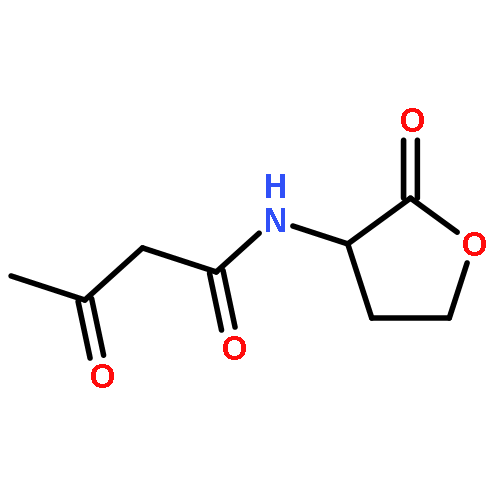
![Benzene, [(3-butynyloxy)methyl]-](http://img.cochemist.com/ccimg/22300/22273-77-4.png)
![Benzene, [(3-butynyloxy)methyl]-](http://img.cochemist.com/ccimg/22300/22273-77-4_b.png)





![Benzene, 1,1'-[(1Z)-1-propene-1,3-diyl]bis-](http://img.cochemist.com/ccimg/1200/1138-83-6.png)
![Benzene, 1,1'-[(1Z)-1-propene-1,3-diyl]bis-](http://img.cochemist.com/ccimg/1200/1138-83-6_b.png)
![1-(4'-Methoxy-[1,1'-biphenyl]-4-yl)ethanone](http://img.cochemist.com/ccimg/13100/13021-18-6.png)
![1-(4'-Methoxy-[1,1'-biphenyl]-4-yl)ethanone](http://img.cochemist.com/ccimg/13100/13021-18-6_b.png)
![Benzene, 1-nitro-2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/4300/4264-29-3.png)
![Benzene, 1-nitro-2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/4300/4264-29-3_b.png)


![Decanamide, 3-oxo-N-[(3S)-tetrahydro-2-oxo-3-furanyl]-](http://img.cochemist.com/ccimg/147800/147795-40-2.png)
![Decanamide, 3-oxo-N-[(3S)-tetrahydro-2-oxo-3-furanyl]-](http://img.cochemist.com/ccimg/147800/147795-40-2_b.png)




![3H-Bis[1]benzopyrano[3,4-b:6',5'-e]pyran-7- (7aH)-one,13,13a-dihydro-7a-hydroxy-9,10- dimethoxy-3,3-dimethyl-,(7aR,13aR)-](http://img.cochemist.com/ccimg/100/76-80-2.png)
![3H-Bis[1]benzopyrano[3,4-b:6',5'-e]pyran-7- (7aH)-one,13,13a-dihydro-7a-hydroxy-9,10- dimethoxy-3,3-dimethyl-,(7aR,13aR)-](http://img.cochemist.com/ccimg/100/76-80-2_b.png)




![β-D-Glucopyranose, cyclic 2,3-[(1S)-4,4',5,5',6,6'-hexahydroxy[1,1'-biphenyl]-2,2'-dicarboxylate] 1-(3,4,5-trihydroxybenzoate)](/data/chemimg/1354400/87392-62-9.png)
![β-D-Glucopyranose, cyclic 2,3-[(1S)-4,4',5,5',6,6'-hexahydroxy[1,1'-biphenyl]-2,2'-dicarboxylate] 1-(3,4,5-trihydroxybenzoate)](/data/chemimg/1354400/87392-62-9_b.png)






![[1,1'-Biphenyl]-2,2'-dicarboxylicacid, 2,2'-diethyl ester](http://img.cochemist.com/ccimg/5900/5807-65-8.png)
![[1,1'-Biphenyl]-2,2'-dicarboxylicacid, 2,2'-diethyl ester](http://img.cochemist.com/ccimg/5900/5807-65-8_b.png)
![Benzene, [(1Z)-2-bromoethenyl]-](/data/chemimg/1121700/588-73-8.png)
![Benzene, [(1Z)-2-bromoethenyl]-](/data/chemimg/1121700/588-73-8_b.png)


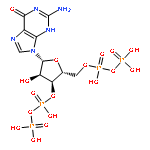
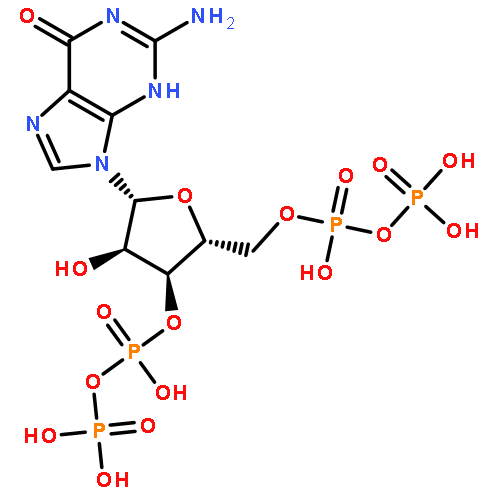
![Ethanol, 2-[2-(2-azidoethoxy)ethoxy]-](/data/chemimg/964400/86520-52-7.png)
![Ethanol, 2-[2-(2-azidoethoxy)ethoxy]-](/data/chemimg/964400/86520-52-7_b.png)
![Dodecanamide, 2,2-difluoro-3-oxo-N-[(1S)-2-oxocyclopentyl]-](http://img.cochemist.com/ccimg/821900/821801-04-1.png)
![Dodecanamide, 2,2-difluoro-3-oxo-N-[(1S)-2-oxocyclopentyl]-](http://img.cochemist.com/ccimg/821900/821801-04-1_b.png)
![Dodecanamide, 2,2-difluoro-3-oxo-N-[(1S)-2-oxocyclohexyl]-](http://img.cochemist.com/ccimg/821900/821801-06-3.png)
![Dodecanamide, 2,2-difluoro-3-oxo-N-[(1S)-2-oxocyclohexyl]-](http://img.cochemist.com/ccimg/821900/821801-06-3_b.png)













![Benzene,1-methyl-4-[(1E)-2-(4-nitrophenyl)ethenyl]-](http://img.cochemist.com/ccimg/24400/24325-70-0.png)
![Benzene,1-methyl-4-[(1E)-2-(4-nitrophenyl)ethenyl]-](http://img.cochemist.com/ccimg/24400/24325-70-0_b.png)
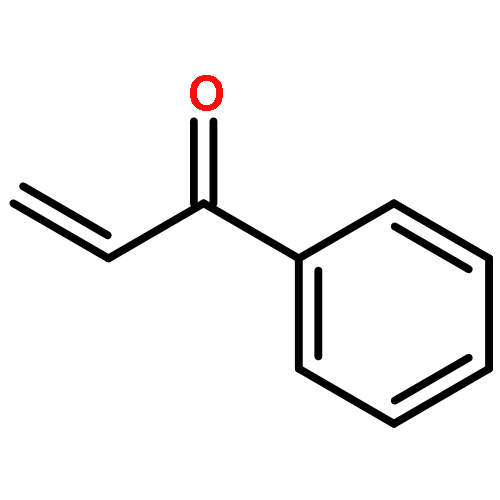
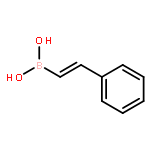
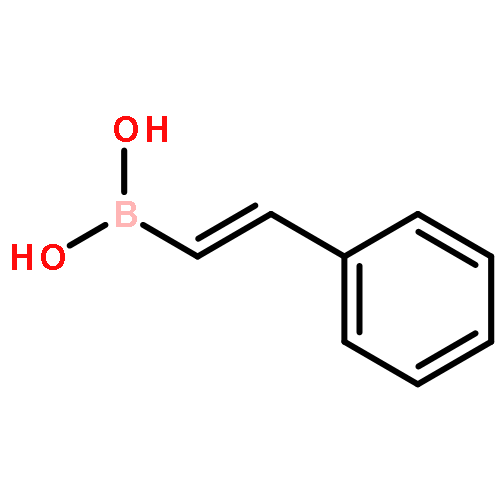



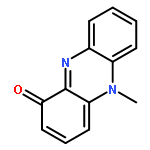
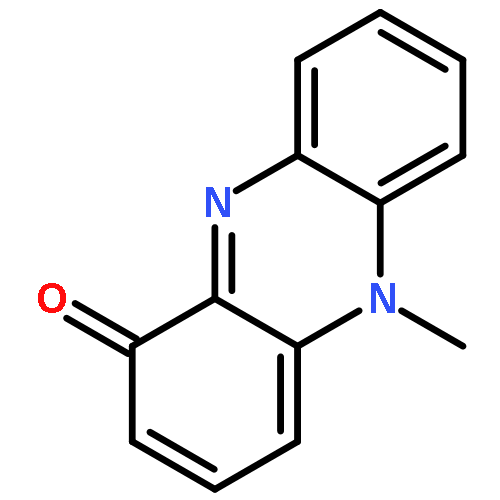

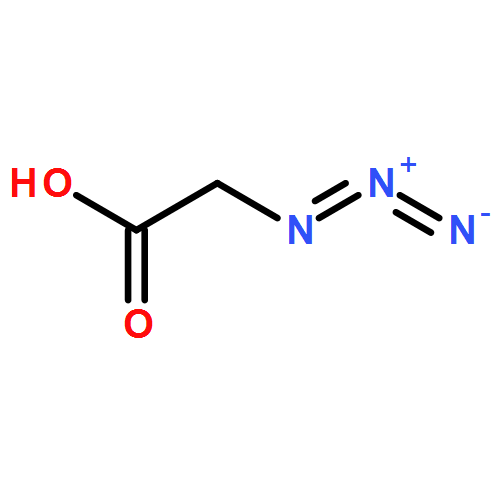







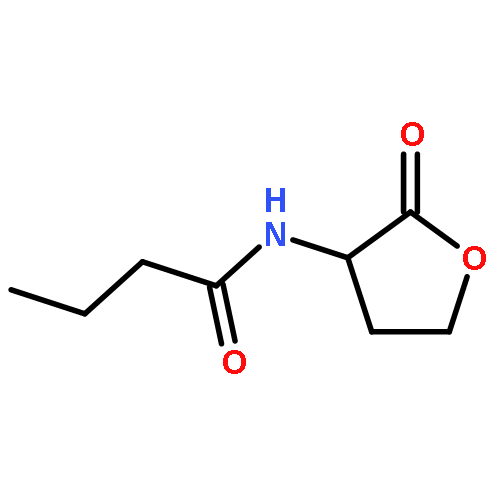
![2,2'-Bi-1H-pyrrole,4-methoxy-5-[(5-methyl-4-pentyl-2H-pyrrol-2-ylidene)methyl]-](http://img.cochemist.com/ccimg/100/82-89-3.png)
![2,2'-Bi-1H-pyrrole,4-methoxy-5-[(5-methyl-4-pentyl-2H-pyrrol-2-ylidene)methyl]-](http://img.cochemist.com/ccimg/100/82-89-3_b.png)


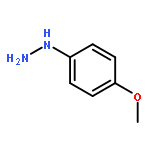
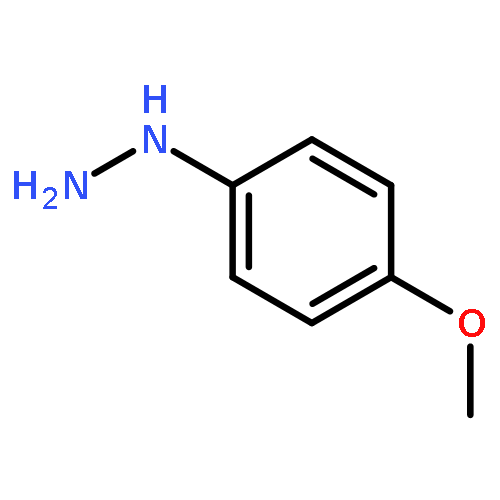







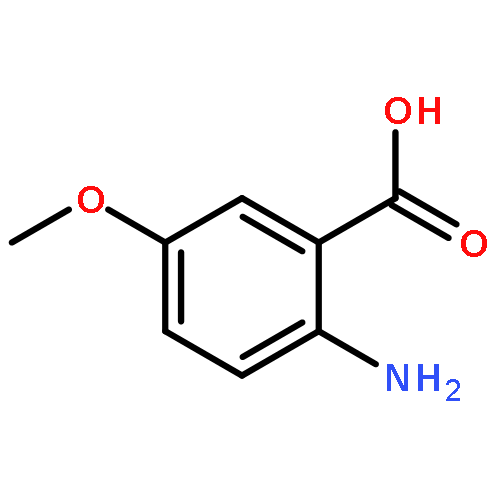
![Octanamide,3-oxo-N-[(3S)-tetrahydro-2-oxo-3-furanyl]-](http://img.cochemist.com/ccimg/147800/147795-39-9.png)
![Octanamide,3-oxo-N-[(3S)-tetrahydro-2-oxo-3-furanyl]-](http://img.cochemist.com/ccimg/147800/147795-39-9_b.png)


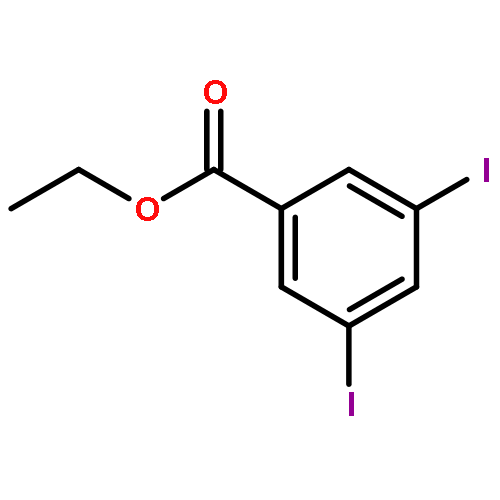






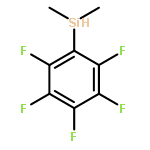

![L-Phenylalanine, 4-azido-N-[(1,1-dimethylethoxy)carbonyl]-](/data/chemimg/538900/33173-55-6.png)
![L-Phenylalanine, 4-azido-N-[(1,1-dimethylethoxy)carbonyl]-](/data/chemimg/538900/33173-55-6_b.png)






![1-[(e)-2-(4-methoxyphenyl)ethenyl]-4-nitrobenzene](http://img.cochemist.com/ccimg/4700/4648-33-3.png)
![1-[(e)-2-(4-methoxyphenyl)ethenyl]-4-nitrobenzene](http://img.cochemist.com/ccimg/4700/4648-33-3_b.png)







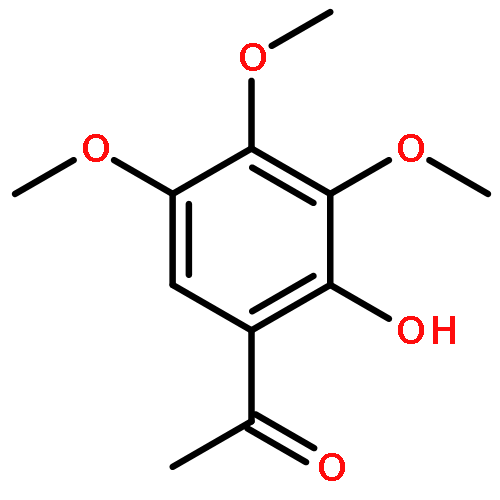







![(2-nonyl-[1,3]dioxolan-2-yl)-acetic acid methyl ester](http://img.cochemist.com/ccimg/109900/109873-29-2.png)
![(2-nonyl-[1,3]dioxolan-2-yl)-acetic acid methyl ester](http://img.cochemist.com/ccimg/109900/109873-29-2_b.png)








![Benzene,1,1'-[1,2-bis(methylene)-1,2-ethanediyl]bis-](http://img.cochemist.com/ccimg/2600/2548-47-2.png)
![Benzene,1,1'-[1,2-bis(methylene)-1,2-ethanediyl]bis-](http://img.cochemist.com/ccimg/2600/2548-47-2_b.png)



![[1,1'-Biphenyl]-2,2'-dicarbonitrile](http://img.cochemist.com/ccimg/4400/4341-02-0.png)
![[1,1'-Biphenyl]-2,2'-dicarbonitrile](http://img.cochemist.com/ccimg/4400/4341-02-0_b.png)


![2-methylbicyclo[2.2.1]hept-5-ene-3-carboxylic Acid](http://img.cochemist.com/ccimg/4400/4397-23-3.png)
![2-methylbicyclo[2.2.1]hept-5-ene-3-carboxylic Acid](http://img.cochemist.com/ccimg/4400/4397-23-3_b.png)
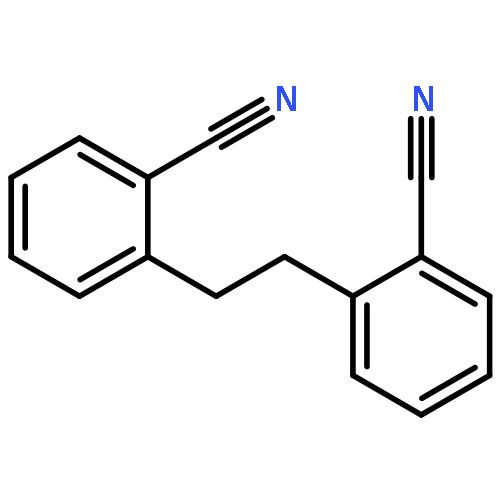
![N-[(3s)-2-oxooxolan-3-yl]decanamide](http://img.cochemist.com/ccimg/177400/177315-87-6.png)
![N-[(3s)-2-oxooxolan-3-yl]decanamide](http://img.cochemist.com/ccimg/177400/177315-87-6_b.png)
![L-Alanine, 3-azido-N-[(9H-fluoren-9-ylmethoxy)carbonyl]-](/data/chemimg/127000/684270-46-0.png)
![L-Alanine, 3-azido-N-[(9H-fluoren-9-ylmethoxy)carbonyl]-](/data/chemimg/127000/684270-46-0_b.png)
![(2-nonyl-[1,3]dioxolan-2-yl)-acetic acid](http://img.cochemist.com/ccimg/596200/596104-60-8.png)
![(2-nonyl-[1,3]dioxolan-2-yl)-acetic acid](http://img.cochemist.com/ccimg/596200/596104-60-8_b.png)






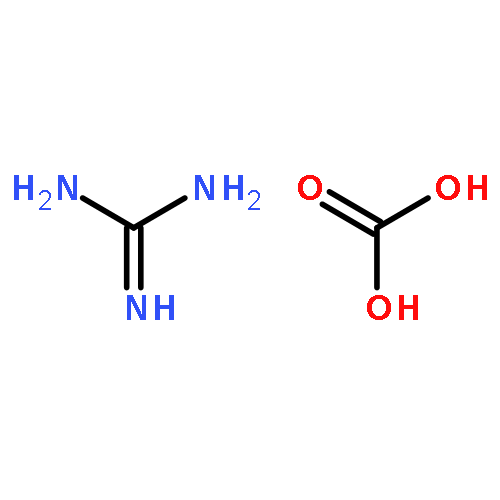


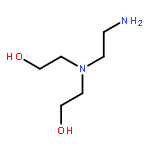
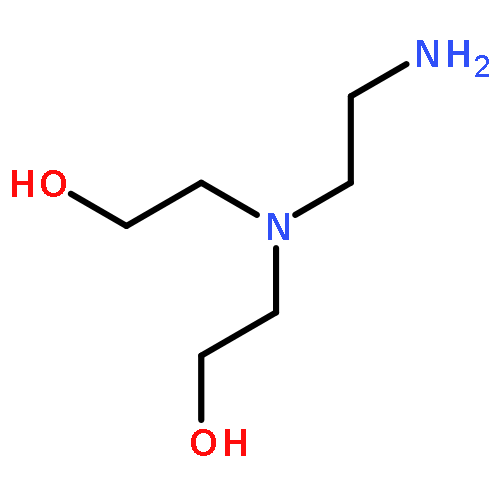
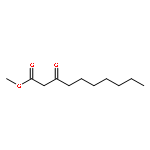
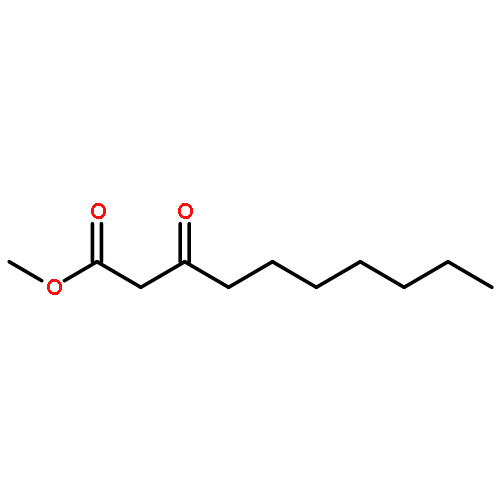
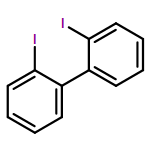

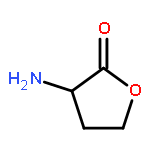
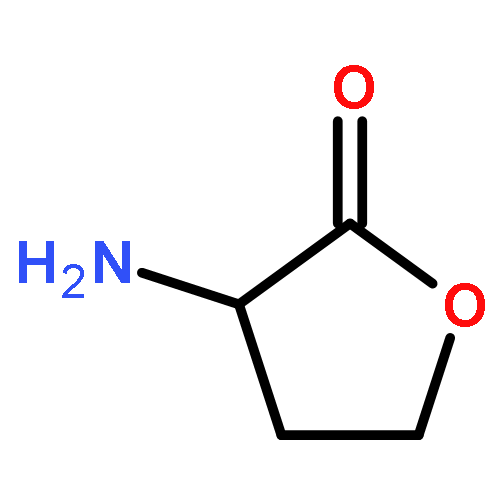
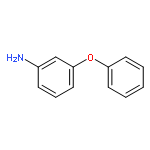



![Hexanamide,3-oxo-N-[(3S)-tetrahydro-2-oxo-3-furanyl]-](http://img.cochemist.com/ccimg/143600/143537-62-6.png)
![Hexanamide,3-oxo-N-[(3S)-tetrahydro-2-oxo-3-furanyl]-](http://img.cochemist.com/ccimg/143600/143537-62-6_b.png)
![Ethanone,1,1'-[1,1'-biphenyl]-2,2'-diylbis-](http://img.cochemist.com/ccimg/24100/24017-95-6.png)
![Ethanone,1,1'-[1,1'-biphenyl]-2,2'-diylbis-](http://img.cochemist.com/ccimg/24100/24017-95-6_b.png)
![3H-Indolium, 2-[5-[1-[6-[(2,5-dioxo-1-pyrrolidinyl)oxy]-6-oxohexyl]-1,3-dihydro-3,3-dimethyl-5-sulfo-2H-indol-2-ylidene]-1,3-pentadien-1-yl]-1-ethyl-3,3-](/data/chemimg/105700/146368-14-1.png)
![3H-Indolium, 2-[5-[1-[6-[(2,5-dioxo-1-pyrrolidinyl)oxy]-6-oxohexyl]-1,3-dihydro-3,3-dimethyl-5-sulfo-2H-indol-2-ylidene]-1,3-pentadien-1-yl]-1-ethyl-3,3-](/data/chemimg/105700/146368-14-1_b.png)








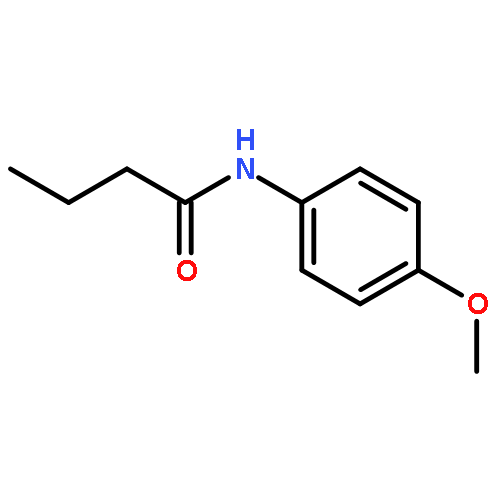



{kind=link}