Co-reporter:Daniel C. Akwaboah, Dimao Wu, and Craig J. Forsyth
Organic Letters 2017 Volume 19(Issue 5) pp:
Publication Date(Web):February 23, 2017
DOI:10.1021/acs.orglett.7b00217
An efficient synthesis of the C1–C9 and the C11–C25 fragments of amphidinolides C, C2, C3, and F from a common intermediate is reported. The construction of the C1–C9 fragment involves an intramolecular hetero-Michael cyclization to form the 3,5-disubstituted trans-tetrahydrofuran moiety. The approach to prepare the C11–C25 fragment utilizes a highly stereoselective aerobic cobalt-catalyzed alkenol cyclization and a chelated Mukaiyama aldol reaction to form the C13–C14 bond and to concomitantly install the C13 hydroxyl group.
Co-reporter:Zhigao Zhang, Yong Chen, Daniel Adu-Ampratwum, Antony Akura Okumu, Nathaniel T. Kenton, and Craig J. Forsyth
Organic Letters 2016 Volume 18(Issue 8) pp:1824-1827
Publication Date(Web):April 4, 2016
DOI:10.1021/acs.orglett.6b00557
An efficient synthesis of the C22–C40 domain of the azaspiracids is described. The synthetic route features a Nozaki–Hiyama–Kishi (NHK) coupling and chelation controlled Mukaiyama aldol reaction to access an acyclic intermediate and a double-intramolecular-hetero-Michael addition (DIHMA) to provide the FG-ring system bridged ketal.
Co-reporter:Dimao Wu and Craig J. Forsyth
Organic Letters 2013 Volume 15(Issue 6) pp:1178-1181
Publication Date(Web):February 26, 2013
DOI:10.1021/ol303515h
Divergent syntheses of the C1–C14 and C15–C25 fragments of amphidinolide C have been achieved. The synthesis of the C15–C25 fragment featured cobalt-catalyzed modified Mukaiyama aerobic alkenol cyclization and sulfur-directed regiocontrolled Wacker oxidation of an internal alkene. The C1–C14 fragment was established by alkenyllithium addition to an aldehyde followed by a challenging olefination of a highly inert C9 ketone.
Co-reporter:Zhigao Zhang, Yue Ding, Jianyan Xu, Yong Chen, and Craig J. Forsyth
Organic Letters 2013 Volume 15(Issue 10) pp:2338-2341
Publication Date(Web):April 29, 2013
DOI:10.1021/ol400487e
An efficient synthesis of the C1–C21 fragment of azaspiracids-1 and −3 is described. This features a Nozaki–Hiyama–Kishi reaction to couple the AB and CD ring precursors and formation of the THF-fused ABCD trioxadispiroketal system under thermodynamic conditions.
Co-reporter:Bo Wang, Po-Hsien Huang, Ching-Shih Chen, and Craig J. Forsyth
The Journal of Organic Chemistry 2011 Volume 76(Issue 4) pp:1140-1150
Publication Date(Web):January 18, 2011
DOI:10.1021/jo102478x
Details of the evolution of strategies toward convergent assembly of the histone deacetylase inhibiting natural product largazole exploiting γ,δ-unsaturated-α,β-epoxy-aldehydes and a thiazole-thiazoline containing ω-amino-acid are described. The initial N-heterocyclic carbene mediated redox amidation exploying these two types of building blocks representing largazole’s structural domains of distinct biosynthetic origin directly afforded the seco-acid of largazole. This was accomplished without any protecting groups resident upon either thioester bearing epoxy-aldehyde or the tetrapeptide. However, the ineffective production of largazole via the final macrolactonization led to an alternative intramolecular esterification/macrolactamization strategy employing the established two building blocks. This provided largazole along with its C2-epimer via an unexpected inversion of the α-stereocenter at the valine residue. The biological evaluation demonstrated that both largazole and 2-epi-largazole led to dose-dependent increases of acetylation of histone H3, indicating their potencies as class I histone deacetylase selective inhibitiors. Enhanced p21 expression was also induced by largazole and its C2 epimer. In addition, 2-epi-largazole displayed more potent activity than largazole in cell viability assays against PC-3 and LNCaP prostate cancer cell lines.
Co-reporter:Yucheng Pang;Chao Fang; Michael J. Twiner;Dr. Christopher O. Miles; Craig J. Forsyth
Angewandte Chemie International Edition 2011 Volume 50( Issue 33) pp:7631-7635
Publication Date(Web):
DOI:10.1002/anie.201101741
Co-reporter:Yucheng Pang;Chao Fang; Michael J. Twiner;Dr. Christopher O. Miles; Craig J. Forsyth
Angewandte Chemie 2011 Volume 123( Issue 33) pp:7773-7777
Publication Date(Web):
DOI:10.1002/ange.201101741
Co-reporter:Bo Wang ; T. Matthew Hansen ; Lynn Weyer ; Dimao Wu ; Ting Wang ; Mathias Christmann ; Yingtao Lu ; Lu Ying ; Mary M. Engler ; Russell D. Cink ; Chi-Sing Lee ; Feryan Ahmed ;Craig J. Forsyth
Journal of the American Chemical Society 2010 Volume 133(Issue 5) pp:1506-1516
Publication Date(Web):December 29, 2010
DOI:10.1021/ja1089099
The phorboxazoles are mixed non-ribosomal peptide synthase/polyketide synthase biosynthetic products that embody polyketide domains joined via two serine-derived oxazole moieties. Total syntheses of phorboxazole A and analogues have been developed that rely upon the convergent coupling of three fragments via biomimetically inspired de novo oxazole formation. First, the macrolide-containing domain of phorboxazole A was assembled from C3−C17 and C18−C30 building blocks via formation of the C16−C18 oxazole, followed by macrolide ring closure involving an intramolecular Still−Genarri olefination at C2−C3. Alternatively, a ring-closing metathesis process was optimized to deliver the natural product’s (2Z)-acrylate with remarkable geometrical selectivity. The C31−C46 side-chain domain was then appended to the macrolide by a second serine amide-derived oxazole assembly. Minimal deprotection then afforded phorboxazole A. This generally effective strategy was then dramatically abbreviated by employing a total synthesis approach wherein both of the natural product’s oxazole moieties were installed simultaneously. A key bis-amide precursor to the bis-oxazole was formed in a chemoselective one-pot, bis-amidation sequence without the use of amino or carboxyl protecting groups. Thereafter, both oxazoles were formed from the key C18 and C31 bis-N-(1-hydroxyalkan-2-yl)amide in a simultaneous fashion, involving oxidation−cyclodehydrations. This synthetic strategy provides a total synthesis of phorboxazole A in 18% yield over nine steps from C3−C17 and C18−C30 synthetic fragments. It illustrates the utility of a synthetic design to form a mixed non-ribosomal peptide synthase/polyketide synthase biosynthetic product based upon biomimetic oxazole formation initiated by amide bond formation to join synthetic building blocks.
Co-reporter:Bo Wang ; T. Matthew Hansen ; Ting Wang ; Dimao Wu ; Lynn Weyer ; Lu Ying ; Mary M. Engler ; Melissa Sanville ; Christopher Leitheiser ; Mathias Christmann ; Yingtao Lu ; Jiehao Chen ; Nicholas Zunker ; Russell D. Cink ; Feryan Ahmed ; Chi-Sing Lee ;Craig J. Forsyth
Journal of the American Chemical Society 2010 Volume 133(Issue 5) pp:1484-1505
Publication Date(Web):December 29, 2010
DOI:10.1021/ja108906e
The phorboxazole natural products are among the most potent inhibitors of cancer cell division, but they are essentially unavailable from natural sources at present. Laboratory syntheses based upon tri-component fragment coupling strategies have been developed that provide phorboxazole A and analogues in a reliable manner and with unprecedented efficiency. This has been orchestrated to occur via the sequential or simultaneous formation of both of the natural product’s oxazole moieties from two serine-derived amides, involving oxidation−cyclodehydrations. The optimized preparation of three pre-assembled components, representing carbons 3−17, 18−30, and 31−46, has been developed. This article details the design and syntheses of these three essential building blocks. The convergent coupling approach is designed to facilitate the incorporation of structural changes within each component to generate unnatural analogues, targeting those with enhanced therapeutic potential and efficacy.
Co-reporter:Chao Fang, Yucheng Pang, and Craig J. Forsyth
Organic Letters 2010 Volume 12(Issue 20) pp:4528-4531
Publication Date(Web):September 17, 2010
DOI:10.1021/ol101833h
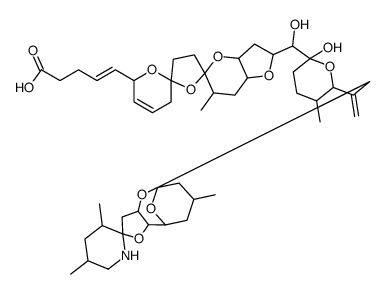
Both C19 and C34 spiroketal domains of okadaic acid were assembled using gold(I) chloride catalyzed spiroketalizations, and the two resulting fragments were coupled to give the C15−C38 fragment of okadaic acid, a known intermediate for the total synthesis of this important natural product.

![Acetaldehyde, [(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/121300/121289-23-4.png)
![Acetaldehyde, [(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/121300/121289-23-4_b.png)