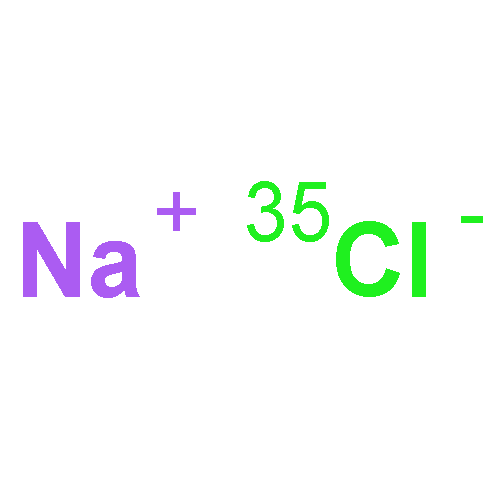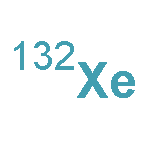Co-reporter:C. J. Smith, Anna K. Huff, Rebecca B. Mackenzie, and Kenneth R. Leopold
The Journal of Physical Chemistry A November 30, 2017 Volume 121(Issue 47) pp:9074-9074
Publication Date(Web):November 3, 2017
DOI:10.1021/acs.jpca.7b09833
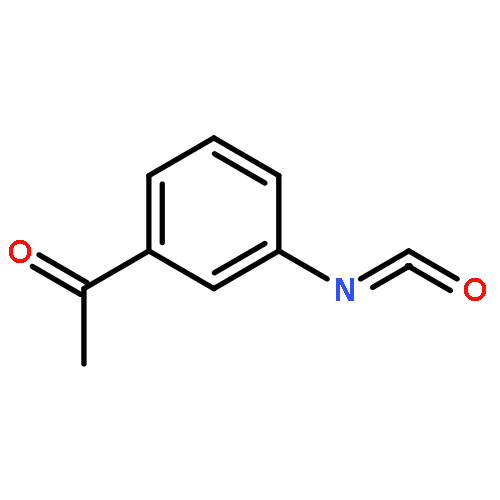
The rotational spectrum of acrylic sulfuric anhydride (CH2═CHCOOSO2OH, AcrSA) has been observed using pulsed-nozzle Fourier transform microwave spectroscopy. The species was produced from the reaction between acrylic acid and sulfur trioxide in a supersonic jet. Spectroscopic constants are reported for both the s-cis- and s-trans-AcrSA conformers of the parent and monodeuterated (OD) isotopologues. Geometries were optimized for both conformers using M06-2X/6-311++G(3df,3pd) methods. Single-point energy calculations at the M06-2X geometries were calculated using the CCSD(T)/complete basis set method with double and triple extrapolation [CBS(D-T)]. Further calculations indicate that the anhydride results from a π2 + π2 + σ2 cycloaddition reaction within the acrylic acid–SO3 complex. Because the C═O double bond of the acrylic acid migrates from one of the COOH oxygens to the other during the reaction, the s-cis form of acrylic acid leads to the s-trans form of the anhydride and vice versa. With zero-point energy corrections applied to the CCSD(T) energies, the s-cis and s-trans forms of CH2═CHCOOSO2OH are 19.0 and 18.8 kcal/mol lower in energy than that of SO3 + their corresponding CH2═CHCOOH precursor conformation. The zero-point-corrected transition state energies for formation of the s-trans and s-cis anhydrides are 0.22 and 0.33 kcal/mol lower than those of the complexes of SO3 with s-cis and s-trans acrylic acid, respectively, indicating that the reaction is essentially barrierless. This system adds to a growing body of examples demonstrating that carboxylic acids readily add to SO3 in the gas phase to produce the corresponding carboxylic sulfuric anhydride.
Co-reporter:Rebecca B. Mackenzie;Christopher T. Dewberry;C. J. Smith;Ryan D. Cornelius
The Journal of Physical Chemistry A February 2, 2017 Volume 121(Issue 4) pp:855-860
Publication Date(Web):January 5, 2017
DOI:10.1021/acs.jpca.6b11255
Aqueous pyridine plays an important role in a variety of catalytic processes aimed at harnessing solar energy. In this work, the pyridine–water interaction is studied by microwave spectroscopy and density functional theory calculations. Water forms a hydrogen bond to the nitrogen with the oxygen tilted slightly toward either of the ortho-hydrogens of the pyridine, and a tunneling motion involving in-plane rocking of the water interconverts the resulting equivalent structures. A pair of tunneling states with severely perturbed rotational spectra is identified and their energy separation, ΔE, is inferred from the perturbations and confirmed by direct measurement. Curiously, values of ΔE are 10404.45 and 13566.94 MHz for the H2O and D2O complexes, respectively, revealing an inverted isotope effect upon deuteration. Small splittings in some transitions suggest an additional internal motion making this complex an interesting challenge for theoretical treatments of large amplitude motion. The results underscore the significant effect of the ortho-hydrogens on the intermolecular interaction of pyridine.
Co-reporter:Christopher T. Dewberry, Jessica L. Mueller, Rebecca B. Mackenzie, Brooke A. Timp, Mark D. Marshall, Helen O. Leung, Kenneth R. Leopold
Journal of Molecular Structure 2017 Volume 1146(Volume 1146) pp:
Publication Date(Web):15 October 2017
DOI:10.1016/j.molstruc.2017.06.015
•The CO2−2,6-difluoropyridine weakly bound complex is structurally characterized.•2,6-fluorination of pyridine−CO2 causes significant structural change.•The microwave spectrum indicates large amplitude angular zero point vibrations.The weakly bound complex formed from CO2 and 2,6-difluoropyridine (2,6-DFP) has been observed by chirped pulse and conventional cavity Fourier transform microwave spectroscopy. As in the related complexes with pyridine and 3,5-difluoropyridine, the carbon of the CO2 approaches the nitrogen of the heterocycle in the plane of the ring. However, the CO2−2,6-DFP complex is found to differ from the pyridine and 3,5-DFP analogues in several respects. First, the data indicate that the N⋯C weak bond distance is 2.9681(32) Å, a value that is ∼0.17 Å longer than that previously determined for CO2−pyridine (2.798 Å) and 0.14 Å longer than that in CO2−3,5-difluoropyridine (2.825 Å). Second, unlike the pyridine and 3,5-difluoropyridine complexes, the CO2 oxygens in CO2−2,6-DFP do not lie in the plane of the ring, i.e., fluorination of the pyridine in the ortho positions causes the CO2 to rotate 90° out of plane. Moreover, the observed rotational constants indicate that the CO2 moiety undergoes large amplitude vibrational motion with an average bending amplitude of ∼31° off this perpendicular geometry. Third, M06-2X/6-311++G(3df,3pd) and MP2/6-311++G(3df,3pd) calculations both indicate that in the equilibrium configuration, the carbon of the CO2 is displaced from the C2 axis of the 2,6-DFP such that axis forms an angle of 20–23° with the line joining the nitrogen and the CO2 carbon. The current results support the previous conjecture that the anomalously short N⋯C distances in CO2−pyridine and CO2−3,5-difluoropyridine result from secondary attractive interactions between the oxygen atoms and the ortho hydrogens. These interactions are eliminated in the 2,6-DFP system by fluorination in the ortho positions, causing the weak bond distance to increase and the CO2 to rotate out of the plane.Download high-res image (113KB)Download full-size image
Co-reporter:Christopher T. Dewberry, Ryan D. Cornelius, Rebecca B. Mackenzie, C.J. Smith, Michael A. Dvorak, Kenneth R. Leopold
Journal of Molecular Spectroscopy 2016 Volume 328() pp:67-72
Publication Date(Web):October 2016
DOI:10.1016/j.jms.2016.08.016
•The CO2-3,5-difluoropyridine van der Waals complex has been studied by microwave spectroscopy and DFT calculations.•Analysis of the observed rotational constants indicates that the N⋯C weak bond distance and the O⋯ortho-H secondary hydrogen bond distance, 2.8245(16) Å and 3.091(2) Å respectively, are slightly longer than those previous observed in pyridine-CO2.•The system is planar and evidence is presented for the importance of secondary hydrogen bonding between the oxygen atoms and the ortho- hydrogen atoms of the pyridine.The rotational spectrum of the weakly bound complex 3,5-difluoropyridine⋯CO2 has been observed using pulsed-nozzle Fourier transform microwave spectroscopy. Spectroscopic constants are reported for the parent and 13CO2 isotopologues. The data indicate a planar structure in which the nitrogen approaches the carbon of the CO2 with either a C2v or effectively C2v geometry in the ground vibrational state. The N⋯C van der Waals bond distance is 2.8245(16) Å and the oxygen⋯ortho-hydrogen distance is 3.091(2) Å. The N⋯C van der Waals bond length is 0.027(8) Å longer than that previously determined for pyridine–CO2, but is still considerably shorter than the 2.998 Å distance in HCN⋯CO2. M06-2X/6-311++G(3df,3pd) calculations place the binding energy of the complex at 4.3 kcal/mol (4.1 kcal/mol with counterpoise correction). The calculations further indicate that a secondary interaction between the ortho-hydrogens of the ring and the CO2 oxygens account for ∼50% of the total binding energy.
Co-reporter:Rebecca B. Mackenzie, Christopher T. Dewberry, and Kenneth R. Leopold
The Journal of Physical Chemistry A 2016 Volume 120(Issue 14) pp:2268-2273
Publication Date(Web):March 29, 2016
DOI:10.1021/acs.jpca.6b01500
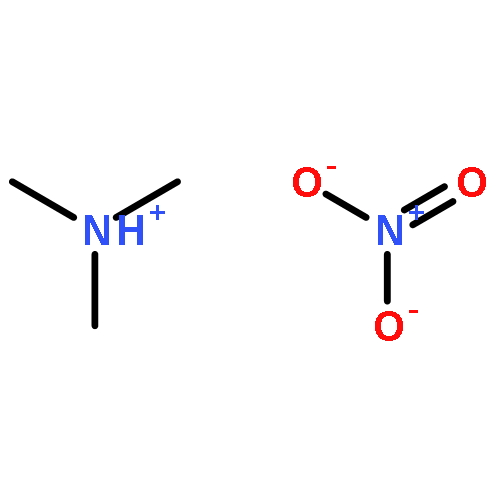
The reactions of amines and carboxylic acids have recently received attention for their possible role in the formation of atmospheric aerosol. Here, we report a microwave study of the trimethylamine–formic acid hydrogen-bonded complex, a simple prototype in which to study amine–carboxylic acid interactions. Spectra of three isotopologues of the system have been observed using a tandem cavity and chirped-pulse Fourier transform microwave spectrometer. The complex has a plane of symmetry, with the acidic proton of the formic acid directed toward the lone pair of the nitrogen. The zero-point-averaged hydrogen bond length is 1.702 Å, and the O–H···N angle is 177°. 14N nuclear quadrupole hyperfine structure has been used to assess the degree of proton transfer from the formic acid to the trimethylamine. Experimental results are supplemented with density functional theory calculations. M06-2X/6-311++G(3df,3pd) calculations indicate a binding energy of 16.8 kcal/mol with counterpoise correction (17.4 kcal/mol without counterpoise correction).
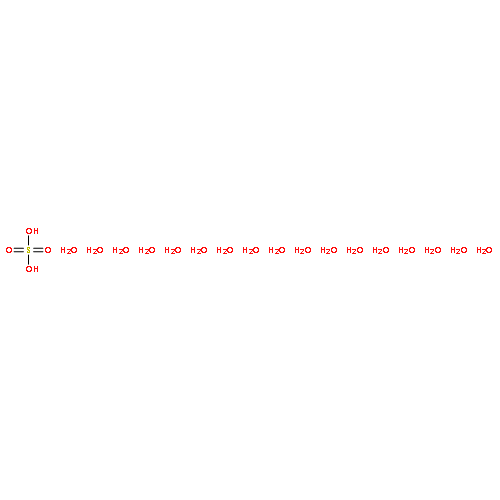
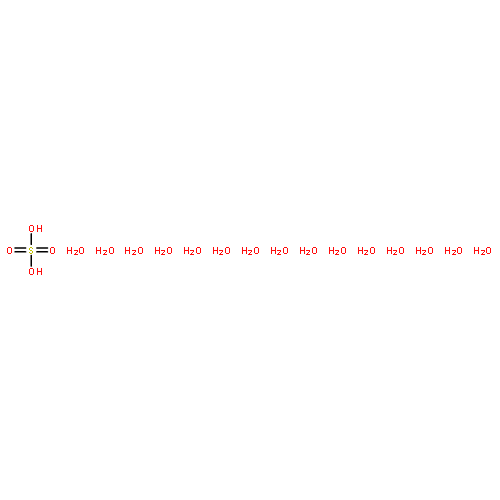
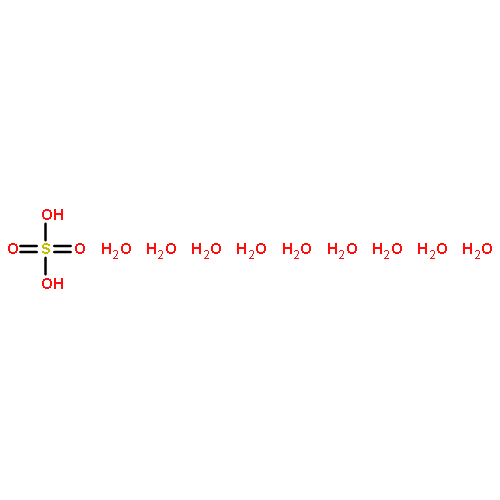
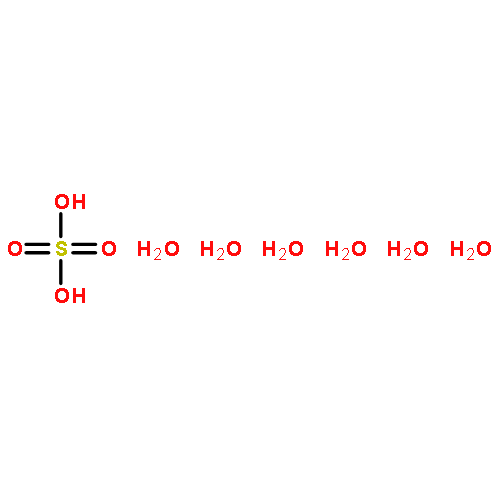
Co-reporter:Christopher T. Dewberry;Rebecca B. Mackenzie
Science 2015 Volume 349(Issue 6243) pp:58-61
Publication Date(Web):03 Jul 2015
DOI:10.1126/science.aaa9704
An unexpected gaseous sulfur species
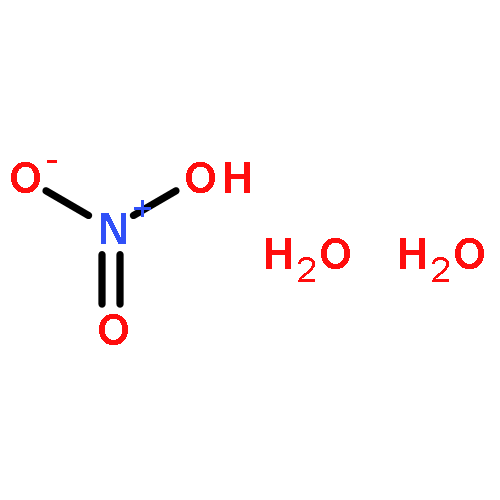
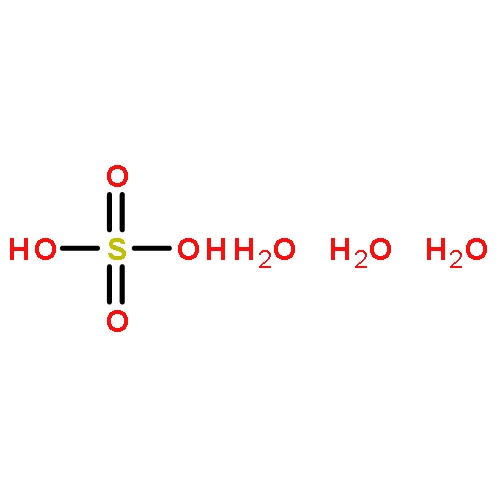
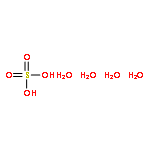
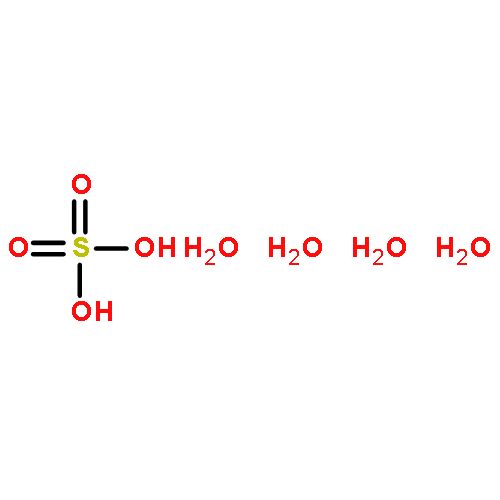
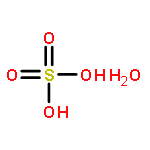
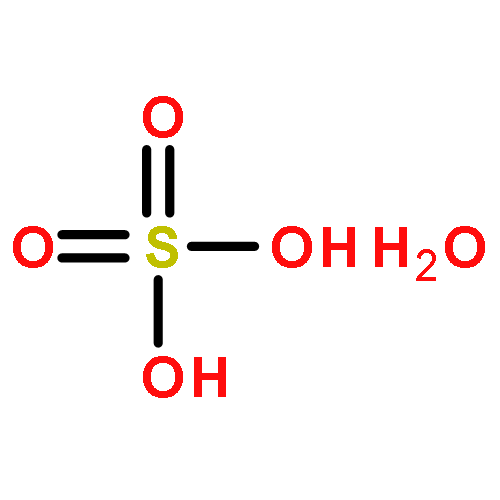
Sulfuric acid plays a central role in both industrial and atmospheric contexts. As such, the behavior of SO3 mixtures in gas phases has been studied for over a century. In gas-phase experiments on wet SO3 and formic acid, Mackenzie et al. discovered a previously unrecognized covalent adduct: formic sulfuric anhydride, or HC(O)OSO3H. The combination of microwave spectroscopy and theoretical calculations reveals its structural properties. The compound may play a role in the nucleation of atmospheric aerosols by serving as an intermediate to H2SO4 formation.
Science, this issue p. 58
Co-reporter:Christopher T. Dewberry, Anna K. Huff, Rebecca B. Mackenzie, Kenneth R. Leopold
Journal of Molecular Spectroscopy 2014 Volume 304() pp:43-46
Publication Date(Web):October 2014
DOI:10.1016/j.jms.2014.09.001
•Rotational spectra of the Xe–SO3 van der Waals complex have been observed in a jet and the Xe–S distance has been determined.•The nuclear quadrupole coupling constant for 131Xe has been obtained.•The electric field gradient at the Xe nucleus is 78% larger than that at the Kr nucleus in Kr–SO3.Nine isotopologues of Xe–SO3 have been observed by pulsed-nozzle Fourier transform microwave spectroscopy. The complex is a symmetric top with a Xe–S van der Waals distance of 3.577(2) Å. The increase in rare gas distance relative to that in Kr–SO3 is equal to the difference in van der Waals radii between Xe and Kr. The 131Xe nuclear quadrupole coupling constant indicates that the electric field gradient at the xenon nucleus is 78% larger than that at the Kr nucleus in Kr–SO3.
Co-reporter:Rebecca B. Mackenzie, Christopher T. Dewberry, and Kenneth R. Leopold
The Journal of Physical Chemistry A 2014 Volume 118(Issue 36) pp:7975-7985
Publication Date(Web):August 21, 2014
DOI:10.1021/jp507060w
Rotational spectra are reported for seven isotopologues of the complex HCOOH–HNO3 in a supersonic jet. The system is planar and bound by a pair of hydrogen bonds, much like the more widely studied carboxylic acid dimers. Double proton exchange interconverts the system between a pair of equivalent structures, as revealed by a splitting of the a-type spectrum that disappears when one of the hydrogen bonding protons is replaced by deuterium. The observation of relative intensities that are consistent with nuclear spin statistics in a symmetric and antisymmetric pair of tunneling states provides additional evidence for such a motion. The observed splittings in the pure rotational spectrum are 1–2 orders of magnitude smaller than those recently reported in the pure rotational spectra of several related carboxylic acid dimers. This is a curious difference, although we note that because the observed spectra do not cross the tunneling doublet, the splittings are a measure of the difference in effective rotational constants for the two states, not the tunneling frequency itself. The observed rotational constants have been used to determine an accurate vibrationally averaged structure for the complex. The two hydrogen bond lengths, 1.686(17) Å and 1.813(10) Å for the hydrogen bonds involving the HNO3 and HCOOH protons, respectively, differ by 0.127(27) Å. Likewise, the associated oxygen–oxygen distances determined for the parent species, 2.631 and 2.794 Å, differ by 0.163 Å. These results suggest that the double proton transfer is necessarily accompanied by substantial motion of the heavy atom frame, and thus this system, in principle, provides an excellent prototype for multidimensional tunneling processes. Ab initio calculations of the binding energy and the barrier height are presented. Excellent agreement between the calculated equilibrium structure and the experimental, vibrationally averaged structure suggests that the vibrational wave function is not highly delocalized in the region between the equivalent potential wells. 14N nuclear quadrupole hyperfine structure is interpreted in terms of the degree to which the HNO3 releases its proton in either of the equivalent potential energy minima.
Co-reporter:Kenneth R. Leopold
Journal of Molecular Spectroscopy 2013 Volumes 293–294() pp:60
Publication Date(Web):November–December 2013
DOI:10.1016/j.jms.2013.09.001
Co-reporter:Brooke A. Timp, Jamie L. Doran, Shyam Iyer, Jens-Uwe Grabow, Kenneth R. Leopold
Journal of Molecular Spectroscopy 2012 Volume 271(Issue 1) pp:20-24
Publication Date(Web):January 2012
DOI:10.1016/j.jms.2011.11.002
The J = 2 ← 1 transitions of the previously unobserved isotopologues 41K79Br and 41K81Br have been recorded with a pulsed-nozzle Fourier transform microwave spectrometer, newly combined with a 532 nm laser ablation source. Aspects of the experimental design are described. Rotational and nuclear quadrupole coupling constants are obtained and combined with published results for 39K79Br and 39K81Br to produce a set of isotopically invariant parameters. New rotational transitions of 23Na35,37Cl, 39K79Br, and 39K127I have also been recorded. Excited vibrational states are not observed, indicating efficient cooling of the metal halides following the initial ablation event.Graphical abstractHighlights► The 41K isotopologs of KBr have been observed by microwave spectroscopy. ► Molecules were produced by laser ablation and the experimental design is described. ► A set of isotopically invariant parameters are determined. ► Previously unobserved transitions of NaCl and KI are also reported.
Co-reporter:Sherri W. Hunt, Denise L. Shelley, Kenneth R. Leopold
Journal of Molecular Spectroscopy 2012 Volume 281() pp:9-12
Publication Date(Web):November 2012
DOI:10.1016/j.jms.2012.09.004
The donor–acceptor complexes (CH3)3N–BF3 and (CH3)3N–B(CH3)3 have been reinvestigated at high resolution by rotational spectroscopy in a supersonic jet. Nuclear hyperfine structure resulting from both nitrogen and boron has been resolved and quadrupole coupling constants have been obtained. The results for both complexes indicate that approximately 0.4 electrons are transferred away from the (CH3)3N moiety upon formation of the dative bond.Graphical abstractElectron transfer in (CH3)3N–BF3 and (CH3)3N–B(CH3)3 is assessed using measured nuclear quadrupole coupling constants.Highlights► Rotational spectra of (CH3)3N−BF3 and (CH3)3N−B(CH3)3 have been observed in a jet. ► Quadrupole coupling constants for 10B, 11B, and 14N have been obtained. ► In both complexes, (CH3)3N donates ∼0.4 electrons to the donor–acceptor bond.
Co-reporter:Kenneth R. Leopold
Journal of Molecular Spectroscopy 2012 Volume 278() pp:27-30
Publication Date(Web):August 2012
DOI:10.1016/j.jms.2012.07.008
Equations are presented for the inertial tensor components of a weakly bound complex in terms of intermolecular coordinates and moments of inertia of the individual moieties. The results are a generalization of similar equations presented in the literature for specific geometries, and allow for the use of up to three angles to specify the orientation of an asymmetric rotor within a complex. The angles chosen are well suited to describing the large amplitude motion characteristic of weakly bound complexes and the resulting expressions should be useful in the analysis of the rotational constants of weakly bound systems with complicated geometries.Graphical abstractHighlights► New equations for the inertial tensor of a weakly bound complex are presented. ► The equations allow an arbitrary orientation of an asymmetric rotor in the complex. ► The results should be useful in the structural analysis of weakly bound systems.
Co-reporter:Jamie L. Doran, Brian Hon, Kenneth R. Leopold
Journal of Molecular Structure 2012 1019() pp: 191-195
Publication Date(Web):
DOI:10.1016/j.molstruc.2012.03.039
Co-reporter:Galen Sedo and Kenneth R. Leopold
The Journal of Physical Chemistry A 2011 Volume 115(Issue 10) pp:1787-1794
Publication Date(Web):February 22, 2011
DOI:10.1021/jp108851t
Microwave spectra have been observed for the gas phase complexes (CH3)314N−H14NO3 and (CH3)315N−H14NO3 and rotational and nuclear quadrupole coupling constants are reported. The structure and binding energy have also been calculated at the MP2 level of theory using the 6-311++G(d,p) and 6-311++G(2df,2pd) basis sets both with and without corrections for basis set superposition error. The HNO3 forms a near-linear hydrogen bond to the amine nitrogen with a rather short hydrogen bond distance of about 1.5−1.6 Å (depending on the basis set and method of computation). The C3 axis of the trimethylamine lies in the plane of the nitric acid. For both the H14NO3 and the (CH3)314N moieties of the parent species, the component of the nuclear quadrupole coupling tensor perpendicular to the molecular symmetry plane, χcc, is sensitive to the electronic structure at the corresponding nitrogen but independent of relative orientation within the plane. Its value, therefore, provides a convenient experimental measure of the degree of proton transfer within the complex. For the HNO3, χcc lies 62% of the way between those of free HNO3 and aqueous NO3−, indicating a substantial degree of proton transfer. A similar comparison of the quadrupole coupling constant of (CH3)3N in the (CH3)3N−HNO3 complex with those of free (CH3)3N and (CH3)3NH+ indicates only about 31% proton transfer, about half that determined from the HNO3 coupling constant. Though surprising at first, this disparity is to be expected if the quadrupole coupling constants vary nonlinearly with the position of the proton relative to the donor and acceptor atoms. Calculations of the 14N nuclear quadrupole coupling constants as a function of proton position using density functional theory are reported and confirm that this is the case. We suggest that when proton transfer is assessed according to changes in individual monomer molecular properties, the overall process may be best described in terms of a dual picture involving proton release by the acid and proton acquisition by the base.
Co-reporter:Galen Sedo, Kenneth R. Leopold
Journal of Molecular Spectroscopy 2010 Volume 262(Issue 2) pp:135-138
Publication Date(Web):August 2010
DOI:10.1016/j.jms.2010.05.009
Eight rotational transitions of the complex (CH3)3CCN–SO3 have been recorded using pulsed-nozzle Fourier transform microwave spectroscopy and a series of ab initio calculations has been performed. The complex is a symmetric top with free or nearly free internal rotation of the SO3 and (CH3)3CCN subunits. The nitrogen–sulfur bond distance is determined to be 2.394(19) Å. Calculations at the MP2/aug-cc-pVTZ level/basis, which are in excellent agreement with the experimental results, give a binding energy of 11.0 kcal/mol relative to (CH3)3CCN and SO3. Physical properties of the system, including N–S bond length, N–S–O angle, binding energy, and the degree of electron transfer (obtained from Townes and Dailey analysis of the 14N nuclear quadrupole coupling constant) are compared with those of similar complexes. The proton affinity of the base is a useful parameter for ordering complexes in the series.
Co-reporter:Shenghai Wu, Galen Sedo, Kenneth R. Leopold
Journal of Molecular Spectroscopy 2009 Volume 253(Issue 1) pp:35-40
Publication Date(Web):January 2009
DOI:10.1016/j.jms.2008.09.012
Microwave spectra of the hydrogen bonded complex 16OD–16OH2 have been recorded using pulsed-nozzle Fourier transform microwave spectroscopy. The potential splitting, ρ, which describes the partial quenching of the OD orbital angular momentum upon complexation, is determined to be −142.703173(65) cm−1. Within the spectroscopic model employed, this value implies an energy difference of 202.46 cm−1 between the ground (2A′) and first excited (2A′′) states of the complex. The observed value of ρ represents a rather large change of 3.85710(11) cm−1 relative to that in the parent complex and implies a 1.30 cm−1 decrease in the 2A′–2A′′ energy spacing relative to the parent species. Comparison with previous results for the 18OH complex suggests that these changes likely arise from changes in vibrationally averaged geometry upon deuteration. Magnetic hyperfine structure from the deuterium and the water protons is analyzed, as is the nuclear electric quadrupole coupling of the deuterium nucleus. Assuming negligible changes in the axial component of the electric field gradient at the deuterium upon complexation, the deuterium quadrupole coupling constant implies an average angular excursion of the OD bond axis from the vibrationally averaged a-inertial axis of the complex of ∼24°.
Co-reporter:Galen Sedo, Jamie L. Doran and Kenneth R. Leopold
The Journal of Physical Chemistry A 2009 Volume 113(Issue 42) pp:11301-11310
Publication Date(Web):September 29, 2009
DOI:10.1021/jp9063033
Four isotopologues of the gas-phase complex HNO3−(H2O)3 have been observed by microwave spectroscopy in a supersonic jet. Rotational and nuclear electric quadrupole coupling constants have been obtained and the experimentally derived inertial defect has been used to infer a near-planar geometry for the complex. The data identify the observed species from among several structures predicted by theory, favoring a 10-membered ring geometry with the HNO3 hydrogen-bonded to the first water, a series of water−water hydrogen bonds, and ring completion with the third water acting as a hydrogen-bond donor to an unprotonated HNO3 oxygen. This structure corresponds to the lowest energy form predicted computationally in several prior studies as well as in this work using the MP2/6-311++G(2df,2pd) level/basis set. Although its observation does not rigorously establish its status as the lowest energy form, the concurrence between the predicted low-energy conformer and that observed in the ultracold supersonic jet strongly suggests that it is indeed the minimum-energy structure. The a-type spectra show evidence of internal dynamics, likely resulting from large amplitude motion of one or more of the water subunits. This complex represents the third step in the sequential hydration of HNO3, and both the theoretical structure and experimental 14N quadrupole coupling constants have been used to track the degree of ionization of the acid as function of hydration number. Based on 14N quadrupole coupling constants, transfer of the HNO3 proton to its nearest water molecule is about one-third complete in the trihydrate.
.gif)